

Abstract
The structures of two response regulators (RRs) from the cyanobacterium Calothrix PCC7601, RcpA and RcpB, were solved to 1.9- and 1.75-Å resolution, respectively. RcpA was found in phosphorylated and RcpB in nonphosphorylated form. Both RRs are members of phytochrome-associated, light-sensing two-component signal transduction pathways, based on histidine kinase-mediated receptor autophosphorylation and phosphorelay to a RR. Despite the overall folding similarity to CheY-type RRs ((β/α)5-motif), RcpA and RcpB form homodimers, irrespective of their phosphorylation state, giving insight into a signal transduction putatively different from that of other known RRs. Dimerization is accomplished by a C-terminal extension of the RR polypeptide chain, and the surface formed by H4, β5, and H5, which constitute a hydrophobic contact area with distinct interactions between residues of either subunit. Sequence alignments reveal that the identified dimerization motif is archetypal for phytochrome-associated RRs, making them a novel subgroup of CheY-type RRs. The protein structures of RcpA and RcpB are compared to the recently presented protein structure of Rcp1 from Synechocystis.
INTRODUCTION
Many bacterial signal transduction systems are based on a central phosphoryl-transfer mechanism involving a minimum of two essential components, a receptor histidine kinase and a response regulator (RR) protein. The fact that also the sensing of light in bacteria employs such a two-component system has only recently been shown through the identification of a now rapidly growing family of photoreceptors in photosynthetic (Hughes et al., 1997; Jiang et al., 1999; Herdman et al., 2000) and nonphotosynthetic (Davis et al., 1999) prokaryotes with structural and functional similarity to the phytochrome photoreceptors of plants. A similar system, involving kinase activity and response regulators of CheY-type, has been identified in the sensory rhodopsins of halobacteria (Hoff et al., 1997). However, the halobacterial light sensor lacks the kinase activity, but instead activates a halobacterial transducer (HTr) that carries a histidine kinase domain via direct protein-protein interactions. HTr then activates CheY-like response regulators with various output functions. The bacterial phytochromes interact directly with response regulators of the well characterized CheY type from the bacterial two-component system, as was shown in in vitro experiments (Yeh et al., 1997; Hübschmann et al., 2001b). This finding, for the first time, links directly the sensing of light to a well characterized signal transduction mechanism. In the cyanobacterium Calothrix PCC7601 we identified two genes encoding for photoreceptors (CphA and CphB) with the phytochrome-specific binding motif in their N-terminal part, and carrying a histidine kinase domain in their C-terminal half. Each of the two genes is followed by an ORF encoding a response regulator, rcpA and rcpB (Jorissen et al., 2002), respectively. Both chromoproteins show red/far red light-dependent photochemistry, undergo light-dependent autophosphorylation, and perform transphosphorylation in an absolutely selective manner upon incubation with their cognate response regulators (RcpA and RcpB) (Hübschmann et al., 2001b). Although the general features of the two-component signal transduction have been identified (e.g., by mutagenizing the key amino acids in the closely related cyanobacterium Synechocystis PCC6803 (Yeh et al., 1997)), the phosphorylated form of cyanobacterial RRs is remarkably stable, in contrast to many other CheY-like proteins. This property is independent of the process of phosphorylation, accomplished either via the cognate photoreceptor or upon direct treatment with small phosphate donors, e.g., acetylphosphate (Hübschmann et al., 2001b). Although cyanobacterial photoreceptors can be homologously expressed and studied (Hübschmann et al., 2001a; B. Quest, N., Tandeau de Marsdac, and W. Gaertner, unpublished), physiological (i.e., in vivo) investigation of the signal transduction (e.g., protein-protein interactions) appears remarkably difficult due to the low concentrations of these molecules per cell (between 10 and 100; T. Lamparter, University of Berlin, personal communication). Analysis of the RRs and of their cognate photoreceptors has revealed not only an extended stability of the phosphorylated forms in contrast to other RRs (Hübschmann et al., 2001b), but also has demonstrated the presence of remarkably stable homodimers (studies on the dynamic behavior of RR-homodimers, including the determination of binding constants, are currently under investigation).
We here present the three-dimensional structures of both response regulators from Calothrix with resolution of 1.9 Å and 1.75 Å, respectively, one (RcpA) in activated, phosphorylated and the other (RcpB) in unphosphorylated form. These two proteins are the first members of the photoreceptor-linked subgroup of CheY proteins that exhibit a specific homodimerization, which is not found in their up-to-now structurally determined eubacterial homologs. We show that contact regions in the formed homodimers of these phytochrome-related RRs are formed by sequence motifs not present in other types of response regulators.
The structure of RcpB (Fig. 1 a, Table 1) has been solved by multiple anomalous wavelength dispersion and that of RcpA by molecular replacement methods using RcpB (53% homology) as a search model. Interestingly, although both response regulators were cloned and expressed in E. coli following the same protocol, RcpA crystallized in phosphorylated and RcpB in unphosphorylated form. A partial phosphorylation of these proteins by the expression host has already been observed during recent phosphorylation studies (Hübschmann et al., 2001b). The high degree of similarity between both proteins thus allows the comparison of both states—active and inactive—of this type of two-component response regulator. Also the recently presented crystal structure of a homologous response regulator from another cyanobacterium, Rcp1 from Synechocystis PCC6803 (Im et al., 2002), reveals the strong sequential and structural homology to the here-presented RRs from Calothrix and indicates the presence of a new subgroup of CheY-type RRs present in cyanobacteria. RcpA and RcpB monomers share the same (β/α)5 architecture with other structurally characterized members of the RR family, e.g, CheY (Stock et al., 1989), as can be deduced from sequence alignments (Figs. 1 a and 2), but they possess extended loop regions and a prolonged C-terminus. The alternating segments of β-strands and α-helices fold into a five-stranded, parallel, β-pleated sheet surrounded by five α-helices.
FIGURE 1.

Overall structure of phospho-RcpA. (a) Overlaid structures of RcpA (dark gray), RcpB (gray), and CheY (light gray) illustrating the overall structural homology and the differences in (1) loop H1/β2, (2) loop β5/H5, and (3) loop H2/β3. In the structure of CheY, H5 is tilted with respect to H5 in RcpA and B. The extended C-termini of the cyanobacterial RRs (4), not present in CheY, participate in the dimer interaction. (b and c) For simplicity, the protein backbones are depicted as ribbons. The illustration shows dimeric phospho-RcpA as viewed from the (b) side and (c) top with the noncrystallographic twofold rotational axis. The active site in each subunit is drawn as ball and stick. The protein N- and C-termini are labeled. Phospho-RcpA crystallized in space group P21212 and diffracted to 1.9 Å. The final model comprises residues 10–148; residues 1–9 and 149 are not visible in the electron density, presumably due to disorder. Apo-RcpB (not shown) formed crystals in space group P41212 which diffracted to 1.6 Å (SeMet) and 1.75 Å (wild-type). The phase information obtained from the SeMet derivative was used to solve and refine the structure of wild-type apo-RcpB to 1.75 Å.
TABLE 1.
Data collection and refinement statistics
| Crystal | RcpA | RcpB |
|---|---|---|
| Space group | P21212 | P41212 |
| Cell dimensions (a, b, c (Å)) | 44.1, 76.7, 85.2 | 72.2, 72.2, 142.8 |
| Resolution limit (Å) | 25.0-1.90 | 20.0-1.75 |
| Wavelength (Å) | 1.050 | 1.009 |
| Measured reflections (total/unique) | 301367/22731 | 260719/39024 |
| Completeness (overall; last shell; %) | 96.9/94.6 | 98.2/98.8 |
| Rmerge* (overall; last shell; %) | 3.7/30.2 | 3.9/12.2 |
| I/σI (overall; last shell; %) | 25.2/3.1 | 27.4/12.2 |
| B-factor (Wilson/overall mean, (Å2))† | 26.4/38.2 | 22.2/26.0 |
| Final R-factor/Rfree (%) | 23.3/27.7 | 19.9/24.3 |
| RMS deviation from ideal geometry | 0.005 | 0.005 |
| Bond (Å) | 1.3 | 1.3 |
| Angles (deg) | 22.7 | 22.9 |
| Dihedrals (deg) |
The resolution range in the highest bin was 1.78–1.75 Å for native RcpB and 1.93–1.90 for RcpA.
The B-factor was estimated from a Wilson plot; for the native datasets over the resolution range 3.9–1.75 Å (RcpB) and 3.22–1.90 Å (RcpA).
FIGURE 2.

Homology alignment of RRs of the CheY type. Conserved active site residues are marked with arrowheads. Asterisks indicate conserved residues involved in dimer formation in RcpA and B. Rcp1 (Synechocystis sp. SLR0474), RcpA and B (Calothrix sp.), and RRs from Anabaena sp. (ALR3158, ALL2898) and D. radiodurans (DRA0049, DRA0204, DR2420) are highly homologous and are also cotranscribed with putative phytochrome homologous receptor histidine kinases. Several other RRs from M. thermoautotrophicum (MTH445), Streptomyces coelicolor (CAB62661) or A. fulgidus, AF1473) were found to also share the dimerization motifs, whereas none of the structurally characterized CheY-homologs such as Spo0A (B. stearothermophilus), Spo0F (Bacillus subtilis), Etr1 (Arabidopsis thaliana), Narl (E. coli), FixJ (S. meloliti), or CheY (E. coli) itself show these conserved residues.
Both proteins carry the conserved phosphate-binding aspartate and the binding-supporting aspartate, glutamate, and lysine residues forming a conserved surface depression that is similar to the active site of phosphorylation in CheY and homologs.
MATERIALS AND METHODS
Protein expression and purification
The coding sequences of the response regulators RcpA (GenBank accession number AF309559) and RcpB (AF309560) from Calothrix sp. PCC7601 were amplified by polymerase chain reaction from genomic DNA and subcloned into the vectors RcpB: pET28(a) (Novagen, Madison, WI) between the NdeI and XhoI restriction sites and RcpA: pMEX8 (Medag, Hamburg) between EcoRI and SalI (Jorissen et al., 2002), placing a HIS6 tag at the amino (pET28(a) or carboxy terminus (pMEX8) of the proteins. The plasmids were transformed into the E. coli strain Bl21(DE3) Gold (Novagen) by electroporation of competent cells and transformed cells were selected on Luria-Bertani agar plates containing 50 μg/ml Kanamycin. Expression was performed in Terrific Broth medium within 6 h at 30°C and with vigorous shaking. For the expression of selenomethionine (SeMet) containing RcpB the E. coli methionine-auxotroph strain B834(DE3) (Novagen) was used. After induction with 0.4 mM isopropylthio-β-galactoside, expression was allowed for 16 h at 30°C in a minimal medium containing 30 mg/l L-selenomethionine (Sigma, St. Louis, MO). In both cases, ∼20–30 mg of recombinant protein was obtained from a 1-L culture. All purification steps were performed below 4–6°C. RcpA was purified in two steps: upon sonication of the cells in a buffer containing 300 mM NaCl, 100 mM Tris-HCl (pH 8.0), 5% (v/v) glycerol, and 10 mM 2-mercaptoethanol, the lysate was clarified by ultracentrifugation (150,000 × g, 40 min, 4°C), and applied to Nickel-NTA resin (Qiagen, Venlo, The Netherlands). Eluent fractions were pooled and purified to homogeneity by gel filtration (150 mM NaCl, 10 mM Tris-HCl, (pH 8.0)) on a Superdex 75 column (Pharmacia, Freiburg, Germany). RcpB and SeMet-RcpB were additionally incubated with thrombin (Pharmacia; overnight, 18°C) and again purified by Nickel-NTA followed by gel filtration. Proteins were concentrated to 2–5 mg/ml by centrifugation with an Amicon centriprep 10 concentrator (Houston, TX) in 150 mM NaCl, 10 mM Tris-HCl (pH 8.0). To protect SeMet-RcpB from oxidation, all aqueous solutions were degassed and contained up to 10 mM 2-mercaptoethanol. Quality and purity of the final samples were monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrospray ionization mass spectrometry (ESI MS). The incorporation of three SeMet residues per RcpB monomer was verified by ESI MS.
Crystallization of RcpA
Crystals were obtained at room temperature with the hanging drop vapor diffusion method using a well solution that contained 1.4 M sodium citrate, 150 mM sodium chloride, 10% (v/v) glycerol, and 100 mM HEPES (pH 7.0). The crystallization droplets contained the RcpA solution described above mixed with an equal volume of the well solution. Crystals appeared after 2 weeks and grew to a size of ∼100 × 100 × 30 microns. RcpA crystallized in space group P21212 with unit-cell dimensions a = 44.1 Å, b = 76.7 Å, and c = 85.2 Å and two molecules in the asymmetric unit (AU) with a solvent content of 50%.
Crystallization of RcpB and SeMet-RcpB
Crystals were grown at 4°C in hanging drops by vapor diffusion against a reservoir of 3.75 M sodium formate, 5% (v/v) glycerol, and 1 mM 2-mercaptoethanol. The droplets contained equal volumes of reservoir solution and protein solution containing 3–5 mg/mL RcpB in 150 mM sodium chloride, 10 mM Tris-HCl (8.0), 5% (v/v) glycerol, and 1 mM 2-mercaptoethanol. Crystals appeared within several days and reached a final size of ∼200 microns in each dimension after 1 week. Both RcpB and SeMet-RcpB formed crystals belonging to the space group P41212 with the unit-cell dimensions a = b = 72.2 Å, c = 142.8 Å and two molecules per AU (solvent content 50%).
Data collection, structure determination, and fefinement
RcpA and RcpB native data were collected to 1.9 Å and 1.75 Å, respectively, at 100 K using a MAR CCD detector (MAR Research, Hamburg, Germany) at the Max-Planck Beamline BW6 (DESY, Hamburg, Germany). A three-wavelength, 1.6 Å multiwavelength anomalous dispersion (MAD) data set of the SeMet-RcpB derivative was collected from a single cryocooled crystal at beamline BW6. Data were processed using DENZO and SCALEPACK (Otwinowski and Minor, 1997).
RcpB
By treating MAD as a special case of MIR, five of the six selenium sites per AU were identified from anomalous-difference Patterson maps of the SeMet-modified crystals calculated with solvent-flattened experimental phases. After phasing with MLPHARE and solvent correction by dodecyl maltoside (1994) a partial model was automatically built into the resulting electron density map using the program wARP (Perrakis et al., 1999). Manual model building in O (Jones et al., 1991) and initial refinement in CNS (Crystallography & NMR system, Brünger et al., 1998) gave a preliminary R-factor of 30.0% and an Rfree of 31.4%. The obtained phases were then combined with the native-RcpB structure factor amplitudes to calculate an initial electron density map from the native data. The native structure was completed by iterative cycles of rebuilding and refinement against data from 20.0–1.75 Å to a final R-factor and Rfree of 20.0% and 24.1%, respectively. Water molecules were built automatically in CNS as well as manually. Except for loop H2/β3 and loop β5/H5, which needed extensive manual building, the interpretation of the electron density maps was straightforward. The final model comprises all residues 1–149 in molecules A and B, with the initiating Met replaced by Ala.
RcpA
The initial phases for RcpA were determined for the native data set by molecular replacement using the structure of SeMet-RcpB as a search model. A two-molecule solution was found with AMoRe (Navaza, 1994) giving a correlation coefficient of 55% and an R-factor of 33%. 2Fobs – Fcalc and Fobs – Fcalc maps were calculated and successive cycles of model building and refinement were performed with 5% of the reflections set aside for the calculation of the free R-factor (Brünger, 1992, 1993). To minimize model bias, composite omit maps were calculated and used for critical inspection of the improving model. The calculated 2Fobs – Fcalc and Fobs – Fcalc maps clearly indicated the presence of additional atoms at residue Asp-70 corresponding to a covalently attached phosphoryl group. It was built by exchanging Asp-70 with a phosphorylated aspartate residue using the respective files from the HIC-Up database (Kleywegt and Jones, 1998). Automated water building, further cycles of rebuilding, and positional refinement to convergence resulted in an R-factor of 23.3% and an Rfree of 27.7%. Except for the terminal residues 1–9, 149, the C-terminal HIS6-tag, and the solvent-exposed loop H1/β2 and loop H2/β3, all residues were clearly visible in the electron density. The final RcpA model covers the residues 10–148.
The crystal structures were deposited in the Research Collaboratory for Structural Bioinformatics data bank, Protein Data Bank (PDB) ID: 1K68 (RcpA) and 1K66 (RcpB).
RESULTS
The crystal structures of two response regulators, RcpA and RcpB, have been solved by multiple anomalous wavelength dispersion (RcpB) and molecular replacement methods (RcpA). The heterologously expressed proteins were purified and subjected to ESI MS to control quality and the extent of selenomethionine incorporation. The average molecular masses were 17125.0/17277.8 amu (wild-type RcpB), 17266.8/17345.2 amu (SeMet-RcpB), and 17634.8/17714.0 amu (RcpA-HIS6), with each sample giving two major mass peaks with a difference of ∼79 amu, corresponding to a covalently attached phosphoryl group 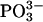 (Fig. 3). The mass difference between wild-type and SeMet-RcpB was determined as 141 amu which perfectly reflects the complete SeMet-substitution of all three methionine residues per molecule. The proteins, eluted from the gel filtration on Superdex 75 columns reproducibly appeared as single, symmetric peaks. In all cases, the eluent volume corresponded to apparent molecular masses of twice the mass we expected from the MS experiments, indicating the presence of stable dimeric forms of both RRs (Fig. 3). A matrix-assisted laser-desorption ionization time-of-flight analysis showed also the presence of the homodimer to a significant extent.
(Fig. 3). The mass difference between wild-type and SeMet-RcpB was determined as 141 amu which perfectly reflects the complete SeMet-substitution of all three methionine residues per molecule. The proteins, eluted from the gel filtration on Superdex 75 columns reproducibly appeared as single, symmetric peaks. In all cases, the eluent volume corresponded to apparent molecular masses of twice the mass we expected from the MS experiments, indicating the presence of stable dimeric forms of both RRs (Fig. 3). A matrix-assisted laser-desorption ionization time-of-flight analysis showed also the presence of the homodimer to a significant extent.
FIGURE 3.

Size exclusion chromatogram of heterologously expressed RcpB. The elution volume (HiLoad 26/60 Superdex 75 prep grade column, flow rate 0.53 ml/min, elution buffer (150 mM NaCl, 5% glycerol, 50 mM Tris-HCl, pH 8.0) corresponds to an apparent molecular weight of ∼35 kD (dimeric RcpB; the same was found for RcpA). Inset: the ESI mass spectroscopic analysis of the same sample reveals that the assumed to be homogenous sample was in fact composed of two main species differing in 79 Da (phosphoryl group).
RcpA crystallized as the phosphorylated form (phospho-RcpA) in space group P21212 as a single crystal that diffracted to 1.9 Å. The final model comprises residues 10–148; residues 1–9 and 149 and the C-terminal HIS6-tag are not visible in the electron density, presumably due to disorder. The apo-form of RcpB formed single crystals in space group P41212 that diffracted to 1.6 Å (SeMet) and 1.75 Å (wild-type). The phase information derived from the SeMet derivative was used to solve and refine the structure of wild-type apo-RcpB. In the following, only the native structure is described, although the structure of the SeMet derivative was refined to a higher resolution. Comparison of the two models showed no significant structural deviation due to the presence of selenium atoms in the molecule.
Overall structure of phospho-RcpA and apo-RcpB
Both RRs contained homodimers within the AU, the respective protomers being related by twofold noncrystallographic rotational axes. RcpA and RcpB monomers share the same (β/α)5 architecture with other structurally characterized members of the RR family, e.g., CheY (Stock et al., 1989) and SpoOF (Madhusudan et al., 1997), as predicted from sequence alignments. Basically they comprise alternating segments of β-strands and α-helices forming a five-stranded, parallel, β-pleated sheet surrounded by five α-helices (Fig. 1).
Active site architecture
Like in other CheY homolog structures, the active site of RcpA and B is an acidic pocket formed by a triad of acidic residues (RcpA: Glu-17, Asp-18, Asp-70; RcpB: Glu-13, Asp-14, Asp-69), a serine (RcpA: Ser-100) or threonine (RcpB: Thr-99), and a lysine (RcpA: Lys-122; RcpB: Lys-121), all together being highly conserved within this family of RR proteins (Figs. 2 and 4).
FIGURE 4.

Active site architecture in RcpA and RcpB. The protein's active site atoms are shown as ball and stick models. Atoms are colored according to type: C (gray), O (red), N (pale blue), P (yellow), and water molecules (turquoise). (a) In the RcpA active site the phosphoryl group occupies the central position and interacts with several groups as described. The sodium ion (black) is coordinated in an octahedral manner involving two water molecules in the first coordination sphere. Interestingly, the crystallographic data show unambiguously that Ser-100 of phospho-RcpA exists in a second alternative rotameric conformation. Whether this represents an inside glimpse into the mechanical details underlying activation is not clear. (b) In RcpB the metal binding site is occupied by a water molecule, H-bridged to side-chain carboxyl groups of Asp-14 and Asp-69 and the backbone carbonyl oxygen of Asn-71.
Phospho-RcpA
During refinement, investigation of 2Fobs – Fcalc and Fobs – Fcalc maps of RcpA revealed a prominent tetrahedral electron density fused with the density of the Asp-70 carboxyl group that could not be accounted for by protein amino acid atoms or solvent molecules (compare Fig. 4, a and b). Due to the biochemical function of this aspartate residue as a phosphoryl accepting group and the remarkable stability of the phosphorylated state, shown in former publications (Hübschmann et al., 2001b), it was clear and unambiguous that this conspicuous density represented a phosphoryl group covalently attached to the carboxyl oxygen of Asp-70. We exchanged Asp-70 with the PDB entry for a phosphorylated aspartic amino acid (PHD) from the Hic-Up database (Kleywegt and Jones, 1998) and the phosphoryl group perfectly fitted into the density. After further steps of refinement and additional water building the overall geometry of the active site and the aspartyl phosphate converged. A detailed view of the aspartyl-phosphate structure is given in Fig. 5. The presence of the phosphorylated form of RcpA was already suggested from data derived from ESI MS spectrometry which reproducibly showed two major masses (17634.8/17714.0 amu; inset in Fig. 3). The difference of 79 amu originates from the presence of a specifically attached phosphoryl group. It has been described previously that other RRs were isolated in their phosphorylated form from E. coli lysates, either due to unspecific phosphotransfer activity or due to small molecule phosphodonors like acetyl phosphate, the latter being constitutively present in the host cells (McCleary and Stock, 1994). In vitro phosphorylation by acetyl phosphate and the remarkable stability of the phosphorylated state that even endures the crystallization procedure had recently been demonstrated by us (Hübschmann et al., 2001b). The phosphoryl moiety is involved in a set of interactions with the surrounding protein mainly of hydrogen bonds and salt bridge contacts (Fig. 4 a). These include amide groups of the peptide backbone of Leu-71, Asn-72, and Thr-101, the side chain hydroxyl group of Ser-100, Thr-101, and the amino group of Lys-121. Interestingly, the electron density of Ser-100 indicates the coexistence of two alternative rotamers (104.4° around the Cα-Cβ bond), one pointing toward the phosphate moiety and forming an H-bond, the other pointing away to the solvent. The putative metal binding site in the acidic surface depression was found to be occupied by an octahedrally coordinated ion. Due to the bond lengths between the ion and its ligands and, after all, due to the crystallization conditions which contained no magnesium, we suggest that this ion is a sodium rather than a magnesium ion, although no direct proof can be given from inspection of the electron density. This sodium emulates the binding interactions normally performed by a bivalent Mg2+ ion. Like in the case of Mg2+-CheY (Bellsolell et al., 1994) the first coordination sphere of this sodium ion consists of the carboxylate oxygens of Asp-18 and Asp-70 (PHD70), the backbone carbonyl oxygen of Asn-72, one phosphoryl oxygen, and two additional water molecules.
FIGURE 5.

Rendered representation of the active-site aspartate PHD70 with the enclosing electron density contoured at 1 σ (mesh). Atoms are colored according to type: C (light gray), O (gray), N (dark gray). The extra density encountered in the experimental map and issuing from the carboxyl group of PHD70 perfectly accommodates the covalently bound phosphoryl group as shown. The correctness of the model was verified by calculating an omit map of the respective region (not shown).
Apo-RcpB
The active site in apo-RcpB is populated by several water molecules (Fig. 4 b). One occupies a position comparable to that of the metal ion in the CheY-Mg2+ structure (Stock et al., 1989), whereas others are involved in the formation of hydrogen bonds with active site residues.
Intermolecular interaction in the homodimers
Altogether, we investigated three different crystal forms grown under different conditions: a tetragonal (P41212) and a monoclinic (P21, data not shown) crystal of apo-RcpB, and one orthorhombic (P21212) crystal of RcpA. In each structure, the molecules were associated as distinct dimers in the unit cell with the respective monomers being related by noncrystallographic twofold rotational symmetries. In both RRs, the formation of these homodimeric complexes is mediated by specific interaction of the surface areas spanned by H4, β5, H5, and the C-terminal part of each subunit. As a consequence, the two active sites in a dimer reside on opposing positions (Fig. 1). The total interface areas were estimated by calculating the differences of Connolly surfaces from two isolated monomers and the dimer (InsightII, probe radius 1.4 Å) to be ∼1000 Å2 in RcpA and 1175 Å2 in RcpB or 16% of the total dimer surface areas. Formation and stabilization of the intermolecular interaction is accomplished mainly by the complementary arrangement of a combination of hydrophobic interactions, hydrogen bonds, and salt bridges (Table 2, Fig. 6 a). A remarkably large part of this interface is dominated by a central, water-excluding hydrophobic core that is surrounded by a ring of polar interactions and several bridging water molecules, tying the rim together. The overall design of this interface represents an archetypal protein-protein contact site in homooligomeric complexes as described, for example, in a survey by Larsen et al. (1998). In RcpA and RcpB, this interface consists of a series of polar and nonpolar residues that are conserved in several other RRs (Fig. 2). Interestingly, these motifs are found—up to now—without exception in all RRs associated with phytochrome-homologous sensor histidine kinases and therefore deserve a more detailed characterization. The most prominent of these conserved motifs, located in the center of the hydrophobic core, is formed by aromatic residues (Tyr-138 and Trp-139 in RcpB; Phe-139 and Trp-140 in RcpA) in both subunits that pack against each other to form an arrangement that could be considered as an aromatic cluster (Fig. 6 b), in the case of RcpB additionally stabilized by one H-bond between the tyrosine hydroxyl group and the NH group of the tryptophan. Another conserved motif, located at the rim of the core, comprises two opposing proline residues (Pro-94 and Pro-146 in RcpB; Pro-95 and Pro-147 in RcpA) from β4 and the C-terminal loop of one subunit that pack against the phenyl ring of Tyr-111 (RcpB; Tyr-112 in RcpA) in H4 of the other subunit (Fig. 6 b). The corresponding PRO residues of the second subunit in turn pack against Tyr-111 in the first subunit.
TABLE 2.
List of interacting residues in RcpA and RcpB
| RcpA | Subunit A | Subunit B | RcpB | Subunit A | Subunit B |
|---|---|---|---|---|---|
| Polar interaction | Lys-92 CE | Asp-113 OD2 | Lys-92 NZ | Glu-108 OG | |
| Arg-93 NE | Phe-109 CE1 | Lys-92 CD | Ser-112 OG | ||
| Arg-93 NH1 | Asp-113 OD2 | Glu-108 OE1 | Ala-147 N | ||
| Tyr-112 OH | Thr-145 O | Glu-108 OE2 | Lys-92 NZ | ||
| Aap-113 OD1 | Arg-93 NH1 | Tyr-111 OH | Val-144 O | ||
| Asn-117 O | Asn-117 ND2 | Ser-114 OG | Lys-85 NZ | ||
| Asn-117 ND2 | Asn-117 O | Ser-116 OG | Ser-116 O | ||
| Tyr-119 O | Thr-144 N | Ser-117 OG | Tyr-138 OH | ||
| Phe-139 CZ | Trp-140 NE1 | Tyr-118 O | Val-144 O | ||
| Trp-140 NE1 | Phe-139 CZ | Arg-127 NH2 | Asp-141 O | ||
| Thr-145 N | Tyr-119 O | Arg-127 NH1 | Asp-141 OD1 | ||
| Thr-145 O | Tyr-112 OH | Tyr-138 OH | Ser-117 OG | ||
| Tyr-138 OH | Trp-139 NE | ||||
| Trp-139 NE | Tyr-138 OH | ||||
| Asp-141 OD1 | Arg-127 NH1 | ||||
| Hydrophobic | Phe-139 | Val-96, LE120, Ile-136, Phe-139 | Pro-94 | Tyr-111 | |
| Pro-95 | Tyr-112 | Ile-97 | Tyr-138 | ||
| Pro-147 | Tyr-112 | Ile-107 | Val-144 | ||
| Tyr-112 | Pro-95, Pro-147 | Tyr-111 | Pro-94, Pro-146 | ||
| Tyr-118 | Val-144 | ||||
| Ile-119 | Ile-142, Tyr-138, Val-143 | ||||
| Val-120 | Val-144 | ||||
| Tyr-138 | Tyr-138, Trp-139 | ||||
| Trp-139 | Tyr-138, Trp-139 | ||||
| Ile-142 | Ile-119 | ||||
| Val-143 | |||||
| Val-144 | Val-120, Ile-107, Tyr-118 | ||||
| Pro-146 | Tyr-111 |
FIGURE 6.

Intersubunit interactions in dimeric RcpB. (a) Interface in the RcpB complex. One monomer is represented by its interacting solvent-accessible surface area colored according to the electrostatic surface potentials. The interacting side chains of the second monomer are shown as ball and stick models and colored according to their physicochemical character: nonpolar and aromatic: violet; polar and charged: atom type C (gray), O (red), N (blue). Interface water molecules are shown as turquoise spheres. (b) Close-up view of the complementary packing of the Pro/Pro/Tyr and the Tyr/Trp motifs in dimeric RcpB. One subunit is represented through its solvent-accessible surface with the interacting residues shown as ball and stick (light gray). The interacting residues of the second subunit are shown as ball and stick models, covered by their van der Waals surface (dark gray). The sequence alignments show the conserved residues depicted. In RcpB, the aromatic cluster is additionally stabilized by two H-bonds as described. The YW/YW (FW/FW in RcpA) arrangement constitutes the central hydrophobic core of the dimer interfaces.
In the homodimeric RcpB the hydrophobic interaction additionally comprises two stretches of nonpolar residues in β5 (Tyr-118, Ile-119, Val-120, Pro-122) and the C-terminus (Ile-142, Val-143, Val-144, Leu-145 (Table 2) flanking the aromatic cluster and packing complementarily in the dimer.
In RcpA the hydrophobic character of the C-terminus is not as strongly pronounced as in RcpB (Fig. 2) and some nonpolar interactions are replaced by polar contacts, such as Thr-145, which is H-bridged to Tyr-109 and Tyr-112 (see Table 2).
The remaining nonpolar and polar interactions are composed mainly of the complementary arrangement of residues in each subunit in both RRs and are listed in Table 2. The surface shape complementarity (Lawrence and Colman, 1998) at the interface regions is excellent and its size lies in the range of typical protein-protein interfaces, Sc (RcpB) = 0.71 and Sc (RcpA) = 0.65 for RcpA (calculated with the program SC (Collaborative Computational Project 4, 1994); waters not included). This additionally reflects the strong surface intimacy in the dimers.
Conformational differences between phospho-RcpA and apo-RcpB
Both proteins form the described dimer in the crystalline state. The purified samples always eluted as symmetric singular peaks with apparent molecular masses of 35 kDa from the size exclusion column, suggesting a homogenous molecular size distribution (Fig. 3). The mass spectrometry analysis revealed that the proteins purified from E. coli always were binary mixtures of both the apo- and the phosphorylated form in a more or less constant ratio.
Phospho-RcpA and apo-RcpB have a very similar active site conformation as depicted in Fig. 7 (the backbone atoms of the complete monomers of RcpA and RcpB can be superimposed with a root-mean square deviation of 1.0 Å). Except for the carboxyl group of the phosphoryl-accepting aspartate, which is rotated by almost 90° around the Cβ-Cγ bond in phospho-RcpA, the other residues (RcpA: Glu-17, Asp-18, Ser-100, and Lys-122; RcpB: Glu-13, Asp-14, Thr-99, and Lys-121) show the same rotameric and spatial orientation. The rotation of the carboxylic group of the phosphate-accepting aspartate side chain upon phosphorylation and the presence of the rather bulky phosphate group in RcpA result mainly in a lateral movement of the side chains of Glu-17, Ser-100, and Lys-121, the latter being hydrogen-bonded to a phosphoryl oxygen atom (2.99 Å) and the side-chain oxygen of Glu-17 (2.45 Å). This in turn leads to a slight drift of the associated backbone chain segments, especially the loop connecting β4 and H4 (0.84 Å) and sheet β5 (1.1 Å) compared to RcpB. Asp-18, due to the water-bridged hydrogen bonding to the phosphate group, also moves ∼0.9 Å upward in direction to the phosphate, and thus affects the position of loop β1/H1 and helix H1.
FIGURE 7.

Comparison of the active-site architectures of phospho-RcpA (dark gray), RcpB (gray), and (a) CheY and (b) activated CheY (Gouet et al., 1999) (light gray) with bound (BeF3)− (with tetragonally ligated central sphere). (a) Thr-87 of CheY is in a remote position compared to the (b) active state. In phospho-RcpA and apo-RcpB the respective side chains (Ser-100 and Thr-99) occupy similar positions comparable to the active form of CheY. The active site of apo-RcpB resembles more the active state of CheY than its inactive state. Due to the presence of the phosphoryl group in RcpA, the side chain of Lys-122, Ser-100, and Glu-17 are laterally shifted with respect to their counterparts in RcpB.
Interestingly, the side chain of Ser-100 was found to exist in two alternative conformations in the structure of RcpB, one pointing toward the phosphate group and forming a H-bridge (2.70 Å) to one of the oxygen atoms, the other pointing away, toward the H4/β5/H5 face in direction to Tyr-119.
DISCUSSION
Bacterial two-component signal response regulators represent the classic paradigm of a molecular switch, being central players in the cell's permanent responsiveness to the ever changing surrounding conditions. Over the past several years, there has been considerable effort to understand the underlying molecular mechanism by which these comparatively small enzymes accomplish their different functions in the diverse contexts of bacterial sensing (for reviews see Stock et al., 2000; Buckler et al., 2000; Stock and Da Re, 2000; Foussard et al., 2001). The structural information on several inactive and phosphorylated homologs of the response regulator superfamily that has been gathered to date has provided a comprehensive yet confusing picture of the signaling system and it seems that every new structure adds additional flavor to a possible RR signal processing mechanism. Apparently, this is also true for the here-presented structures of cyanobacterial phytochrome-associated RRs which, once more, add variation to the well known theme.
Active versus inactive state
The central question for response regulator functions undoubtedly focuses on the structural changes accompanying the transition from the inactive to the active state, and their interaction with downstream components. The literature provides vast information concerning the mechanics involved in the switching event. Common to all is the conformational change that is undergone by a highly conserved threonine or serine residue in β-sheet 4 (Thr-87 in CheY) upon phosphorylation. But from this point on, the picture seems to blur in the different systems. For CheY it is proposed that the threonine flip generates space for a conserved aromatic residue in β5, which then can occupy a more buried rotatory conformation and thus enables downstream components (FliM, CheZ) to bind to the H4/β5/H5 surface (Zhu et al., 1997; Lee et al., 2001a,b). On the other hand, the same conformational changes were not detected in the constitutively active mutant CheYD13K (Jiang et al., 1997) and another mutant, CheYT87A, can still be activated by phosphorylation even though the key residue Thr-87 is replaced by a nonpolar alanine (Appleby and Bourret, 1998). Receiver domains of other two-component systems, such as FixJN and Spo0A show very different effects upon phosphorylation. Whereas the receiver domain FixJN undergoes significant conformational changes like CheY and homodimerizes to finally become part of a transcriptional activation complex (Birck et al., 1999), the structure of Spo0A remains relatively unperturbed (Lewis et al., 1999). One might in fact speculate that the two rotamers which have been detected in RcpA might result from two populations of molecules (phosphorylated and unphosphorylated, which were not determined in quantitative amounts) that—as outlined above—only deviate significantly at position Ser-100. This then would indicate a role of the serine residue in the release of the phosphate group similar to that found for the threonine residue in CheY. However, the low concentration of these molecules in the cyanobacterial cell makes an isolation of Rcps from the host cell in different states of activation (to learn about the conformational consequences) practically impossible.
The x-ray structures of phospho-RcpA and apo-RcpB reveal yet another, slightly altered picture. Primarily, both response regulators seem to be constitutively dimeric and dimerization occurs via the tight interaction of both H4/β5/H5 surfaces, a feature as yet unobserved in other receiver domains. This is in contrast to above mentioned cases where activation/deactivation affects the aggregation of RRs. Apparently, this is in contrast to the other structurally characterized cyanobacterial RR, Rcp1 (Im et al., 2002). For this protein, the authors suggest a phosphorylation-induced monomer-dimer conversion, based on an increase of dimer content upon phosphorylation. Although an unchanged dimer arrangement is found in the Calothrix proteins, as is strongly supported by our gel filtration experiments (elution of the RRs of Calothrix in dimeric form), one has to keep in mind that the packing of the protein in the crystal, where unphysiologically high concentrations are required, does not necessarily show the conformation during functional activity.
In fact, the conformational changes within the active site and associated regions in phospho-RcpA compared to apo-RcpB are only of a minor nature, comparable to, for example, Spo0A. The major conformational differences between the apo- and the phospho-form predominantly concern displacements in the region forming the acidic crevice at the surface as described in the previous section. In the present state of this work we are only able to compare the inactive state of RcpA with the activated state of RcpB. Nevertheless, a number of characteristic features of these regulatory domains can still be deduced. Our results indicate that the specificity of dimer formation is driven by a highly conserved complementarity of the interface region, generating a tightly interacting homodimer. The interfaces in dimeric RcpA and B bury a surface of 1000 Å2 and 1175 Å2, respectively, which is in the range of areas observed in the complexes of CheY and FliM (1110 Å2; Lee et al., 2001b), CheY and CheA (P2 domain, 1200 Å2; Welch et al., 1998), or dimeric FixJ (880 Å2; Birck et al., 1999). Nevertheless, the number of specific interactions between subunit residues and the extent to which water is involved is different and very specific in RcpA and B. According to the data presented, formation of the “ON” state of RcpA and B apparently does not involve dimer formation in dependence of the phosphorylation state. However, if the dimeric form is constitutive as promoted by our data, it remains to be investigated what the characteristic features of the activated species of these receiver domains are and how the signal is processed to further downstream partners. So far, unfortunately, there is nearly no biochemical data available on RcpA/B downstream interacting components that relates our system to a better understood signaling mechanism, except of the recently reported red/far-red light-mediated changes of the cAMP-level in Anabaena by Ohmori et al. (2002).
Dimerization of RcpA and B
The most predominant difference between RcpA or B and other structurally characterized receiver domains is their intrinsic ability for homodimer formation. As we could show, this ability can be ascribed to a set of highly conserved residues, some of which are part of the extended (with respect to CheY and homologs) C-terminal region in the proteins. Among other receiver domains of the CheY type that are structurally characterized, only four others—Etr1 (Muller-Dieckmann et al., 1999), FixJN or Spo0A (Lewis et al., 1999; Birck et al., 1999; Gouet et al., 1999), and Rcp1 (Im et al., 2002)—also seem to form homodimers in a phosphorylation-dependent manner but the interaction of the respective protomers is essentially different to that found here. For the unphosphorylated N-terminal receiver domain of the sporulation response regulator N-Spo0A (Bacillus stearothermophilus) it was shown that dimer formation is mediated by the swapping of a helical domain. The receiver domain of the transcriptional activator FixJ from Sinorhizobium meliloti associates via the β4/H5 regions, also driven by phosphorylation. This analysis appears difficult to perform. For Rcp1 only a change in the relative content of dimer, but no detailed structural change upon phosphorylation, has been described.
Comparison of the amino acid sequences shows that RcpA shares a higher homology with Rcp1 from Synechocystis PCC6803 than with its paralogue RcpB. According to that, many of the nonconserved residues mediating the stabilization and formation of the dimer are equivalent in RcpA and Rcp1 but different in RcpB, a feature that might enable the correct recognition of the cognate partners, although a phosphorylation-dependent monomer/dimer conversion appears to take place in Rcp1, but not in RcpA (Im et al., 2002). A general sequence comparison of known cyanobacterial phytochrome-associated RRs with other more CheY-homologous RRs gives rise to the suggestion that the unique C-terminal extensions in the proteins not only stabilize the formation of the dimeric protein, but also play a role in the specific recognition among the protomers.
Comparison with other receiver domains
To inspect the degree of relationship to other proteins of the RR-type, the sequence information available on bacterial phytochromes and their associated phosphate receiver modules were collected and BLAST searches were performed with the sequences of RcpA and B (Fig. 2). Strikingly, the alignments of different RRs revealed a subset of residues involved in the homodimer formation that is highly conserved among RRs associated with phytochrome-like receptor-histidine kinases such as cyanobacterial Rcp1 (Synechocystis PCC6803), RcpA and RcpB (Calothrix sp. PCC7102), or homologs from Anabaena PCC7120, Deinococcus radiodurans, and Agrobacterium tumefaciens (Fig. 2). Additionally we found the dimerization motif in several other bacterial RRs, some of which are cotranscribed with yet uncharacterized receptor histidine kinases (D. radiodurans, Methanobacterium thermoautotrophicum, Archeoglobus fulgidus). We therefore deduce that dimer formation of the phytochrome-associated RR type is a highly conserved characteristic and is promoted by a set of invariant residues common to a certain subgroup of RRs, some of which are involved in light-driven signaling events.
CONCLUSIONS
The cyanobacterial response regulators RcpA and RcpB are members of a phytochrome-associated, light-sensing signal transduction pathway, involving histidine-kinase mediated autophosphorylation of the receptor and phosphotransfer to the response regulator. Both cyanobacterial RRs fold into a common (β/α)5 structural motif, which is also found in other CheY-type RRs; however, they remain in a homodimeric arrangement, irrespective of their phosphorylation state. From the here-presented results, it appears plausible that RcpA and B and homologs constitute a novel subgroup of receiver domains with a different functional principle, yet adressing the questions concerning the nature, function, and dynamic of dimer formation. The nature and specificity of the described interactions is not found in other CheY RRs, but is exclusively present in phytochrome-associated RRs. The interaction sites reside in a C-terminal extension and the surface spanned by H4, β5, and H5 of RcpA and RcpB. The novel structural motif of prokaryotic, phytochrome-associated RRs makes them archetypal for a rapidly growing subgroup of CheY-type signal transduction components.
The presented crystallographic results suggest that dimerization is independent of the phosphorylation state of Rcps, but, as discussed, this apparent property might as well be imposed by the experimental condition during crystallization. The possibility for a phosphorylation-dependent dimerization as described by Im et al. (2002) still remains possible and needs to be further investigated.
Acknowledgments
The authors thank H. Bartunik and G. Bourenkov from the BW6 Beamline (DESY (the German Synchrotron Research Centre), Hamburg) for greatly assisting in MAD data collection.
C.B. is a recipient of a PhD grant from SFB533 (Light-induced dynamics of biopolymeres).
References
- Appleby, J. L., and R. B. Bourret. 1998. Proposed signal transduction role for conserved CheY residue Thr87, a member of the response regulator active-site quintet. J. Bacteriol. 180:3563–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellsolell, L., J. Prieto, and M. Coll. 1994. Magnesium binding to the bacterial chemotaxis protein CheY results in large conformational changes involving its functional surface. J. Mol. Biol. 238:489–495. [DOI] [PubMed] [Google Scholar]
- Birck, C., L. Mourey, P. Gouet, B. Fabry, J. Schumacher, P. Rousseau, D. Kahn, and J. Samama. 1999. Conformational changes induced by phosphorylation of the FixJ receiver domain. Struct. Fold. Des. 7:1505–1515. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T. 1992. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 355:472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T. 1993. Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr. D Biol. Crystallogr. 49:24–36. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D. 54:905–921. [DOI] [PubMed] [Google Scholar]
- Buckler, D. R., G. S. Anand, and A. M. Stock. 2000. Response-regulator phosphorylation and activation: a two-way street? Trends Microbiol. 8:153–156. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 50:760–763. [DOI] [PubMed] [Google Scholar]
- Davis, S. J., A. V. Vener, and R. D. Vierstra. 1999. Bacteriophytochromes: Phytochrome-like photoreceptors from nonphotosynthetic eubacteria. Science. 286:2517–2520. [DOI] [PubMed] [Google Scholar]
- Foussard, M., S. Cabantous, J. Pedelacq, V. Guillet, S. Tranier, L. Mourey, C. Birck, and J. Samama. 2001. The molecular puzzle of two-component signaling cascades. Microbes Infect. 3:417–424. [DOI] [PubMed] [Google Scholar]
- Gouet, P., B. Fabry, V. Guillet, C. Birck, L. Mourey, D. Kahn, and J. Samama. 1999. Structural transitions in the FixJ receiver domain. Struct. Fold. Des. 7:1517–1526. [DOI] [PubMed] [Google Scholar]
- Herdman, M., T. Coursin, R. Rippka, J. Houmard, and N. Tandeau de Marsac. 2000. A new appraisal of the prokaryotic origin of eukaryotic phytochromes. J. Mol. Evol. 51:205–213. [DOI] [PubMed] [Google Scholar]
- Hoff, W. D., K.-H. Jung, and J. L. Spudich. 1997. Molecular mechanism of photosignaling by archaeal sensory rhodopsins. Annu. Rev. Biophys. Biomol. Struct. 26:223–258. [DOI] [PubMed] [Google Scholar]
- Hughes, J., F. Mittmann, A. Wilde, W. Gärtner, T. Börner, E. Hartmann, and T. Lamparter. 1997. A prokaryotic phytochrome. Nature. 386:663. [DOI] [PubMed] [Google Scholar]
- Hübschmann, T., T. Börner, E. Hartmann, and T. Lamparter. 2001a. Characterization of the Cph1 holo-phytochrome from Synechocystis sp PCC 6803. Eur. J. Biochem. 268:2055–2063. [DOI] [PubMed] [Google Scholar]
- Hübschmann, T., H. J. M. M. Jorissen, T. Börner, W. Gärtner, and N. Tandeau de Marsac. 2001b. Phosphorylation of proteins in the light-dependent signalling pathway of a filamentous cyanobacterium. Eur. J. Biochem. 268:3383–3389. [DOI] [PubMed] [Google Scholar]
- Im, Y. J., S. H. Rho, C. M. Park, S. S. Yang, J. G. Kang, J. Y. Lee, P.-S. Song, and S. H. Eom. 2002. Crystal structure of a cyanobacterial phytochrome response regulator. Protein Sci. 11:614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, M., R. B. Bourret, M. I. Simon, and K. Volz. 1997. Uncoupled phosphorylation and activation in bacterial chemotaxis. The 2.3 Å structure of an aspartate to lysine mutant at position 13 of CheY. J. Biol. Chem. 272:11850–11855. [DOI] [PubMed] [Google Scholar]
- Jiang, Z. Y., L. R. Swem, B. G. Rushing, S. Devanathan, G. Tollin, and C. E. Bauer. 1999. Bacterial photoreceptor with similarity to photoactive yellow protein and plant phytochromes. Science. 285:406–409. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- Jorissen, H. J. M. M., B. Quest, A. Remberg, T. Coursin, S. E. Braslavsky, K. Schaffner, N. Tandeau de Marsac, and W. Gärtner. 2002. Two independent light-sensing two-component systems in a filamentous cyanobacterium. Eur. J. Biochem. 269:2662–2671. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J., and T. A. Jones. 1998. Databases in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 54:1119–1131. [DOI] [PubMed] [Google Scholar]
- Larsen, T. A., A. J. Olson, and D. S. Goodsell. 1998. Morphology of protein-protein interfaces. Struct. Fold. Des. 6:421–427. [DOI] [PubMed] [Google Scholar]
- Lawrence, M. C., and P. M. Colman. 1998. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234:946–950. [DOI] [PubMed] [Google Scholar]
- Lee, S. Y., H. S. Cho, J. G. Pelton, D. Yan, E. A. Berry, and D. E. Wemmer. 2001a. Crystal structure of activated CheY. Comparison with other activated receiver domains. J. Biol. Chem. 276:16425–16431. [DOI] [PubMed] [Google Scholar]
- Lee, S. Y., H. S. Cho, J. G. Pelton, D. Yan, R. K. Henderson, D. S. King, L. Huang, S. Kustu, E. A. Berry, and D. E. Wemmer. 2001b. Crystal structure of an activated response regulator bound to its target. Nat. Struct. Biol. 8:52–56. [DOI] [PubMed] [Google Scholar]
- Lewis, R. J., J. A. Brannigan, K. Muchova, I. Barak, and A. J. Wilkinson. 1999. Phosphorylated aspartate in the structure of a response regulator protein. J. Mol. Biol. 294:9–15. [DOI] [PubMed] [Google Scholar]
- Madhusudan, M., J. Zapf, J. A. Hoch, J. M. Whiteley, N. H. Xuong, and K. I. Varughese. 1997. A response regulatory protein with the site of phosphorylation blocked by an arginine interaction: crystal structure of SPo0F from Bacillus subtilis. Biochemistry. 36:12739–12745. [DOI] [PubMed] [Google Scholar]
- McCleary, W. R., and J. B. Stock. 1994. Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 269:31567–31572. [PubMed] [Google Scholar]
- Muller-Dieckmann, H. J., A. A. Grantz, and S. H. Kim. 1999. The structure of the signal receiver domain of the Arabidopsis thaliana ethylene receptor ETR1. Struct. Fold. Des. 7:1547–1556. [DOI] [PubMed] [Google Scholar]
- Navaza, J. 1994. AMoRe: an automated package for molecular replacement. Acta Crystallogr. A. 50:157–163. [Google Scholar]
- Ohmori, M., K. Terauchi, S. Okamoto, and M. Watanabe. 2002. Regulation of cAMP-mediated photosignaling by a phytochrome in the cyanobacterium Anabaena cylindrica. Photochem. Photobiol. 75:675–679. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z., and W. Minor. 1997. Processing of x-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, A. C. W. Carter and R. M. Sweet, editors. Academic Press, New York. 307–326. [DOI] [PubMed]
- Perrakis, A., R. Morris, and V. S. Lamzin. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6:458–463. [DOI] [PubMed] [Google Scholar]
- Stock, A. M., J. M. Mottonen, J. B. Stock, and C. E. Schutt. 1989. Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature. 337:745–749. [DOI] [PubMed] [Google Scholar]
- Stock, A. M., V. L. Robinson, and P. N. Goudreau. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183–215. [DOI] [PubMed] [Google Scholar]
- Stock, J. B., and S. Da Re. 2000. Signal transduction: response regulators on and off. Curr. Biol. 10:R420–R422. [DOI] [PubMed] [Google Scholar]
- Welch, M., N. Chinardet, L. Mourey, C. Birck, and J. Samama. 1998. Structure of the CheY-binding domain of histidine kinase CheA in complex with CheY. Nat. Struct. Biol. 5:25–29. [DOI] [PubMed] [Google Scholar]
- Yeh, K.-C., S.-H. Wu, J. T. Murphy, and J. C. Lagarias. 1997. A cyanobacterial phytochrome two-component light sensory system. Science. 277:1505–1508. [DOI] [PubMed] [Google Scholar]
- Zhu, X., K. Volz, and P. Matsumura. 1997. The CheZ-binding surface of CheY overlaps the CheA- and FliM-binding surfaces. J. Biol. Chem. 272:23758–23764. [DOI] [PubMed] [Google Scholar]