

Abstract
Molecular mechanism of the binding of neuropeptide achatin-I (Gly-d-Phe-Ala-Asp) to large unilamellar vesicles of zwitterionic egg-yolk phosphatidylcholine (EPC) was investigated by means of natural-abundance 13C and high-resolution (of 0.01 Hz order) 1H NMR spectroscopy. The binding equilibrium was found to be sensitive to the ionization state of the N-terminal 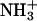 group in achatin-I; the de-ionization of
group in achatin-I; the de-ionization of 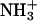 decreases the bound fraction of the peptide from ∼15% to nearly none. The electrostatic attraction between the N-terminal positive
decreases the bound fraction of the peptide from ∼15% to nearly none. The electrostatic attraction between the N-terminal positive 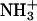 group and the negative
group and the negative 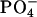 group in the EPC headgroup plays an important role in controlling the equilibrium. Analysis of the 13C chemical shifts (δ) of EPC showed that the binding location of the peptide within the bilayer is the polar region between the glycerol and ester groups. The binding caused upfield changes Δδ of the 13C resonance for almost all the carbon sites in achatin-I. The changes Δδ for the ionic Asp at the C-terminus are more than five times as large as those for the other residues. The drastic changes for Asp result from the dehydration of the ionic
group in the EPC headgroup plays an important role in controlling the equilibrium. Analysis of the 13C chemical shifts (δ) of EPC showed that the binding location of the peptide within the bilayer is the polar region between the glycerol and ester groups. The binding caused upfield changes Δδ of the 13C resonance for almost all the carbon sites in achatin-I. The changes Δδ for the ionic Asp at the C-terminus are more than five times as large as those for the other residues. The drastic changes for Asp result from the dehydration of the ionic 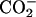 groups, which are strongly hydrated by electrostatic interactions in bulk water. The side-chain conformational equilibria of the aromatic d-Phe and ionic Asp residues were both affected by the binding, and the induced changes in the equilibria appear to reflect the peptide-lipid hydrophobic interactions.
groups, which are strongly hydrated by electrostatic interactions in bulk water. The side-chain conformational equilibria of the aromatic d-Phe and ionic Asp residues were both affected by the binding, and the induced changes in the equilibria appear to reflect the peptide-lipid hydrophobic interactions.
INTRODUCTION
Binding of bioactive peptides to phospholipid bilayers is an important physical phenomenon, which relates to a variety of biological functions in living cellular systems. However, much of the molecular mechanism of the membrane-binding remains unrevealed, coupled with the practical difficulties of handling the complex system of peptides and bilayers. The elucidation of this subject is thus one of the current aims of both theoretical and experimental biophysicists as a preliminary but essential step for the total understanding of the biological functions (Simon and McIntosh, 2002). In this study, we investigate the molecular mechanism of the binding of a neuropeptide achatin-I (Gly-d-Phe-Ala-Asp; for the structure, see Fig. 1) to phosphatidylcholine lipid (PC) bilayers through dealing with the problems of fundamental significance; factors controlling the binding equilibrium, peptide location within the bilayer, and binding effect on the peptide side-chain conformations are examined by means of natural-abundance 13C and high-resolution (of 0.01 Hz order) 1H NMR spectroscopy.
FIGURE 1.

Molecular structure of a neuropeptide achatin-I with the ionization state corresponding to the neutral pH. Arrows denote the side-chain conformations studied in this work.
Achatin-I was first isolated in the ganglia of an African snail, Achatina fulica Férussac, and it is known as the neuropeptide containing d-amino acid (Kamatani et al., 1989). A subsequent discovery of the sequence analogs, such as Gly-d-Phe-Gly-Asp from the brain extracts of a different species, has shown the wide importance of the sequence family in the nerve actions (Iwakoshi et al., 2000). Achatin-I controls a cardiac activity in the snail (Fujimoto et al., 1991), and excites heart-regulatory neurons in the central nervous system by inducing inward current due to Na+ ions (Kamatani et al., 1989). The finding of the stereoisomeric specificity for the neuroexcitation implied the existence of a particular receptor for achatin-I (Kamatani et al., 1989), whereas the modulatory effect of achatin-I was also discovered upon membrane-current induced specifically by the other neurotransmitters, such as oxytocin and acetylcholine (Liu and Takeuchi, 1993). To understand the molecular mechanism of those complicated biological functions of achatin-I (Kamatani et al., 1989; Fujimoto et al., 1991; Kim et al., 1991; Liu and Takeuchi, 1993; Emaduddin et al., 1996; Poteryaev et al., 1998; Satake et al., 1999), it seems to be necessary to consider carefully the role of microenvironment around the receptors other than the simple binding of achatin-I to its specific receptor. It is likely that the today's lack of knowledge on the association of the peptide with lipids, which constitute the majority of membrane-environment of receptors, is a significant disadvantage for elucidating the problem. In this study, we thus focus on probing the binding of achatin-I to the membrane using the model lipid bilayer.
Achatin-I contains both the hydrophobic d-Phe and hydrophilic Asp within the short sequence of four residues. The ionic 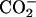 groups of Asp are expected to contribute to the binding equilibrium through the peptide-lipid electrostatic interactions. The phenyl ring of d-Phe, on the other hand, can contribute through the hydrophobic interactions. In this work, we focus on the electrostatic effect on the binding equilibrium. Besides the negatively charged
groups of Asp are expected to contribute to the binding equilibrium through the peptide-lipid electrostatic interactions. The phenyl ring of d-Phe, on the other hand, can contribute through the hydrophobic interactions. In this work, we focus on the electrostatic effect on the binding equilibrium. Besides the negatively charged 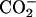 groups in the C-terminal Asp, achatin-I involves positively charged
groups in the C-terminal Asp, achatin-I involves positively charged 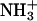 in the N-terminal Gly. Since PC, which is a major lipid constituent of biological membranes, also involves both positive
in the N-terminal Gly. Since PC, which is a major lipid constituent of biological membranes, also involves both positive 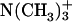 and negative
and negative 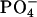 moieties in the hydrophilic headgroup, the electrostatic attractions between the opposite charges are probable factors controlling the binding of achatin-I within the bilayer. We can examine this point by changing the ionization states of the peptide through the control of pH (pD). If the electrostatic interactions are important, the loss of charge on either the
moieties in the hydrophilic headgroup, the electrostatic attractions between the opposite charges are probable factors controlling the binding of achatin-I within the bilayer. We can examine this point by changing the ionization states of the peptide through the control of pH (pD). If the electrostatic interactions are important, the loss of charge on either the 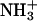 or
or 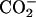 is to decrease the binding affinity.
is to decrease the binding affinity.
To gain further insight into the binding equilibrium, information on the peptide location within the bilayer is indispensable. In view of the electrostatic effect on the binding, possible locations of the N-terminal positive 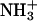 and C-terminal negative
and C-terminal negative 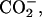 respectively, are near the
respectively, are near the 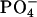 and
and 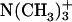 in the headgroup of PC. The location of the hydrophobic phenyl ring of d-Phe is expected to be closer to the hydrophobic core of the acyl chains rather than to the charged headgroup. In a previous work, we have demonstrated that the high-resolution NMR of natural-abundance 13C within phospholipid is powerful in determining the binding location of peptides and proteins within the bilayers and emulsions (Okamura et al., 2001); the high ability of discriminating a wide variety of carbon sites in the lipid molecule provides a novel method of the location analysis (Okamura and Nakahara, 2001) in addition to the traditional methods, such as ESR, fluorescence quenching, x-ray and neutron scattering, and solid-state NMR (see the reviews in Simon and McIntosh, 2002, for the details of the techniques). Here, we apply the method of 13C NMR to examine the location of achatin-I within the PC bilayer.
in the headgroup of PC. The location of the hydrophobic phenyl ring of d-Phe is expected to be closer to the hydrophobic core of the acyl chains rather than to the charged headgroup. In a previous work, we have demonstrated that the high-resolution NMR of natural-abundance 13C within phospholipid is powerful in determining the binding location of peptides and proteins within the bilayers and emulsions (Okamura et al., 2001); the high ability of discriminating a wide variety of carbon sites in the lipid molecule provides a novel method of the location analysis (Okamura and Nakahara, 2001) in addition to the traditional methods, such as ESR, fluorescence quenching, x-ray and neutron scattering, and solid-state NMR (see the reviews in Simon and McIntosh, 2002, for the details of the techniques). Here, we apply the method of 13C NMR to examine the location of achatin-I within the PC bilayer.
When achatin-I binds to the phospholipid bilayers as a transfer from water to the bilayer, the most drastic change that occurs in the molecular environment around the peptide is dehydration due to the interactions with the lipid molecules. It is then obvious that the understanding of the hydration in bulk water is essential to elucidate the binding mechanism. The hydration effect on achatin-I in bulk water was featured, for example, by the observation that the hydration on the ionic 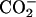 groups at the C-terminal Asp almost cancels out the gas-phase energy differences of tens of kJ mol−1 among the Asp side-chain conformers (Kimura et al., 2004). Our particular interest here is what happens to this strong hydration upon binding to the bilayers. The 13C NMR chemical shifts of achatin-I give important information on this, since they are a sensitive probe for the molecular environment around the peptide. It should also be noted that the analysis of the relative line widths of the 13C signals of achatin-I provides insight into the changes in the dynamics of the environment in response to the binding.
groups at the C-terminal Asp almost cancels out the gas-phase energy differences of tens of kJ mol−1 among the Asp side-chain conformers (Kimura et al., 2004). Our particular interest here is what happens to this strong hydration upon binding to the bilayers. The 13C NMR chemical shifts of achatin-I give important information on this, since they are a sensitive probe for the molecular environment around the peptide. It should also be noted that the analysis of the relative line widths of the 13C signals of achatin-I provides insight into the changes in the dynamics of the environment in response to the binding.
Finally, we examine the effect of membrane-binding on the conformational equilibrium of achatin-I. In this report, the conformations in the side chains are focused on as an extension of our previous work on the side-chain conformational equilibria of achatin-I in bulk water (Kimura et al., 2004). How do the dehydration and interactions with the phospholipid molecules upon binding affect the peptide conformations? The analysis of the dihedral-angle dependent vicinal 1H-1H couplings from the fine structures of NMR signals makes it possible to examine the side-chain conformational equilibria. Although the side-chain conformations of peptides have been a subject of structural biology for the past decades, the researches were limited mainly to the systems of isotropic solution including our previous study of achatin-I in bulk water. To the best of our knowledge, a comparative study of the free and membrane-bound conformations of the side chains, which discloses the effect of binding, has not been done. There are a few reports concerning the conformational equilibria within phospholipid bilayers; for example, the equilibrium of Val-1 residue in gramicidin A was studied by performing numerical analysis to simulate the solid-state NMR spectra (Lee and Cross, 1994). The analysis of the vicinal J couplings in aqueous solution gives us more direct information on the conformational problem, when we can observe the fine structures of the signals. To provide insight into the binding effect, it is of interest to compare with the effect of transfer from water to a less polar solvent, such as methanol.
METHODS
Experimental
Materials
Achatin-I (Gly-d-Phe-Ala-Asp) monoammonium salt was purchased from Sigma Chemical (St. Louis, MO). Egg-yolk phosphatidylcholine (EPC), used in this work as phosphatidylcholine lipid, was purchased from NOF (Tokyo, Japan). Solvent heavy water (D2O; 99.90% D) was from Euriso-top (Saint Aubin, France), and D2O solutions of DCl (18 wt %) and NaOD (40 wt %) used to control the solution pD (pH) were from Aldrich Chemical (Milwaukee, WI). Solvent methanol-d4 (99.8% D) was also from Aldrich.
Achatin-I solutions in water and methanol were prepared at the concentrations of 2 and 1 mM, respectively. Large unilamellar vesicles (LUV) of EPC with a diameter of 100 nm were prepared at the lipid concentration of 20 mM in water, corresponding to the EPC/achatin-I molar ratio of 10. When we measured the natural-abundance 13C NMR, the concentrations of achatin-I and EPC were increased to 5 and 50 mM, respectively, at the fixed EPC/achatin-I ratio of 10. Here, we neglect the difference in the binding equilibrium due to the increased concentrations, and argue the peptide location within the bilayer and molecular environment around the peptide on the basis of the 13C NMR.
The procedure for the preparation of LUV was as follows: a required amount of EPC was dissolved in chloroform (Wako Pure Chemical, Osaka, Japan), and the solvent was evaporated by a rotary vacuum evaporator. The sample was then dried overnight under vacuuming. Lipid film thus formed was vortex mixed with D2O for ∼5 min to prepare a homogenous suspension of multilamellar vesicles. To obtain LUV, the multilamellar vesicles underwent 10 cycles of freezing and thawing, and the resultant was extruded 21 times through a polycarbonate filter with the pore diameter of 100 nm. Mixture of achatin-I and LUV was prepared by dissolving the solid achatin-I in the LUV sample.
To examine the electrostatic effect of peptide-lipid interactions on the binding equilibrium, peptide-LUV mixtures were prepared at different pD conditions. By controlling the pD at the acidic 3.2, neutral 5.9, and basic 11.6, the ionization states of the α-amino group in the N-terminal Gly, and α- and β-carboxyl groups in the C-terminal Asp of achatin-I, were changed; the ionization states at the pDs are (α-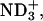 α-
α-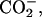 β-CO2D) for the acidic, (α-
β-CO2D) for the acidic, (α-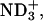 α-
α-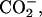 β-
β-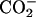 ) for the neutral, and (α-ND2, α-
) for the neutral, and (α-ND2, α-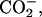 β-
β-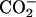 ) for the basic condition. It should be noted that the acidification of the solution de-ionizes the side-chain β-carboxyl group in Asp, corresponding to the pKa of 3.9 (Dawson et al., 1986). pKa of the α-carboxyl group in Asp, on the other hand, is ∼2 units smaller than that of the β-one. We did not decrease the pD below 3.2 to control the ionization state of the α-carboxyl group, because it does not assure the full ionization of the lipid
) for the basic condition. It should be noted that the acidification of the solution de-ionizes the side-chain β-carboxyl group in Asp, corresponding to the pKa of 3.9 (Dawson et al., 1986). pKa of the α-carboxyl group in Asp, on the other hand, is ∼2 units smaller than that of the β-one. We did not decrease the pD below 3.2 to control the ionization state of the α-carboxyl group, because it does not assure the full ionization of the lipid 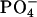 according to the intrinsic pKa = 0.8 in phosphatidylcholine (Moncelli et al., 1994). The ionization state of the EPC headgroup was thus kept constant throughout the examined pD range in the zwitterionic form of
according to the intrinsic pKa = 0.8 in phosphatidylcholine (Moncelli et al., 1994). The ionization state of the EPC headgroup was thus kept constant throughout the examined pD range in the zwitterionic form of 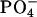 and
and 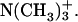 At the examined pDs, the LUV was confirmed, by the NMR analysis of the 1H signals, to be stable during the experiments. Binding characteristics of achatin-I to EPC LUV can be examined by comparing with the peptide in bulk water, so that the aqueous solutions of achatin-I were prepared at the acidic pD = 3.2, neutral pD = 7.5 (results taken from Kimura et al., 2004), and basic pD = 13.1, where the ionization states are identical to the corresponding systems of the achatin-LUV mixtures. The ionization state of membrane-bound achatin-I was confirmed to be identical with that in bulk water according to the chemical shifts and geminal spin-spin couplings in 1H NMR. The pD values were obtained by an addition of 0.4 units to the readings of the pH meter with a glass electrode (HORIBA D-21 or B-212, Kyoto, Japan) (Glasoe and Long, 1960). The solution pD was controlled by D2O solutions of DCl or NaOD.
At the examined pDs, the LUV was confirmed, by the NMR analysis of the 1H signals, to be stable during the experiments. Binding characteristics of achatin-I to EPC LUV can be examined by comparing with the peptide in bulk water, so that the aqueous solutions of achatin-I were prepared at the acidic pD = 3.2, neutral pD = 7.5 (results taken from Kimura et al., 2004), and basic pD = 13.1, where the ionization states are identical to the corresponding systems of the achatin-LUV mixtures. The ionization state of membrane-bound achatin-I was confirmed to be identical with that in bulk water according to the chemical shifts and geminal spin-spin couplings in 1H NMR. The pD values were obtained by an addition of 0.4 units to the readings of the pH meter with a glass electrode (HORIBA D-21 or B-212, Kyoto, Japan) (Glasoe and Long, 1960). The solution pD was controlled by D2O solutions of DCl or NaOD.
Determination of binding equilibrium
Binding equilibria of achatin-I to EPC LUV at the acidic, neutral, and basic pDs were examined by ultracentrifugation method; the bound peptides were separated with the lipids (as a top fraction) from the free peptides (dissolving in the bottom fraction). The ultracentrifugation was carried out on a Beckman (Fullerton, CA) TL-100 instrument, which was equipped with a TLA-100.2 rotor. The experimental condition was 230 000 × g at 30°C for 2 h. The amount of free peptides was determined by measuring the concentration in the bottom fraction. The concentrations were measured by 1H NMR according to the relative peak area to that of a reference compound of a known concentration; for the independent samples of the bottom fraction and the reference solution, the spectra were recorded at the same instrument conditions, such as the gain parameter for the detection of free-induction decay (FID) signals. The above NMR method of concentration measurement is particularly useful for peptides without an inherent fluorescent probe of tryptophan residue.
NMR measurements
To investigate the location of achatin-I within the bilayers and binding effect on the molecular environment around the peptide, 13C NMR spectra of 1), the mixture of achatin-I and EPC LUV, 2), EPC LUV, and 3), achatin-I aqueous solution were measured on a JEOL (Tokyo, Japan) 600 MHz spectrometer (JNM-ECA600, equipped with a superconducting magnet of 14.10 T) operating at the frequency of 151 MHz for the 13C nuclei. When we measured the 13C NMR, a probe for the sample tube of 10 mm OD was installed. The temperature was controlled at 30.0 ± 0.1°C during the measurements. The data points of 32 768 were sampled over the spectrum range of 250 ppm, and the corresponding digital resolution was 0.01 ppm (1.2 Hz). FID signals were accumulated 17,000–87,000 times. The 13C chemical shifts (δ) relative to the DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate) methyl carbons were obtained by referring to the absorption frequency of the solvent deuteron monitored as the lock signal. 13C-signal assignment for achatin-I was carried out by consulting the previous reports on the 13C NMR of amino acid systems (see Wüthrich, 1976, and Howarth and Lilley, 1978). For the nonterminal residues, the assignment can simply be done according to the reported δ-values for the nonterminal residues in peptides. For the terminal residues, it was done on the basis of δ for monomeric amino acids by adopting the effect of a peptide-bond formation on either side of α-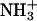 or α-
or α-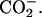 13C-signal assignment for EPC was also carried out by referring to the reported results (Li et al., 1993).
13C-signal assignment for EPC was also carried out by referring to the reported results (Li et al., 1993).
1H NMR spectra were recorded at 30.0 ± 0.1°C on a JEOL 500 MHz spectrometer (JNM-ECA500, wide-bore type with a magnet of 11.75 T) or the 600 MHz spectrometer. A probe for the sample tube of 5 mm OD was used for the 1H measurements. FID signals were accumulated 8–256 times. For each sample, the measurement was repeated two times. The 1H chemical shifts δ, relative to the DSS methyl protons, were obtained by referring to the HDO and OH signals of 4.712 ppm in water and 4.783 ppm in methanol, respectively. The correction of δ-values according to the magnetic susceptibility of methanol was not made, since the δ-values in methanol were used only as the differences Δδ among the signals to identify the ionization state of achatin-I in methanol. When the binding effect on the side-chain conformational equilibria was examined from the fine structures of the 1H signals, the digital frequency resolution was set to 0.01 Hz. Otherwise, the resolution was set to 0.34 Hz. To achieve the former resolution, 524,288 data points were sampled over the spectrum range of 10 ppm. The high resolution made it possible to detect sensitively the binding effect on the conformational equilibria. 1H signals of achatin-I were assigned according to a previous work on the aqueous solution system (Kimura et al., 2004). Assignment of 1H signals of EPC was done by referring to the reported results (Li et al., 1993).
Conformational analysis
For the amino-acid residues of ABX-type spin system in 1H NMR, the vicinal (Jαβ1 and Jαβ2) and geminal (Jβ1β2) coupling constants for the three protons can be determined explicitly by the analysis of the spin Hamiltonian (Pople et al., 1959). In achatin-I (Gly-d-Phe-Ala-Asp), d-Phe and Asp residues correspond to ABX-type spin system. Since a vicinal coupling constant exhibits a dihedral-angle dependence (Karplus, 1959, 1963), the side-chain conformational equilibria of d-Phe and Asp with respect to the Cα-Cβ bond can be studied according to the observed Jαβ1 and Jαβ2. In the presence of EPC LUV, the J values of achatin-I were observed as the average for the membrane-bound and free states due to fast exchanges on the NMR timescale. Thus, the exclusive determination of J for the bound state (Jb) requires the separate observations for both in the presence (Jav) and in the absence (Jf) of LUV according to
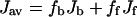 |
(1) |
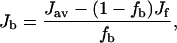 |
(2) |
where fb and ff are the fractions of the bound and free peptides, respectively. In this work, fb was determined by the ultracentrifugation method mentioned above.
On the basis of the obtained Jαβ1 and Jαβ2 for the membrane-bound state, the conformational equilibria within the membrane can be analyzed. As shown previously (Kimura et al., 2004), Jαβ1 and Jαβ2 in achatin-I in bulk water can be expressed as the probability-weighted averages over the staggered conformers shown in Fig. 2 (Pachler, 1964). The three-staggered-state model was adopted here to analyze the conformational equilibria in membrane. Thus, for both the free and bound states, the Jαβ1 and Jαβ2 values can be expressed as:
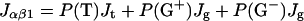 |
(3a) |
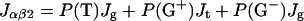 |
(3b) |
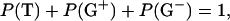 |
(3c) |
where P(T), P(G+), and P(G−) are the probabilities of conformers T, G+, and G−, and Jt and Jg are the vicinal coupling constants between the α- and β-protons in the trans and gauche conformations, respectively. The conformer probabilities are obtained by solving Eqs. 3. The conformers are named as above by focusing on the dihedral angle of C-Cα-Cβ-C; T is for the trans conformer, G+ and G− are for the gauche. In G+ and G−, respectively, the β-carboxyl group in Asp (or β-phenyl group in d-Phe) is trans and gauche to the α-ND in the peptide bond.
FIGURE 2.

Staggered side-chain conformers of the d-Phe and Asp residues in achatin-I (Gly-d-Phe-Ala-Asp) with respect to the analyzed dihedral angles on the Cα-Cβ bond.
To determine the conformer probabilities, a parameter set of Jt and Jg is necessary. In this work, we apply Pachler's parameter set of Jt = 13.56 and Jg = 2.60 Hz (Pachler, 1964), which is generally used to analyze the side-chain conformational equilibrium of amino-acid systems; for the parameter choice, see the references cited elsewhere (Kimura et al., 2002, 2004). It is assumed here that Pachler's parameter set, determined in aqueous solution, is applicable to the membrane-bound peptide. This unavoidable approximation is expected to provide good results, since a through-bond coupling between the vicinal protons at a fixed conformation mainly reflects the electronic state. As described previously (Kimura et al., 2004), the assignment of the β-proton signals of Asp residue to the β1- and β2-protons in Fig. 2 was done on the basis of the selective deuterium labeling experiments of the monomer (Kainosho and Ajisaka, 1975). The β-proton signals of d-Phe were assigned according to the labeling experiments for Ac-l-Phe-NDMe (Kobayashi et al., 1979); note that the β1- and β2- protons in d-Phe correspond to those in the mirror image of l-Phe.
RESULTS AND DISCUSSION
Effect of the peptide ionization state on the binding equilibrium
Let us consider how the binding equilibrium of peptides to lipid bilayers is controlled by the electrostatic and/or hydrophobic effects. To provide insight into the electrostatic effect on the binding of achatin-I (Gly-d-Phe-Ala-Asp) to zwitterionic PC bilayers, here we investigated the binding equilibrium by changing the ionization states of the peptide amino and carboxyl groups through the control of pH (pD). The equilibrium was examined by using EPC LUV as the PC bilayer at the molar ratio of peptide/lipid = 1 (2 mM):10 (20 mM).
Shown in Fig. 3 are the bound fraction of achatin-I obtained at the acidic (pD = 3.2), neutral (pD = 5.9), and basic (pD = 11.6) conditions. At the neutral pD, the α-amino group of the N-terminal Gly is positively ionized, and the α- and β-carboxyl groups of the C-terminal Asp are negatively ionized; see the ionization state in Fig. 1. The bound fraction of achatin-I with this ionization state was determined to amount to 16%. When the pD changed from the neutral to the basic, the de-ionization of the α-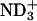 in Gly decreased the bound fraction drastically to almost none. When the pD changed from the neutral to the acidic, on the other hand, the neutralization of the β-
in Gly decreased the bound fraction drastically to almost none. When the pD changed from the neutral to the acidic, on the other hand, the neutralization of the β-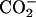 group in Asp decreased the bound fraction to 10%. Thus, both the positive and negative charges in achatin-I are found to play an important role in determining the binding equilibrium.
group in Asp decreased the bound fraction to 10%. Thus, both the positive and negative charges in achatin-I are found to play an important role in determining the binding equilibrium.
FIGURE 3.

pD dependence of the bound fraction (%) of achatin-I to EPC LUV in the mixture of 2 mM achatin-I and 20 mM EPC. The acidic pD = 3.2, neutral pD = 5.9, and basic pD = 11.6. The ionization states of the amino and carboxyl groups in achatin-I at each pD are shown in the parentheses. The 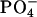 and
and 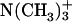 in EPC are fully ionized throughout the examined pD range.
in EPC are fully ionized throughout the examined pD range.
In the binding process at the neutral pD, the N-terminal 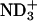 and C-terminal
and C-terminal 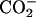 in achatin-I are expected to be attracted electrostatically by the
in achatin-I are expected to be attracted electrostatically by the 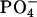 and
and 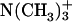 in the headgroup of EPC, respectively. The de-ionization of the
in the headgroup of EPC, respectively. The de-ionization of the 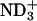 or
or 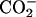 in the peptide loses the attractive interactions. Thus, the observed effect of ionization state can be ascribed to the peptide-lipid electrostatic attractions. The marked decrease in the bound fraction upon de-ionization of the N-terminal
in the peptide loses the attractive interactions. Thus, the observed effect of ionization state can be ascribed to the peptide-lipid electrostatic attractions. The marked decrease in the bound fraction upon de-ionization of the N-terminal 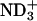 suggests the key importance of the attraction between the
suggests the key importance of the attraction between the 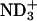 in achatin-I and
in achatin-I and 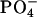 in EPC. As a result of the electrostatic attraction with the N-terminal
in EPC. As a result of the electrostatic attraction with the N-terminal 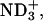 we observed in the presence of achatin-I downfield changes Δδ by ∼0.05 ± 0.01 ppm in the 31P NMR of
we observed in the presence of achatin-I downfield changes Δδ by ∼0.05 ± 0.01 ppm in the 31P NMR of 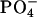 ; the spectrum showed a typical signal, a sharp peak with a broad shoulder peak on the higher field (Seelig, 1978). The change Δδ is not so drastic due probably to the strong hydration of the
; the spectrum showed a typical signal, a sharp peak with a broad shoulder peak on the higher field (Seelig, 1978). The change Δδ is not so drastic due probably to the strong hydration of the 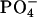 in the absence of the peptide, due in addition to a low fraction of the peptide-binding lipids. At the examined concentrations, we could not detect the low-sensitive 14N signal of the
in the absence of the peptide, due in addition to a low fraction of the peptide-binding lipids. At the examined concentrations, we could not detect the low-sensitive 14N signal of the 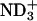 group to discuss the binding mechanism. Further discussions concerning the peptide location within bilayer will be made in the later section on the basis of 13C NMR.
group to discuss the binding mechanism. Further discussions concerning the peptide location within bilayer will be made in the later section on the basis of 13C NMR.
The role of an N-terminal 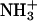 group in the peptide-lipid electrostatic attraction was also examined previously for opioid peptides, Met-enkephalin (Tyr-Gly-Gly-Phe-Met) and Leu-enkephalin (Tyr-Gly-Gly-Phe-Leu), which were isolated from the mammalian brain extracts (Hughes et al., 1975). It was shown that the enkephalins bind to negative lipids, such as lysophosphatidylglycerol in micelles (Deber and Behnam, 1984), phosphatidylserine in small (Jarrell et al., 1980) and large (Milon et al., 1990) unilamellar vesicles. In this article, we present that the in-membrane electrostatic attractions between the N-terminal
group in the peptide-lipid electrostatic attraction was also examined previously for opioid peptides, Met-enkephalin (Tyr-Gly-Gly-Phe-Met) and Leu-enkephalin (Tyr-Gly-Gly-Phe-Leu), which were isolated from the mammalian brain extracts (Hughes et al., 1975). It was shown that the enkephalins bind to negative lipids, such as lysophosphatidylglycerol in micelles (Deber and Behnam, 1984), phosphatidylserine in small (Jarrell et al., 1980) and large (Milon et al., 1990) unilamellar vesicles. In this article, we present that the in-membrane electrostatic attractions between the N-terminal 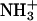 in achatin-I and
in achatin-I and 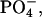 even in the zwitterionic PC bilayer, play an important role in controlling the binding equilibrium. The finding indicates that once achatin-I encounters with the neutral bilayer surface through diffusion, the electrostatic attraction operates at atomic resolution between the charged groups of the peptide and lipid for their binding.
even in the zwitterionic PC bilayer, play an important role in controlling the binding equilibrium. The finding indicates that once achatin-I encounters with the neutral bilayer surface through diffusion, the electrostatic attraction operates at atomic resolution between the charged groups of the peptide and lipid for their binding.
Location within bilayer
In the previous section, we showed the importance of the electrostatic attraction between achatin-I and the headgroup of PC in determining the binding equilibrium. It is then expected that the binding location of achatin-I within the bilayer at physiological pH is near the hydrophilic surface rather than the hydrophobic core of the acyl chains. To scrutinize this expectation, the 13C NMR of EPC molecules in LUV at the neutral pD was investigated both with and without achatin-I. The chemical shifts (δ) and line widths (ν1/2) of the 13C signals for the two systems were compared to see how the carbon sites on the lipid are perturbed by the peptide binding.
The observed 13C spectrum of the 1:10 molar mixture of achatin-I (5 mM) and EPC (50 mM) LUV is shown in Fig. 4. The signals from the peptide can be easily distinguished from those of the lipid, since the peptide signals are obviously sharper than the relatively broad lipid signals (the peaks for the peptide are marked with asterisks). The number of the peptide signals observed is equal to the number of the carbon sites within the peptide. Hence, the peptide signals were observed as the average of the membrane-bound and free states due to the rapid exchange between the two states on the NMR timescale. According to the analysis of the lipid signals, we discuss below the peptide-binding location. The free-induction decay signals were accumulated until we made sure that the signal/noise ratios were high enough to analyze the peak positions of the lipid signals within the uncertainties due to the digital frequency resolution. With this accumulation, the peak positions of the sharper peptide signals were also determined within the ambiguity due to the digital resolution.
FIGURE 4.

13C NMR spectrum of the mixture of 5 mM achatin-I and 50 mM EPC LUV observed by the 600 MHz spectrometer operating at 151 MHz for the 13C nuclei. Signals with the asterisk correspond to the carbons in the peptide and are assigned as shown.
Fig. 5 illustrates the chemical-shift changes Δδ of the lipid signals induced by the presence of achatin-I. The changes Δδ are relatively large in the ranges between the glycerol carbons and the α-carbons in the acyl chains. The change Δδ for the methylene carbons of the acyl chains is comparable to those for the α-carbons. The line widths ν1/2 of the 13C signals, on the other hand, did not exhibit any obvious effect of the presence of achatin-I at the examined concentrations. The above observations indicate that the large body of achatin-I molecule locates between the polar glycerol and ester groups in PC. The notable upfield changes Δδ of the glycerol and ester-carbonyl 13C chemical shifts are attributed to the dehydration due to the binding of achatin-I. On the other hand, the marked downfield change Δδ for the α-carbon next to the ester groups can be explained by the magnetic deshielding effect from the phenyl ring (Johnson and Bovey, 1958) of d-Phe residue, which is expected to locate around the ester group at the internal site of the peptide-binding region. The reason is not obvious for the downfield change Δδ for the methylene carbons. It is considered to result from the secondary effect of the binding at the glycerol to ester groups, since the methylene carbons are significantly separated from the polar groups in terms of the molecular size of achatin-I.
FIGURE 5.

13C chemical-shift changes Δδ of EPC (50 mM) within LUV induced by the presence of achatin-I (5 mM) at the neutral pD. Signals from the glycerol C1 and C3 carbons were observed inseparably, and the same value Δδ is given for the C1 and C3. Chemical shifts of glycerol C2 and β-CH2 were not obtained due to the broad signal and signal overlap with =CHCH2CH=, respectively.
The binding equilibrium of achatin-I to the PC bilayer is markedly controlled by the electrostatic effect due to the attractions between the ionic groups of the peptides and lipids, particularly between the peptide N-terminal 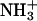 and lipid
and lipid 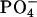 in the headgroup. In contrast, the hydrophobic effect due to the phenyl ring of d-Phe is less apparent in view of its location at the polar ester group within the bilayer. The hydrophobic effect is thus likely to assist cooperatively the electrostatic effect on the achatin-I binding through modulating rather than determining the peptide location. Trapping of achatin-I at the membrane surface increases the local peptide concentration around the receptors, and this increase may facilitate the access to the binding sites of the receptors.
in the headgroup. In contrast, the hydrophobic effect due to the phenyl ring of d-Phe is less apparent in view of its location at the polar ester group within the bilayer. The hydrophobic effect is thus likely to assist cooperatively the electrostatic effect on the achatin-I binding through modulating rather than determining the peptide location. Trapping of achatin-I at the membrane surface increases the local peptide concentration around the receptors, and this increase may facilitate the access to the binding sites of the receptors.
Changes in the peptide molecular environment
When achatin-I is transferred from bulk water to the bilayer, many of the hydrating water molecules on the peptide need to be released due to a restricted space for hydration by the nearby lipid molecules. Such changes in the molecular environment around achatin-I are discussed here on the basis of the observed changes in the 13C NMR of the peptide.
The chemical-shift changes Δδ for all the carbon-atom sites in achatin-I due to the binding to EPC LUV are illustrated in Fig. 6. Almost all the changes Δδ are toward upfield. The upfield changes result from the dehydration upon binding to the bilayers. Noticeably, Δδ for the ionic Asp at the C-terminus is more than five times as large as those for the other residues. The marked changes Δδ for Asp show that the dehydration effect is most significant on this residue. In a previous article, we demonstrated that the hydration in bulk water on the C-terminal Asp in achatin-I is so strong that the gas-phase (absence-of-solvent) energy differences of tens of kJ mol−1 among the Asp side-chain conformers are almost cancelled out (Kimura et al., 2004). In other words, the hydration competes against the enormous intramolecular electrostatic energy due to the vicinal 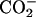 groups. The changes Δδ for the Asp carbons shown in Fig. 6 are induced by the decreased hydration of the
groups. The changes Δδ for the Asp carbons shown in Fig. 6 are induced by the decreased hydration of the 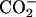 groups, which have a large hydration shell in bulk water due to the strong electrostatic solute-solvent interactions.
groups, which have a large hydration shell in bulk water due to the strong electrostatic solute-solvent interactions.
FIGURE 6.

13C chemical-shift changes Δδ of achatin-I (5 mM) induced by the binding to EPC (50 mM) LUV at the neutral pD.
The presence of EPC LUV increased the line widths ν1/2 of the peptide signals for all the carbon sites ranging between +0.5 and +5.1 Hz. The site-selective line broadening is a consequence of the membrane-binding, which causes slower molecular motions around the peptide accompanying the dehydration. In contrast to the binding effect on the chemical shifts, the effect on the line widths was not prominent for the Asp residue. The dynamic similarity is understandable because the Asp in the bound achatin-I is hydrated at the water-abundant bilayer surface (Wiener and White, 1992) and the EPC bilayer is in the liquid-crystalline phase at 30°C (Szoka and Papahadjopoulos, 1980). The notable broadening was for the α-carbon of Ala (+5.1 Hz), the ring and β-carbons of d-Phe (+2.9 Hz for Ring (4), +2.4 Hz for Ring (2, 6), +2.3 Hz for Ring (3, 5), and +2.6 Hz for β); for the notation of the carbon sites, see Fig. 1. These broadening for Ala and d-Phe can be explained in terms of the effective decrease in the water-accessibility for the residues at the nonterminal sequence positions (Kimura et al., 2004). The largest ν1/2 increase in d-Phe at Ring (4) carbon at the end of the side chain is in accord with the anchoring of the hydrophobic ring to the membrane interior. The 13C information thus obtained provides the peptide binding-location model given later.
Changes in the peptide side-chain conformations
In this section, the dihedral-angle-dependent vicinal 1H-1H couplings in NMR are examined to see how the membrane-binding affects the side-chain conformational equilibria of the hydrophobic d-Phe and hydrophilic Asp residues in achatin-I. How are the side-chain conformational equilibria in the membrane-bound state different from those in bulk water? What is the controlling factor? The conformational equilibria within the PC bilayer are expected to depend on the binding location and the molecular environment therein examined above.
1H NMR spectrum
1H NMR spectrum of the 1:10 molar mixture of achatin-I (2 mM) and EPC (20 mM) LUV at the neutral pD is presented in Fig. 7 a. The signals from all of the residues in achatin-I, appearing as the average over the membrane-bound and free states, show clear fine structures and can be distinguished from the lipid signals. Since the peptide signals are much sharper than the lipid signals, we can obtain the chemical shifts and coupling constants of achatin-I from the peak positions.
FIGURE 7.

1H NMR spectra observed for (a) the mixture of 2 mM achatin-I and 20 mM EPC LUV at the neutral pD and (b) 1 mM achatin-I in methanol-d4.
1H-1H Coupling Constants
The signals from the d-Phe and Asp residues give the typical 12-line ABX-type fine structures due to the spin-spin couplings among the one α- and two β-protons (Kimura et al., 2004). In Fig. 7 a, the α-proton of d-Phe gave four-line signals with the average chemical shift of 4.516 ppm, whereas the β-protons of d-Phe gave eight-line signals with the average chemical shift of 3.064 ppm. The α-proton of Asp gave four-line signals at 4.332 ppm, whereas the β-protons of Asp gave eight-line signals at 2.641 ppm. According to the ABX-type fine structures, we determined the two vicinal coupling constants (Jαβ1 and Jαβ2) and one geminal coupling constant (Jβ1β2), according to the analysis of the spin Hamiltonian of the three coupling protons (Pople et al., 1959).
The obtained Jαβ1, Jαβ2, and Jβ1β2 in achatin-I in the presence of EPC LUV at the acidic, neutral, and basic pDs are summarized in Table 1 with those observed in the absence of LUV. The membrane-binding effect on Js was evident for both the d-Phe and Asp at the acidic and neutral pDs; remember that the binding of achatin-I was significant at these pDs. Since the 1H-1H coupling constants can be sensitively detected in the order of 0.01 Hz (Kimura et al., 2002, 2004), many of the changes are of one order of magnitude larger than the resolution limit. As there is nearly no bound fraction at the basic pD, on the other hand, the J values are almost identical irrespective of the presence of LUV.
TABLE 1.
Vicinal (Jαβ1 and Jαβ2) and geminal (Jβ1β2) 1H-1H coupling constants (Hz) among α- and β-protons of the d-Phe and Asp residues in 2 mM achatin-I in bulk water (observed), in the mixture with 20 mM EPC LUV (observed), and within EPC LUV (estimated from the observed J values and the bound fractions according to Eq. 2 in the text)
| In bulk water (observed)
|
In the mixture with EPC LUV (observed)
|
Within EPC LUV (estimated)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| pD | Ionization state | J | d-Phe | Asp | d-Phe | Asp | d-Phe | Asp | |
| Acidic | (α-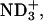 α- α-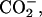 β-CO2D) β-CO2D) |
Jαβ1 | 8.49 ± 0.03 | 7.03 ± 0.00 | 8.51 ± 0.00 | 6.86 ± 0.05 | 8.68 ± 0.27 | 5.38 ± 0.49 | |
| Jαβ2 | 7.39 ± 0.04 | 5.13 ± 0.01 | 6.90 ± 0.01 | 5.02 ± 0.01 | 2.63 ± 0.45 | 4.06 ± 0.09 | |||
| Jβ1β2 | 13.50 ± 0.02 | 16.81 ± 0.01 | 14.95 ± 0.05 | 16.40 ± 0.01 | 27.58 ± 0.66 | 12.83 ± 0.10 | |||
| Neutral | (α-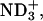 α- α-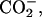 β- β-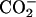 ) ) |
Jαβ1 | 9.26 ± 0.03* | 9.87 ± 0.06* | 9.10 ± 0.00 | 9.44 ± 0.00 | 8.25 ± 0.11 | 7.17 ± 0.32 | |
| Jαβ2 | 6.94 ± 0.08* | 3.73 ± 0.04* | 6.99 ± 0.02 | 3.89 ± 0.02 | 7.25 ± 0.54 | 4.74 ± 0.08 | |||
| Jβ1β2 | 13.20 ± 0.03* | 15.88 ± 0.01* | 13.43 ± 0.03 | 15.81 ± 0.01 | 14.65 ± 0.25 | 15.44 ± 0.12 | |||
| Basic | (α-ND2, α-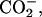 β- β-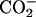 ) ) |
Jαβ1 | 7.89 ± 0.05 | 9.63 ± 0.01 | 7.92 ± 0.01 | 9.64 ± 0.01 | †— | †— | |
| Jαβ2 | 7.56 ± 0.04 | 3.95 ± 0.01 | 7.52 ± 0.03 | 3.93 ± 0.01 | †— | †— | |||
| Jβ1β2 | 13.59 ± 0.02 | 15.65 ± 0.01 | 13.59 ± 0.02 | 15.64 ± 0.01 | †— | †— | |||
Values taken from the results in Kimura et al. (2004).
Bound fraction at this pD was negligible as shown in Fig. 3.
Conformational equilibria
Before going to the side-chain conformational equilibria within the bilayer, we first see those in bulk water. Based on the observed vicinal Jαβ1 and Jαβ2 in Table 1, conformer probabilities (P) on the Cα-Cβ bond were determined by solving Eqs. 3. The upper traces in Fig. 8 show the obtained conformer distributions for the free peptide at the acidic, neutral, and basic pDs. The most notable trend is that at any ionization state, the β-functional groups of both d-Phe and Asp are most probable at the trans conformation to the α-CO group in the backbone (conformer T) (Kimura et al., 2004).
FIGURE 8.

Probabilities P of the side-chain conformer in Fig. 2 of d-Phe and Asp residues in achatin-I in the free state (upper traces) and in the membrane-bound state (lower traces). The ionization states of the amino and carboxyl groups in achatin-I at each pD are shown in the parentheses.
We estimated from Eq. 2 the J values of achatin-I in the membrane-bound state, and the results are summarized in Table 1. Based on the estimated vicinal Jαβ1 and Jαβ2, the side-chain conformer distributions were determined as shown by the lower traces in Fig. 8. At the neutral pD, where both the amino and carboxyl groups in achatin-I are ionized, the population of trans conformer T decreases and those of gauche conformers G+ and G− increase in response to the binding to the bilayers. This trend is common to d-Phe and Asp. At the acidic pD, where the β-carboxyl group in the Asp side chain is un-ionized, the conformational changes due to the binding are drastic for d-Phe between G+ and G−, whereas the changes for Asp are smaller. The distributed conformer populations in the bound achatin-I at the neutral pD indicate a loose packing around the peptide with large conformational fluctuations in the side chains; the steric effect due to the presence of the PC molecules is not so significant as to constrain the side chains to take a specific conformer within the bilayer. Such a soft binding is advantageous to the catalytic activity of membrane to accommodate the peptide conformation required for the receptor binding (Gysin and Schwyzer, 1983; Behnam and Deber, 1984; Deber and Behnam, 1985; Sargent and Schwyzer, 1986).
To comprehend the membrane-binding effect on the conformational equilibria, it is insightful to compare the changes (ΔP) in the conformer probabilities with those due to the transfer from water to an alcohol solvent with hydrophobic moiety. The 1H NMR spectrum of 1 mM achatin-I monoammonium salt dissolved in methanol-d4 is shown in Fig. 7 b. The α-proton chemical shift δ of the C-terminal Asp is in the downfield relative to that of Ala by 0.054 ppm. This observation corresponds to the ionization state of Asp in aqueous solution at neutral pH, where the two carboxyl groups are ionized; the de-ionization of these groups leads the α-proton δ of Asp to ∼0.6 ppm downfield relative to that of Ala (Kimura et al., 2004). The ionic counterparts, the N-terminal amino group and free ammonia, are hence positively ionized. The presence of 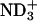 at the N-terminus is further confirmed by the α-proton δ of the N-terminal Gly, which is in the downfield relative to the β-proton δ of d-Phe by ∼0.6 ppm; the de-ionization of the N-terminal
at the N-terminus is further confirmed by the α-proton δ of the N-terminal Gly, which is in the downfield relative to the β-proton δ of d-Phe by ∼0.6 ppm; the de-ionization of the N-terminal 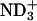 changes the Gly δ to ∼0.2 ppm downfield of the β-proton δ of d-Phe . From the ABX-type fine structures observed for d-Phe and Asp, we determined the couplings among the α- and β-protons (see Supplementary Material for the J values).
changes the Gly δ to ∼0.2 ppm downfield of the β-proton δ of d-Phe . From the ABX-type fine structures observed for d-Phe and Asp, we determined the couplings among the α- and β-protons (see Supplementary Material for the J values).
The changes ΔP in the conformer probabilities in d-Phe and Asp induced by the membrane binding at the neutral pD are illustrated in Fig. 9 together with the ΔP values induced by the transfer from water to methanol. We can compare the two systems to analyze the conformational equilibria at the identical ionization states described above. The changes ΔP upon membrane-binding show a similar trend to those upon transfer to methanol. A decrease in trans conformer T and increase in gauche conformers G+ and G− were observed in both d-Phe and Asp. The similarity suggests that the side-chain conformational changes upon membrane-binding are attributed to the hydrophobic interactions with lipid molecules. The decrease of conformer T, where the β-functional group is trans to the bulky α-carbonyl group, is reasonable in view of the expected decrease of the solvent accessible surface area.
FIGURE 9.

Changes ΔP in the probabilities of the side-chain conformers of d-Phe and Asp in achatin-I upon membrane-binding at the neutral pD (left) and upon transfer to methanol (right). The ionization states of achatin-I are shown in the parentheses.
The changes ΔP due to the membrane-binding and the transfer from water to methanol are the same for the d-Phe with the differences of <0.01. For the Asp, on the other hand, the changes accompanying the binding are smaller than those accompanying the transfer by up to 0.15. These observations are in agreement with the binding location, i.e., the aromatic ring of d-Phe locates at the ester group near the acyl chain, whereas the ionic 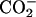 groups of Asp locate around the choline
groups of Asp locate around the choline 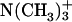 group near the edge of the membrane environment. Note that water is more abundant around the ionic headgroup than around the ester group (Wiener and White, 1992).
group near the edge of the membrane environment. Note that water is more abundant around the ionic headgroup than around the ester group (Wiener and White, 1992).
CONCLUSIONS
We studied the molecular mechanism of the binding of neuropeptide achatin-I (Gly-d-Phe-Ala-Asp) to EPC LUV used as a zwitterionic PC bilayer. The binding model in Fig. 10 is constructed on the basis of the major characteristics of the chemical shifts, line widths, and spin-spin couplings in the 13C and 1H NMR data. Achatin-I binds in the polar region between the glycerol and ester groups of PC. The binding equilibrium is markedly controlled by the in-membrane electrostatic attraction between the N-terminal 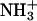 in achatin-I and
in achatin-I and 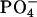 in the headgroup of PC. By taking the binding location into account, it is considered that when the N-terminal
in the headgroup of PC. By taking the binding location into account, it is considered that when the N-terminal 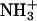 group locates close to the
group locates close to the 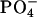 of a given EPC molecule, the C-terminal
of a given EPC molecule, the C-terminal 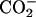 groups are attracted more effectively by the
groups are attracted more effectively by the 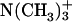 of the other EPC molecule(s). Upon binding to the bilayer, the change in the molecular environment around the peptide is drastic for the ionic Asp, which has the strong electrostatic hydration. It should be emphasized that the most water-exposed Asp residue in the membrane-bound achatin-I encounters the most notable effect of dehydration. Membrane-binding induces the changes in the side-chain conformational equilibria of both the aromatic d-Phe and ionic Asp residues. The conformational changes are not controlled by the steric effect due to the packing of the peptide within the bilayer, but reflect the hydrophobic interactions with lipids. Thus, achatin-I bound near the membrane surface involves large conformational fluctuations in the side chains.
of the other EPC molecule(s). Upon binding to the bilayer, the change in the molecular environment around the peptide is drastic for the ionic Asp, which has the strong electrostatic hydration. It should be emphasized that the most water-exposed Asp residue in the membrane-bound achatin-I encounters the most notable effect of dehydration. Membrane-binding induces the changes in the side-chain conformational equilibria of both the aromatic d-Phe and ionic Asp residues. The conformational changes are not controlled by the steric effect due to the packing of the peptide within the bilayer, but reflect the hydrophobic interactions with lipids. Thus, achatin-I bound near the membrane surface involves large conformational fluctuations in the side chains.
FIGURE 10.

Molecular model for the binding of achatin-I to the surface of zwitterionic PC bilayer on the basis of the 13C and 1H NMR experiments. The colors represent the atomic element as follows: light blue (H), gray (C), blue (N), red (O), and yellowish green (P).
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
T.K. is grateful for the fellowship from the 21st Century Center of Excellence Program and Creative Scientific Research. We thank Professor Yukio Sugiura of our institute for use of the 600 MHz NMR spectrometer, and Dr. Takashi Aoyama and Professor Atsuhiro Oka of our institute for use of the ultracentrifugation apparatus and advice on the operation.
This work is supported by the Grant-in-Aid for Scientific Research (Nos. 14540531, 15205004, and 15076205) from the Japan Society for the Promotion of Science, the Grant-in-Aid for Creative Scientific Research (No. 13NP0201) from the Ministry of Education, Culture, Sports, Science, and Technology, and by the CREST (Core Research for Evolutional Science and Technology) of Japan Science and Technology Corporation (J.S.T.).
References
- Behnam, B. A., and C. M. Deber. 1984. Evidence for a folded conformation of methionine- and leucine-enkephalin in a membrane environment. J. Biol. Chem. 259:14935–14940. [PubMed] [Google Scholar]
- Dawson, R. M. C., D. C. Elliott, W. H. Elliott, and K. M. Jones. 1986. Data for Biochemical Research, 3rd ed. Oxford Science Publications, Oxford, UK.
- Deber, C. M., and B. A. Behnam. 1984. Role of membrane lipids in peptide hormone function: binding of enkephalins to micelles. Proc. Natl. Acad. Sci. USA. 81:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deber, C. M., and B. A. Behnam. 1985. Transfer of peptide hormones from aqueous to membrane phases. Biopolymers. 24:105–116. [Google Scholar]
- Emaduddin, M., G. J. Liu, H. Takeuchi, and E. Munekata. 1996. Multiple intracellular signal transduction pathways mediating inward current produced by the neuropeptide, achatin-I. Eur. J. Pharmacol. 302:129–139. [DOI] [PubMed] [Google Scholar]
- Fujimoto, K., I. Kubota, Y. Yasuda-Kamatani, H. Minakata, K. Nomoto, M. Yoshida, A. Harada, Y. Muneoka, and M. Kobayashi. 1991. Purification of achatin-I from the atria of the African giant snail, Achatina fulica, and its possible function. Biochem. Biophys. Res. Commun. 177:847–853. [DOI] [PubMed] [Google Scholar]
- Glasoe, P. K., and F. A. Long. 1960. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64:188–190. [Google Scholar]
- Gysin, B., and R. Schwyzer. 1983. Head group and structure specific interactions of enkephalins and dynorphin with liposomes: investigation by hydrophobic photolabeling. Arch. Biochem. Biophys. 225:467–474. [DOI] [PubMed] [Google Scholar]
- Howarth, O. W., and D. M. Lilley. 1978. Carbon-13-NMR of peptides and proteins. In Progress in Nuclear Magnetic Resonance Spectroscopy. J. W. Emsley, J. Feeney, and L. H. Sutcliffe, editors. Pergamon Press, Oxford, UK. 1–40.
- Hughes, J., T. W. Smith, H. W. Kosterlitz, L. A. Fothergill, B. A. Morgan, and H. R. Morris. 1975. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 258:577–579. [DOI] [PubMed] [Google Scholar]
- Iwakoshi, E., M. Hisada, and H. Minakata. 2000. Cardioactive peptides isolated from the brain of a Japanese octopus, Octopus minor. Peptides. 21:623–630. [DOI] [PubMed] [Google Scholar]
- Jarrell, H. C., R. Deslauriers, W. H. McGregor, and I. C. P. Smith. 1980. Interaction of opioid peptides with model membranes. A carbon-13 nuclear magnetic study of enkephalin binding to phosphatidylserine. Biochemistry. 19:385–390. [DOI] [PubMed] [Google Scholar]
- Johnson, C. E., Jr., and F. A. Bovey. 1958. Calculation of nuclear magnetic resonance spectra of aromatic hydrocarbons. J. Chem. Phys. 29:1012–1014. [Google Scholar]
- Kainosho, M., and K. Ajisaka. 1975. Conformational analysis of amino acids and peptides using specific isotope substitution. II. Conformation of serine, tyrosine, phenylalanine, aspartic acid, asparagine, and aspartic acid β-methyl ester in various ionization states. J. Am. Chem. Soc. 97:5630–5631. [DOI] [PubMed] [Google Scholar]
- Kamatani, Y., H. Minakata, P. T. M. Kenny, T. Iwashita, K. Watanabe, K. Funase, X. P. Sun, A. Yongsiri, K. H. Kim, P. Novales-Li, E. T. Novales, C. G. Kanapi, H. Takeuchi, and K. Nomoto. 1989. Achatin-I, an endogenous neuroexcitatory tetrapeptide from Achatina fulica Férussac containing a D-amino acid residue. Biochem. Biophys. Res. Commun. 160:1015–1020. [DOI] [PubMed] [Google Scholar]
- Karplus, M. 1959. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 30:11–15. [Google Scholar]
- Karplus, M. 1963. Vicinal proton coupling in nuclear magnetic resonance. J. Am. Chem. Soc. 85:2870–2871. [Google Scholar]
- Kim, K. H., H. Takeuchi, Y. Kamatani, H. Minakata, and K. Nomoto. 1991. Structure-activity relationship studies on the endogenous neuroactive tetrapeptide achatin-I on giant-neurons of Achatina fulica Fërussac. Life Sci. 48:91–96. [DOI] [PubMed] [Google Scholar]
- Kimura, T., N. Matubayasi, and M. Nakahara. 2004. Side-chain conformational thermodynamics of aspartic acid residue in the peptides and achatin-I in aqueous solution. Biophys. J. 86:1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, T., N. Matubayasi, H. Sato, F. Hirata, and M. Nakahara. 2002. Enthalpy and entropy decomposition of free-energy changes for side-chain conformations of aspartic acid and asparagine in acidic, neutral, and basic aqueous solutions. J. Phys. Chem. B. 106:12336–12343. [Google Scholar]
- Kobayashi, J., U. Nagai, T. Higashijima, and T. Miyazawa. 1979. Nuclear magnetic resonance study of side-chain conformation of phenylalanine residue in [Met5]-enkephalin: solvent, pH, and temperature dependence. Biochim. Biophys. Acta. 577:195–206. [DOI] [PubMed] [Google Scholar]
- Lee, K.-C., and T. A. Cross. 1994. Side-chain structure and dynamics at the lipid-protein interface: Val-1 of the gramicidin A channel. Biophys. J. 66:1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K.-L., C. A. Tihal, M. Guo, and R. E. Stark. 1993. Multinuclear and magic-angle spinning NMR investigations of molecular organization in phospholipid-triglyceride aqueous dispersions. Biochemistry. 32:9926–9935. [DOI] [PubMed] [Google Scholar]
- Liu, G. J., and H. Takeuchi. 1993. Modulation of neuropeptide effects by achatin-I, an Achatina endogenous tetrapeptide. Eur. J. Pharmacol. 240:139–145. [DOI] [PubMed] [Google Scholar]
- Milon, A., T. Miyazawa, and T. Higashijima. 1990. Transferred nuclear Overhauser effect analyses of membrane-bound enkephalin analogues by 1H nuclear magnetic resonance: correlation between activities and membrane-bound conformations. Biochemistry. 29:65–75. [DOI] [PubMed] [Google Scholar]
- Moncelli, M. R., L. Becucci, and R. Guidelli. 1994. The intrinsic pKa values for phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine in monolayers deposited on mercury electrodes. Biophys. J. 66:1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura, E., T. Kimura, M. Nakahara, M. Tanaka, T. Handa, and H. Saito. 2001. 13C NMR method for the determination of peptide and protein binding sites in lipid bilayers and emulsions. J. Phys. Chem. B. 105:12616–12621. [Google Scholar]
- Okamura, E., and M. Nakahara. 2001. NMR studies on lipid bilayer interfaces coupled with anesthetics and endocrine disrupters. In Lipid Interfaces in Chemical, Biological, and Pharmaceutical Applications. A. G. Volkov, editor. Marcel Dekker, New York. 775–805.
- Pachler, K. G. R. 1964. Nuclear magnetic resonance study of some α-amino acids. II. Rotational isomerism. Spectrochim. Acta. 20:581–587. [Google Scholar]
- Pople, J. A., W. G. Schneider, and H. J. Bernstein. 1959. High-Resolution Nuclear Magnetic Resonance. McGraw-Hill, New York.
- Poteryaev, D. A., I. S. Zakharov, P. M. Balaban, and A. V. Belyavsky. 1998. A novel neuropeptide precursor gene is expressed in the terrestrial snail central nervous system by a group of neurons that control mating behavior. J. Neurobiol. 35:183–197. [PubMed] [Google Scholar]
- Sargent, D. F., and R. Schwyzer. 1986. Membrane lipid phase as catalyst for peptide-receptor interactions. Proc. Natl. Acad. Sci. USA. 83:5774–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake, H., Y. Yasuda-Kamatani, K. Takuwa, K. Nomoto, H. Minakata, T. Nagahama, K. Nakabayashi, and O. Matsushima. 1999. Characterization of a cDNA encoding a precursor polypeptide of a D-amino acid-containing peptide, achatin-I and localized expression of the achatin-I and fulicin genes. Eur. J. Biochem. 261:130–136. [DOI] [PubMed] [Google Scholar]
- Seelig, J. 1978. 31P Nuclear magnetic resonance and the head group structure of phospholipids in membranes. Biochim. Biophys. Acta. 515:105–140. [DOI] [PubMed] [Google Scholar]
- Simon, S. A., and T. J. McIntosh. 2002. Peptide-Lipid Interactions. Academic Press, San Diego, CA.
- Szoka, F., Jr., and D. Papahadjopoulos. 1980. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 9:467–508. [DOI] [PubMed] [Google Scholar]
- Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data: III. Complete structure. Biophys. J. 61:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich, K. 1976. NMR in Biological Research: Peptides and Proteins. North-Holland Publishing Company, Amsterdam, The Netherlands.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.