

Abstract
We report the backbone dynamics of monomeric phospholamban in dodecylphosphocholine micelles using 1H/15N heteronuclear NMR spectroscopy. Phospholamban is a 52-amino acid membrane protein that regulates Ca-ATPase in cardiac muscle. Phospholamban comprises three structural domains: a transmembrane domain from residues 22 to 52, a connecting loop from 17 to 21, and a cytoplasmic domain from 1 to 16 that is organized in an “L”-shaped structure where the transmembrane and the cytoplasmic domain form an angle of ∼80° (Zamoon et al., 2003; Mascioni et al., 2002). T1, T2, and 1H/15N nuclear Overhauser effect values measured for the amide backbone resonances were interpreted using the model-free approach of Lipari and Szabo. The results point to the existence of four dynamic domains, revealing the overall plasticity of the cytoplasmic helix, the flexible loop, and part of the transmembrane domain (residues 22–30). In addition, using Carr-Purcell-Meiboom-Gill-based experiments, we have characterized phospholamban dynamics in the μs-ms timescale. We found that the majority of the residues in the cytoplasmic domain, the flexible loop, and the first ten residues of the transmembrane domain undergo dynamics in the μs-ms range, whereas minimal dynamics were detected for the transmembrane domain. Hydrogen/deuterium exchange factors measured at different temperatures support the existence of slow motion in both the loop and the cytoplasmic helix. We propose that these dynamic properties are critical factors in the biomolecular recognition of phospholamban by Ca-ATPase and other interacting proteins such as protein kinase A and protein phosphatase 1.
INTRODUCTION
PLB is an integral membrane protein that regulates intracellular calcium transport in cardiac muscle. Recent studies in transgenic mice have suggested a significant role for PLB in cardiac disease (Chu et al., 2002). In fact, a single mutation of PLB was directly implicated in human dilated cardiomyopathy with consequent heart failure (Schmitt et al., 2003). As new research highlights the importance of this protein for healthy cardiac function, it will become an increasingly prominent therapeutic target in the treatment of cardiac disease (MacLennan and Kranias, 2003).
PLB decreases the rate of cardiac relaxation by inhibiting the ability of the Ca-ATPase to pump calcium into the SR (MacLennan and Kranias, 2003). In the SR membrane, PLB is thought to exist as a pentamer (Arkin et al., 1997) and to undergo depolymerization upon association with Ca-ATPase (Reddy et al., 1999). In its enzymatic cycle, Ca-ATPase is proposed to exist in two distinct conformations: high Ca2+ affinity (E1) and low Ca2+ affinity (E2), with both ATP binding and autophosphorylation controlling the calcium translocation process (Stokes and Green, 2003). It is proposed that PLB interacts with both the cytoplasmic and transmembrane sites of the enzyme, binding with higher affinity to the E2 conformation. Upon β-adrenergic stimulation of the cardiac myocyte (Tada and Kadoma, 1989), PLB is phosphorylated at Ser-16 by cAMP-dependent protein kinase and at Thr-17 by Ca/calmodulin-dependent kinase. This reverses the inhibition of the Ca-ATPase, increasing the rate of calcium transport across the SR membrane and, thus, cardiac relaxation (Simmerman and Jones, 1998).
Since the monomeric form of PLB is the active form for Ca-ATPase inhibition (Kimura et al., 1997; Cornea et al., 1997), we have focused our investigation on a fully functional, monomeric PLB mutant in which the three transmembrane cysteines have been replaced by A36, F41, and A46, respectively (Karim et al., 1998, 2000). The solution NMR structure of this monomeric mutant of PLB (AFA-PLB) in DPC micelles has recently been determined (Zamoon et al., 2003). AFA-PLB is comprised of three structural domains: a hydrophobic helix, an amphipathic helix, and a five-residue connecting β-turn loop. The paramagnetic quenching experiments reported in that study show that the amphipathic helix, ranging from residues 2 to 16, is partially embedded in the detergent micelle, with the hydrophobic residues (A15, A11, L7, and V4) pointing toward the hydrocarbon chains, whereas the more hydrophilic residues (T17, S16, R13, and R9) are exposed to the bulk solvent, rendering the two phosphorylation sites, Ser-16 and Thr-17 adjacent to the loop region, more accessible to kinases. The hydrophobic helix, comprising residues 22–52, is embedded in the micellar core between residues 35 and 50. These data corroborate the solid-state NMR study of PLB in oriented lipid bilayers, which found that one helix spanned the lipid bilayer at an ∼10° angle with the lipid plane (transmembrane helix) and one lay on the surface of the lipid bilayer (an amphipathic helix) (Mascioni et al., 2002).
Determining the structure of PLB and its topology in lipid bilayers is only the first step toward understanding PLB's role in the complex molecular mechanisms that regulate muscle contractility. The importance of this small membrane protein in the regulation of muscle contractility stems from its ability to interact with several proteins in vivo: Ca-ATPase, protein kinase A, Ca/calmodulin-dependent kinase, and protein phosphatase 1. Therefore, the recognition of PLB by these proteins must be encoded not only in PLB's primary and secondary structure but also in its ability to mold itself to each of these proteins, making the characterization of PLB dynamics critical to determining how these conformational changes originate.
Analyzing 15N relaxation parameters by solution NMR is a powerful means of characterizing protein backbone dynamics and identifying the domains involved in protein-protein interactions (Ishima and Torchia, 2000). In this article, we report the analysis of the backbone dynamics of AFA-PLB in DPC micelles. The relaxation values are interpreted using the model-free approach of Lipari and Szabo (1982a,b). The dynamic properties of each domain in AFA-PLB are also correlated with its thermodynamic stability as assessed by hydrogen/deuterium exchange factors (Veglia et al., 2002). Our results indicate that whereas PLB has three distinct structural domains, it has four dynamic domains. In addition to the amphipathic helix and the loop, our data show that there are two distinct dynamic behaviors in the transmembrane helix, dividing it into a more flexible region (residues 22–30) and a rigid region (residues 31–52). Our results corroborate early sequence analysis studies, which predicted that the region spanning residues 22–30 would be hydrophilic and hence separate from the hydrophobic transmembrane domain (Fujii et al., 1987). Though this inherent difference is undetectable form the static NMR structure, it appears when the dynamic behavior of the backbone is probed. The mobility observed in residues 22–30 may have strong implications for the function of PLB, giving PLB the flexibility it needs to conform to each of the proteins it must interact with to fulfill its physiological purpose.
MATERIALS AND METHODS
Expression and purification of 15N AFA-PLB
15N AFA-PLB was expressed and purified as previously described (Buck et al., 2003). 15N AFA-PLB was expressed in Escherichia coli BL21DE3 cells with an MBP fusion partner and a TEV protease cleavage site incorporated between them. Purification of the fusion protein was accomplished by affinity chromatography on an amylose resin column, followed by cleavage with recombinant TEV protease. Final separation was achieved by fast protein liquid chromatography, followed by dialysis and lyophilization, resulting in a white powder.
TEV protease was expressed and purified as previously described (Parks et al., 1994) in E. coli BL21DE3 cells with an MBP fusion partner and a 6x-His tag. TEV protease self-cleaves in vivo to remove the fusion partner. Purification was accomplished using a Ni-nitriloacetic acid resin (Qiagen, Germantown, MD) affinity chromatography column.
NMR spectroscopy
Samples were prepared by dissolving uniformly 15N-labeled AFA-PLB in 300 μL phosphate-buffered saline, pH 4, containing 600 mM deuterated DPC and 10% D2O. All experiments were carried out at 50°C on a Varian INOVA spectrometer equipped with a triple resonance probe operating at 1H Larmor frequency of 600.48, except for the CPMG-based experiments designed to elucidate μs-ms timescale motions, which were carried out on a similarly equipped Varian INOVA spectrometer operating at 1H Larmor frequency of 800.23 MHz.
15N relaxation measurements were acquired using two-dimensional, proton-detected heteronuclear NMR experiments, implementing standard pulse sequences based on Farrow et al. (1994).
T1 and T2 spectra were recorded with spectral widths of 7000 Hz sampled over 896 complex points in the ω2 (1H) dimension, and 1700 Hz over 64 complex points in the ω1 (15N) dimension with 64 scans for each increment in the indirect dimension. The heteronuclear steady-state NOE spectra were acquired with a spectral width of 7000 Hz over 1024 complex points in the ω2 (1H) dimension and 1700 Hz over 96 complex points in the ω1 (15N) dimension. 15N decoupling during acquisition was achieved using a GARP-1 pulse sequence (Shaka et al., 1985). The field strength of the CPMG refocusing train was ∼3.3 kHz and a 1.2-ms delay was used between the refocusing pulses (Carr and Purcell, 1954; Meiboom and Gill, 1958). The effects of cross-relaxation between 1H-15N dipolar and 15N chemical shift anisotropy were removed applying 1H 180° pulses during relaxation delays (Palmer et al., 1992). The relaxation delay for both T1 and T2 measurements was 1.5 s.
T1 values were measured in a series of spectra with relaxation delays of 10, 20, 40, 180, 300, 500, 800, 1000, and 1100 ms. T2 measurements were taken with relaxation delays of 10, 30, 50, 70, 90, 110, and 150 ms. To allow NOE evolution, 1H-15N steady-state NOE values were measured with two different data sets, one collected with no initial proton saturation and a second with initial proton saturation. The proton saturation period was 3 s.
To characterize the μs-ms timescale dynamics of AFA-PLB, we analyzed the conformational exchange broadening using a semiquantitative approach recently described by Wang et al. (2001). First, R2 was measured at 800 MHz using a composite refocusing pulse during the R2 period, while a WALTZ-16 proton decoupling was active during the R2 evolution period to eliminate the effects of 15N-H1 scalar coupling, 15N chemical shift anisotropy, and 15N-1H dipole cross-correlation. The R2 measurement was repeated with a CPMG pulse train during R2 time 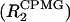 to suppress the contributions to the relaxation arising from chemical or conformational exchange. Spectra were acquired with 64 points in the ω1 (15N) dimension and 2456 complex points in the ω2 (1H) dimension, with spectral widths of 1700 and 9000 Hz, respectively. The repetition rates within the CPMG pulse train (τp) used were 0.4, 0.8, and 1.6 ms. The relaxation delays for both R2 and
to suppress the contributions to the relaxation arising from chemical or conformational exchange. Spectra were acquired with 64 points in the ω1 (15N) dimension and 2456 complex points in the ω2 (1H) dimension, with spectral widths of 1700 and 9000 Hz, respectively. The repetition rates within the CPMG pulse train (τp) used were 0.4, 0.8, and 1.6 ms. The relaxation delays for both R2 and 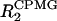 experiments were 10, 30, 50, 70, 90, 110, and 150 ms.
experiments were 10, 30, 50, 70, 90, 110, and 150 ms.
For the determination of the H/D exchange factors, AFA-PLB samples were lyophilized and resuspended in solutions containing 10%, 30%, 50%, and 70% D2O, respectively, and the HSQC spectra were obtained after a fixed 30-min incubation period at different temperatures (i.e., 35, 37, 40, 45, 50, and 55°C). The intensities of the amide resonances were measured using NMRView software and the values were normalized to those found in the sample with 10% D2O. Peak intensities were plotted as a function of the mole fraction of H2O in the solution, and the exchange factors, χ, were determined using the following equation:
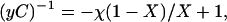 |
where y is the peak volume, C is a normalization factor, and X is the mole fraction of H2O in the solution (Veglia et al., 2002).
Data processing and analysis
The spectra were processed using NMRPipe software (Delaglio et al., 1995). Peak intensities were measured and analyzed using the peak picking routine built into NMRView (Johnson and Blevins, 1994). The T1 and T2 spectra were processed using a sine bell apodization function shifted by 90° for both dimensions. The final sizes of the matrices were 1024 × 128 real points after zero filling in both dimensions and the Fourier transformation. An automated baseline correction was applied in both dimensions, and a linear prediction of 64 points in the ω1 (15N) dimension. The spectra were referenced to the DSS signal (Wishart et al., 1995).
T1 and T2 values were obtained by fitting peak intensities using single exponential decay:
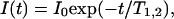 |
where I(t) is the peak intensity, t is the time, and I0 is the intensity at time 0.
The analysis of the uncertainties of the T1 and T2 values was carried out in several different ways: 1), by estimating the root-mean-square baseplane noise (σt); 2), by comparing the peak heights on duplicate spectra at 10 ms (shortest value of relaxation delay) (σh); and 3), by using the “jackknife” method (Skelton et al., 1993; Quenouille, 1956). Under our experimental conditions, methods 1 and 2 gave approximately the same results with an average uncertainty of <1%, whereas method 3 gave an average uncertainty of ∼1.5% for T1. Due to the lower S/N ratio in our T2 experiments, the uncertainty for these experiments is somewhat higher. The estimated error from the base plane noise is ∼6%. A comparable error was obtained using the other two methods.
The analysis of the conformational exchange broadening experiments was carried out using the above equation for the peak intensities. Since no duplicate experiments were available, the errors for R2 and 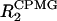 were estimated, using the “jackknife” method, to be between 1 and 2%. These errors were propagated for the ΔR2 calculations.
were estimated, using the “jackknife” method, to be between 1 and 2%. These errors were propagated for the ΔR2 calculations.
The heteronuclear steady-state 15N-1H NOE values were obtained from the ratios of peak intensities in the saturated spectrum to those in the unsaturated spectrum. Errors were estimated by evaluating the standard deviation of the NOE, σNOE:
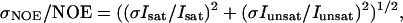 |
where σIsat and σIunsat are the standard deviations of the noise in the spectra (Farrow et al., 1994).
Model-free analyses (Lipari and Szabo, 1982a,b) were performed using Modelfree software (Palmer et al., 1991). A sum squared error was minimized for each residue in five different models: 1), S2, 2), S2 and τe, 3), S2 and Rex, 4), S2, τe, and Rex, and 5), S2s, S2f, and τe. S2 is the generalized order parameter. τe, the effective internal correlation time, is on the picosecond timescale. Rex is a chemical exchange term. S2s and S2f are terms that result from splitting the generalized order parameter into two order parameters. S2s reflects slower motions and S2f, faster motions. Model selection was performed as previously described according to Mandel et al. (1995). The calculations were repeated for two diffusion models, isotropic and local.
For the isotropic model, an initial τM of 11.5 ns was assumed. For the local diffusion model, in addition to the model-free parameters a nonlinear least squares fit was used to optimize a local correlation time, τloc, at each residue. In this model, the local correlation time, τloc, replaces the global correlation time, τm, and no global rotational diffusion model is assumed. For both the isotropic and local models, 500 synthetic data sets were generated using Monte Carlo simulations and used for error estimation. The uncertainties in the model-free parameters were reported as the standard deviation of the simulated parameters.
Separate calculations were also carried out using the local diffusion model assuming that the N- and C-terminal domains reorient independently. The PLB molecule was divided into two regions, the first spanning from residue 2 through 22, and the second from 23 through 52. This procedure is reminiscent of the dynamics analysis carried out with calmodulin (Tjandra et al., 1995a,b; Evenas et al., 1999). The results obtained show that S2, τe, and Rex terms are identical to dynamics analysis carried out with the full-length protein (data not shown).
Given the “L”-shape topology of PLB (Zamoon et al., 2003), axial diffusion should represent a valid alternative model for interpreting the relaxation data (Lee et al., 1997; Bruschweiler et al., 1995). In general, the diffusion tensor can be calculated using molecular Cartesian coordinates and the T1/T2 ratios for each residue (Lee et al., 1997). The accuracy of the diffusion tensor relies on the exclusion of spins that undergo slow motion (Tjandra et al., 1995a), and requires the exclusion of 1), residues with NOE < 0.65, and 2), residues that do not satisfy the following condition:
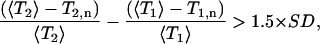 |
where T2,n is the T2 value for residue n, and 〈T2〉 is the average T2 value, and SD is the standard deviation of the first member of the above inequality (Tjandra et al., 1995a). In the case of AFA-PLB, we would need to exclude all the residues of the cytoplasmic domain from 2 through 30 with the exception of residue 14, leaving only the residues in the transmembrane helix suitable for the calculation of the rotational diffusion tensor. Moreover, the residues suitable for these calculations adopt an α-helix conformation with the NH vectors approximately parallel to the helix axis. Thus, these residues would provide only redundant angular information, biasing the determination of the rotational diffusion tensor. Finally, it should be noted that AFA-PLB is embedded in a micelle and the overall correlation time is affected by the presence of the micelle. Given all the above, we concluded that under our conditions the axial diffusion model is not applicable.
RESULTS AND DISCUSSION
NMR relaxation data
The backbone dynamics of AFA-PLB have been determined through solution NMR measurements of relaxation parameters T1, T2, and the steady-state NOE of the amide resonances. Fig. 1 represents the results of the relaxation measurements, showing the measured values plotted against residue number. Note that the recombinant AFA-PLB contains a residual Ala at the N-terminus from the incorporated cleavage site (Buck et al., 2003), so the first observed residue is Met-1 of the native PLB sequence. Residues for which relaxation measurements could not be assigned included the N-terminal A, P21, and overlapping residues A15 and M20, R25 and N27, and R14 and L39.
FIGURE 1.

15N backbone relaxation measurements for AFA-PLB in DPC micelles. T1 (A), T2 (B), and 1H/15N heteronuclear NOE experiments (C) performed on a 600 MHz spectrometer (see Materials and Methods for experimental details).
The values of the three sets of relaxation measurements (NOE, T1, and T2) are correlated with the mobility of each amide in the protein backbone. Internal motions affect the rate at which an excited nucleus may sample the fluctuating fields around it to exchange energy and relax. Examination of the experimental data immediately reveals that AFA-PLB has distinctly different motional regimes across the protein backbone (Fig. 1). In all three data sets, the amphipathic helix shows a pattern of decreasing internal motions from the N-terminus to the loop (residues 1–16). The loop (residues 17–21) has much faster motions, with heteronuclear NOE values as low as 0.2. The transmembrane helix (residues 22–52) can clearly be divided into two motional regimes, discounting the fast motion observed at the terminus. Beyond approximately residue 33, the transmembrane domain reveals the slowest internal motions of AFA-PLB, with heteronuclear NOE values approaching 0.8. Residues 22–32, however, have much faster motions. Residue 22 has the fastest internal motion of the protein, as revealed by its T2 value, which is the longest in the protein, and its heteronuclear NOE value, which is comparable to those in the loop. Both the heteronuclear NOE and T1 values slowly increase from the loop-like residue 22 to the transmembrane-like residue 32. The T2 values reveal a two-tiered transmembrane domain, with residues 22–32 demonstrating, on the average, longer T2 values than residues 32–52 (Fig. 1).
Fig. 2 reveals that there is a significant difference between the T1/T2 ratios of the two helices of AFA-PLB. Table 1 presents the results of the T1/T2 averages taken for the entire protein as well as when separated into cytoplasmic and transmembrane domains. The ratio of T1/T2 is maximal when a helix is aligned with the major principle component of the diffusion tensor and minimal when aligned with the minor principle component (Bruschweiler et al., 1995; Campos-Olivas and Summers, 1999). The different T1/T2 ratios indicate that the two helices are not collinear, but aligned differently with the principle components of the diffusion tensor. This is in agreement with both the solution structure of AFA-PLB and the solid-state NMR study which determined the orientation of the two helices of PLB to be approximately perpendicular to each other (Zamoon et al., 2003; Mascioni et al., 2002). The ratio of T1/T2 can be also be used to estimate the overall rotational correlation time of a protein, τm, based on the spectral density function. (Lee et al., 1997). Although the 80° angle between the two domains may explain the differences in the average T1/T2 ratios of the two helices of AFA-PLB, this cannot account for the low values of T1/T2 ratios observed in the first ten residues of the transmembrane helix. These residues show increasing T1/T2 ratios going from 5 to 10, whereas the average value of the remaining residues is ∼15. This provides further evidence that these residues are less restricted and undergo different dynamics from the rest of the transmembrane domain.
FIGURE 2.

Plot of T1/T2 ratios obtained from data in Fig. 1.
TABLE 1.
Estimated τm values (ns) based on T1/T2 ratios
| Standard deviation | ||
|---|---|---|
| τm (overall) | 11.5 | 3.2 |
| τm (domains I, the loop, and Ib) | 8.2 | 0.4 |
| τm (domain II) | 15.4 | 0.8 |
Model-free data analysis
The model-free analysis of AFA-PLB was carried out with Modelfree software (Palmer et al., 1991) and the model was selected according to the criteria described by Mandel et al. (1995). Five parameter sets were fit to both the isotropic and the local diffusion models, as described in the Materials and Methods section. The isotropic diffusion model gave rise to order parameters in the transmembrane domain residues with values between 0.4 and 0.5, which is inconsistent with the structure determined by Zamoon et al. (2003). Therefore, the local diffusion model was chosen to describe the dynamics of AFA-PLB. Table 2 summarizes the models chosen for each residue. The best fit for residues undergoing chemical exchange was obtained with a three-parameter fit (model 4). In particular, the residues of the cytoplasmic helix, 1 through 11, were fit with model 4, with the exception of E2, K3, and R9. It should be noted that R9 and R13 were fitted using model 1 and appear to be more motionally restricted. This may be due to their interactions with the headgroups of the DPC micelle. Residues involved in the loop and part of domain Ib were fitted with model 3, whereas the remainder of domain Ib was better interpreted using model 4. The relaxation data for the most hydrophobic portion of PLB (residues 32–54) have been interpreted using models 2 and 1, with the exception of residues 35–37 and 40, and the C-terminal residues 50–52. Residues that could not be fit using any model included I12 and L41.
TABLE 2.
Summary of model-free results
| Residue | Model | S2 | dS2 | τe (ps) | dτe | Rex (s−1) | dRex |
|---|---|---|---|---|---|---|---|
| Met-1 | 4 | 0.74 | 0.04 | 108 | 78 | 3.5 | 2.2 |
| Glu-2 | 2 | 0.78 | 0.05 | 435 | 139 | ||
| Lys-3 | 3 | 0.69 | 0.06 | 7.3 | 2.8 | ||
| Val-4 | 4 | 0.79 | 0.07 | 87 | 91 | 1.9 | 1.7 |
| Gln-5 | 4 | 0.89 | 0.04 | 134 | 34 | 1.0 | 1.5 |
| Tyr-6 | 4 | 0.90 | 0.04 | 105 | 70 | 1.5 | 1.5 |
| Leu-7 | 4 | 0.95 | 0.05 | 167 | 100 | 0.6 | 1.6 |
| Thr-8 | 4 | 0.90 | 0.02 | 132 | 23 | 3.3 | 2.1 |
| Arg-9 | 1 | 1.00 | 0.07 | ||||
| Ser-10 | 4 | 0.90 | 0.03 | 45 | 45 | 4.0 | 2.1 |
| Ala-11 | 4 | 0.95 | 0.04 | 155 | 93 | 1.3 | 1.6 |
| Arg-13 | 1 | 1.00 | 0.06 | ||||
| Ser-16 | 3 | 0.54 | 0.04 | 10.5 | 2.3 | ||
| Thr-17 | 3 | 0.66 | 0.05 | 16.0 | 2.5 | ||
| Ile-18 | 2 | 0.79 | 0.07 | 295 | 106 | ||
| Glu-19 | 3 | 0.61 | 0.03 | 7.8 | 2.4 | ||
| Gln-22 | 3 | 0.63 | 0.05 | 5.6 | 3.0 | ||
| Gln-23 | 3 | 0.75 | 0.05 | 11.1 | 2.3 | ||
| Ala-24 | 4 | 0.78 | 0.01 | 341 | 32 | 7.8 | 2.0 |
| Gln-26 | 4 | 0.84 | 0.01 | 153 | 24 | 5.0 | 2.4 |
| Leu-28 | 4 | 0.89 | 0.03 | 178 | 67 | 2.8 | 2.3 |
| Gln-29 | 4 | 0.95 | 0.04 | 171 | 86 | 0.8 | 1.9 |
| Asn-30 | 4 | 0.90 | 0.01 | 128 | 19 | 4.3 | 1.9 |
| Leu-31 | 4 | 0.95 | 0.04 | 236 | 108 | 1.4 | 2.1 |
| Phe-32 | 2 | 0.95 | 0.06 | 21 | 16 | ||
| Ile-33 | 2 | 0.95 | 0.07 | 21 | 38 | ||
| Asn-34 | 1 | 1.00 | 0.05 | ||||
| Phe-35 | 4 | 0.95 | 0.04 | 66 | 24 | 2.6 | 2.9 |
| Ala-36 | 4 | 0.95 | 0.02 | 72 | 18 | 4.6 | 2.8 |
| Leu-37 | 4 | 0.95 | 0.02 | 50 | 17 | 3.6 | 2.8 |
| Ile-38 | 1 | 1.00 | 0.07 | ||||
| Ile-40 | 4 | 0.95 | 0.03 | 48 | 17 | 2.5 | 2.6 |
| Leu-42 | 2 | 0.95 | 0.05 | 21 | 23 | ||
| Leu-3 | 2 | 0.95 | 0.07 | 21 | 16 | ||
| Leu-44 | 1 | 1.00 | 0.05 | ||||
| Ile-45 | 2 | 0.95 | 0.06 | 21 | 17 | ||
| Ala-46 | 1 | 1.00 | 0.05 | ||||
| Ile-47 | 2 | 0.95 | 0.06 | 21 | 21 | ||
| Ile-48 | 4 | 0.95 | 0.02 | 65 | 18 | 4.6 | 3.2 |
| Val-49 | 1 | 1.00 | 0.05 | ||||
| Met-50 | 4 | 0.95 | 0.02 | 47 | 16 | 3.8 | 2.8 |
| Leu-51 | 4 | 0.89 | 0.03 | 110 | 66 | 4.1 | 2.8 |
| Leu-52 | 4 | 0.90 | 0.05 | 161 | 48 | 0.3 | 1.3 |
Overlapped residues and residues that could not be fit with any model are excluded from the table.
Fig. 3 shows the order parameters, the internal correlation times, and the exchange terms as functions of the residue number. Order parameters reveal relatively restricted motions across the protein, but with patterns similar to those of the experimental relaxation data. Order parameters increase steadily across the amphipathic helix to residue 13. A decrease is observed starting at residue 16 to a minimum order parameter of 0.65 at residue 22. The values then steadily increase to about residue 30, where very restricted motions are then observed throughout the transmembrane helix. The effective internal correlation time, τe, reveals long correlation times up to residue 33 and short correlation times in the transmembrane domain starting at 34. Though the τe values do not distinguish between the hydrophilic sections of the protein, the order parameters clearly delineate the four dynamic domains of AFA-PLB. Domain Ia comprises the most rigid cytoplasmic section (residues 1–16). The loop contains the least rigid residues of the protein (residues 17–21). Domain Ib represents the hydrophilic residues of the transmembrane helix (residues 22–30). Finally, domain II covers the membrane-spanning residues 31–52. The calculated exchange terms are higher in both the loop and domain Ib, with lower values in domains Ia and II.
FIGURE 3.

Model-free analysis of 15N relaxation data of AFA-PLB. Order parameter (S2) (top), internal correlation time (τe) (middle), and exchange terms (Rex) (bottom) plotted as a function of the residue number for AFA-PLB. The four dynamics domains are color-coded and mapped onto a representative structure of PLB determined in micelles using solution NMR experiments (Zamoon et al., 2003). The color scheme is blue, S2 ≥ 0.85, red, S2 ≥ 0.75, and yellow, S2 < 0.75.
It should be noted that separate calculations carried out assuming that the N- and C-terminal domains of PLB reorient independently gave identical results for the S2 and for the Rex terms (data not shown).
Conformational exchange analysis
Slow motion in the NMR timescale (μs-ms dynamics) can be investigated using either T1ρ measurements with the maximum effective radio frequency field strength or CPMG-based experiments (Palmer et al., 2001). To assess μs-ms dynamics for AFA-PLB, we used a fast method that was recently developed by Zuiderweg and co-workers (Wang et al., 2001). This method is based on the differences between R2 (relaxation rates) values measured using a pulse sequence with a single, composite refocusing pulse during the R2 evolution period, and 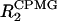 values, where a CPMG train of pulses substitutes for the single pulse. The differences between these
values, where a CPMG train of pulses substitutes for the single pulse. The differences between these  and R2 values are directly correlated with the Rex terms and are reported in Fig. 4 A. Values close to zero correspond to an absence of dynamics in the μs-ms timescale, whereas higher values of ΔR2 indicate residues that are involved in conformational exchange. The C-terminal residues 50 and 52 display higher values of ΔR2, whereas the ΔR2 values of the remaining residues embedded in the membrane bilayer (40–49) do not differ significantly, indicating that this portion of AFA-PLB is not affected by conformational exchange. In contrast, a substantial difference in ΔR2 values is observed for residues 1–30, which constitute domain Ia, the loop, and domain Ib of AFA-PLB. In addition, higher ΔR2 values are present in residues 36 and 37, indicating that their dynamics in the μs-ms timescale substantially modulate their chemical shifts.
and R2 values are directly correlated with the Rex terms and are reported in Fig. 4 A. Values close to zero correspond to an absence of dynamics in the μs-ms timescale, whereas higher values of ΔR2 indicate residues that are involved in conformational exchange. The C-terminal residues 50 and 52 display higher values of ΔR2, whereas the ΔR2 values of the remaining residues embedded in the membrane bilayer (40–49) do not differ significantly, indicating that this portion of AFA-PLB is not affected by conformational exchange. In contrast, a substantial difference in ΔR2 values is observed for residues 1–30, which constitute domain Ia, the loop, and domain Ib of AFA-PLB. In addition, higher ΔR2 values are present in residues 36 and 37, indicating that their dynamics in the μs-ms timescale substantially modulate their chemical shifts.
FIGURE 4.

(A) Plot of the difference between 15N 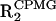 and R2, performed with and without CPMG pulse train, respectively. Large values of ΔR2 correspond to conformational exchange broadening caused by motion in the μs-ms timescale. (B) Comparison of the experimental exchange terms (black bars) versus exchange terms calculated from model-free analysis (white bars).
and R2, performed with and without CPMG pulse train, respectively. Large values of ΔR2 correspond to conformational exchange broadening caused by motion in the μs-ms timescale. (B) Comparison of the experimental exchange terms (black bars) versus exchange terms calculated from model-free analysis (white bars).
Fig. 4 B shows a comparison of the chemical exchange values (Rex) obtained from CPMG experiments and those calculated from the model-free approach. It is clear that model-free analysis overestimates the exchange terms. This apparent discrepancy is most likely due to the presence of anisotropic diffusion of the PLB/micelle complex. In fact, changes in R2 relaxation time can be misinterpreted as conformational exchange effects in model-free analysis, so that particular care must be taken when interpreting the Rex terms (Schurr et al., 1994; Tjandra et al., 1996; Evenäs et al., 1999; Osborne and Wright, 2001). As explained in the Materials and Methods section, the majority of the residues in the cytoplasmic region of AFA-PLB do not meet the selection criteria necessary to define the rotation diffusion tensor, rendering the axial diffusion model inapplicable. The local diffusion model used in our interpretation of PLB dynamics may likewise cause the overestimation of the conformational exchange terms. Nonetheless, the qualitative agreement between the Rex experimental data and that calculated as shown in Fig. 4 B confirms the presence of dynamics in the μs-ms range for domains Ia, Ib, and the flexible loop.
These slow exchange dynamics are also supported by the proton/deuterium exchange factors (χ) (Veglia et al., 2002) measured at different temperatures (Fig. 5). Small values for these exchange factors have been correlated with the most thermodynamically stable (rigid) regions of membrane proteins, whereas higher exchange factors are indicative of residues that are more exposed to solvent exchange, and less thermodynamically stable (mobile) (Veglia et al., 2002). As shown in Fig. 5, at 35°C the exchange factors reveal two distinct regions: one with higher exchange factors spanning residues 1–33, and a second with lower exchange factors, from 34 to 50. At 37°C, the profile delineated by the exchange factors changes and the region from 34 through 38 shows higher values of χ. These changes are more marked as the temperature increases. This supports the additional dynamics found in this region using CPMG-based experiments.
FIGURE 5.

Plot of the exchange factors for AFA-PLB solubilized in DPC micelles as carried out at 35, 37, 40, 45, 50, and 55°C.
In sum, it is possible to conclude that 1), the most exchange-protected region is limited to residues 40–50 (more dynamically restricted or motionally correlated); 2), domains Ia, the loop, and Ib are more solvent-exposed and prone to hydrogen bonding opening (more mobile); and 3), the first part of domain II spanning residues 29–38 is slightly less resistant to exchange than the rest of domain II. These results are in remarkable agreement with cysteine accessibility studies carried out on single-cysteine PLB mutants reconstituted in lipid membranes (Cornea et al., 2002), showing that our investigation is directly relevant to the structural dynamics of PLB in lipid membranes.
Comparison of PLB dynamics with the dynamics of other single-pass membrane proteins
15N backbone dynamics data have been obtained and analyzed for several monotopic membrane proteins similar in structure and topology to AFA-PLB. IKe major coat protein (Williams et al., 1996), fd coat protein (Almeida and Opella, 1997), and M13 coat protein (Papavoine et al., 1997) are all comprised of a single transmembrane helix connected to an amphipathic helix by a hinge region. Though the structures of these proteins are similar, the dynamics data and analyses are markedly different for all three proteins, as well as for AFA-PLB. AFA-PLB shows clear differences between its amphipathic and transmembrane helices, as well as sharp differences in its loop region in the NOE, T1, and T2 values. fd coat protein has slightly slower motions in its transmembrane domain than in the amphipathic helix and loop regions. IKe major coat protein shows a small dip in T1 value in its loop region, but no change for T2 and NOE between the terminal regions. M13 coat protein shows drastic changes between domains: the amphipathic helix has much faster motions than the transmembrane helix, and there is a slight dip in the loop region. These differences are reflected in the model-free analyses.
The internal rotational dynamics of AFA-PLB was fit with one-, two-, and three-parameter models in which S2, τe, and Rex characterize the subnanosecond internal dynamics of different residues. fd coat protein was fit with S2 and τe, and then an additional order parameter and correlation time for nanosecond motions of the amphipathic helix were modeled as diffusion in a cone. IKe major coat protein was fit with S2 and Rex. M13 coat protein was fit with a combination of models: S2, S2 and τe, and S2s, S2f, and τe. The order parameters for these proteins all approach unity in helical regions, except for M13 coat protein. This protein has order parameters around 0.5 through the amphipathic helix and around 1 in the transmembrane helix. Although there are structural, sequence, and topological similarities, the different dynamics of these membrane proteins, rather than their structure and topology, may account for their different functionalities.
Dynamics of PLB and its functional implications
Using electron paramagnetic resonance and time-resolved phosphorescence anisotropy Thomas and co-workers have shown that protein rotational dynamics play a critical role in the functional interaction of PLB and the Ca-ATPase (Cornea et al., 1997; Thomas et al., 1998; Kirby et al., 2004). The common model proposed by these studies is that PLB undergoes large conformational and dynamical changes upon interaction with Ca-ATPase, and that these in turn affect the internal dynamics of the Ca-ATPase itself. In particular, the cytoplasmic domain seems to be the most affected by the presence of the enzyme, which appears to pull domain Ia up and away from the membrane surface. Since these insightful studies were carried out on single-site labeled PLB, they do not provide a collective view of the protein dynamics. Therefore, the aim of our investigation is to completely characterize the PLB backbone dynamics to understand the implications of PLB dynamics for the recognition process by several different proteins, including Ca-ATPase, protein kinases, and protein phosphatase 1.
The most important results from our investigations are 1), the identification of four different dynamical domains in PLB, with two transmembrane subdomains that are distinguishable only when dynamics is taken into account; and 2), the elucidation of the dynamics of the cytoplasmic helix of PLB in the μs-ms timescale. Although our conclusions are based on studies in detergent micelles, the dynamic features of the different domains in AFA-PLB have been recently confirmed by the site-directed spin-labeling of AFA-PLB in lipid bilayers (Karim et al., 2004). These latter results confirm two key conclusions of this study: the cytoplasmic domain of PLB is substantially more dynamic than the transmembrane domain, and the cytoplasmic domain contains at least two distinct dynamical subdomains. This study's results are thus directly relevant to the structural dynamics of PLB in lipid membranes.
The plasticity of the flexible portion of the transmembrane helix of PLB has also been hypothesized in a recent molecular modeling article, where to fulfill the “biochemical constraints” obtained from coimmunoprecipitation assays, the authors modeled an “unwound” structure of PLB positioned in the groove between M3 and M4 of the Ca-ATPase (Toyoshima et al., 2003). In this article, upon binding to the Ca-ATPase, residues 22–30 are predicted to lose their helical structure, whereas the transmembrane-spanning portion remains helical. This model is also supported by recent fluorescence experiments, demonstrating that a monomeric PLB mutant labeled at A24C shows increased local motion upon interaction with Ca-ATPase (Li et al., 2003). In a recent article, Hughes and Middleton published evidence that contradicts “unwound” conformation of PLB in the bound form; instead, they provide some evidence for the formation of a continuous 52-amino acid helix of PLB bound to Ca-ATPase (Hughes and Middleton, 2003). These results will remain inconclusive until the full structure of PLB bound to Ca-ATPase is elucidated. Nonetheless, it is worth mentioning that the dramatic structural and dynamical changes occurring between the E1 and E2 forms of the enzyme suggest that the structure of PLB needs to be rather flexible and in an unfavorable, high-energy state to allow the enzyme turnover to take place. To this extent, the elucidation of the μs-ms dynamics of the cytoplasmic, amphipathic helix supports the hypothesis that changes in both the dynamics and the conformation of PLB are required for the inhibitory process to take place. Since the μs-ms dynamics have been generally associated with the regions of proteins involved in protein-protein interactions (Columbus and Hubbell, 2002), our results may explain the eclectic behavior of PLB, which is able to interact with Ca-ATPase, PKA, protein phosphatase 1, as well as the lipid membranes.
CONCLUSIONS
Although PLB has three structural domains, its backbone dynamics reveal a more complicated picture. PLB cannot simply be viewed as two rigid helices connected by a hinge-like loop; rather, our dynamics analysis identifies four dynamics domains on the ps-ns timescale: domain Ia (residues 1–16), the loop (residues 17–22), domain Ib (residues 23–30), and domain II (31–52). The relaxation analysis of the slow dynamics shows the presence of μs-ms motion in domain Ia, the loop, and domain Ib, whereas domain II is more motionally restricted. The μs-ms dynamics of domain Ia and the loop were an expected result, but the flexibility of residues 22–30 (domain Ib) was not. We propose that the plasticity of this region is a key factor that allows this small membrane protein to adapt its conformation to several different targets by facilitating PLB recognition by several different proteins, including Ca-ATPase, protein kinase A, Ca/calmodulin-dependent kinase, and protein phosphatase 1.
Acknowledgments
The authors acknowledge Francesca Massi for many helpful discussions, and David Live and Beverly Ostrowski for help with the NMR experiments. We also benefited from the Minnesota High-Field NMR Facility in the Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota.
NMR instrumentation was provided with funds from the National Science Foundation (BIR-961477) and the University of Minnesota Medical School. This work was supported in part by grants to G.V. (National Institutes of Health grant GM64742; American Heart Association grant 0160465Z), and D.D.T. (National Institutes of Health grant GM27906, University of Minnesota Academic Health Center).
Abbreviations used: PLB, phospholamban; CPMG, Carr-Purcell-Meiboom-Gill; MBP, maltose binding protein; NOE, nuclear Overhauser effect; TEV, tobacco etch virus; DPC, dodecylphosphocholine; SR, sarcoplasmic reticulum.
References
- Almeida, F. C. L., and S. J. Opella. 1997. fd coat protein structure in membrane environments: Structural dynamics of the loop between the hydrophobic trans-membrane helix and the amphipathic in-plane helix. J. Mol. Biol. 270:481–495. [DOI] [PubMed] [Google Scholar]
- Arkin, I. T., P. D. Adams, A. T. Brunger, S. O. Smith, and D. M. Engelman. 1997. Structural perspectives of phospholamban, a helical transmembrane pentamer. Annu. Rev. Biophys. Biomol. Struct. 26:157–179. [DOI] [PubMed] [Google Scholar]
- Bruschweiler, R., X. Liao, and P. E. Wright. 1995. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science. 268:886–889. [DOI] [PubMed] [Google Scholar]
- Buck, B., J. Zamoon, T. Kirby, T. M. Da Silva, D. D. Thomas, and G. Veglia. 2003. Overexpression, purification, and characterization of recombinant Ca-ATPase regulators for high-resolution solution and solid-state NMR studies. Protein Express Purif.. 30:253–261. [DOI] [PubMed] [Google Scholar]
- Campos-Olivas, R., and M. F. Summers. 1999. Backbone dynamics of the N-terminal domain of the HIV-1 capsid protein and comparison with the G94D mutant conferring cyclosporin resistance/dependence. Biochemistry. 38:10262–10271. [DOI] [PubMed] [Google Scholar]
- Carr, H. Y., and E. M. Purcell. 1954. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 94:630–638. [Google Scholar]
- Chu, G., K. Haghighi, and E. G. Kranias. 2002. From mouse to man: understanding heart failure through genetically altered mouse models. J. Card. Fail. 8(Suppl):S432–S449. [DOI] [PubMed] [Google Scholar]
- Columbus, L., and W. L. Hubbell. 2002. A new spin on protein dynamics. Trends Biochem. Sci. 27:288–295. [DOI] [PubMed] [Google Scholar]
- Cornea, R. L., Z. Chen, and L. R. Jones. 2002. Defining the membrane delimitation of phospholamban. Biophys. J. 82:227a. (Abstr.) [Google Scholar]
- Cornea, R. L., L. R. Jones, J. M. Autry, and D. D. Thomas. 1997. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry. 36:2960–2967. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- Evenäs, J., S. Forsén, A. Malmendal, and M. Akke. 1999. Backbone dynamics and energetics of a calmodulin domain mutant exchanging between closed and open conformations. J. Mol. Biol. 289:603–617. [DOI] [PubMed] [Google Scholar]
- Farrow, N. A., R. Muhandiram, A. U. Singer, S. M. Pascal, C. M. Kay, G. Gish, S. E. Shoelson, T. Pawson, J. D. Forman-Kay, and L. E. Kay. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 33:5984–6003. [DOI] [PubMed] [Google Scholar]
- Fujii, J., A. Ueno, K. Kitano, S. Tanaka, M. Kadoma, and M. Tada. 1987. Complete complementary DNA-derived amino acid sequence of canine cardiac phospholamban. J. Clin. Invest. 79:301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, E., and D. A. Middleton. 2003. Solid-state NMR reveals structural changes in phospholamban accompanying the functional regulation of the Ca+2-ATPase. J. Biol. Chem. 278:20835–20842. [DOI] [PubMed] [Google Scholar]
- Johnson, B. A., and R. A. Blevins. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 4:603–614. [DOI] [PubMed] [Google Scholar]
- Ishima, R., and D. A. Torchia. 2000. Protein dynamics from NMR. Nat. Struct. Biol. 7:740–743. [DOI] [PubMed] [Google Scholar]
- Karim, C. B., T. L. Kirby, M. G. Paterlini, Z. Zhang, F. Nitu, Y. Nesmelov, and D. D. Thomas. 2004. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc. Nat. Acad. Sci. USA. In press. [DOI] [PMC free article] [PubMed]
- Karim, C. B., C. G. Marquardt, J. D. Stamm, G. Barany, and D. D. Thomas. 2000. Synthetic null-cysteine phospholamban analogue and the corresponding transmembrane domain inhibit the Ca-ATPase. Biochemistry. 39:10892–10897. [DOI] [PubMed] [Google Scholar]
- Karim, C. B., J. D. Stamm, J. Karim, L. R. Jones, and D. D. Thomas. 1998. Cysteine reactivity and oligomeric structures of phospholamban and its mutants. Biochemistry. 37:12074–12081. [DOI] [PubMed] [Google Scholar]
- Kimura, Y., K. Kurzydlowski, M. Tada, and D. H. MacLennan. 1997. Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 272:15061–15064. [DOI] [PubMed] [Google Scholar]
- Kirby, T. L., C. B. Karim, and D. D. Thomas. 2004. EPR reveals a large-scale conformational change in the cytoplasmic domain of PLB upon binding to the SR CaATPase. Biochemistry. 43:5842–5852. [DOI] [PubMed] [Google Scholar]
- Lee, L. K., M. Rance, W. J. Chazin, and A. G. Palmer III.1997. Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13Cα nuclear spin relaxation. J. Biomol. NMR. 9:287–298. [DOI] [PubMed] [Google Scholar]
- Li, J., D. J. Bigelow, and T. C. Squire. 2003. Phosphorylation by cAMP-dependent protein kinase modulates the structural coupling between the transmembrane and cytosolic domains of phospholamban. Biochemistry. 36:10674–10682. [DOI] [PubMed] [Google Scholar]
- Lipari, G., and A. Szabo. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104:4546–4559. [Google Scholar]
- Lipari, G., and A. Szabo. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104:4559–4570. [Google Scholar]
- MacLennan, D. H., and E. G. Kranias. 2003. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4:566–578. [DOI] [PubMed] [Google Scholar]
- Mandel, A. M., M. Akke, and A. G. Palmer. 1995. Backbone dynamics of Escherichia coli ribonuclease H1: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246:144–163. [DOI] [PubMed] [Google Scholar]
- Mascioni, A., C. Karim, J. Zamoon, D. D. Thomas, and G. Veglia. 2002. Solid-state NMR and rigid body molecular dynamics to determine domain orientations of monomeric phospholamban. J. Am. Chem. Soc. 124:9392–9393. [DOI] [PubMed] [Google Scholar]
- Meiboom, S., and D. Gill. 1958. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instr. 29:688–691. [Google Scholar]
- Osborne, M. J., and P. E. Wright. 2001. Anisotropic rotational diffusion in model-free analysis for a ternary DHFR complex. J. Biomol. NMR. 19:209–230. [DOI] [PubMed] [Google Scholar]
- Parks, T. D., K. K. Leuther, E. D. Howard, S. A. Johnson, and W. G. Dougherty. 1994. Release of peptides and proteins from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216:413–417. [DOI] [PubMed] [Google Scholar]
- Palmer, A. G., 3rd, C. D. Kroenke, and J. P. Loria. 2001. NMR methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339:204–238. [DOI] [PubMed] [Google Scholar]
- Palmer, A. G. 3rd, M. Rance, and P. E. Wright. 1991. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J. Am. Chem. Soc. 113:4371–4380. [Google Scholar]
- Palmer, A. G. 3rd, N. J. Skelton, W. J. Chazin, P. E. Wright, and M. Rance. 1992. Suppression of the effects of cross-relaxation between dipolar and anisotropic chemical shift relaxation mechanisms in the measurements of spin-spin relaxation rates. Mol. Phys. 75:699–711. [Google Scholar]
- Papavoine, C. H. M., M. Remerowski., L. M. Horstink, R. N. H. Konings, C. W. Hilbers, and F. J. M. van de Ven. 1997. Backbone dynamics of the major coat protein of the bacteriophage M13 in detergent micelles by 15N magnetic resonance relaxation measurements using the model-free approach and reduced spectral density mapping. Biochemistry. 36:4015–4026. [DOI] [PubMed] [Google Scholar]
- Quenouille, M. H. 1956. Notes on bias in estimation. Biometrika. 43:353–360. [Google Scholar]
- Reddy, L. G., L. R. Jones, and D. D. Thomas. 1999. Depolymerization of phospholamban in the presence of calcium pump: a fluorescence energy transfer study. Biochemistry. 38:3954–3962. [DOI] [PubMed] [Google Scholar]
- Schmitt, J. P., M. Kamisago, M. Asahi, G. H. Li, F. Ahmad, U. Mende, E. G. Kranias, D. H. MacLennan, J. G. Seidman, and C. E. Seidman. 2003. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 299:1410–1413. [DOI] [PubMed] [Google Scholar]
- Schurr, J. M., H. P. Babcock, and B. Fujimoto. 1994. A test of the model free formulas. Effects of anisotropic rotational diffusion and dimerization. J. Magn. Reson. B. 105:211–224. [DOI] [PubMed] [Google Scholar]
- Shaka, A. J., P. B. Barker, and R. Freeman. 1985. Computer-optimized decoupling scheme for wideband applications and low-level operation. J. Magn. Reson. 64:547–552. [Google Scholar]
- Simmerman, H. K., and L. R. Jones. 1998. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 78:921–947. [DOI] [PubMed] [Google Scholar]
- Skelton, N. J., A. G. Palmer III, M. Akke, J. Kördel, M. Rance, and W. Chazin. 1993. Practical aspects of two-dimensional proton detected 15N spin relaxation measurements. J. Magn. Res. Ser. B. 102:253–264. [Google Scholar]
- Stokes, D. L., and N. M. Green. 2003. Structure and function of the calcium pump. Annu. Rev. Biophys. Biomol. Struct. 32:445–468. [DOI] [PubMed] [Google Scholar]
- Tada, M., and M. Kadoma. 1989. Regulation of the Ca2+ pump ATPase by cAMP-dependent phosphorylation of phospholamban. Bioessays. 10:157–163. [DOI] [PubMed] [Google Scholar]
- Thomas, D. D., L. G. Reddy, C. B. Karim, M. Li, R. Cornea, J. M. Autry, L. R. Jones, and J. Stamm. 1998. Direct spectroscopic detection of molecular dynamics and interactions of the calcium pump and phospholamban. Ann. N. Y. Acad. Sci. 853:186–194. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., S. E. Feller, R. W. Pastor, and A. Bax. 1995a. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117:12562–12566. [Google Scholar]
- Tjandra, N., H. Kuboniwa, H. Ren, and A. Bax. 1995b. Rotational dynamics of calcium-free calmodulin studied by 15N NMR relaxation measurements. Eur. J. Biochem. 230:1014–1024. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., A. Szabo, and A. Bax. 1996. Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J. Am. Chem. Soc. 118:6986–6991. [Google Scholar]
- Toyoshima, C., M. Asahi, Y. Sugita, R. Khanna, T. Tsuda, and D. H. MacLennan. 2003. Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. USA. 100:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia, G., A. C. Zeri, C. Ma, and S. J. Opella. 2002. Deuterium/hydrogen exchange factors measured by solution nuclear magnetic resonance spectroscopy as indicators of the structure and topology of membrane proteins. Biophys. J. 82:2176–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., Y. Pang, T. Holder, J. R. Brender, A. V. Kurochkin, and E. R. Zuiderweg. 2001. Functional dynamics in the active site of the ribonuclease binase. Proc. Natl. Acad. Sci. USA. 98:7684–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K. A., N. A. Farrow, C. M. Deber, and L. E. Kay. 1996. Structure and dynamics of bacteriophage IKe major coat protein in MPG micelles by solution NMR. Biochemistry. 35:5145–5157. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., C. G. Bigam, J. Yao, F. Abildgaard, H. J. Dyson, E. Oldfield, J. L. Markley, and B. D. Sykes. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 6:135–140. [DOI] [PubMed] [Google Scholar]
- Zamoon, J., A. Mascioni, D. D. Thomas, and G. Veglia. 2003. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys. J. 85:2589–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]