

Abstract
The peptide bond quenches tryptophan fluorescence by excited-state electron transfer, which probably accounts for most of the variation in fluorescence intensity of peptides and proteins. A series of seven peptides was designed with a single tryptophan, identical amino acid composition, and peptide bond as the only known quenching group. The solution structure and side-chain χ1 rotamer populations of the peptides were determined by one-dimensional and two-dimensional 1H-NMR. All peptides have a single backbone conformation. The φ-, ψ-angles and χ1 rotamer populations of tryptophan vary with position in the sequence. The peptides have fluorescence emission maxima of 350–355 nm, quantum yields of 0.04–0.24, and triple exponential fluorescence decays with lifetimes of 4.4–6.6, 1.4–3.2, and 0.2–1.0 ns at 5°C. Lifetimes were correlated with ground-state conformers in six peptides by assigning the major lifetime component to the major NMR-determined χ1 rotamer. In five peptides the χ1 = −60° rotamer of tryptophan has lifetimes of 2.7–5.5 ns, depending on local backbone conformation. In one peptide the χ1 = 180° rotamer has a 0.5-ns lifetime. This series of small peptides vividly demonstrates the dominant role of peptide bond quenching in tryptophan fluorescence.
INTRODUCTION
The environmental sensitivity of tryptophan fluorescence is reasonably well understood in simple model systems (Callis, 1997; Chen and Barkley, 1998). Ideally, this knowledge could be used to interpret or predict intrinsic protein fluorescence in terms of protein structure around the tryptophan residues. Individual tryptophans in polypeptides often have multiexponential fluorescence decays. Although excited-state reactions are still invoked (Hudson et al., 1999; Ladokhin, 1999; Moncrieffe et al., 2000; Nanda and Brand, 2000), the multiple fluorescence lifetimes are generally attributed to ground-state heterogeneity (Donzel et al., 1974; Dahms et al., 1995; Hellings et al., 2003). The emitting state of the indole chromophore has several nonradiative decay channels, including intersystem crossing (Volkert et al., 1977; Robbins et al., 1980), solvent quenching (McMahon et al., 1992), excited-state proton transfer (Vander Donckt, 1969; Saito et al., 1984; Yu et al., 1992), and excited-state electron transfer (Petrich et al., 1983; Shizuka et al., 1988; Chen et al., 1996; Chen and Barkley, 1998). Neither the ground-state conformations causing the multiple lifetimes nor the quenching mechanisms governing those lifetimes are known for proteins. Recent efforts have focused on the peptide bond as the major quenching group in peptides and proteins (Sillen et al., 2000; Ababou and Bombarda, 2001; Laboulais et al., 2001; Adams et al., 2002; Callis and Vivian, 2003; Hellings et al., 2003). The peptide bond is thought to quench by electron transfer from the excited indole ring (donor) to the carbonyl group (acceptor), forming a dark radical ion pair (Callis and Vivian, 2003).
According to transition state theory, the electron transfer rate depends on three factors: driving force of reaction, reorganization energy of solute and surrounding solvent molecules, and electronic coupling matrix between reactant and product states (Marcus and Sutin, 1985). Assuming that the driving force and reorganization energy are invariant and that the electronic coupling matrix decays exponentially with distance between donor and acceptor, the electron transfer rate may be estimated by an exponential function dependent on distance (Hopfield, 1974; McLendon, 1988). Several groups have used distance-dependent electron transfer rates to interpret tryptophan fluorescence lifetimes in peptides (Adams et al., 2002) and proteins (Sillen et al., 2000; Laboulais et al., 2001; Hellings et al., 2003). In contrast, other groups have used the energetic contributions to electron transfer rates to explain tryptophan lifetimes and quantum yields in proteins (Ababou and Bombarda, 2001; Callis and Vivian, 2003). Callis and Vivian (2003) correlated the sum of the driving force and reorganization energy with the average transition energy between the emitting 1La and charge-transfer states. This analysis ignored the distance dependence, because electron transfer between the indole ring and the carbonyl group occurs over short distances (Beratan et al., 1992). Evaluation of different approaches to calculating the electron transfer rate by comparison with experimental data is hampered by the complex quenching environment in proteins.
It would be very helpful to have a model system, in which the local environments of tryptophan are relatively simple, but the fluorescence properties are sufficiently diverse, to test the developing hypotheses about the mechanism of electron transfer. We have been using analogs of the cyclic hexapeptides first studied by Kessler et al. (1988a,b), because the cyclic backbone has a single predominant conformation and the side chains are fully exposed to solvent. Our previous study of c[d-PpYTFWF] assigned the three fluorescence lifetimes to the corresponding χ1 rotamers of the tryptophan side chain by comparing fluorescence decay amplitudes and NMR-determined χ1 rotamer populations (Adams et al., 2002). We also estimated distance-dependent electron transfer rates and used these values to calculate fluorescence lifetimes for each rotamer, in reasonable agreement with the experimental lifetimes. Rooted in this preliminary success, we designed and synthesized a series of cyclic peptides with the same amino acid composition but with the single tryptophan at different positions in the sequence. This varies the local backbone conformation of the tryptophan residue, which is expected to vary the χ1 rotamer populations (Dunbrack and Karplus, 1994) as well as the electron transfer rates. Here, we determine the solution structures of the peptides by 1H-NMR and characterize the tryptophan fluorescence by steady-state and time-resolved techniques. Assigning fluorescence lifetimes to χ1 rotamers yields a set of lifetime values associated with known peptide structures.
MATERIALS AND METHODS
Materials
Oxime resin, N-α-t-Boc-d-proline, -l-phenylalanine, -l-tryptophan, -l-threonine, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, and N-hydroxybenzotriazole·H2O were purchased from Novabiochem (San Diego, CA). N-α-t-Boc-l-phosphotyrosine was purchased from Advanced Chemtech (Louisville, KY). O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate was purchased from Perceptive Biosystems (Hamburg, Germany). All other reagents and solvents were highest grade available.
Peptide synthesis
Peptides were synthesized manually by Boc chemistry on oxime resin followed by on-resin cyclization with cleavage as described previously (Adams et al., 2002). Synthesis was started by attaching the following amino acids to the resin: Trp2, Thr3, Phe4, and Phe6 for c[d-PWTFpYF], Phe4 for c[d-PWTFFpY], and Phe5 for c[d-PWTpYFF]; Trp3 for c[d-PpYWTFF]; Thr3 for c[d-PpYTWFF] and Phe3 for c[d-PpYFWFT]; Thr3 for c[d-PpYTFWF]; and Phe4 for c[d-PpYTFFW]. Crude products contained ∼60–70% cyclic peptide, except for c[d-PWTFpYF]. This sequence gave no major cyclized product, presumably an example of the high sequence dependence of the oxime resin in cyclization (Nishino et al., 1992). Peptides were purified by RP-HPLC on a Vydac C-18 semiprep column (Grace Vydac, Columbia, MD): mobile phase A, CH3CN; mobile phase B, 22% CH3OH/H2O with 0.1% trifluoroacetic acid; gradient A/B, 25–50% at 3 mL/min. Retention times were usually 10–13 min for cyclic peptides. Eluted peptides were lyophilized, and molecular weights were verified by negative-ion electrospray ionization mass spectrometry (MicroMass Quattro II) in the Cleveland Mass Spectrometry Facility, Cleveland State University. All cyclic peptides showed a single peak at m/z 920 (M–H). Peptide purity was estimated to be >99%.
1H-NMR
Solution structures of cyclic peptides were determined at 5°C using a Varian Inova 600 MHz spectrometer (Palo Alto, CA). For experiments in H2O, 1 mg of peptide was dissolved in 600 μL of 10% D2O (Cambridge Isotope, Andover, MA), 5 mM phosphate buffer, and the pH was adjusted to 4.5–6. Chemical shifts were referenced to 3-(trimethylsilyl)-propanate-2,2,3,3-d4. For experiments in D2O, 1 mg of peptide sample was dissolved in 600 μL of D2O. High-resolution one-dimensional experiments were acquired with spectral width of 8000 Hz and 20,480 or 24,576 data points of 64 transients. The temperature dependence of the chemical shifts δ of amide protons was measured in 5° increments from 5 to 40°C. Temperature coefficients −dδ/dT were determined from the slope of a plot of chemical shift versus temperature by linear regression.
Total correlation spectroscopy (TOCSY) spectra (Bax and Davis, 1985) were acquired with a mixing time of 80 ms, spectral width of 8000 Hz, and 4096 data points in 256 t1 increments of 32 transients. Water suppression for TOCSY was performed by low-power irradiation with a presaturation delay pulse of 1.5 s. Nuclear Overhauser effect spectroscopy (NOESY) spectra (Kumar et al., 1980) were acquired with mixing times up to 300 ms and 4096 data points in 256-t1 increments of 64 transients. Water suppression for NOESY was done with either WATERGATE (Piotto et al., 1992) or WET (Smallcombe et al., 1995) pulse sequences. Interproton distances were calculated from NOESY spectra using the isolated two-spin approximation theory. A reference distance of 2.82 Å from the indole NH to H7 of the indole ring was taken from the crystal structure. Coupling constants were measured from high-resolution one-dimensional spectra. Double-quantum filtered-correlated spectroscopy (DQF-COSY) spectra acquired with a spectral width of 8000 Hz and 8192 data points in 256-t1 increments of 64 transients were used in cases of overlapping resonances.
NMR data were processed using Felix 2000 (Accelrys, San Diego, CA) on INDY or OCTANE workstations (Silicon Graphics, Mountain View, CA). Convolution difference water deconvolution was used to remove the residual water signal from the spectra. Data were routinely zero-filled to twice the number of data points in both dimensions. Sine-bell squared and sine-bell apodization functions were shifted and used to achieve the best resolution and sensitivity in the D1 and D2 dimensions, respectively. The data were then phase-corrected and baseline-corrected in D1 dimension.
Structure determination was carried out using the NMR-Refine module of InsightII 2000 (Accelrys) as before (Adams et al., 2002). Ensembles of 30 structures were generated in the DGII submodule from NOE-derived distance restraints, hydrogen-bond restraints, 3J dihedral-angle restraints, and six chiral restraints (Havel et al., 1983). The intensities of NOE cross-peaks were grouped as strong (1.8–2.7 Å), medium (1.8–3.3 Å), and weak (1.8–5.0 Å). The hydrogen-bond restraints were defined as O···N distance ranges of 2.6–3.2 Å. The 3J dihedral-angle restraints were derived from the Karplus equation. The DGII calculation was done using 20,000 steps of simulated annealing and 500 steps of conjugate gradient optimization. Final structures were computed from the average of the ensemble. A 500-step conjugate gradient optimization with all restraints was used to release the conformational distortion of side chains caused by the averaging step.
Fluorescence spectroscopy
Absorption and fluorescence measurements were performed on cyclic peptides in 0.01 M phosphate buffer, pH 6.9 at 5°C. Absorption spectra were measured on a Varian Cary 3E UV-vis spectrophotometer. Fluorescence measurements were made under magic-angle conditions. Samples were excited at 300 nm to avoid excitation of phosphotyrosine. Absorbance of samples was <0.05 at 300 nm. Steady-state fluorescence was measured on a SLM 8000 photon counting spectrofluorometer (SLM, Urbana, IL) in ratio mode with single excitation and emission monochromators set at 4-nm bandpass. Emission spectra I(λ) were corrected using correction factors determined with a standard lamp from Optronics (Orlando, FL). Centers of gravity of emission spectra λcg were calculated from
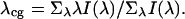 |
(1) |
Fluorescence quantum yields Φ were determined relative to tryptophan (Sigma, St. Louis, MO; recrystallized four times from 70% ethanol) in H2O using a quantum yield of 0.14 at 25°C (Chen, 1967).
Fluorescence lifetimes were measured by time-correlated single photon counting as described in detail elsewhere (Zawadzki et al., 2003). The excitation source was a Tsunami mode-locked Ti-sapphire laser pumped by a Millennia diode-pumped Nd-YVO4 laser (Spectra-Physics, Mountain View, CA). Emission wavelength was selected by a monochromator with a 4-nm band pass. The instrumental response is ∼80 ps full width at half-maximum. Fluorescence decay data with ≥2 × 104 counts in the peak were acquired from 325 to 400 nm emission wavelength in 5-nm increments. Decay data were stored in 1024 channels of 7.3, 9.8, and 24.4 ps per channel, corresponding to 7.5-, 10-, and 25-ns timescales.
Decay curves were deconvolved using the Beechem global program (Beechem, 1989), assuming a sum of exponentials
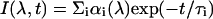 |
(2) |
with relative amplitudes αi(λ) and lifetimes τi of component i at wavelength λ. Lifetimes but not amplitudes were constrained to be the same in the global analysis. Goodness of fit was judged by reduced chi-square, 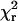 , and the autocorrelation function of the weighted residuals. The radiative rate constant was calculated from
, and the autocorrelation function of the weighted residuals. The radiative rate constant was calculated from
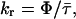 |
(3) |
where 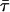 is the amplitude-weighted lifetime
is the amplitude-weighted lifetime
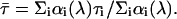 |
(4) |
Decay-associated emission spectra Ii(λ) of component i were calculated from the lifetime data and steady-state emission spectrum as
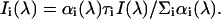 |
(5) |
Centers of gravity of decay-associated emission spectra λcgi were calculated using Ii(λ) in Eq. 1.
RESULTS AND DISCUSSION
Peptide design
The salient features of the peptide c[d-PpYTFWF] used in our previous study are (Adams et al., 2002): 1), a single tryptophan; 2), no amino-acid side chains that quench 3-methylindole fluorescence (Chen and Barkley, 1998); and 3), a single predominant backbone conformation. In addition, this peptide scaffold offers a range of φ-, ψ-angles at different positions in the sequence. Side chain χ1 rotamer populations depend on the φ-, ψ-angles due to repulsive steric interactions with the peptide backbone (Dunbrack and Karplus, 1993, 1994). Therefore, we surmised that moving the tryptophan to different positions in the sequence would vary the χ1 rotamer populations. Tryptophan fluorescence decays should respond in at least two ways to changes in local backbone conformation. First, the decay amplitudes should change because of the correlation between amplitude and rotamer population (Adams et al., 2002). Second, the fluorescence lifetimes should change because the electron transfer rates depend on peptide structure. The latter is the subject of a future study.
The peptide sequences are summarized in Table 1. Peptides are named by the position of tryptophan in the amino acid sequence. Peptides 4a and 6 were designed by simply switching tryptophan at position 5 in c[d-PpYTFWF] with phenylalanine at positions 4 and 6. The analogous design of switching tryptophan with phosphotyrosine at position 2 yielded a linear peptide, independent of starting residue. Peptides 2a and 2b with phosphotyrosine at positions 4 and 6 instead of position 5 were synthesized instead. The design of peptide 4b will be discussed later.
TABLE 1.
Tryptophan backbone conformation in cyclic peptides, 5°C, and predicted χ1 rotamer populations from backbone-dependent rotamer library
| Predicted population
|
|||||
|---|---|---|---|---|---|
| Peptide | Sequence | φ, ψ | χ1 = −60° | χ1 = 180° | χ1 = 60° |
| 2a | c[d-PWTFFpY] | −70°, −32° | 0.62 | 0.22 | 0.16 |
| 2b | c[d-PWTpYFF] | −70°, −29° | 0.62 | 0.22 | 0.16 |
| 3 | c[d-PpYWTFF] | −100°, 164° | 0.85 | 0.08 | 0.07 |
| 4a | c[d-PpYTWFF] | −67°, 5° | 0.52 | 0.01 | 0.47 |
| 4b | c[d-PpYFWFT] | −48°, −48° | 0.25 | 0.71 | 0.04 |
| 5* | c[d-PpYTFWF] | −104°, −46° | 0.68 | 0.32 | 0 |
| 6 | c[d-PpYTFFW] | −155°, 139° | 0.01 | 0.75 | 0.24 |
25°C, Adams et al., 2002.
Peptide structure
The structure of peptide 5 was determined previously by 1H-NMR spectroscopy at 25°C (Adams et al., 2002). The other peptide structures were determined at 5°C, because the NOESY spectra had better signal/noise at the lower temperature, especially in the NH-Hα region. Chemical shifts were assigned using sequential assignments in NOESY spectra and spin-type assignments in TOCSY spectra. The patterns of NOE cross-peaks in the NH-Hα region of the NOESY spectra depend more on the position in the peptide sequence than on the amino acid present at that position. The 3JNH-Hα coupling constants also show position, but little amino acid, dependence, implying that the global backbone conformation is similar in the series of cyclic peptides. The average structures of the peptides were computed from ensembles of 23–29 structures with no NOE distance restraint violations >0.5 Å. The backbone RMSDs are 0.16–0.30 Å and the all-atom RMSDs are 1.26–2.03 Å, consistent with a single predominant backbone conformation and multiple side-chain conformations in each peptide. The aromatic side chains extend out from the cyclic backbone, whereas the threonine hydroxyl hovers over the center of the backbone ring (Kessler et al., 1988a). Fig. 1 shows the backbone and major χ1 rotamer of the tryptophan side chain of all the peptides. The peptide backbones superpose best at positions 3 and 6.
FIGURE 1.

Backbones and tryptophan side chains of cyclic hexapeptides 2a (green), 2b (yellow green), 3 (magenta), 4a (tan), 4b (brown), 5 (black), and 6 (purple). Backbone HN, N, Cα, and CO atoms superposed best at positions 3 and 6.
As noted previously (Kessler et al., 1988a; Adams et al., 2002), the small temperature dependence of the chemical shifts of Xaa3- and Xaa6-NH indicates that two hydrogen bonds form between Xaa3-CO···NH-Xaa6 and Xaa6-CO···NH-Xaa3 in the cyclic peptides (see Table S1 in Supplementary Material). The two hydrogen bonds between Xaa3 and Xaa6 lock the backbone in two tight turn-like structures comprising Xaa6–d-Pro1–Xaa2–Xaa3 and Xaa3–Xaa4–Xaa5–Xaa6. Comparing the φ-, ψ-angles of the peptides (Table 2) with the characteristic values of turn structures identifies a βII′ turn around d-Pro1–Xaa2 in peptides 2b, 3, 4a, 5, and 6 and a βI turn around Xaa4–Xaa5 in peptides 2a, 2b, 3, 4b, 5, and 6. Long-distance interresidue NOEs and 3JNH-Hα coupling constants support these assignments. Interresidue NOEs for βII′ turns are observed between Xaa2-NH and Xaa3-NH, and for βI turns between Xaa4-NH and Xaa5-NH and between Xaa5-NH and Xaa6-NH. The broad single NH resonance for Xaa4 and the 3JNH-Hα = 7.1–8.7 Hz for Xaa5 indicate φ-angles of ∼−60° and −90°, respectively (Wuthrich, 1986; Kessler et al., 1988b).
TABLE 2.
Backbone conformation in cyclic peptides, 5°C
| Peptide | d-Pro1φ, ψ | Xaa2φ, ψ | Xaa3φ, ψ | Xaa4φ, ψ | Xaa5φ, ψ | Xaa6φ, ψ |
|---|---|---|---|---|---|---|
| 2a | 70°, −86° | −70°, −32° | −98°, 162° | −53°, −38° | −78°, −41° | −127°, 85° |
| 2b | 65°, −115° | −70°, −29° | −99°, 164° | −52°, −34° | −75°, −53° | −143°, 132° |
| 3 | 58°, −112° | −65°, −27° | −100°, 164° | −53°, −42° | −73°, −48° | −137°, 124° |
| 4a | 75°, −114° | −91°, 54° | −151°, 176° | −67°, 5° | −144°, −58° | −108°, 107° |
| 4b | 61°, −94° | −73°, −38° | −94°, 172° | −48°, −48° | −120°, 37° | −154°, 72° |
| 5* | 70°, −132° | −87°, 67° | −174°, 156° | −50°, −29° | −104°, −46° | −140°, 123° |
| 6 | 85°, −109° | −111°, 57° | −149°, 158° | −56°, −23° | −83°, −53° | −155°, 139° |
25°C, Adams et al., 2002.
In similar cyclic hexapeptides, a major backbone conformation with all trans peptide bonds and a minor conformation with a cis peptide bond was observed for five of seven sequences (Kessler et al., 1988b, 1990). The other two sequences as well as our series of cyclic peptides have all trans peptide bonds. Sequence comparisons suggest that d-Pro1–Xaa2, where Xaa2 is an aromatic amino acid, confers a single backbone conformation on the cyclic hexapeptide. In general, moving tryptophan to different positions in the sequence does not change the global backbone conformation dramatically. Overlapping the peptide backbones shows some twisting of the backbone ring, particularly at positions 2 and 3 (Table 2). This might be caused by side-chain repulsion given that four of the six amino acids have bulky aromatic side chains. In peptide 4b, the smaller threonine side chain at position 6 folds further into the center of the cyclic backbone, twisting the backbone at positions 5 and 6.
Tryptophan rotamer populations
The χ1 rotamer populations were determined using coupling constants measured from high-resolution one-dimensional or DQF-COSY spectra at 5°C. The 3JHα–Hβ coupling constants of aromatic side chains in the peptides range from 4.0–11.7 Hz, indicating multiple χ1 rotamer conformations (Tables 3, 4, and see Table S2 in Supplementary Material). The NOESY spectra of all the peptides showed a single set of cross-peaks between Hα and Hβ, consistent with fast averaging of side-chain conformations on the NMR timescale. The relative populations of the canonical χ1 = ±60° and 180° rotamers were calculated from the 3JHα–Hβ coupling constants using the Pachler equations (Pachler, 1963). Stereospecific assignment of the major χ1 rotamer was made for aromatic side chains with resolved cross-peaks between Hα–Hβ and NH–Hβ. The rotamer populations of tryptophan as well as the conformation of the major rotamer are given in Table 3. The populations for peptide 5 at 5°C are close to the previous values at 25°C (Adams et al., 2002). The rotamer populations are similar in peptides 2a, 2b, and 6 and in peptides 4a and 4b. The major rotamer of tryptophan is the χ1 = −60° rotamer in peptides 2a, 2b, 3, 4b, and 5 and the χ1 = 180° rotamer in peptide 6. The three χ1 rotamers are about equally populated in peptide 4a. For comparison, Table 1 shows the predicted populations for the three canonical χ1 rotamers of tryptophan in the backbone-dependent rotamer library (Dunbrack and Karplus, 1993, 1994). This library is based on crystal structures of 850 polypeptide chains. Not surprisingly, the predicted and NMR-determined rotamer populations differ somewhat for given φ-, ψ-angles, but agree on the major rotamer except in the case of peptide 4b.
TABLE 3.
Tryptophan coupling constants and χ1 rotamer populations in cyclic peptides, 5°C
| Peptide | 3JHα–Hβr (Hz) | 3JHα–Hβs (Hz) | pI | Major rotamer | pII | pIII |
|---|---|---|---|---|---|---|
| 2a | 9.1 | 4.3 | 0.59 | χ1 = −60° | 0.25 | 0.16 |
| 2b | 8.8 | 4.6 | 0.56 | χ1 = −60° | 0.26 | 0.18 |
| 3 | 4.9 | 11.7 | 0.83 | χ1 = −60° | 0.21 | −0.04 |
| 4a | 6.4* | 5.8* | 0.36 | 0.34 | 0.30 | |
| 4b | 6.3 | 7.3 | 0.43 | χ1 = −60° | 0.34 | 0.23 |
| 5 | 10.4 | 4.3 | 0.72 | χ1 = −60° | 0.14 | 0.14 |
| 6 | 8.8 | 4.0 | 0.57 | χ1 = 180° | 0.30 | 0.13 |
Stereospecificity unresolved.
TABLE 4.
Coupling constants and χ1 rotamer populations in c[d-PpYTWFF], 4a, and c[d-PpYFWFT], 4b, 5°C
| Residue | 3JHα–Hβr (Hz) | 3JHα–Hβs (Hz) | pI | Major rotamer | pII | pIII |
|---|---|---|---|---|---|---|
| c[d-PpYTWFF], 4a | ||||||
| d-Pro1 | ||||||
| pTyr2 | 10.3 | 4.3 | 0.70 | χ1 = −60° | 0.16 | 0.14 |
| Thr3 | ||||||
| Trp4 | 6.4* | 5.8* | 0.36 | 0.34 | 0.30 | |
| Phe5 | 7.8 | 6.8 | 0.47 | 0.39 | 0.14 | |
| Phe6 | 9.6 | 4.9 | 0.64 | 0.21 | 0.15 | |
| c[d-PpYFWFT], 4b | ||||||
| d-Pro1 | ||||||
| pTyr2 | 11.3 | 4.4 | 0.79 | χ1 = −60° | 0.16 | 0.05 |
| Phe3 | 11.2 | 5.4 | 0.79 | χ1 = −60° | 0.25 | −0.04 |
| Trp4 | 6.3 | 7.3 | 0.43 | χ1 = −60° | 0.34 | 0.23 |
| Phe5 | 7.3 | 5.9 | 0.43 | χ1 = −60° | 0.30 | 0.27 |
| Thr6 | 3.9 | |||||
Stereospecificity unresolved.
For the tryptophan in peptide 4a, the NMR results indicate approximately equal populations of the three χ1 rotamers, whereas the backbone-dependent rotamer library predicts approximately equal populations of the χ1 = ±60° rotamers but no χ1 = 180° rotamer. Inspection of the structure of peptide 4a reveals that the ψ = 5° backbone conformation results in less steric hindrance for the χ1 = 60° rotamer relative to other backbone conformations at position 4 and no steric hindrance by either backbone or adjacent side chains for the χ1 = 180° rotamer. In an attempt to increase the population of one tryptophan rotamer, threonine at position 3 was switched with the phenylalanine at position 6 in peptide 4b. Sandwiching tryptophan at position 4 between bulky phenylalanine side chains had only modest effect, although a major χ1 = −60° rotamer could be deciphered (Table 4). The ψ = −48° backbone conformation in peptide 4b results in steric repulsion between NH of Phe5 and the indole side chain for the χ1 = 60° rotamer, possibly disfavoring it. On the other hand, the rotamer populations and assignments for other aromatic amino acids in peptides 4a and 4b suggest an explanation for the design failure (Table 4). The major rotamer of Phe3 in peptide 4b is χ1 = −60°, so that the phenyl ring points toward pTyr2 instead of Trp4 and the expected conformational constraint on Trp4 fails to occur. The increase in population of the χ1 = −60° rotamer of pTyr2 in peptide 4b, which points the phenol phosphate ring away from Phe3, is consistent with this explanation. In contrast, the three χ1 rotamers of Phe5 are more equally populated, when threonine is placed at position 6.
As in the previous study of peptide 5, the presence of multiple χ1 rotamers makes it impossible to determine the χ2 rotamer populations for each χ1 rotamer. NOE intensities alone can be interpreted at best semiquantitatively. NOE cross-peaks between two Hβ protons and H2 and H4 in the indole ring appear with comparable intensities, suggesting little χ2 preference for tryptophan in any of the peptides. The presence of cross-peaks between Hα with both H2 and H4, which indicate primarily (χ1, χ2) = −60°, −90°; 180°, 90° and (χ1, χ2) = −60°, 90°; 180°, −90°, respectively, excludes correlation between χ1 and χ2.
Fluorescence lifetimes
Fluorescence experiments were performed at 5°C to compare with the NMR data. The absorption spectra of the peptides are identical above 280 nm (data not shown). The slightly higher shoulder at ∼270 nm in the peptides compared to tryptophan and N-acetyl tryptophanamide (NATA) is likely due to phosphotyrosine. This shoulder is a little different in each peptide. The fluorescence emission maxima vary among the peptides from 350 to 355 nm (Table 5). The wavelength of maximum emission λmax depends on the local electrostatic environment surrounding the indole ring (Callis and Burgess, 1997; Vivian and Callis, 2001). The small emission spectral shifts reveal subtle differences in electrostatic environment of the solvent-exposed tryptophan in this series of peptides. The fluorescence quantum yields vary sixfold from 0.04 to 0.24. The changes in quantum yield indicate different extents of peptide bond quenching in the peptides.
TABLE 5.
Fluorescence data, 5°C
| Peptide | λmax (nm) | λcg (nm) | Φ |
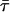 (ns) (ns) |
kr × 10−7 (s−1) |
|---|---|---|---|---|---|
| 2a | 353 | 361 | 0.14 ± 0.01 | 3.48 | 4.0 ± 0.3 |
| 2b | 354 | 361 | 0.13 ± 0.01 | 3.27 | 4.0 ± 0.3 |
| 3 | 355 | 361 | 0.18 ± 0.01 | 3.28 | 5.5 ± 0.3 |
| 4a | 351 | 360 | 0.19 ± 0.01 | 4.58 | 4.1 ± 0.2 |
| 4b | 352 | 360 | 0.14 ± 0.01 | 2.84 | 4.9 ± 0.3 |
| 5 | 350 | 358 | 0.24 ± 0.01 | 4.68 | 5.1 ± 0.2 |
| 6 | 351 | 359 | 0.04 ± 0.002 | 0.97 | 4.1 ± 0.2 |
| NATA | 353 | 0.21 ± 0.01 | 3.83 | 5.5 ± 0.3 |
The fluorescence decays give good fits to four exponential functions (Table 6). The fourth component of ultrashort lifetime τ4 = 4–90 ps probably represents scattered light. The decay-associated emission spectrum I4(λ) has a Raman peak at 330 nm and decreases monotonically at longer wavelengths (not shown). The maximum contribution at 330 nm corresponds to <0.8% of total intensity for peptides 2a–5 and 1.3% for peptide 6. Recently, a 20-ps component with red-shifted emission spectrum was reported for tryptophan and two peptides (Larsen et al., 2003; Pandit et al., 2003). The decay-associated emission spectra of the 20-ps component include a Raman peak at 320–360 nm and a small broad peak centered around 420 nm. Although our measurements do not extend above 400 nm, I4(λ) of the cyclic peptides showed no signs of increased intensity above 380 nm. The relative amplitudes of the three lifetime components corrected for scattered light are given in Table 6.
As expected, both the relative amplitudes and the lifetimes depend on the peptide. The long lifetime τ1 ranges from 4.4 to 6.6 ns, the intermediate lifetime τ2 from 1.4 to 3.2 ns, and the short lifetime τ3 from 0.2 to 1.0 ns. Four peptides have a salient lifetime component: τ2 = 2.7 ns of peptide 3, τ1 = 6.6 ns and τ1 = 5.5 ns of peptides 4a and 5, and τ3 = 0.5 ns of peptide 6. The amplitude α1 = 0.74 of peptide 5 at 5°C is somewhat larger than the previous value at 25°C (Adams et al., 2002). In the other three peptides, the major lifetime component appears to be τ1, although the assignment is less compelling. Two lifetime components, τ1 and τ2, have large amplitudes in peptides 2a and 2b, and the three lifetime components have roughly equal amplitudes in peptide 4b. The amplitudes of all peptides show slight wavelength dependence. The centers of gravity of the decay-associated emission spectra Ii(λ) are given in Table 6. In five peptides, the λcgi values of the emission spectra shift progressively to the red with increasing lifetime. However, two peptides do not follow this trend. In peptide 2b, the λcgi values of the 4.4- and 2.4-ns components are the same and 10 nm to the red of the 1.0-ns component. In peptide 5, the λcgi values of the 5.5- and 0.8-ns components are red-shifted compared to the 3.2-ns component. If the multiexponential fluorescence decays of the peptides were due to solvent relaxation (Lakowicz, 2000), then all the peptides should show progressively red-shifted decay-associated emission spectra with increasing lifetime. Moreover, solvent relaxation of tryptophan zwitterion has a 1.2-ps decay time in aqueous solution at room temperature (Shen and Knutson, 2001). The relaxation time is unlikely to be three orders-of-magnitude slower for solvent-exposed tryptophans in small peptides.
TABLE 6.
Fluorescence decay parameters, 5°C
| Peptide | α1(350) | τ1 (ns) | λcg1 (nm) | α2(350) | τ2 (ns) | λcg2 (nm) | α3(350) | τ3 (ns) | λcg3 (nm) | α4(350) | τ4 (ns) | λcg4 (nm) | 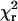 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 0.35 | 4.66 | 362 | 0.34 | 2.76 | 360 | 0.07 | 0.82 | 352 | 0.24 | 0.07 | 354 | 1.11 |
| 0.46 | 0.44 | 0.10 | |||||||||||
| 2b | 0.41 | 4.39 | 360 | 0.29 | 2.36 | 360 | 0.09 | 1.04 | 349 | 0.21 | 0.08 | 355 | 1.05 |
| 0.52 | 0.37 | 0.11 | |||||||||||
| 3 | 0.21 | 6.03 | 363 | 0.48 | 2.70 | 359 | 0.10 | 0.53 | 356 | 0.21 | 0.06 | 356 | 1.08 |
| 0.26 | 0.61 | 0.13 | |||||||||||
| 4a | 0.20 | 6.59 | 361 | 0.12 | 2.03 | 355 | 0.05 | 0.19 | 355 | 0.63 | 0.004 | 355 | 1.09 |
| 0.55 | 0.32 | 0.13 | |||||||||||
| 4b | 0.19 | 5.21 | 361 | 0.17 | 2.04 | 358 | 0.14 | 0.49 | 355 | 0.50 | 0.01 | 353 | 1.05 |
| 0.38 | 0.33 | 0.29 | |||||||||||
| 5 | 0.58 | 5.54 | 358 | 0.09 | 3.20 | 353 | 0.10 | 0.75 | 357 | 0.23 | 0.09 | 354 | 1.12 |
| 0.74 | 0.13 | 0.13 | |||||||||||
| 6 | 0.04 | 4.94 | 361 | 0.18 | 1.38 | 359 | 0.45 | 0.52 | 357 | 0.33 | 0.02 | 352 | 1.07 |
| 0.06 | 0.27 | 0.67 |
Global analysis of data from 325- to 400-nm emission wavelength, 5-nm increments on 7.5-, 10-, and 25-ns timescales. Σiαi(350) = 1. Second line gives relative amplitudes without scattered light.
Radiative rates kr were calculated from fluorescence quantum yields and amplitude-weighted lifetimes using Eqs. 3 and 4 (Table 5). The values for peptides 3, 4b, and 5 and for NATA are the same within error, whereas the values for peptides 2a, 2b, 4a, and 6 are ∼25% lower. An apparent decrease in radiative rate in Pro–Trp and Phe–Trp dipeptides was attributed to intramolecular static quenching (Chen et al., 1991). The ultrafast lifetime component attributed to scattered light could include contributions from statically quenched species. If so, the fractional intensity of the ultrafast component 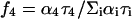 should be higher in peptides evidencing static quenching. However, this trend is not discernable in the data of Table 6.
should be higher in peptides evidencing static quenching. However, this trend is not discernable in the data of Table 6.
Correlation of lifetimes and rotamers
The rotamer model of tryptophan photophysics proposes that the multiexponential fluorescence decay represents multiple ground-state conformations (Donzel et al., 1974; Szabo and Rayner, 1980; Ross et al., 1981). Differences among conformers in the local environment of the indole ring presumably cause different fluorescence lifetimes. A number of workers have proposed that the multiple fluorescence lifetimes of single tryptophans in small peptides represent ground-state χ1 rotamers. For example, the three fluorescence lifetimes of [Trp2]oxytocin were assigned to canonical χ1 rotamers of tryptophan by comparing the decay amplitudes with the NMR-determined χ1 rotamer populations (Ross et al., 1992). Double and triple exponential decays of single tryptophans in α-helix-forming peptides were attributed to χ1 rotamers (Willis et al., 1994; Clayton and Sawyer, 1999). However, the possibility that multiple backbone conformations also contribute to the lifetime heterogeneity could not be ruled out in these studies. Because each of our cyclic hexapeptides has a single predominant backbone conformation, side-chain rotamers become the only source of conformational heterogeneity. Moreover, the peptide bond is the only known quenching group in the peptides. Therefore, the three fluorescence lifetimes τi can be credited to tryptophan rotamers with different excited-state electron transfer rates.
The amplitudes αi of the fluorescence decay should equal the NMR-determined χ1 rotamer populations under the following conditions: interconversion of χ1 rotamers is slow compared to the fluorescence timescale, and χ2 rotamers do not generate lifetime heterogeneity. This would apply if 1), each χ1 rotamer has a major χ2 rotamer; 2), interconversion of χ2 rotamers is fast compared to the fluorescence timescale (Gordon et al., 1992); or 3), the two canonical χ2 rotamers of a χ1 rotamer have the same lifetime. For all peptides except 4a, the αi value of the major lifetime component (Table 6) is in reasonable agreement with the population pI of the major χ1 rotamer (Table 3). The small discrepancies between amplitude and rotamer population could be due to experimental error, χ1 rotamer interconversion on the fluorescence timescale, or χ2 rotamers. Therefore, we associate the major lifetime component with the corresponding χ1 rotamer conformation. For peptides 2a, 2b, 3, 4b, and 5, the major lifetime component is assigned to the χ1 = −60° rotamer with τi values ranging from 2.7 to 5.5 ns. For peptide 6, the major lifetime component is assigned to the χ1 = 180° rotamer with τ3 = 0.5 ns.
The above correlations support the following conclusions. First, the lifetime of a given χ1 rotamer varies with the conformation of the peptide backbone, resulting from differences in local environment around the indole ring. Second, by dint of proximity, the peptide bond is the most important quencher of tryptophan fluorescence in peptides and proteins. Simply permuting the position of tryptophan in our series of peptides, which lack quenching side chains, changes the fluorescence quantum yield by a factor of six. Third, the effect of backbone conformation on tryptophan fluorescence is manifest in two ways: amplitude or χ1 rotamer population and lifetime.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Dr. Dale Ray for assistance with NMR spectroscopy and Professor Frank D. Sönnichsen for helpful discussions about peptide structure determination.
References
- Ababou, A., and E. Bombarda. 2001. On the involvement of electron transfer reactions in the fluorescence decay kinetics heterogeneity of proteins. Protein Sci. 10:2102–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, P. D., Y. Chen, K. Ma, M. G. Zagorski, F. D. Sönnichsen, M. L. McLaughlin, and M. D. Barkley. 2002. Intramolecular quenching of tryptophan fluorescence by the peptide bond in cyclic hexapeptides. J. Am. Chem. Soc. 124:9278–9286. [DOI] [PubMed] [Google Scholar]
- Bax, A., and D. G. Davis. 1985. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65:355–360. [Google Scholar]
- Beechem, J. M. 1989. A second generation global analysis program for the recovery of complex inhomogeneous fluorescence decay kinetics. Chem. Phys. Lipids. 50:237–251. [DOI] [PubMed] [Google Scholar]
- Beratan, D. N., J. N. Betts, and J. N. Onuchic. 1992. Tunneling pathway and redox-state-dependent electronic couplings at nearly fixed distance in electron-transfer proteins. J. Phys. Chem. 96:2852–2855. [Google Scholar]
- Callis, P. R. 1997. 1La and 1Lb Transitions of tryptophan: applications of theory and experimental observations to fluorescence of proteins. Met. Enzymol. 278:113–151. [DOI] [PubMed] [Google Scholar]
- Callis, P. R., and B. K. Burgess. 1997. Tryptophan fluorescence shifts in proteins from hybrid simulations: an electrostatic approach. J. Phys. Chem. B. 101:9429–9432. [Google Scholar]
- Callis, P. R., and J. T. Vivian. 2003. Understanding the variable fluorescence quantum yield of tryptophan in proteins using QM–MM simulations. Quenching by charge transfer to the peptide backbone. Chem. Phys. Lett. 369:409–414. [Google Scholar]
- Chen, R. F. 1967. Fluorescence quantum yields of tryptophan and tyrosine. Anal. Lett. 1:35–42. [Google Scholar]
- Chen, R. F., J. R. Knutson, H. Ziffer, and D. Porter. 1991. Fluorescence of tryptophan dipeptides: correlations with the rotamer model. Biochemistry. 30:5184–5195. [DOI] [PubMed] [Google Scholar]
- Chen, Y., and M. D. Barkley. 1998. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 37:9976–9982. [DOI] [PubMed] [Google Scholar]
- Chen, Y., B. Liu, H.-T. Yu, and M. D. Barkley. 1996. The peptide bond quenches indole fluorescence. J. Am. Chem. Soc. 118:9271–9278. [Google Scholar]
- Clayton, A. H., and W. H. Sawyer. 1999. Tryptophan rotamer distributions in amphipathic peptides at a lipid surface. Biophys. J. 76:3235–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahms, T. E. S., K. J. Willis, and A. G. Szabo. 1995. Conformational heterogeneity of tryptophan in a protein crystal. J. Am. Chem. Soc. 117:2321–2326. [Google Scholar]
- Donzel, B., P. Gaudchon, and P. Wahl. 1974. Study of the conformation in the excited state of two tryptophanyl diketopiperazines. J. Am. Chem. Soc. 96:801–808. [Google Scholar]
- Dunbrack, R. L., Jr., and M. Karplus. 1993. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 230:543–574. [DOI] [PubMed] [Google Scholar]
- Dunbrack, R. L., Jr., and M. Karplus. 1994. Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains. Nat. Struct. Biol. 1:334–340. [DOI] [PubMed] [Google Scholar]
- Gordon, H. L., H. C. Jarrell, A. G. Szabo, K. J. Willis, and R. L. Somorjai. 1992. Molecular-dynamics simulations of the conformational dynamics of tryptophan. J. Phys. Chem. 96:1915–1921. [Google Scholar]
- Havel, T. F., I. D. Kuntz, and G. M. Crippen. 1983. The combinatorial distance geometry method for the calculation of molecular conformation. I. A new approach to an old problem. J. Theor. Biol. 104:359–381. [DOI] [PubMed] [Google Scholar]
- Hellings, M., M. De Maeyer, S. Verheyden, Q. Hao, E. J. M. Van Damme, W. J. Peumans, and Y. Engelborghs. 2003. The dead-end elimination method, tryptophan rotamers, and fluorescence lifetimes. Biophys. J. 85:1894–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield, J. J. 1974. Electron transfer between biological molecules by thermally activated tunneling. Proc. Natl. Acad. Sci. USA. 71:3640–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, B. S., J. M. Huston, and G. Soto-Campos. 1999. A reversible “dark state” mechanism for complexity of the fluorescence of tryptophan in proteins. J. Phys. Chem. A. 103:2227–2234. [Google Scholar]
- Kessler, H., U. Anders, and M. Schudok. 1990. An unexpected cis peptide bond in the minor conformation of a cyclic hexapeptide containing only secondary amide bonds. J. Am. Chem. Soc. 112:5908–5916. [Google Scholar]
- Kessler, H., J. W. Bats, C. Griesinger, S. Koll, M. Will, and K. Wagner. 1988a. Peptide conformations. 46. Conformational analysis of a superpotent cytoprotective cyclic somatostatin analog. J. Am. Chem. Soc. 110:1033–1049. [Google Scholar]
- Kessler, H., M. Klein, and K. Wagner. 1988b. Peptide conformation. 48. Conformation and biological activity of proline containing cyclic retro-analogues of somatostatin. Int. J. Pept. Protein Res. 31:481–498. [PubMed] [Google Scholar]
- Kumar, A., R. R. Ernst, and K. Wüthrich. 1980. A two-dimensional nuclear Overhauser enhancement (two-dimensional NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 95:1–6. [DOI] [PubMed] [Google Scholar]
- Laboulais, C., E. Deprez, H. Leh, J.-F. Mouscadet, J.-C. Brochon, and M. Le Bret. 2001. HIV-1 integrase catalytic core: molecular dynamics and simulated fluorescence decays. Biophys. J. 81:473–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin, A. S. 1999. Red-edge excitation study of nonexponential fluorescence decay of indole in solution and in a protein. J. Fluoresc. 9:1–9. [Google Scholar]
- Lakowicz, J. R. 2000. On spectral relaxation in proteins. Photochem. Photobiol. 72:421–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, O. F. A., I. H. M. van Stokkum, A. Pandit, R. van Grondelle, and H. van Amerongen. 2003. Ultrafast polarized fluorescence measurements on tryptophan and a tryptophan-containing peptide. J. Phys. Chem. B. 107:3080–3085. [Google Scholar]
- Marcus, R. A., and N. Sutin. 1985. Electron transfers in chemistry and biology. Biochim. Biophys. Acta. 811:265–322. [Google Scholar]
- McLendon, G. 1988. Long-distance electron transfer in proteins and model systems. Acc. Chem. Res. 21:160–167. [Google Scholar]
- McMahon, L. P., W. J. Colucci, M. L. McLaughlin, and M. D. Barkley. 1992. Deuterium isotope effects in constrained tryptophan derivatives: implications for tryptophan photophysics. J. Am. Chem. Soc. 114:8442–8448. [Google Scholar]
- Moncrieffe, M. C., N. Juranic, M. D. Kemple, J. D. Potter, S. Macura, and F. G. Prendergast. 2000. Structure-fluorescence correlations in a single tryptophan mutant of carp parvalbumin: solution structure, backbone and side-chain dynamics. J. Mol. Biol. 297:147–163. [DOI] [PubMed] [Google Scholar]
- Nanda, V., and L. Brand. 2000. Aromatic interactions in homeodomains contribute to the low quantum yield of a conserved, buried tryptophan. Proteins Struct. Funct. Genet. 40:112–125. [DOI] [PubMed] [Google Scholar]
- Nishino, N., M. Xu, H. Mihara, T. Fujimoto, Y. Ueno, and H. Kumagai. 1992. Sequence dependence in solid-phase synthesis cyclization-cleavage for cyclo(arginyl-glycyl-aspartyl-phenylglycyl). Tetrahedron Lett. 33:1479–1482. [Google Scholar]
- Pachler, K. G. R. 1963. Nuclear magnetic resonance (NMR) study of some α-amino acids. I. Coupling constants in alkaline and acidic medium. Spectrochim. Acta. 19:2085–2092. [Google Scholar]
- Pandit, A., O. F. A. Larsen, I. H. M. van Stokkum, R. van Grondelle, R. Kraayenhof, and H. van Amerongen. 2003. Ultrafast polarized fluorescence measurements on monomeric and self-associated melittin. J. Phys. Chem. B. 107:3086–3090. [Google Scholar]
- Petrich, J. W., M. C. Chang, D. B. McDonald, and G. R. Fleming. 1983. On the origin of nonexponential fluorescence decay in tryptophan and its derivatives. J. Am. Chem. Soc. 105:3824–3832. [Google Scholar]
- Piotto, M., V. Saudek, and V. Sklenar. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 2:661–665. [DOI] [PubMed] [Google Scholar]
- Robbins, R. J., G. R. Fleming, G. S. Beddard, G. W. Robinson, P. J. Thistlewhaite, and G. J. Woolfe. 1980. Photophysics of aqueous tryptophan: pH and temperature effects. J. Am. Chem. Soc. 102:6271–6279. [Google Scholar]
- Ross, J. B. A., K. W. Rousslang, and L. Brand. 1981. Time-resolved fluorescence and anisotropy decay of tryptophan in adrenocorticotropin-(1–24). Biochemistry. 20:4361–4369. [DOI] [PubMed] [Google Scholar]
- Ross, J. B. A., H. R. Wyssbrod, R. A. Porter, G. P. Schwartz, C. A. Michaels, and W. R. Laws. 1992. Correlation of tryptophan fluorescence intensity decay parameters with 1H NMR-determined rotamer conformations: [tryptophan2]oxytocin. Biochemistry. 31:1585–1594. [DOI] [PubMed] [Google Scholar]
- Saito, I., H. Sugiyama, A. Yamamoto, S. Muramatsu, and T. Matsuura. 1984. Photoinduced reactions. 158. Photochemical hydrogen-deuterium exchange reaction of tryptophan. The role of nonradiative decay of singlet tryptophan. J. Am. Chem. Soc. 106:4286–4287. [Google Scholar]
- Shen, X., and J. R. Knutson. 2001. Subpicosecond fluorescence spectra of tryptophan in water. J. Phys. Chem. B. 105:6260–6265. [Google Scholar]
- Shizuka, H., M. Serizawa, T. Shimo, I. Saito, and T. Matsuura. 1988. Fluorescence-quenching mechanism of tryptophan. Remarkably efficient internal proton-induced quenching and charge-transfer quenching. J. Am. Chem. Soc. 110:1930–1934. [Google Scholar]
- Sillen, A., J. F. Diaz, and Y. Engelborghs. 2000. A step toward the prediction of the fluorescence lifetimes of tryptophan residues in proteins based on structural and spectral data. Protein Sci. 9:158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallcombe, S. H., S. L. Patt, and P. A. Keifer. 1995. WET solvent suppression and its applications to LC NMR and high-resolution NMR spectroscopy. J. Magn. Reson. A. 117:295–303. [Google Scholar]
- Szabo, A. G., and D. M. Rayner. 1980. Fluorescence decay of tryptophan conformers in aqueous solution. J. Am. Chem. Soc. 102:554–563. [Google Scholar]
- Vander Donckt, E. 1969. Fluorescence solvent shifts and singlet excited state pKs of indole derivatives. Bull. Soc. Chim. Belg. 78:69–75. [Google Scholar]
- Vivian, J. T., and P. R. Callis. 2001. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 80:2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkert, W. A., R. R. Kuntz, C. A. Ghiron, R. F. Evans, R. Santus, and M. Bazin. 1977. Flash photolysis of tryptophan and N-acetyl-L-tryptophanamide: the effect of bromide on transient yields. Photochem. Photobiol. 26:3–9. [Google Scholar]
- Willis, K. J., W. Neugebauer, M. Sikorska, and A. G. Szabo. 1994. Probing α-helical secondary structure at a specific site in model peptides via restriction of tryptophan side-chain rotamer conformation. Biophys. J. 66:1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuthrich, K. 1986. NMR of Proteins and Nucleic Acids. American Chemical Society, Boca Raton, FL.
- Yu, H. T., W. J. Colucci, M. L. McLaughlin, and M. D. Barkley. 1992. Fluorescence quenching in indoles by excited-state proton transfer. J. Am. Chem. Soc. 114:8449–8454. [Google Scholar]
- Zawadzki, K. M., C.-P. Pan, M. D. Barkley, D. Johnson, and S. S. Taylor. 2003. Endogenous tryptophan residues of cAPK regulatory subunit type IIβ reveal local variations in environments and dynamics. Proteins Struct. Funct. Genet. 51:552–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.