

Abstract
We used fluorescence correlation spectroscopy (FCS) to analyze the binding of fluorescently labeled peptides to lipid vesicles and compared the deduced binding constants to those obtained using other techniques. We used a well-characterized peptide corresponding to the basic effector domain of myristoylated alanine-rich C kinase substrate, MARCKS(151–175), that was fluorescently labeled with Alexa488, and measured its binding to large unilamellar vesicles (diameter ∼100 nm) composed of phosphatidylcholine and phosphatidylserine or phosphatidylinositol 4,5-bisphosphate. Because the large unilamellar vesicles are significantly larger than the peptide, the correlation times for the free and bound peptide could be distinguished using single color autocorrelation measurements. The molar partition coefficients calculated from the FCS measurements were comparable to those obtained from binding measurements of radioactively labeled MARCKS(151–175) using a centrifugation technique. Moreover, FCS can measure binding of peptides present at very low concentrations (1–10 nmolar), which is difficult or impossible with most other techniques. Our data indicate FCS can be an accurate and valuable tool for studying the interaction of peptides and proteins with lipid membranes.
INTRODUCTION
The binding of peripheral proteins to membranes is crucial for biological processes such as signal transduction and vesicle trafficking (DiNitto et al., 2003). Translocation of proteins from the cytoplasm to the plasma membrane increases their local concentration in the membrane phase ∼1000-fold, as discussed in detail elsewhere (Adam and Delbrück, 1968; Berg and Purcell, 1977; Kholodenko et al., 2000; McCloskey and Poo, 1986; McLaughlin and Aderem, 1995) . For example, the C1 and C2 domains of protein kinase C (PKC) mediate its translocation to the plasma membrane, where it is exposed to a significantly higher effective concentration of its membrane-bound substrates (Cullen, 2003; Mellor and Parker, 1998; Newton, 2003). The myristoylated alanine-rich C kinase substrate (MARCKS) protein, the main substrate of PKC in many cell types (Anderem, 1992; Arbuzova et al., 2002; Blackshear, 1993), is anchored to the membrane by an N-terminal myristate and a cluster of basic residues in its effector domain (McLaughlin and Aderem, 1995). PKC phosphorylation of three serines in the MARCKS effector domain weakens its electrostatic attraction to acidic lipids in the plasma membrane, resulting in translocation of MARCKS to the cytoplasm in living cells (Ohmori et al., 2000).
Several different laboratories have investigated the membrane binding of the proteins in the calcium/phospholipid second messenger system (i.e., G-proteins, phosphoinositide-specific phospholipase C, PKC, MARCKS) using a variety of techniques. A recent monograph on peptide-lipid interactions discusses biophysical techniques such as isothermal titration calorimetry (ITC), fluorescence resonance energy transfer, spin labeling, and centrifugation (Simon and McIntosh, 2002). Fluorescence correlation spectroscopy (FCS) is an alternative technique that has definite advantages for studying protein-membrane (and protein-protein) interactions (Elson and Magde, 1974; Magde et al., 1972, 1974). FCS measures fluctuations in the emission spectra of a small number of fluorescent molecules diffusing into and out of the focus volume of an excitation laser. Statistical analysis of these fluctuations yields correlation curves carrying information about diffusion, chemical reactions, and other processes (Elson, 2001). Although FCS was developed 30 years ago, recent technical advances have led to a renaissance of interest in this technique; several recent brief reviews (Hess et al., 2002; Krichevsky and Bonnet, 2002; Muller et al., 2003; Schwille and Haustein, 2002; Thompson et al., 2002; Van Craenenbroeck and Engelborghs, 2000) and a monograph (Rigler and Elson, 2001) describe these developments and their application to a variety of model and cellular systems.
Before using FCS for detailed studies of protein-membrane interactions, however, we need to demonstrate that the technique provides accurate measurements. Hence these studies focus on the binding of simple, well-characterized peptides to phospholipid vesicles, which allows us to compare the measurements to values determined by conventional, well-established techniques. Large unilamellar vesicles (LUVs) formed by extrusion through 100 nm diameter pores are particularly well-suited for these experiments because they are both thermodynamically stable (in contrast to sonicated vesicles) and uniform in size (in contrast to both ethanol-injected vesicles and multilamellar vesicles); these 100 nm vesicles have been characterized by a number of techniques (Hope et al., 1985). Moreover, LUVs are significantly larger than typical proteins and peptides, permitting separation of two correlation times in single-color autocorrelation measurements. Although FCS has been used previously to measure the binding of proteins to vesicles (Dorn et al., 1998; Takakuwa et al., 1999), no detailed comparisons of results obtained with FCS results and those from more conventional techniques have appeared in the literature. We chose to work with a peptide that corresponds to the effector domain of the MARCKS protein, MARCKS(151–175). This 25-mer peptide has 13 basic and five aromatic residues. The membrane binding of both MARCKS and the effector domain peptide has been studied by several different techniques, as reviewed elsewhere (Arbuzova et al., 2002; McLaughlin and Aderem, 1995; McLaughlin et al., 2002; Qin and Cafiso, 1996). The peptide binds only weakly to electrically neutral vesicles (e.g., those formed from phosphatidylcholine, PC) through its five aromatic residues; the binding increases exponentially with increasing mole fraction of acidic lipid (e.g., phosphatidylserine, PS), as expected for electrostatic interactions (Arbuzova et al., 2000). This allows us to measure the binding of a single peptide to LUVs over the entire range accessible with FCS measurements (effective dissociation constant of peptide with lipid is ∼1 mM to 10 nM) by simply changing the fraction of acidic lipid in the vesicles.
The binding of MARCKS and its effector domain to phospholipid vesicles is also of biological interest. Although MARCKS is present at high concentrations in cells, its physiological role is unknown. One hypothesis is that MARCKS reversibly sequesters phosphatidylinositol 4,5-bisphosphate (PIP2) through its effector domain, then releases PIP2 upon PKC phosphorylation or binding of calcium/calmodulin (Ca/CaM) (Gambhir et al., 2004). There is also much interest in using single molecule techniques to study the interaction of Ca/CaM with biological effectors. Thus, our FCS studies of MARCKS peptide binding to PC/PIP2 vesicles provide the basis for single molecule studies of calmodulin. Finally, several recent theoretical articles describing the binding of basic peptides to membranes consider the possible redistribution of monovalent acidic lipids when a basic peptide binds to the membrane (Haleva et al., 2004; May et al., 2000) whereas other reports assume the redistribution is negligible (Wang et al., 2004). Thus, an accurate description of how the partitioning depends on the mole fraction of acidic lipid in the membrane is useful for testing different theoretical approaches.
We demonstrate that FCS can accurately determine the binding constants of fluorescently labeled peptides to vesicles. We labeled a peptide corresponding to the effector domain of MARCKS, MARCKS(151–175) with Alexa488, then measured its binding to PC/PS vesicles using FCS. We compared the FCS results to those available from other measurements (e.g., centrifugation experiments with radioactively labeled MARCKS(151–175)) and observed excellent agreement over the entire five orders of magnitude range of the partition coefficient. This suggests that FCS can be used with confidence to study the membrane binding of more complicated proteins to LUVs. We conclude with a discussion of the advantages of the FCS approach over more conventional techniques (e.g., centrifugation, equilibrium dialysis, ITC).
EXPERIMENTAL SETUP
Fig. 1 illustrates the principle of FCS and the binding mechanism of fluorescently labeled MARCKS(151–175) to negatively charged lipid membranes. Fig. 1 A is a sketch of a free and vesicle-bound peptide undergoing Brownian motion in the laser focus volume. We used the one-color autocorrelation method to measure the diffusion of the labeled peptide into and out of the illuminated volume. A photodiode detects the fluctuation in fluorescence, which is analyzed by a digital correlator. These measurements are expressed as a correlation time for the free peptide, i.e., the time it takes for the peptide to diffuse in and out of the ∼300 nm diameter detection region. We then add unlabeled large unilamellar vesicles, LUVs (monodisperse 100 nm diameter phospholipid vesicles), and monitor the autocorrelation function as the vesicle concentration increases and fluorescent peptides bind to the vesicles. Because the bound peptides diffuse more slowly, they have a significantly higher (∼25-fold) correlation time (τbound ≫ τfree); hence the free and bound species can be distinguished easily. Fig. 1 B shows an idealized autocorrelation function.
FIGURE 1.

Determination of binding affinities of peptides for large unilamellar vesicles (LUVs) with FCS. (A) Sketch of the detection volume of the FCS instrument. A free fluorescent peptide and a 100 nm diameter LUV, shown with an adsorbed peptide, diffuse in and out of the volume, producing fluctuations in the fluorescence intensity. A free fluorescent peptide has a correlation time τfree ∼0.1 ms, and the peptide bound to the vesicle has a longer correlation time, τbound ∼2 ms, because its diffusion coefficient is lower. If the peptide concentration is 10 nM, the ∼0.3 fl detection volume contains approximately one free peptide. The detection volume (diameter ∼300 nm), LUV (diameter ∼100 nm), and peptide (length ∼7 nm) are drawn approximately to scale. (B) Plot of an idealized two-component autocorrelation function for τfree ≪ τbound. The correlation times are indicated with dashed vertical lines. When the fractions of free and bound peptide are identical, the molar partition coefficient, K = 1/[lipid]. Modified from Van Craenenbroeck and Engelborghs (2000). (C) Sketch of a peptide corresponding to the basic effector domain of MARCKS, MARCKS(151–175), bound to a negatively charged PC/PS lipid membrane. The peptide has 13 basic residues (indicated by +; the sequence is acetyl-CKKKKKRFSFKKSFKLSGFSFKKNKK-amide); the fluorescent probe Alexa488 is attached covalently to the Cys residue at the N-terminus. As described in the text, several different measurements indicate the peptide is in an extended conformation with the five Phe residues penetrating to the level of the acyl side chains. Most of the binding energy comes from nonspecific electrostatic interactions between the basic residues and the acidic lipids in the LUV.
The apparent dissociation constant, Kd, of the peptide with lipid is, by definition, the lipid concentration at which 50% of the peptide is bound. Describing the interaction with a Kd would be appropriate if the peptide formed a 1:1 complex with a lipid. However, the peptide actually partitions onto the vesicle because of nonspecific electrostatic and hydrophobic interactions (Arbuzova et al., 2000). Thus we describe the binding with a molar partition coefficient defined as K = 1/Kd (see Eq. 4 below). Fig. 1 C shows the basic effector domain of MARCKS(151–175) binding to a negatively charged PC/PS lipid membrane: the 25-residue peptide is in an extended (∼7 nm) conformation both in solution and bound to PC/PS and PC/PIP2 membranes (see Fig. 1 in Gambhir et al., 2004 and references therein). The 13 basic residues (indicated with plus signs) interact electrostatically with the acidic lipids in the membrane. The five aromatic phenylalanine residues, which insert to the level of the acyl chain region of the lipids, are not illustrated (Ellena et al., 2003; Zhang et al., 2003). The relatively weak binding of the peptide to electrically neutral bilayers illustrates that these hydrophobic interactions provide only a minor contribution to the strong binding observed with the peptide to negatively charged vesicles (Arbuzova et al., 2000).
We used the hydrophilic fluorophore Alexa488, which was covalently bound via a Cys at the N-terminus of the peptide; ESR measurements show that the highly basic amino terminal region of the MARCKS(151–175) peptide does not penetrate the polar headgroup region of the bilayer (Qin and Cafiso, 1996). Thus, attaching a hydrophilic fluorophore to this region of the peptide should not significantly affect the molar partition coefficient of the peptide. In contrast, hydrophobic fluorescent probes partition into the membrane and enhance the value of K (e.g., attaching TexasRed to most small basic peptides increases K ∼100-fold for negatively charged vesicles). In brief, Alexa488 is an excellent fluorophore for FCS measurements: it has low triplet state excitation, a high quantum yield, and high photostability (Schwille and Haustein, 2002).
FCS measurements were performed with an Axiovert 200 microscope with a ConfoCor2 unit equipped with an 40×/1.2 water immersion objective (Apochromat) and a continuous argon ion laser (Zeiss, Jena, Germany). The excitation of the fluorophore was at 488 nm with an incident laser power of 120 μW for all experiments. The ConfoCor2 software controlled the experimental setup and produced the autocorrelation data. The samples were measured in LabTek II chamber slides with eight wells (Nunc, Wiesbaden, Germany). We minimized the adsorption of the peptide to the chamber during the experiment by precoating the chambers with a neutral lipid bilayer. Briefly, we added a low concentration of sonicated unilamellar vesicles formed from PC to the chambers and incubated them overnight. Before use, we rinsed the chambers gently five times with buffer to remove free sonicated unilamellar vesicles. The laser focus was positioned in solution ∼200 μm above the top surface of the cover slide. The focus volume was determined by calibration with a 30 nM Rhodamine 6G solution (diffusion constant DRh6G = 2.8 × 10−10 m2 s−1; Magde et al., 1974).
MATERIALS
We used Rhodamine 6G in buffer solution containing 100 mM NaCl, 10 mM HEPES, pH 7 (Merck, Darmstadt, Germany) for system calibration. PC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine) and PS (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine) were obtained from Avanti Polar Lipids (Alabaster, AL). The ammonium salt of L-α-phosphatidylinositol 4,5-bisphosphate (PIP2) was purchased from Boehringer (Mannheim, Germany). LUVs were prepared by drying the lipid mixture on a rotary evaporator, hydrating the lipids in a solution containing 100 mM KCl, 10 mM HEPES, pH 7, then taking the multilamellar vesicles through five cycles of freezing and thawing followed by 10 extrusion cycles through a stack of two polycarbonate filters (100 nm pore size diameter) using the mini-extruder from Avanti Polar Lipids (Hope et al., 1985). As discussed in detail elsewhere (Wang et al., 2002), several steps in the preparation of PIP2 containing vesicles require special attention: 1), PIP2 is less soluble in chloroform than most lipids and the formation of mixed lipid films requires rapid evaporation; 2), PIP2 is less stable than conventional lipids; and 3), PIP2 can be lost more readily than most lipids during the extrusion procedure. Thus, we regard the comparison with data in the literature obtained with PC/PS vesicles as the best test of the utility of the FCS technique.
The peptide corresponding to the basic effector domain of bovine MARCKS (residues 151–175) was obtained from the Proteomics Center, State University of New York (Stony Brook, NY). The amino acid sequence of the effector domain is
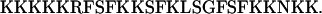 |
The peptide was synthesized with an extra Cys residue at the N-terminus to facilitate labeling with Alexa488 (Molecular Probes, Eugene, OR). The ends were blocked with acetyl and amide groups, producing a peptide with 13 positive and zero negative charges. Alexa488, however, has one positive and three negative charges at pH 7; in principle, these charges could affect the electrostatic binding of the peptide to the vesicles. The probe was attached to the region of peptide that is most distal to the membrane when the peptide binds (Fig. 1 C), assuming that it would not affect the value of the partition coefficient, K, significantly. Independent centrifugation measurements confirm that Alexa-labeled and radioactively NEM-labeled MARCKS(151–175) bind with similar values of K to PC/PS (10:1) vesicles (data not shown). The purity of the final labeled peptide was checked with mass spectroscopy and HPLC; we estimate the labeled peptide was >95% pure. All buffer solutions were prepared with deionized water (conductivity <0.1 10−6 S/m). Solutions were degassed before use to minimize air bubble formation during the measurements.
DATA ANALYSIS
The signal I(t) generated by fluorescent molecules diffusing through the detection volume fluctuates around a mean value I(t) = 〈I(t)〉 +δI(t). The normalized time correlation function is defined as G(τ) = 〈δI(t)δI(t + τ)〉/〈I(t)〉2. For identical fluorescent particles undergoing Brownian motion in a three-dimensional Gaussian focus volume element, the autocorrelation curve can be described with the equation (Hess et al., 2002)
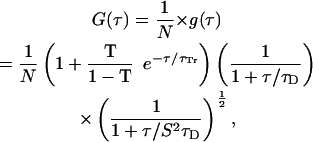 |
(1) |
where N is the average number of the fluorescent molecules in the laser focus and τD is the correlation time of the particles. The correlation time represents the diffusion time, or the average passage time of the molecule through the focus volume, and is defined by the Einstein equation τD = ω2/4D, where ω2 is the square of the radius of the laser focus and D is the diffusion constant. The structural parameter S, the ratio of the distances from the center of the laser beam focus in the radial and axial directions, respectively, was determined from measurements with Rhodamine 6G to be S = 5.2 and the focus volume was 0.13 fl. The fraction of fluorophores in the triplet state T and the triplet lifetime τTr are fitting parameters for the triplet characteristics in autocorrelation curves. For Alexa488, they were determined by fit of the data in Fig. 2 A.
FIGURE 2.

(A) Autocorrelation curves of Alexa-labeled MARCKS(151–175) and NBD-labeled PC/PS vesicles (5:1), which have a diameter of 100 nm. The diffusion time, which correlates with the particle size, is ∼70 μs and 1700 μs for peptides and vesicles, respectively. (B and C) Measurement range and detection limit. The number of particles in the effective volume is plotted versus the concentration of vesicles (B) and peptides (C) in the sample. Error bars (mean ± SD) are not shown for data points when they are smaller than the size of symbols.
In the case of a multicomponent system, the measured correlation function G(τ) is a weighted sum of the autocorrelation functions of each component Gi(τ) (i = 1, 2, …M) (Clamme et al., 2003; Thompson, 1991) as
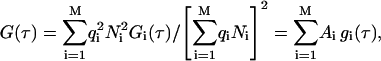 |
(2) |
where Ni is the mean particle number and qi is the ratio of the fluorescence yield of the ith component (given by the product of the detection efficiency, absorption cross section, and fluorescence quantum yield) to that of the first component. We consider only two diffusing species: the fluorescently labeled peptides (index P) and LUVs with bound fluorescently labeled peptides (index V). We used Eq. 2 for the data analysis with the amplitudes given by
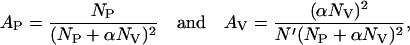 |
(3) |
where NP and NV denote the number of free peptides and vesicles, respectively. N′ is the number of slowly diffusing particles that can be detected (i.e., vesicles with ≥1 bound fluorescent peptide). The value α is the ratio of the fluorescence yield of vesicles with peptides bound to that of free peptide and represents the average number of peptides bound per vesicle if the binding does not change the fluorescence quantum yield and the detection efficiency. (Indeed, the binding of our peptide to vesicles does not change the fluorescence intensity of the Alexa-488 fluorophore, as measured with a conventional spectrometer (data not shown), provided the number of bound peptides is sufficiently low that self-quenching does not occur.) When the average number of peptides bound per vesicle, α, is ≤1, the number of particles that can be detected with the slow diffusion time (i.e., vesicles) is equal to the number of vesicle-bound peptides: N′ = αNv. For most of our measurements (e.g., low lipid concentration, molar partition coefficient K > 104 M−1), the average number of peptides bound per vesicle, α, is ≥ 1; in this case, the number of particles with the slow (vesicle) diffusion is equal to the total number of vesicles present: N′ = Nv.
Under our experimental conditions, we were not able to determine α accurately with these types of measurements. Our calculation of α from the amplitudes AP and AV yielded values that are much smaller than expected. A reason for this discrepancy might be the signal/noise ratio that makes the determination of AV difficult when many peptides are bound to a small number of vesicles (<0.1 nM vesicle concentration; squares and inverted triangles in Fig. 3 D). Quenching effects could also play a role in determining Av accurately if a large number of peptides are bound per vesicle (Clamme et al., 2003), but this effect should not be important under our experimental conditions. Thus, to calculate the fraction of peptide bound and deduce the molar partition coefficient, we only use the behavior of the free peptide as discussed below.
FIGURE 3.

FCS measurements of MARCKS(151–175) binding to PC/PS LUVs. (A–C) Time-resolved count rates from solutions containing Alexa-labeled MARCKS(151–175) and 5:1 PC/PS vesicles. (A) The solution contains 2 nM peptide and no vesicles. (B) The solution contains 2 nM peptide and 2 × 10−7 M lipid. Peaks with different amplitudes appear in the plot because ∼15% of the peptide has bound to the vesicles. Peaks represent the diffusion of a single LUV with ∼10 bound peptides through the illuminated volume. (C) The solution contains 2 nM peptide and 10−6 M lipid. The number of peaks increases significantly because 57% of the peptides are now bound to LUVs. (D) The effect of vesicle (lipid) concentration on the autocorrelation curves obtained from a solution containing 2 nM peptide. Autocorrelation curves (bottom to top) are shown for 0 M, 2 × 10−7 M, 1 × 10−6 M, 2 × 10−6 M, and 4 × 10−6 M accessible lipid concentrations, corresponding to 0%, 15%, 57%, 74%, and 90% peptide bound, respectively. The lines through the data represent the best fit of the two-component model, Eq. 2.
Kinetic considerations
In using Eq. 2, we assume that a bound fluorescent peptide dissociates only rarely during its diffusion through the illuminated focus volume. FCS detects two components: the rapidly diffusing peptide free in solution and the slowly diffusing peptide bound to vesicles (Elson, 2001). The conditions we used for the FCS measurements permit us to ignore kinetic effects based on data from stopped-flow kinetics experiments (Arbuzova et al., 1997). Specifically, the diffusion time of the vesicle/peptide complex (∼1700 μs) is shorter than the binding lifetime of the peptide to the vesicle (approximately the reciprocal of the off constant). For example, the measured dissociation rate constant for 15:1 PC/PS vesicles is 100 s−1, and the corresponding lifetime of the peptide-vesicle complex (10 ms) is longer than the time for the vesicle-bound peptide to diffuse across the beam waist of the laser focus (∼2 ms). The lifetime of the peptide-vesicle complex is >10 ms for vesicles containing >6% PS because binding of the MARCKS(151–175) to PC/PS vesicles is a diffusion-limited process (Arbuzova et al., 1998). Thus the lifetime of the peptide-vesicle complex is directly proportional to the molar partition coefficient, which increases with the mole fraction of PS in the vesicle. Even for a diffusion-limited forward rate constant, the experimental results we report for vesicles containing 6% PS in Fig. 4 (K = 103 M−1) agree well with independent equilibrium measurements, as expected from the above kinetic analysis. Thus FCS measurements should be useful for monitoring protein or peptide binding to 100 nm LUVs if K ≥ 103 M−1.
FIGURE 4.

FCS measurements of peptide binding to PC/PS vesicles. (A) The percentage of peptide bound, deduced from data similar to those illustrated in Fig. 3 D for 5:1 PC/PS vesicles, is plotted versus the accessible lipid concentration. The solutions contained 100 mM KCl, 10 mM HEPES, pH 7, 2 nM Alexa-labeled MARCKS(151–175) and PC/PS vesicles comprised of 4.8 (○), 9.1 (∇), 12.5 (×), 16.7 (□), and 25 (+) mol % PS. The curves are the least-square fits of Eq. 4 to the data. (B) The molar partition coefficient, K, is plotted as a function of mol % PS for 2 nM (○) and 10 nM (▵) peptide concentrations. The error bars are not shown in the graph because they are smaller than the size of the symbols. The straight line is the least-squares best fit to the data obtained with 2 nM peptide. The FCS data agree well with centrifugation binding measurements conducted with 2 nM radioactively labeled NEM-MARCKS(151–175) (▪), reported by Arbuzova et al. (2000).
When proteins or peptides bind only weakly to vesicles, however, kinetics may affect FCS measurements; e.g., the molar partition coefficient of MARCKS(151–175) for PC vesicles is ∼100 M−1, so the lifetime of the peptide-vesicle complex is only ∼100 μs. This weak binding cannot be described with Eq. 2. (The kinetics of peptide binding to PC/PIP2 vesicles becomes complicated when the vesicles contain <1% PIP2: the forward rate constant is no longer diffusion-limited because three PIP2 must diffuse together to form the appropriate binding site; Wang et al., 2002.)
Determination of the binding constant
The binding of peptides to lipid bilayers can be described by defining a molar partition coefficient or binding constant, K, as discussed elsewhere (Ben-Tal et al., 1997; Murray et al., 1998; Peitzsch and McLaughlin, 1993). K is the proportionality constant between the fraction of peptide bound to the membrane and the molar concentration of the peptide in the bulk aqueous phase, [P]. Our measurements used conditions where the molar concentration of accessible lipid [L]acc is much greater than the molar concentration of peptides bound to the membrane [P]mem. If [L]acc ≫ [P]mem, the relationship can be written as [P]mem = K[P][L]. The total molar concentration of peptide is the sum of the bound and free peptide concentrations: [P]tot = [P]mem + [P]. Combining these two expressions we obtain
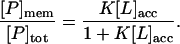 |
(4) |
[L]acc is ∼50% of the total lipid concentration because the peptide is added to a solution of LUVs and binds only to the outer leaflet of the membrane. We demonstrated that the peptide cannot penetrate the vesicles in experiments on giant unilamellar vesicles using a conventional epifluorescence microscopy (Gambhir et al., 2004). The molar partition coefficient deduced from Eq. 4 is the reciprocal of the lipid concentration that binds 50% of the peptide.
The FCS data provide the amplitudes AP and AV of the two component correlation function using Eq. 3. Furthermore, the conservation of mass requires that the total number of peptides
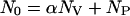 |
(5) |
is constant. Using Eqs. 3 and 5, we find AP = NP/(NO)2. The fraction of peptide bound to vesicles is the number density of bound peptides with respect to the total number of peptides or
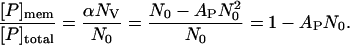 |
(6) |
Thus, the fraction of peptide bound illustrated in Fig. 4 A is determined from the amplitude of the free peptide, AP, and the total number of peptides, N0. AP is calculated from the two-component autocorrelation function fit of Eq. 2 to data of the type illustrated in Fig. 3 D. N0 is calculated from the one-component autocorrelation function fit of Eq. 1 to data obtained when only peptide is present (e.g., Fig. 2 A or 3 D). The fraction of peptide bound is plotted versus accessible lipid concentration (Fig. 4 A) and the binding constant, K, is determined from fitting these data with Eq. 4. Error bars for the fraction of peptide bound shown in Fig. 4 A are calculated using the standard Gaussian formula for error propagation applied to Eq. 6. For low values of fraction peptide bound, the error is dominated by the standard deviation of N0, and for high values, the error is dominated by the standard deviation of AP. The standard deviations are not shown in Fig. 4 because they are approximately the same size as the symbols.
RESULTS
Fig. 2 A shows autocorrelation curves of Alexa-labeled MARCKS(151–175) and NBD-labeled PC/PS LUVs; the data were obtained from separate experiments where only one component was present in solution. We fit the autocorrelation data using Eq. 1 (the one-component model) to deduce diffusion times of Alexa-labeled MARCKS(151–175) and PC/PS vesicles of 66 μs and 1700 μs, respectively. The corresponding hydrodynamic radii of the peptide and the vesicle are 2.2 nm and 55 nm, respectively. These values agree well with the theoretical predictions. The extruded vesicles exhibited an ideal autocorrelation function, indicating they are monodisperse.
Determining the binding constant K requires accurate measurement of relative molar concentrations, so we investigated the effects of signal/noise ratio and concentration on the FCS measurements. We used dilution experiments, measuring the number of particles in the effective focus volume as a function of the sample concentration for PC/PS vesicles (Fig. 2 B) and Alexa-labeled MARCKS(151–175) (Fig. 2 C). Data points were fit with the function N = c Veff(1 + ν/c)2 NA, where N denotes the measured particle number, c the particle concentration, Veff the effective detection volume, ν the relative background, and NA the Avogadro constant (Langowski et al., 2000). These experiments indicate FCS measurements are suitable in the linear concentration ranges of 10−6 M–10−3 M accessible lipid (corresponding to 10−11 M–10−8 M vesicle concentration for 100 nm diameter LUVs) and 10−9 M–10−6 M peptide concentration. The fluctuation of the fluorescence signal becomes comparable to the background at lower concentrations.
Fig. 3 shows binding experiments using 2 nM Alexa-labeled MARCKS(151–175) with unlabeled 5:1 PC/PS vesicles. Fig. 3, A–C, show a typical fluorescence signal versus time records plotted for different lipid concentrations. (The count rate is the number of photons detected by the photodiode per time interval and corresponds to the fluorescence intensity of all fluorescent particles diffusing simultaneously through the laser focus volume. The software provides the count rate as an online record during data acquisition, giving qualitative information about the peptide binding to the vesicles.) These records show that the system is behaving as expected. To resolve single vesicles with bound peptides, we chose a binning time of ∼6 ms, which is comparable to the average diffusion time of a vesicle through the detection volume. Fig. 3 A shows the record of 2 nM Alexa-labeled MARCKS with a count rate of ∼5–10 kHz; at this low peptide concentration the average number of particles in the focus volume (0.13 fl) is only ∼0.38 and single peptides cannot be resolved. When vesicles are added to the peptide solution, we observe peaks with different amplitudes (Fig. 3 B): the slower random walk of the peptide-vesicle complex through the laser focus volume and the number of peptides bound per vesicle produce higher peaks. Increasing the vesicle concentration increases the number of peaks significantly (Fig. 3 C), but decreases the average height of the peaks because the bound peptides are distributed over a larger number of vesicles. These results are similar to those obtained in FCS measurements of the complexation of DNA with fluorescently labeled polycations (Clamme et al., 2003) or a mixture of fluorescein and fluorescein-coated beads containing many fluorophores (Van Craenenbroeck and Engelborghs, 2000).
Fig. 3 D shows autocorrelation curves for experiments where 0%, 15%, 57%, 74%, or 90% (bottom to top) of the peptide was bound. The lower autocorrelation curve (solid diamonds) represents the single particle population obtained when all of the peptides are free. Data were fit by Eq. 1 with a predetermined structural parameter of S = 5.2, yielding a diffusion time τD of 66 μs for the free peptide. When the peptide binds to the vesicles, the autocorrelation curves exhibit two characteristic diffusion times. The data shown in Fig. 3 D were fit according to Eq. 2, keeping the diffusion time of the free peptide fixed at the predetermined value. The amplitudes AP and AV can be calculated with the two-component model of Eq. 2. The population of bound peptides grows as the lipid concentration increases, and becomes more and more dominant in the autocorrelation curve. The amplitude G(0) increases simultaneously, indicating that the total number of fluorescent particles in the focus volume has decreased. This can be explained by the fact that several peptides bind to each vesicle.
Fig. 4 A shows the binding of MARCKS(151–175) to PC/PS vesicles with different ratios of PS, plotting the percentage of peptide bound as a function of the accessible lipid concentration. The curves are the least-square fits of Eq. 4 to the data. The peptide binds strongly to vesicles with a high fraction of PS and weakly to those with a low fraction of PS. Fig. 4 B shows the molar partition coefficients as a function of mol % PS in the vesicles, deduced from the data in Fig. 4 A. We repeated some binding measurements using 10 nM peptide as a control; the values obtained (triangles) agree well with those from experiments using 2 nM peptide (circles). These data argue strongly that 1), the peptide is adsorbing as a monomer and 2), the binding of the peptide does not change significantly the surface charge density of the vesicles. The molar partition coefficient, K, increases exponentially with the mole fraction of PS in the vesicles, a result that agrees qualitatively with theoretical calculations based on the Poisson-Boltzmann equation and the assumption that the monovalent acidic lipids do not redistribute (Arbuzova et al., 2000).
One key objective of this report is to compare FCS measurements of peptide binding to vesicles with those obtained using other well-established techniques. The solid squares in Fig. 4 B show measurements of the binding of radioactively labeled MARCKS(151–175) to PC/PS LUVS using a centrifugation technique (Arbuzova et al., 2000): the data agree well, indicating that FCS can be used to measure the binding of peptides and proteins to phospholipid vesicles.
Fig. 5 A illustrates FCS measurements of the binding of Alexa-labeled MARCKS (151–175) to vesicles containing the multivalent acidic lipid PIP2 rather than PS (net charge −1). These experiments used 2 nM peptide and PC/PIP2 vesicles containing either 0.5 (triangles) or 1.0 (squares) mol % PIP2. The percentage of peptide bound is plotted versus the accessible lipid concentration; the curves indicating the molar partition coefficients are the least-square fits of Eq. 4. Fig. 5 B shows the molar partition coefficient determined from FCS measurements plotted as a function of mol % PIP2 when the peptide concentration was 2 nM (open circles) or 10 nM (open triangles). We have included the molar partition coefficients determined from centrifugation measurements with 2 nM radioactively labeled MARCKS(151–175) (solid symbols) for comparison with the FCS data. The FCS measurements with 2 nM peptide (open circles) agree qualitatively with these independent centrifugation measurements (solid symbols). When the peptide was present at a concentration of 10 nM, we observed weaker binding with the FCS technique (open triangles), in agreement with centrifugation measurements (not shown). The molar partition coefficient decreases at higher peptide concentrations because when a significant number of peptides bind to the vesicle the concentration of free PIP2 is lower. Previous work has shown that ∼3 PIP2 molecules diffuse together to form a binding site for the peptide (Rauch et al., 2002; Wang et al., 2002). The molar partition coefficient of MARCKS(151–175) and many other basic peptides for LUVs containing 1% PIP2 is approximately the same as for 15% PS (Wang et al., 2002). In both cases, the binding is due to nonspecific electrostatic interactions and will be affected by the free concentration of charged lipid on the vesicles. Fig. 5 B shows that if the peptide binds strongly to the charged lipid, the true value of the partition coefficient can be determined only in the presence of very low peptide concentrations (∼nM in this case). These measurements are difficult (e.g., with radioactive labels) or impossible (e.g., with spin labels, most fluorescent labels, ITC, and NMR) to make using most conventional techniques. The FCS approach, however, is ideally suited to measure binding in the range of 1–10 nM.
FIGURE 5.

FCS measurements of MARCKS(151–175) binding to PC/PIP2 LUVs. (A) Binding of 2 nM Alexa-labeled MARCKS(151–175) to PC/PIP2 vesicles comprising 0.5 (▵) and 1 (□) mol % PIP2; binding was determined from FCS data (not shown) similar to those illustrated in Fig. 3 D. The curves are the least-square fits of Eq. 4 to the data. (B) The molar partition coefficient plotted versus the mol % PIP2 for 2 nM (○, deduced from the data in A) and 10 nM (▵) peptide concentrations. The error bars are not shown because they are smaller than the size of the symbols. For comparison, we have included the molar partition coefficients measured with 2 nM radioactively labeled MARCKS(151–175) using a centrifugation technique (▴, Arbuzova et al., 2000; ▪, Wang et al., 2001); the agreement between the FCS and centrifugation data is satisfactory.
DISCUSSION
We used FCS to measure the binding of MARCKS(151–175) to PC/PS vesicles and compared our results to those obtained with other conventional techniques (Fig. 4 B). We chose the basic effector domain peptide of MARCKS because 1), its binding to PC/PS vesicles has been measured by centrifugation, fluorescence, electrophoretic mobility, and EPR techniques and 2), the structure of the peptide and its location in the membrane have been determined. EPR measurements of spin-labeled peptides, MAS NMR, circular dichroism, and monolayer penetration experiments show that the peptide exists at the membrane interface in a nonhelical extended conformation with its five Phe groups inserted into the acyl chain region (Ellena et al., 2003; Qin and Cafiso, 1996; Victor et al., 1999; Wang et al., 2001; Zhang et al., 2003). Fig. 4 B shows that FCS measurements of the molar partition coefficient (open symbols) agree well with the centrifugation measurements (solid symbols). This demonstrates the potential of using FCS to study the binding of more complicated proteins to LUVs. The advantages and disadvantages of different techniques for measuring peptide-lipid interactions are described in a recent monograph by Simon and McIntosh (2002); Berney and Danuser (2003) review critically the limitations of the fluorescence resonance energy transfer approach to study interactions.
We conclude by discussing briefly the limitations and advantages of the FCS technique to monitor the binding of peptides/proteins to LUVs. One obvious limitation of FCS is in measuring weak peptide-membrane interactions: if the lifetime of the peptide on the vesicles is short, i.e., less than the time for the vesicle to diffuse through the illuminated volume, FCS is not appropriate. For typical diffusion-limited binding, this corresponds to K values of <103 M−1 for our peptide. This is not a serious limitation because other techniques (e.g., equilibrium dialysis, centrifugation) can easily be used in this range. In contrast, we showed that the FCS technique produces reliable binding measurements for nanomolar peptide concentrations. It is very difficult, in our experience, to make experiments in this range using conventional methods (equilibrium dialysis, centrifugation, filtration) because both the peptides and lipids typically adsorb onto the walls of containers, pipettes, etc. Because FCS permits direct measurement of peptide concentration in solution, any peptide loss can be monitored during the experiment. Furthermore, FCS measurements can be carried out rapidly and require only small quantities of peptide (or protein), an important consideration when measuring proteins that are difficult to obtain and/or label. Finally, FCS can measure a large number of parameters in a small volume element: it can determine not only concentrations and mobility constants, but also dynamic and photophysical parameters, as discussed in several recent reviews (Hess et al., 2002; Krichevsky and Bonnet, 2002; Schwille and Haustein, 2002; Thompson et al., 2002).
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft grant SFB486-B10 to J.R., and by the National Health Institutes' grant GM24971 to S.M.
References
- Adam, G., and M. Delbrück. 1968. Reduction of dimensionality in biological diffusion processes. In Structural Chemistry and Molecular Biology. A. Rich and N. Davidson, editors. W.H. Freeman and Company, San Francisco. 198–215.
- Anderem, A. 1992. The MARCKS brothers: a family of protein kinase C substrates. Cell. 71:713–716. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., D. Murray, and S. McLaughlin. 1998. MARCKS, membranes, and calmodulin: kinetics of their interaction. Biochim. Biophys. Acta. 1376:369–379. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., A. A. P. Schmitz, and G. Vergeres. 2002. Cross-talk unfolded: MARCKS proteins. Biochem. J. 362:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuzova, A., J. Wang, D. Murray, J. Jacob, D. S. Cafiso, and S. McLaughlin. 1997. Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J. Biol. Chem. 272:27167–27177. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., L. Wang, J. Wang, G. Hangyas-Mihalyne, D. Murray, B. Honig, and S. McLaughlin. 2000. Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry. 39:10330–10339. [DOI] [PubMed] [Google Scholar]
- Ben-Tal, N., B. Honig, C. Miller, and S. McLaughlin. 1997. Electrostatic binding of proteins to membranes. Theoretical predictions and experimental results with charybdotoxin and phospholipid vesicles. Biophys. J. 73:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, H. C., and E. M. Purcell. 1977. Physics of chemoreception. Biophys. J. 20:193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney, C., and G. Danuser. 2003. Fluorescence resonance energy transfer or no fluorescence resonance energy transfer: a quantitative comparison. Biophys. J. 84:3992–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshear, P. J. 1993. The MARCKS family of cellular protein kinase C substrates. J. Biol. Chem. 268:1501–1504. [PubMed] [Google Scholar]
- Clamme, J. P., J. Azoulay, and Y. Mely. 2003. Monitoring of the formation and dissociation of polyethylene-amine/DNA complexes by two-photon fluorescence correlation spectroscopy. Biophys. J. 84:1960–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen, P. J. 2003. Calcium signalling: the ups and downs of protein kinase C. Curr. Biol. 13:R699–R701. [DOI] [PubMed] [Google Scholar]
- DiNitto, J. P., T. C. Cronin, and D. G. Lambright. 2003. Membrane recognition and targeting by lipid-binding domains. Sci. STKE. Dec 16;2003(213):re16. Review. [DOI] [PubMed]
- Dorn, I. T., K. R. Neumaier, and R. Tampe. 1998. Molecular recognition of histidine-tagged molecules by metal-chelating lipids monitored by fluorescence energy transfer and correlation spectroscopy. J. Am. Chem. Soc. 120:2753–2763. [Google Scholar]
- Ellena, J. F., M. C. Burnitz, and D. S. Cafiso. 2003. Location of the myristoylated alanine-rich C-kinase substrate (MARCKS) effector domain in negatively charged phospholipid bicelles. Biophys. J. 85:2442–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson, E., and D. Magde. 1974. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers. 13:1–27. [DOI] [PubMed] [Google Scholar]
- Elson, E. L. 2001. Fluorescence correlation spectroscopy measures molecular transport in cells. Traffic. 2:789–796. [DOI] [PubMed] [Google Scholar]
- Gambhir, A., G. Mihalyne, I. Zaitseva, D. S. Cafiso, J. Wang, D. Murray, S. N. Pentyala, S. O. Smith, and S. McLaughlin. 2004. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86:2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haleva, E., N. Ben Tal, and H. Diamant. 2004. Increased concentration of polyvalent phospholipids in the adsorption domain of a charged protein. Biophys. J. 86:2165–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, S. T., S. Huang, A. A. Heikal, and W. W. Webb. 2002. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 41:697–705. [DOI] [PubMed] [Google Scholar]
- Hope, M. J., M. B. Bally, G. Webb, and P. R. Cullis. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure: characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta. 812:55–65. [DOI] [PubMed] [Google Scholar]
- Kholodenko, B. N., J. B. Hoek, and H. V. Westerhoff. 2000. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 10:173–178. [DOI] [PubMed] [Google Scholar]
- Krichevsky, O., and G. Bonnet. 2002. Fluorescence correlation spectroscopy: the technique and its applications. Rep. Prog. Phys. 65:251–297. [Google Scholar]
- Langowski, J., M. Wachsmuth, K. Rippe, and M. Tewes. 2000. Biomolecular shape and interactions determined by fluorescence correlation spectroscopy. In Energies et Forces de l‘Interaction entre Macromolécules Biologiques: l’Aspect Quantitatif. J.-P. Frénoy, editor. Publications CNRS, Paris. 65–77.
- Magde, D., E. Elson, and W. W. Webb. 1972. Thermodynamic fluctuations in a reacting system-measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 64:2026–2029. [Google Scholar]
- Magde, D., E. L. Elson, and W. W. Webb. 1974. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 13:29–61. [DOI] [PubMed] [Google Scholar]
- May, S., D. Harries, and A. Ben-Shaul. 2000. Lipid demixing and protein-protein interactions in the adsorption of charged proteins on mixed membranes. Biophys. J. 79:1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey, M. A., and M. Poo. 1986. Rates of membrane-associated reactions: reduction of dimensionality revisited. J. Cell Biol. 102:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin, S., and A. Aderem. 1995. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20:272–276. [DOI] [PubMed] [Google Scholar]
- McLaughlin, S., J. Wang, A. Gambhir, and D. Murray. 2002. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31:151–175. [DOI] [PubMed] [Google Scholar]
- Mellor, H., and P. J. Parker. 1998. The extended protein kinase C superfamily. Biochem. J. 332:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, J., Y. Chen, and E. Gratton. 2003. Fluorescence correlation spectroscopy. In Methods in Enzymology. G. Marriott and I. Parker, editors. Academic Press, New York. 69–92. [DOI] [PubMed]
- Murray, D., L. H. Matsumoto, C. A. Buser, J. Tsang, C. T. Sigal, N. Ben-Tal, B. Honig, M. D. Resh, and S. McLaughlin. 1998. Electrostatics and the membrane association of SRC: theory and experiment. Biochemistry. 37:2145–2159. [DOI] [PubMed] [Google Scholar]
- Newton, A. C. 2003. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J. 370:361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori, S., N. Sakai, Y. Shirai, H. Yamamoto, E. Miyamoto, N. Shimizu, and N. Saito. 2000. Importance of protein kinase C targeting for the phosphorylation of its substrates, myristoylated alanine-rich C-kinase substrate. J. Biol. Chem. 275:26449–26457. [DOI] [PubMed] [Google Scholar]
- Peitzsch, R. M., and S. McLaughlin. 1993. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry. 32:10436–10443. [DOI] [PubMed] [Google Scholar]
- Qin, Z., and D. S. Cafiso. 1996. Membrane structure of the protein kinase C and calmodulin binding domain of myristoylated alanine-rich C kinase substrate determined by site-directed spin labeling. Biochemistry. 35:2917–2925. [DOI] [PubMed] [Google Scholar]
- Rauch, M. E., C. G. Ferguson, G. D. Prestwich, and D. S. Cafiso. 2002. Myristoylated alanine-rich C kinase substrate (MARCKS) sequesters spin-labeled phosphatidylinositol-4,5-bisphosphate in lipid bilayers. J. Biol. Chem. 277:14068–14076. [DOI] [PubMed] [Google Scholar]
- Rigler, R., and E. S. Elson. Editors .2001. Fluorescence Correlation Spectroscopy. Springer Verlag, Berlin, Heidelberg, New York.
- Schwille, P., and E. Haustein. 2002. Fluorescence Correlation Spectroscopy: A Tutorial for the Biophysics Textbook Online (BTOL), http://www.biophysics.org/btol/index.html.
- Simon, S. A., and T. J. McIntosh. Editors .2002. Peptide-Lipid Interactions. Academic Press, San Diego, CA.
- Takakuwa, Y., C. G. Pack, X. L. An, S. Manno, E. Ito, and M. Kinjo. 1999. Fluorescence correlation spectroscopy analysis of the hydrophobic interactions of protein 4.1 with phosphatidyl serine liposomes. Biophys. Chem. 82:149–155. [DOI] [PubMed] [Google Scholar]
- Thompson, N. L. 1991. Fluorescence correlations spectroscopy. In Topics in Fluorescence Spectroscopy. R. Lakowicz, editor. Plenum Press, New York. 337–378.
- Thompson, N. L., A. M. Lieto, and N. W. Allen. 2002. Recent advances in fluorescence correlation spectroscopy. Curr. Opin. Struct. Biol. 12:634–641. [DOI] [PubMed] [Google Scholar]
- Van Craenenbroeck, E., and Y. Engelborghs. 2000. Fluorescence correlation spectroscopy: molecular recognition at the single molecule level. J. Mol. Recognit. 13:93–100. [DOI] [PubMed] [Google Scholar]
- Victor, K., J. Jacob, and D. S. Cafiso. 1999. Interactions controlling the membrane binding of basic protein domains: phenylalanine and the attachment of the myristoylated alanine-rich C-kinase substrate protein to interfaces. Biochemistry. 38:12527–12536. [DOI] [PubMed] [Google Scholar]
- Wang, J., A. Arbuzova, G. Hangyás-Mihályné, and S. McLaughlin. 2001. The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 276:5012–5019. [DOI] [PubMed] [Google Scholar]
- Wang, J., A. Gambhir, G. Hangyas-Mihalyne, D. Murray, U. Golebiewska, and S. McLaughlin. 2002. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277:34401–34412. [DOI] [PubMed] [Google Scholar]
- Wang, J., A. Gambhir, S. McLaughlin, and D. Murray. 2004. A computational model for the electrostatic sequestration of PI(4,5)P2 by membrane-adsorbed basic peptides. Biophys. J. 86:1969–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W., E. Crocker, S. McLaughlin, and S. O. Smith. 2003. Binding of peptides with basic and aromatic residues to bilayer membranes: phenylalanine in the MARCKS effector domain penetrates into the hydrophobic core of the bilayer. J. Biol. Chem. 278:21459–21466. [DOI] [PubMed] [Google Scholar]