

Abstract
We have monitored the membrane-bound channel and nonchannel conformations of gramicidin utilizing red-edge excitation shift (REES), and related fluorescence parameters. In particular, we have used fluorescence lifetime, polarization, quenching, chemical modification, and membrane penetration depth analysis in addition to REES measurements to distinguish these two conformations. Our results show that REES of gramicidin tryptophans can be effectively used to distinguish conformations of membrane-bound gramicidin. The interfacially localized tryptophans in the channel conformation display REES of 7 nm whereas the tryptophans in the nonchannel conformation exhibit REES of 2 nm which highlights the difference in their average environments in terms of localization in the membrane. This is supported by tryptophan penetration depth measurements using the parallax method and fluorescence lifetime and polarization measurements. Further differences in the average tryptophan microenvironments in the two conformations are brought out by fluorescence quenching experiments using acrylamide and chemical modification of the tryptophans by N-bromosuccinimide. In summary, we report novel fluorescence-based approaches to monitor conformations of this important ion channel peptide. Our results offer vital information on the organization and dynamics of the functionally important tryptophan residues in gramicidin.
INTRODUCTION
Ion channels are transmembrane proteins that regulate ionic permeability in cell membranes. They represent an important class of molecules due to their ability to serve as key elements in signaling and sensing pathways and to connect the inside of the cell to its outside in a selective fashion. They are crucial for normal functioning of cells since a defective ion channel can lead to diseases (Cooper and Jan, 1999) such as cystic fibrosis (Stutts et al., 1995). The recent success in crystallographic analyses of ion channels, starting with the Streptomyces lividans K+ channel (KcsA) (Doyle et al., 1998), constitutes an exciting development in contemporary membrane biology (Rees et al., 2000).
The linear peptide gramicidin forms prototypical ion channels specific for monovalent cations and has been extensively used to study the organization, dynamics, and function of membrane-spanning channels (Killian, 1992; Koeppe and Andersen, 1996; Wallace, 2000). The transmembrane gramicidin channel is formed by the head-to-head dimerization of β6.3 helices (O'Connell et al., 1990). The channel interior is lined by the polar carbonyl and amide moieties of the peptide backbone, a feature shared with the selectivity filter of the bacterial KcsA K+ channel (Wallace, 2000). An important aspect of this conformation is the membrane interfacial location of the tryptophan residues, a common feature of many transmembrane helices (Reithmeier, 1995; Yau et al., 1998; Ulmschneider and Sansom, 2001).
Gramicidins are linear pentadecapeptide antibiotics with a molecular weight of ∼1900. They are produced by the soil bacterium Bacillus brevis, and consist of alternating l- and d-amino acids (Sarges and Witkop, 1965). The natural mixture of gramicidins, often denoted as gramicidin A′ (termed gramicidin D in older literature), consists of ∼85% of gramicidin A, which has four tryptophan residues at positions 9, 11, 13, and 15. Gramicidin A′ is readily available commercially and is fluorescent, due to the presence of tryptophan residues (Mukherjee and Chattopadhyay, 1994). It has one of the most hydrophobic sequences known and has been widely used as a model peptide for membrane-spanning regions of intrinsic membrane proteins (Andersen et al., 1999; Martinac and Hamill, 2002).
The unique sequence of alternating l- and d-chirality renders gramicidin sensitive to the environment in which it is placed. Gramicidin therefore adopts a wide range of environment-dependent conformations. Two major folding motifs have been identified for gramicidin in various media: 1), the single-stranded helical dimer (the “channel” form), and 2), the double-stranded intertwined helix (collectively known as the “nonchannel” form) (Urry, 1971; Ramachandran and Chandrasekaran, 1972; Veatch et al., 1974). The head-to-head (amino terminal-to-amino terminal) single-stranded β6.3 helical dimer form is the cation-conducting channel conformation of gramicidin in membranes, originally termed the 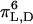 helix (Urry, 1971; Urry et al., 1971; Ramachandran and Chandrasekaran 1972; Veatch et al., 1974, 1975). This dimer form is stabilized by six intermolecular hydrogen bonds and has been characterized by NMR, circular dichroism (CD), and fluorescence spectroscopy. In this conformation, the carboxy terminus of the peptide is exposed to the membrane interface and the amino terminus is buried in the lipid bilayer. The interior of the channel is formed by the polar peptide backbone and side chains project outward in contact with neighboring lipid fatty acyl chains and help in modulating the channel conductance. The first well-resolved structure of gramicidin in a membrane-mimetic environment, which confirmed and extended the above structural eatures, was obtained by solution NMR (Arseniev et al., 1985). Later, the high resolution structure of the membrane-bound gramicidin A in a lipid bilayer was deduced by solid-state NMR (Cornell et al., 1988; Ketchem et al., 1993). Other conformations (which can be collectively termed as nonchannel conformations) have been shown to exist in membranes with polyunsaturated lipids (Sychev et al., 1993), and in membranes with increased acyl chain lengths (Killian et al., 1988; Galbraith and Wallace, 1998; Zein and Winter, 2000).
helix (Urry, 1971; Urry et al., 1971; Ramachandran and Chandrasekaran 1972; Veatch et al., 1974, 1975). This dimer form is stabilized by six intermolecular hydrogen bonds and has been characterized by NMR, circular dichroism (CD), and fluorescence spectroscopy. In this conformation, the carboxy terminus of the peptide is exposed to the membrane interface and the amino terminus is buried in the lipid bilayer. The interior of the channel is formed by the polar peptide backbone and side chains project outward in contact with neighboring lipid fatty acyl chains and help in modulating the channel conductance. The first well-resolved structure of gramicidin in a membrane-mimetic environment, which confirmed and extended the above structural eatures, was obtained by solution NMR (Arseniev et al., 1985). Later, the high resolution structure of the membrane-bound gramicidin A in a lipid bilayer was deduced by solid-state NMR (Cornell et al., 1988; Ketchem et al., 1993). Other conformations (which can be collectively termed as nonchannel conformations) have been shown to exist in membranes with polyunsaturated lipids (Sychev et al., 1993), and in membranes with increased acyl chain lengths (Killian et al., 1988; Galbraith and Wallace, 1998; Zein and Winter, 2000).
The detailed molecular mechanism of gramicidin adopting various conformations in membranes is not known. However, it has been reported that the initial conformation that gramicidin adopts when incorporated into membranes is dependent on the nature of the solvent in which it was dissolved before incorporation in membranes (LoGrasso et al., 1988; Killian et al., 1988). Thus, when gramicidin is dissolved in solvents such as chloroform/methanol, benzene/methanol, or ethanol before incorporation into membranes, it tends to adopt a nonchannel conformation. Upon sonication and incubation at 65°C, this is converted to the characteristic channel conformation. On the other hand, if gramicidin is dissolved in solvents such as trifluoroethanol before membrane incorporation, it appears to adopt the channel conformation even without sonication and incubation at elevated temperature. Gramicidin conformation in membranes therefore appears to be dependent on its “solvent history” (LoGrasso et al., 1988). The channel conformation of gramicidin in membranes has been characterized in molecular detail by solid-state NMR (Ketchem et al., 1993; Kovacs et al., 1999). However, the nonchannel forms in membranes have not been characterized well although various crystal structures of the nonchannel conformations adopted by gramicidin in organic solvents have been deduced (Wallace and Ravikumar, 1988; Langs, 1988; Langs et al., 1991). Interestingly, the crystal structure of the membrane-bound channel form has not yet been solved.
In membranes, the single-stranded β6.3 dimer channel conformation is the most preferred (thermodynamically stable) conformation (Killian et al., 1988), and this conformation has been shown to be maintained in membranes with a range of acyl chain lengths (Cornell et al., 1989) and upon ion occupation (Smith et al., 1990). The rate of interconversion from the nonchannel to the channel form can be modulated by sonication and prolonged incubation at elevated temperatures (LoGrasso et al., 1988; Killian et al., 1988). The membrane-interface-seeking property of tryptophan is implicated in the thermodynamic stability of the single-stranded channel form in membranes (Arumugam et al., 1996; Wimley and White, 1996; Chattopadhyay et al., 1997; Yau et al., 1998).
Spectroscopic methods provide valuable information about the peptide conformation in membranes in absence of x-ray crystallographic data. Moreover, information obtained from spectroscopic measurements inherently contains useful dynamic aspects, often missing from static crystallographic structures. For gramicidin, CD spectroscopy has served as a valuable tool to distinguish the channel conformation from other forms (LoGrasso et al., 1988; Killian et al., 1988; Woolley and Wallace, 1994). Furthermore, size exclusion chromatography has provided information about the equilibrium ratio of single-stranded and double-stranded conformation existing in various environments (Bañó et al., 1991; Salom et al., 1998). However, none of these techniques provide information about the dynamics and the molecular properties such as polarity and viscosity of the immediate microenvironment of the peptide.
In this article, we have investigated the organization and dynamics of the functionally important tryptophan residues of gramicidin in the channel and nonchannel conformations utilizing a combination of red-edge excitation shift (REES) and other fluorescence approaches including membrane penetration depth analysis using the parallax method (Chattopadhyay and London, 1987), and CD spectroscopy. A shift in the wavelength of maximum fluorescence emission toward higher wavelengths, caused by a shift in the excitation wavelength toward the red edge of the absorption band, is termed REES. This effect is mostly observed with polar fluorophores in motionally restricted media such as very viscous solutions or condensed phases where the dipolar relaxation time for the solvent shell around a fluorophore is comparable to or longer than its fluorescence lifetime (Demchenko, 1988, 2002; Mukherjee and Chattopadhyay, 1995; Chattopadhyay, 2003; Raghuraman et al., 2003). REES arises from slow rates of solvent relaxation (reorientation) around an excited-state fluorophore which depends on the motional restriction imposed on the solvent molecules in the immediate vicinity of the fluorophore. Utilizing this approach, it becomes possible to probe the mobility parameters of the environment itself (which is represented by the relaxing solvent molecules) using the fluorophore merely as a reporter group. Further, since the ubiquitous solvent for biological systems is water, the information obtained in such cases will come from the otherwise “optically silent” water molecules.
The unique feature about REES is that whereas all other fluorescence techniques (such as fluorescence quenching, energy transfer, and polarization measurements) yield information about the fluorophore (either intrinsic or extrinsic) itself, REES provides information about the relative rates of solvent (water in biological systems) relaxation dynamics which is not possible to obtain by other techniques. This makes REES extremely useful since hydration plays a crucial modulatory role in a large number of important cellular events including protein folding, lipid-protein interactions, and ion transport (Mentré, 2001; Kouyama et al., 2004). An in-depth discussion of the photophysical framework for REES is provided in recent reviews (Chattopadhyay, 2003; Raghuraman et al., 2003).
We have previously shown that REES and related techniques (wavelength-selective fluorescence approach) serve as a powerful tool to monitor the organization and dynamics of probes and peptides bound to membranes (Chattopadhyay and Mukherjee, 1993, 1999a,b; Mukherjee and Chattopadhyay, 1994; Ghosh et al., 1997; Kelkar et al., 2003; Mukherjee et al., 2004) and membrane-mimetic media such as micelles and reverse micelles (Rawat et al., 1997; Rawat and Chattopadhyay, 1999; Chattopadhyay et al., 2002; Raghuraman and Chattopadhyay, 2003, 2004; Raghuraman et al., 2004). In addition, we have previously used the wavelength-selective fluorescence approach to analyze the organization and dynamics of tryptophans in the soluble hemolytic protein α-toxin (Raja et al., 1999) and the cytoskeletal proteins tubulin (Guha et al., 1996) and spectrin (Chattopadhyay et al., 2003).
MATERIALS AND METHODS
Materials
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-(5-doxyl)stearoyl-sn-glycero-3-phosphocholine (5-PC), and 1-palmitoyl-2-(12-doxyl)stearoyl-sn-glycero-3-phosphocholine (12-PC) were obtained from Avanti Polar Lipids (Alabaster, AL). 2-(9-anthroyloxy)stearic acid (2-AS), and 12-(9-anthroyloxy)stearic acid (12-AS) were from Molecular Probes (Eugene, OR). Gramicidin A′ (from Bacillus brevis), dimyristoyl-sn-glycero-3-phosphocholine (DMPC), and N-bromosuccinimide (NBS) were purchased from Sigma Chemical (St. Louis, MO). Ultrapure-grade acrylamide was from Invitrogen Life Technologies (Carlsbad, CA). The purity of acrylamide was checked from its absorbance using its molar extinction coefficient (ɛ) of 0.23 M−1 cm−1 at 295 nm and optical transparency beyond 310 nm (Eftink, 1991a). Gramicidin A′, as obtained, is a mixture of gramicidins A, B, and C. Lipids were checked for purity by thin layer chromatography on silica gel precoated plates (Sigma) in chloroform/methanol/water (65:35:5, v/v/v) and were found to give only one spot in all cases with a phosphate-sensitive spray and subsequent charring (Dittmer and Lester, 1964). The concentration of phospholipids was determined by phosphate assay subsequent to total digestion by perchloric acid (McClare, 1971). DMPC was used as an internal standard to assess lipid digestion. All other chemicals used were of the highest purity available. Solvents used were of spectroscopic grade. Water was purified through a Millipore (Bedford, MA) Milli-Q system and used for all experiments.
Sample preparation
All experiments were done using unilamellar vesicles (ULV) of POPC containing 2% (mol/mol) gramicidin A′. The channel conformation of gramicidin was generated essentially as described earlier (Mukherjee and Chattopadhyay, 1994). To generate the nonchannel conformation of gramicidin in membranes, ULVs were prepared by the ethanol injection method (Kremer et al., 1977). In general, 1280 nmol of POPC (640 nmol for the nonchannel form) in chloroform/methanol was mixed with 25.6 nmol of gramicidin (12.8 nmol for the nonchannel form) in methanol. A few drops of chloroform were added to this solution. The solution was mixed well and dried under a stream of nitrogen while warming gently (∼40°C) and dried further under a high vacuum for at least 12 h. To prepare the channel form, the dried film was swelled in 1.5 ml of 10 mM sodium phosphate, 150 mM sodium chloride, pH 7.2 buffer, and samples were vortexed for 3 min to uniformly disperse the lipids. The samples were sonicated to clarity under argon (∼30 min in short bursts while being cooled in an ice/water mixture) using a Branson model 250 sonifier (Branson Ultrasonics, Danbury, CT) fitted with a microtip. The sonicated samples were centrifuged at 15,000 rpm for 15 min to remove any titanium particles shed from the microtip during sonication, and incubated for 12 h at 65°C with continuous shaking to completely convert to the channel conformation (Killian et al., 1988; LoGrasso et al., 1988). For the nonchannel conformation, the dried film was dissolved in ethanol to give a final concentration of 40 mM lipid in ethanol. The ethanolic solution was then injected into 1.5 ml of 10 mM sodium phosphate, 150 mM sodium chloride, pH 7.2 buffer, and samples were vortexed to uniformly disperse the lipids.
Two sets of samples were prepared to monitor the conformational transition of the nonchannel form to the channel form. In one set, nonchannel samples prepared by the above protocol were incubated at 65°C for 12 h. Alternatively, sonication has been suggested to accelerate the conformational transformation (LoGrasso et al., 1988). A separate set of samples were therefore prepared by sonicating the ULVs formed by the ethanol injection method. The samples were sonicated for 30 min, followed by centrifugation and overnight incubation at 65°C as described above.
All samples were incubated in dark at room temperature for 1 h before fluorescence or CD measurements. Background samples were prepared the same way except that gramicidin was omitted. All experiments were done with multiple sets of samples at 25°C.
Depth measurements using the parallax method
The actual spin (nitroxide) content of the spin-labeled phospholipids (5- and 12-PC) was assayed using fluorescence quenching of anthroyloxy-labeled fatty acids (2- and 12-AS) as described earlier (Abrams and London, 1993). For depth measurements using the parallax method, ULVs were prepared by sonication followed by incubation at 65°C for 12 h (channel conformation) and by the ethanol injection method (nonchannel conformation) as described above. These samples were made by drying 160 nmol of POPC containing 15 mol % spin-labeled phospholipid (5- or 12-PC) and 3.2 nmol of gramicidin under a stream of nitrogen while being warmed gently (35°C) and then under a high vacuum for at least 3 h. Duplicate samples were prepared in each case except for samples lacking the quencher (5- or 12-PC) where triplicates were prepared. Background samples lacking the fluorophore (gramicidin) were prepared in all experiments, and their fluorescence intensity was subtracted from the respective sample fluorescence intensity.
Steady-state fluorescence measurements
Steady-state fluorescence measurements were performed with a Hitachi F-4010 steady-state spectrofluorometer (Tokyo, Japan) using 1-cm-pathlength quartz cuvettes. Excitation and emission slits with a nominal bandpass of 5 nm were used. Background intensities of samples in which gramicidin was omitted were subtracted from each sample spectrum to cancel out any contribution due to the solvent Raman peak and other scattering artifacts. The spectral shifts obtained with different sets of samples were identical in most cases. In other cases, the values were within ±1 nm of the ones reported. Fluorescence polarization measurements were performed using a Hitachi polarization accessory. Polarization values were calculated from the equation (Lakowicz, 1999)
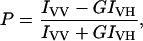 |
(1) |
where IVV and IVH are the measured fluorescence intensities (after appropriate background subtraction) with the excitation polarizer vertically oriented and emission polarizer vertically and horizontally oriented, respectively. G is the grating correction factor and is the ratio of the efficiencies of the detection system for vertically and horizontally polarized light, and is equal to IHV/IHH. All experiments were done with multiple sets of samples and average values of polarization are shown in Fig. 4.
FIGURE 4.

Fluorescence polarization (open bar) and apparent rotational correlation time (shaded bar) of various forms of gramicidin. The apparent rotational correlation times were calculated using Eq. 7 (see text for other details). The ratio of gramicidin to POPC was 1:50 (mol/mol). All other conditions are as in Fig. 1. The excitation wavelength was 280 nm and emission was monitored at 331 nm. Data shown are the mean ± standard error of multiple measurements. See Materials and Methods for other details.
Fluorescence quenching measurements
Acrylamide quenching experiments of gramicidin tryptophan fluorescence were carried out by measurement of fluorescence intensity in separate samples containing increasing amounts of acrylamide taken from a freshly prepared 4 M stock solution in water. Samples were kept in dark for at least 1 h before measuring fluorescence. The excitation wavelength used was 295 nm and emission was monitored at 334 nm. Corrections for inner filter effect were made using the equation (Lakowicz, 1999)
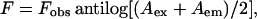 |
(2) |
where F is the corrected fluorescence intensity and Fobs is the background-subtracted fluorescence intensity of the sample. The values Aex and Aem are the measured absorbances at the excitation and emission wavelengths. The absorbances of the samples were measured using a Hitachi U-2000 UV-visible absorption spectrophotometer. Quenching data were analyzed by fitting to the Stern-Volmer equation (Lakowicz, 1999),
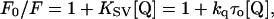 |
(3) |
where F0 and F are the fluorescence intensities in the absence and presence of the quencher, respectively; [Q] is the molar quencher concentration; and KSV is the Stern-Volmer quenching constant. The Stern-Volmer quenching constant KSV is equal to kqτo where kq is the bimolecular quenching constant and τo is the lifetime of the fluorophore in the absence of quencher.
N-Bromosuccinimide modification
Gramicidin in the channel or nonchannel conformation was subjected to NBS modification by serial addition of small aliquots from a freshly prepared 2 mM stock solution of NBS in water to a stirred solution of the sample. The sample was incubated in the sample compartment of the fluorimeter in dark (shutters closed) for 5 min before measurement of fluorescence intensity. The background-subtracted fluorescence intensity was corrected for dilution. The excitation wavelength used was 280 nm and emission was monitored at 331 nm for gramicidin in the channel conformation and at 335 nm for the nonchannel conformation. The fluorescence intensity of gramicidin in absence of NBS served as a control. All experiments were done with multiple sets of samples and average values are shown in Fig. 7.
FIGURE 7.

Residual fluorescence of gramicidin after NBS modification plotted as a function of molar ratio of NBS to gramicidin tryptophan in the nonchannel (○) and channel (•) conformations. F0 is the fluorescence in absence of NBS and F is the fluorescence in presence of increasing concentrations of NBS. Concentration of POPC was 0.21 mM and the ratio of gramicidin to POPC was 1:50 (mol/mol). The excitation wavelength was fixed at 280 nm and emission was monitored at 331 and 335 nm for the channel and nonchannel conformations, respectively. Data shown represent the mean ± standard error of multiple measurements. See Materials and Methods for other details.
Time-resolved fluorescence measurements
Fluorescence lifetimes were calculated from time-resolved fluorescence intensity decays using a Photon Technology International (London, Western Ontario, Canada) LS-100 luminescence spectrophotometer in the time-correlated single-photon counting mode. This machine uses a thyratron-gated nanosecond flash lamp filled with nitrogen as the plasma gas (17 ± 1 inches of mercury vacuum) and is run at 22–25 kHz. Lamp profiles were measured at the excitation wavelength using Ludox (colloidal silica) as the scatterer. To optimize the signal/noise ratio, 5000 photon counts were collected in the peak channel. All experiments were performed using excitation and emission slits with a nominal bandpass of 8 nm or less. The sample and the scatterer were alternated after every 10% acquisition to ensure compensation for shape and timing drifts occurring during the period of data collection. This arrangement also prevents any prolonged exposure of the sample to the excitation beam thereby avoiding any possible photodamage to the fluorophore. The data stored in a multichannel analyzer was routinely transferred to an IBM PC for analysis. Fluorescence intensity decay curves so obtained were deconvoluted with the instrument response function and analyzed as a sum of exponential terms,
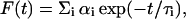 |
(4) |
where F(t) is the fluorescence intensity at time t and αi is a pre-exponential factor representing the fractional contribution to the time-resolved decay of the component with a lifetime τi such that Σi αi = 1. The decay parameters were recovered using a nonlinear least-squares iterative fitting procedure based on the Marquardt algorithm (Bevington, 1969). The program also includes statistical and plotting subroutine packages (O'Connor and Phillips, 1984). The goodness of the fit of a given set of observed data and the chosen function was evaluated by the reduced χ2 ratio, the weighted residuals (Lampert et al., 1983), and the autocorrelation function of the weighted residuals (Grinvald and Steinberg, 1974). A fit was considered acceptable when plots of the weighted residuals and the autocorrelation function showed random deviation about zero with a minimum χ2 value (generally not >1.2). Mean (average) lifetimes 〈τ〉 for biexponential decays of fluorescence were calculated from the decay times and pre-exponential factors using the equation (Lakowicz, 1999),
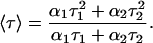 |
(5) |
Circular dichroism measurements
CD measurements were carried out at room temperature (25°C) on a JASCO J-715 spectropolarimeter (Tokyo, Japan) which was calibrated with (+)-10-camphorsulfonic acid (Chen and Yang, 1977). The spectra were scanned in a quartz optical cell with a path length of 0.1 cm. All spectra were recorded in 0.5 nm wavelength increments with a 4-s response and a band width of 1 nm. For monitoring changes in secondary structure, spectra were scanned in the far-UV range from 200 to 280 nm at a scan rate of 100 nm/min. Each spectrum is the average of 12 scans with a full scale sensitivity of 10 mdeg. All spectra were corrected for background by subtraction of appropriate blanks and were smoothed making sure that the overall shape of the spectrum remains unaltered. Data are represented as mean residue ellipticities and were calculated using the formula
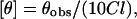 |
(6) |
where θobs is the observed ellipticity in mdeg, l is the pathlength in cm, and C is the concentration of peptide bonds in mol/L.
RESULTS
Gramicidin conformations monitored by circular dichroism spectroscopy
Circular dichroism spectroscopy has been previously utilized to distinguish various conformations of gramicidin (Killian et al., 1988; LoGrasso et al., 1988; Cox et al., 1992; Abdul-Manan and Hinton, 1994). We therefore used characteristic CD spectroscopic features to confirm the conformations generated by our experimental protocols (as outlined in Materials and Methods). The CD spectra of various gramicidin conformations are shown in Fig. 1. The CD spectrum for the channel conformation has two characteristic peaks of positive ellipticity at ∼218 and 235 nm, a valley at ∼230 nm, and negative ellipticity below 208 nm. These are characteristic of the single-stranded β6.3 conformation. The nonchannel form, on the other hand, is characterized by a large negative peak at 229 nm, a weaker positive peak at 218 nm, and positive ellipticity below 208 nm. As shown in Fig. 1, the CD spectrum of the gramicidin conformation induced by sonication followed by prolonged heat incubation at 65°C, is representative of the channel conformation and that induced by ethanol injection resembles the nonchannel form (Killian et al., 1988; LoGrasso et al., 1988; Cox et al., 1992). We will term samples prepared this way as the channel and nonchannel conformations, respectively.
FIGURE 1.

Far-UV CD spectra of the nonchannel (- - -), intermediate I (- · -), intermediate II (· · ·), and channel (—) forms of gramicidin in vesicles of POPC. The various forms of gramicidin were generated as described in the text. The channel conformation was prepared by sonication followed by heat incubation at 65°C for 12 h and the nonchannel conformation was generated by the ethanol injection method. Intermediate I was generated by ethanol injection followed by 65°C incubation for 12 h. Intermediate II was formed by ethanol injection followed by sonication and 65°C incubation for 12 h. The ratio of gramicidin to POPC was 1:50 (mol/mol). See Materials and Methods for other details.
Although the initial conformation of gramicidin in membranes is influenced by its solvent history (Killian et al., 1988; LoGrasso et al., 1988), the thermodynamically stable conformation of gramicidin is the channel conformation (Killian et al., 1988). To monitor the conversion of the nonchannel form to the channel form, we generated two intermediate forms and monitored their CD spectroscopic features. Intermediate I was generated by ethanol injection (which gives rise to the nonchannel form) followed by prolonged heat incubation at 65°C. Intermediate II, on the other hand, was generated by ethanol injection followed by sonication in addition to prolonged heat incubation at 65°C. As seen in Fig. 1, the spectral features of both the intermediates are different from the features observed for the nonchannel form (shown in Fig. 1), and have increased characteristics of the channel conformation. The extent of conversion from nonchannel to the channel form is more pronounced in the case of intermediate II, i.e., when the nonchannel form prepared by ethanol injection was subjected to prolonged heating at high temperature along with sonication.
Red-edge excitation shift of tryptophans as an indicator of gramicidin conformation
The fluorescence emission spectra of various conformations of gramicidin are shown in Fig. 2. Tryptophans in the channel form of gramicidin exhibit an emission maximum of 333 nm, when excited at 280 nm. The emission maximum of the nonchannel form, however, displays a slight red shift and is at 335 nm, in agreement with previous literature (Cox et al., 1992). Fig. 2 also shows the emission spectra of intermediates I and II. The emission maximum of intermediates I and II are 335 and 334 nm, respectively. It is apparent from the figure that the conversion from the nonchannel to the channel form via intermediates I and II is accompanied by a progressive decrease in fluorescence intensity. The shift in the maxima of fluorescence emission of the tryptophan residues of gramicidin in the channel conformation as a function of excitation wavelength is shown in Fig. 3. (We have used the term maximum of fluorescence emission in a somewhat wider sense here. In every case, we have monitored the wavelength corresponding to maximum fluorescence intensity, as well as the center of mass of the fluorescence emission. In most cases, both these methods yielded the same wavelength. In cases where minor discrepancies were found, the center of mass of emission has been reported as the fluorescence maximum.) As the excitation wavelength is changed from 280 to 310 nm, the emission maximum is shifted from 333 to 340 nm which corresponds to a REES of 7 nm. It is possible that there could be further red shift if excitation is carried out beyond 310 nm. We found it difficult to work in this wavelength range due to low signal/noise ratio and artifacts due to the solvent Raman peak that sometimes remained even after background subtraction. Such dependence of the emission maximum on excitation wavelength is characteristic of the red-edge excitation shift. This implies that the tryptophans in the gramicidin channel conformation are localized in a motionally restricted region of the membrane. This is consistent with the interfacial localization of the channel tryptophans in the membrane (O'Connell et al., 1990; Cox et al., 1992; Ketchem et al., 1993; Mukherjee and Chattopadhyay, 1994). The membrane interface is characterized by unique motional and dielectric characteristics distinct from both the bulk aqueous phase and the more isotropic hydrocarbon-like deeper regions of the membrane (Ashcroft et al., 1981; Perochon et al., 1992; Slater et al., 1993; White and Wimley, 1994). This specific region of the membrane exhibits slow rates of solvent relaxation and is also known to participate in intermolecular charge interactions (Yeagle, 1987) and hydrogen bonding through the polar headgroup (Boggs, 1987). These structural features which slow down the rate of solvent reorientation have previously been recognized as typical features of microenvironments giving rise to significant REES effects. It is therefore the membrane interface which is most likely to display red-edge effects (Chattopadhyay, 2003).
FIGURE 2.

Fluorescence emission spectra of the nonchannel (- - -), intermediate I (- · -), intermediate II (· · ·), and channel (—) forms of gramicidin in vesicles of POPC. The excitation wavelength was 280 nm. All other conditions are as in Fig. 1.
FIGURE 3.

Effect of changing excitation wavelength on the wavelength of maximum emission for the nonchannel (○), intermediate I (▵), intermediate II (□), and channel (•) forms of gramicidin. The ratio of gramicidin to POPC was 1:50 (mol/mol). All other conditions are as in Fig. 1. See Materials and Methods for other details.
In contrast, in the case of the nonchannel conformation, the emission maximum was shifted from 335 to 337 nm which corresponds to a REES of 2 nm, when the excitation wavelength was shifted from 280 to 310 nm (Fig. 3). This suggests that the average environment of the tryptophan residues in the nonchannel conformation is more dynamic (less ordered) than what is experienced in the channel conformation. These results show that REES is sensitive to the conformation of membrane-bound gramicidin and could potentially be used to distinguish these two conformations. The double-helical antiparallel form of gramicidin is a predominant form in ethanol, the organic solvent used in the present study for preparation of the nonchannel conformation (Killian, 1992). This form is retained in the membrane-bound form in the absence of heating at high temperature (Killian et al., 1988). In this conformation, some of the tryptophan residues are localized in the deeper regions of the membrane bilayer (see Fig. 8). This region of the bilayer is more isotropic (bulk hydrocarbon-like), and is characterized by a lower polarity. In addition, the deeper hydrophobic region of the bilayer is more dynamic (less restrictive) due to the motional gradient that exists in the bilayer (Seelig, 1977; Chattopadhyay and Mukherjee, 1999a). Interestingly, we have previously shown, using anthroyloxy and nitrobenzoxadiazol (NBD)-labeled membrane probes, that the rates of solvent relaxation in membranes are depth-dependent, and the deeper regions of the membrane display less pronounced red-edge effects (Chattopadhyay and Mukherjee, 1999a,b). We attribute the reduction in REES for gramicidin tryptophans in the nonchannel conformation to differential rates of solvent reorientation as a function of tryptophan depth in the membrane. This is reinforced by fluorescence polarization results (see below). Interestingly, Fig. 3 shows that intermediates I and II display REES of 3 and 4 nm, respectively. The fluorescence spectral features of the intermediates are therefore indicative of the progressive conversion of the nonchannel form to the channel form.
FIGURE 8.

Schematic representation of the nonchannel and channel conformations of gramicidin indicating the location of various tryptophans along the vertical axis (z axis) of the bilayer in these two forms (modified from Arumugam et al., 1996).
Fluorescence polarization and lifetime of tryptophans in various forms of gramicidin
The fluorescence polarization of the gramicidin tryptophans in various conformations is shown in Fig. 4. The lowest polarization was observed in the case of the nonchannel conformation. This is possibly due to the relatively isotropic and less restrictive nature of the average environment of the tryptophans in this conformation, in agreement with earlier reports (Cox et al., 1992). To monitor the conversion of the nonchannel form to the channel form, we measured fluorescence polarization of the intermediate forms. Fig. 4 shows that the polarization values are progressively higher for intermediates I and II. The highest polarization is observed in the case of the channel conformation due to the interfacial localization of the tryptophans since the interface is a motionally restricted region of the membrane (see above). The fluorescence polarization results therefore further support our REES results.
Fluorescence lifetime serves as a sensitive indicator of the local environment and polarity in which a given fluorophore is placed (Prendergast, 1991). A typical decay profile of gramicidin tryptophans with its biexponential fitting and the statistical parameters used to check the goodness of the fit is shown in Fig. 5. Table 1 shows the gramicidin tryptophan lifetimes in various conformations. As can be seen from the table, all the fluorescence decays could be fitted well with a biexponential function. The mean fluorescence lifetimes of gramicidin tryptophans were calculated using Eq. 5 and are shown in Table 1. The mean fluorescence lifetime for gramicidin tryptophans are found to be longer in the nonchannel form (2.76 ns) than the channel form (1.72 ns). Table 1 also shows the mean fluorescence lifetime for the tryptophans in the intermediates I and II. An increase in polarity of the tryptophan environment is known to reduce the lifetime of tryptophans due to fast deactivating processes in polar environments (Kirby and Steiner, 1970; Ho and Stubbs, 1992). The reduction in mean fluorescence lifetime of the tryptophans in the channel conformation can be attributed to the polarity of the interfacial region where the channel tryptophans are localized.
FIGURE 5.

Time-resolved fluorescence intensity decay of the channel conformation of gramicidin in POPC vesicles. Excitation wavelength was at 297 nm which corresponds to a peak in the spectral output of the nitrogen lamp. Emission was monitored at 340 nm. The sharp peak on the left is the lamp profile. The relatively broad peak on the right is the decay profile, fitted to a biexponential function. The two lower plots show the weighted residuals and the autocorrelation function of the weighted residuals. All other conditions are as in Fig. 1. See Materials and Methods for other details.
TABLE 1.
Lifetimes of various forms of membrane-bound gramicidin
| Gramicidin conformation | α1 | τ1 (ns) | α2 | τ2 (ns) | 〈τ〉* (ns) |
|---|---|---|---|---|---|
| Nonchannel | 0.78 | 1.05 | 0.22 | 4.25 | 2.76 |
| Intermediate I | 0.87 | 0.65 | 0.13 | 3.73 | 2.07 |
| Intermediate II | 0.83 | 0.64 | 0.17 | 2.92 | 1.74 |
| Channel | 0.89 | 0.87 | 0.11 | 3.46 | 1.72 |
To ensure that the polarization values measured for various gramicidin conformations (see Fig. 4, white bars) are not influenced by lifetime-induced artifacts, the apparent (average) rotational correlation times for gramicidin tryptophans were calculated using Perrin's equation (Lakowicz, 1999),
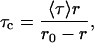 |
(7) |
where r0 is the limiting anisotropy of tryptophan, r is the steady-state anisotropy (derived from the polarization values using r = 2P/(3–P)), and 〈τ〉 is the mean fluorescence lifetime taken from Table 1. The values of the apparent rotational correlation times, calculated this way using a value of r0 of 0.09 (Weber, 1960), are shown in Fig. 4 (shaded bars). As is evident from the figure, the overall trend in fluorescence polarization observed with the transition of gramicidin from the nonchannel to the channel form (through the intermediate forms), is preserved when the change in the apparent rotational correlation times of gramicidin tryptophans is considered. This clearly shows that the observed changes in polarization values are free from lifetime-induced artifacts.
Acrylamide quenching and N-bromosuccinimide modification of gramicidin tryptophan fluorescence
Acrylamide quenching of tryptophan fluorescence is widely used to monitor tryptophan environments in proteins (Eftink, 1991b). Fig. 6 shows a representative Stern-Volmer plot of acrylamide quenching of gramicidin tryptophans in the channel and nonchannel conformations. The slope (KSV) of such a plot is related to the accessibility (degree of exposure) of the tryptophans to the quencher. The quenching parameter obtained by analyzing the Stern-Volmer plot is shown in Table 2. The Stern-Volmer constant (KSV) for acrylamide quenching of the nonchannel form of gramicidin was found to be 1.41 M−1 whereas the value for the channel form was found to be 0.95 M−1. However, interpretation of the Stern-Volmer constant is complicated this way due to its intrinsic dependence on fluorescence lifetime (see Eq. 3). The bimolecular quenching constant (kq) for acrylamide quenching is therefore a more accurate measure of the degree of exposure since kq takes into account differences in fluorescence lifetime. The bimolecular quenching constants, calculated using Eq. 3, are shown in Table 2. The kq values show that the tryptophans in the channel conformation are relatively more accessible to acrylamide although the difference is small. In case of membrane-bound peptides with a heterogenous distribution of tryptophans such as found in the nonchannel conformation of gramicidin, if acrylamide preferentially quenched the exposed tryptophans, a blue shift (i.e., toward lower wavelength) of the residual fluorescence emission maximum would be expected (Caputo and London, 2003). Interestingly, Fig. 6 shows that the fluorescence emission maximum of gramicidin in either conformation remains invariant and does not exhibit any blue shift of the emission maximum upon quenching by acrylamide.
FIGURE 6.

Representative data for Stern-Volmer analysis of acrylamide quenching of gramicidin fluorescence in the nonchannel (○) and channel (•) conformations. F0 is the fluorescence in the absence of quencher, and F is the corrected fluorescence in the presence of quencher. The excitation wavelength was fixed at 295 nm and emission was monitored at 334 nm. The emission maxima of the nonchannel (□) and channel (▪) conformations, when excited at 295 nm, in the presence of increasing concentrations of acrylamide are also shown. Concentration of POPC was 0.11 mM and the ratio of gramicidin to POPC was 1:50 (mol/mol). See Materials and Methods for other details.
TABLE 2.
Acrylamide quenching of gramicidin fluorescence
| Gramicidin conformation | KSV* (M−1) | kq (×10−9)† (M−1 s−1) |
|---|---|---|
| Nonchannel | 1.41 ± 0.11 | 0.51 |
| Channel | 0.95 ± 0.06 | 0.55 |
Concentration of POPC was 0.11 mM and the ratio of gramicidin to POPC was 1:50 (mol/mol). The excitation wavelength was 295 nm; emission was monitored at 334 nm. See Materials and Methods for other details.
Further analysis of this observation in terms of contribution of individual tryptophan residues is complicated due to the heterogeneity in fluorescence parameters (such as quantum yield and lifetime) of individual tryptophans in multitryptophan peptides such as gramicidin because of environmental sensitivity (Eftink, 1991c). In addition, the presence of the membrane bilayer could complicate the interpretation of fluorescence quenching data since quenchers are usually polar in nature and their access to the fluorophores may be influenced by the solubility of the quencher in the membrane (Moro et al., 1993). Acrylamide has earlier been reported to be able to permeate the membrane bilayer (Moro et al., 1993). Nonetheless, in the case of fluorophores embedded in the hydrocarbon-like deeper regions of the membrane, fluorescence quenching depends on a number of factors such as the nature of the molecular interaction between the quencher and membrane components and not merely on the ability of the quencher to permeate the membrane.
The accessibility of the gramicidin tryptophans was further explored by chemical modification using NBS as an oxidant. NBS oxidation of the indole moiety of tryptophan to the nonfluorescent oxindole (Spande and Witkop, 1967) can be used to assess the relative accessibility of tryptophan residues in proteins and peptides (Raja et al., 1999; Verza and Bakás, 2000; Schibli et al., 2002). Fig. 7 shows the residual fluorescence intensity of gramicidin after NBS modification of tryptophans plotted as a function of molar ratio of NBS to gramicidin tryptophan in the channel and nonchannel conformations. It is apparent from the figure that the tryptophans in the nonchannel conformation are more sensitive to NBS modification than those in the channel conformation. For example, at a molar ratio of NBS to gramicidin tryptophan of 0.2, fluorescence intensity was reduced to 50% in the nonchannel conformation. The corresponding value in the case of the channel conformation was 0.7.
In case of soluble proteins, NBS is unable to modify tryptophan residues that are deeply buried (Raja et al., 1999; Verza and Bakás, 2000). Fig. 7 shows that all the tryptophans of gramicidin in either conformation are susceptible to NBS modification and it is possible to completely abolish tryptophan fluorescence with ∼3 molar excess of NBS (less for the nonchannel conformation). This implies that the membrane bilayer does not act as a barrier to NBS modification and resistance to modification may arise due to decreased accessibility of the tryptophan residues due to packing constraints. Interestingly, the location, depth, orientation, and distribution of the tryptophan residues of gramicidin are markedly different in the channel and nonchannel conformations. In the channel conformation, the gramicidin tryptophans are clustered at the membrane-water interface and Trp-9 and Trp-15 are involved in stacking (aromatic-aromatic) interaction (Hu et al., 1993; Ketchem et al., 1993; Mukherjee and Chattopadhyay, 1994; Separovic et al., 1999). In the nonchannel conformation, however, there is a distribution of tryptophans along the membrane axis (Arumugam et al., 1996; see Fig. 8). The increased accessibility of tryptophans in the nonchannel conformation to NBS may be attributed to the relatively shallow location of Trp-15 and Trp-13 in the membrane as opposed to the interfacial location of the tryptophans in the channel conformation. The absence of stacking interaction in the nonchannel conformation may also play a role in the increased accessibility to NBS.
Membrane penetration depths of gramicidin tryptophans
Membrane penetration depth is an important parameter in the study of membrane structure and organization (Chattopadhyay, 1992; London and Ladokhin, 2002). Knowledge of the precise depth of a membrane-embedded group or molecule often helps define the conformation and topology of membrane probes and proteins. In addition, properties such as polarity, fluidity, segmental motion, ability to form hydrogen bonds, and the extent of solvent penetration are known to vary in a depth-dependent manner. To gain a better insight of gramicidin conformations in membranes, especially in terms of the localization of the tryptophan residues, average penetration depths of the gramicidin tryptophan residues were determined in the channel and nonchannel conformations. Average depths of the tryptophan residues in membrane-bound gramicidin were calculated by the parallax method (Chattopadhyay and London, 1987) using the equation
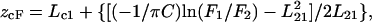 |
(8) |
where zcF = the depth of the fluorophore from the center of the bilayer, Lc1 = the distance of the center of the bilayer from the shallow quencher (5–PC in this case), L21 = the difference in depth between the two quenchers (i.e., the transverse distance between the shallow and the deep quencher), and C = the two-dimensional quencher concentration in the plane of the membrane (molecules/Å2). Here F1/F2 is the ratio of F1/F0 and F2/F0 in which F1 and F2 are fluorescence intensities in the presence of the shallow (5–PC) and deep quencher (12–PC), respectively, both at the same quencher concentration C; F0 is the fluorescence intensity in the absence of any quencher. All the bilayer parameters used were the same as described previously (Chattopadhyay and London, 1987). The average depths of penetration of the tryptophan residues for the channel and the nonchannel conformations are shown in Table 3. The tryptophans in the channel conformation are, on the average, positioned at a relatively shallow location in the membrane as evidenced from the average depth of 11 Å for the channel conformation (Table 3). This value is consistent with the interfacial localization of tryptophans in this conformation (see Fig. 8) and with a β6.3 helical dimer of 25 Å length (Ketchem et al., 1993). The average distance from the center of the bilayer of tryptophans in the nonchannel conformation, on the other hand, is 7.3 Å, indicating deeper localization. Although these are average depths, they are still useful since they represent the minimum depths of penetration in each case.
TABLE 3.
Average membrane penetration depths of gramicidin tryptophans in the channel and nonchannel forms by the parallax method
| Gramicidin conformation | Depth from the center of the bilayer zcF (Å) |
|---|---|
| Nonchannel | 7.3 |
| Channel | 11.0 |
Depths were calculated from fluorescence quenchings obtained with samples containing 15 mol % of 5-PC and 12-PC and using Eq. 8. Samples were excited at 280 nm, and emission was monitored at 331 nm. The ratio of gramicidin to total lipid was 1:50 (mol/mol). See Materials and Methods for other details.
DISCUSSION
Gramicidin serves as an excellent model for transmembrane channels due to its small size, ready availability, and the relative ease with which chemical modifications can be performed. This makes gramicidin unique among small membrane-active peptides and provides the basis for the use of gramicidin to explore the principles that govern the folding and function of membrane-spanning channels in particular, and membrane proteins in general. It is interesting to note that gramicidin represents a useful model for realistic determination of conformational preference in a lipid bilayer environment despite the alternating sequence of l–d chirality generally not encountered in naturally occurring peptides and proteins. This is due to the fact that the dihedral angle combinations generated in the conformation space by various gramicidin conformations are “allowed” according to the Ramachandran plot (Andersen et al., 1996). Gramicidin channels share important structural features such as membrane interfacial localization of tryptophan residues, the channel interior being made of the peptide backbone, and ion selectivity arising out of backbone interactions, with other naturally occurring channel proteins such as the bacterial KcsA K+ channel (Wallace, 2000).
The tryptophan residues in gramicidin channels are believed to be crucial for maintaining the structure and function of the channel (Hu et al., 1993; Salom et al., 1998; Andersen et al., 1998). The importance of the tryptophans has been demonstrated by the observation that the cation conductivity of the channel decreases upon substitution of one or all of the tryptophan residues by phenylalanine, tyrosine, or naphthylalanine (Prasad et al., 1983; Daumas et al., 1989; Becker et al., 1991; Fonseca et al., 1992), and also upon ultraviolet irradiation or chemical modification of the tryptophan side chains (Barth and Stark, 1991; Andersen et al., 1998). In fact, it has been proposed that the tryptophans increase ion permeability by electrostatic interactions (Andersen et al., 1998; Busath, 1993).
The distributions and depths of the gramicidin tryptophans represent major differences between the two forms (shown in Fig. 8). The reason that the nonchannel conformation is not the thermodynamically stable form in membranes is due to the fact that in this form some of the tryptophan residues are buried in the low dielectric nonpolar region of the membrane which is energetically unfavorable (Wimley and White, 1996). The preferred localization of the tryptophan residues at the membrane interface has been ascribed to the ability of the tryptophan-NH groups to form hydrogen bonds with the hydrogen bond acceptors near the lipid headgroups, one of the strongest candidates for such acceptors being the lipid carbonyls (Ippolito et al., 1990). Interfacial water molecules present near the lipid headgroup region also form a distinct class of possible hydrogen bond acceptors and could give rise to REES effects due to suitable timescale of reorientation. The proposition that the tryptophan residues of membrane-bound gramicidin channels are indeed hydrogen-bonded to the neighboring hydrogen bond acceptors is further supported by the fact that the gramicidin analog in which all the four tryptophan residues are replaced by phenylalanines, which are more hydrophobic and cannot act as hydrogen bond donors, appears to preferentially adopt the alternate antiparallel double-stranded helical dimer conformation (Cotten et al., 1997; Salom et al., 1998), and exhibits drastically reduced channel activity (Fonseca et al., 1992). In the absence of any tryptophan residues, the double-stranded helical dimer nonchannel conformation becomes the energetically favored state in the membrane.
The solvent history dependence of gramicidin conformations in membranes has been previously studied by nuclear magnetic resonance (Zhang et al., 1992; Arumugam et al., 1996; Abdul-Manan and Hinton, 1994; Bouchard et al., 1995), circular dichroism (LoGrasso et al., 1988; Killian et al., 1988), high performance liquid chromatography (Bañó et al., 1991), and vibrational spectroscopy (Bouchard and Auger, 1993). In this work, we have utilized red-edge excitation shift (REES), and related fluorescence parameters to distinguish the membrane-bound channel and nonchannel conformations of gramicidin. More specifically, we have used fluorescence lifetime, polarization, quenching, chemical modification, and membrane penetration depth analysis, in addition to REES measurements to address this issue. Our results show that REES, which is sensitive to rate of solvent reorientation and hence a good reporter of the fluorophore environment (rather than the fluorophore itself), can detect gramicidin conformation and could potentially be used to distinguish these two conformations. We have previously shown, using anthroyloxy and NBD-labeled membrane probes, that depth-dependent solvent relaxation in membranes can be used as a “dipstick” (Chattopadhyay and Mukherjee, 1999a,b). We show here that this approach can also be used to distinguish conformations of a membrane-bound peptide that differ in the distributions and depths of the tryptophan residues. In summary, we report here novel fluorescence-based approaches to monitor conformations of this important ion channel peptide. Our results offer vital information on the organization and dynamics of the functionally important tryptophan residues in gramicidin.
Acknowledgments
We gratefully acknowledge R. Rukmini for help with the membrane penetration depth experiments, and H. Raghuraman and Thomas Pucadyil for helpful discussions. We thank Y. S. S. V. Prasad and G. G. Kingi for technical help and members of our laboratory for critically reading the manuscript.
S.S.R. thanks the Council of Scientific and Industrial Research, and D.A.K. thanks the University Grants Commission for the award of Senior Research Fellowships. This work was supported by the Department of Science and Technology, and the Council of Scientific and Industrial Research, Government of India.
Satinder S. Rawat and Devaki A. Kelkar have contributed equally to the work.
Satinder S. Rawat's present address is Room 211, Building 469, Laboratory of Experimental and Computational Biology, NCI-FCRDC, National Institutes of Health, Frederick, MD 21702-1201 USA.
References
- Abdul-Manan, N., and J. F. Hinton. 1994. Conformation states of gramicidin A along the pathway to the formation of channels in model membranes determined by 2D NMR and circular dichroism spectroscopy. Biochemistry. 33:6773–6783. [DOI] [PubMed] [Google Scholar]
- Abrams, F. S., and E. London. 1993. Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipid polar headgroup. Biochemistry. 32:10826–10831. [DOI] [PubMed] [Google Scholar]
- Andersen, O. S., G. Saberwal, D. V. Greathouse, and R. E. Koeppe. 1996. Gramicidin channels—a solvable membrane “protein” folding problem. Ind. J. Biochem. Biophys. 33:331–342. [PubMed] [Google Scholar]
- Andersen, O. S., C. Nielsen, A. M. Maer, J. A. Lundbæk, M. Goulian, and R. E. Koeppe. 1999. Ion channels as tools to monitor lipid bilayer-membrane protein interactions: gramicidin channels as molecular force transducers. Methods Enzymol. 294:208–224. [DOI] [PubMed] [Google Scholar]
- Andersen, O. S., D. V. Greathouse, L. L. Providence, M. D. Becker, and R. E. Koeppe. 1998. Importance of tryptophan dipoles for protein function: 5-fluorination of tryptophans in gramicidin A channels. J. Am. Chem. Soc. 120:5142–5146. [Google Scholar]
- Arseniev, A. S., I. L. Barsukov, V. F. Bystrov, A. L. Lomize, and Y. A. Ovchinnikov. 1985. 1H-NMR study of gramicidin A transmembrane ion channel. Head-to-head right-handed, single-stranded helices. FEBS Lett. 186:168–174. [DOI] [PubMed] [Google Scholar]
- Arumugam, S., S. Pascal, C. L. North, W. Hu, K.-C. Lee, M. Cotten, R. R. Ketchem, F. Xu, M. Brenneman, F. Kovacs, F. Tian, A. Wang, S. Huo, and T. A. Cross. 1996. Conformational trapping in a membrane environment: a regulatory mechanism for protein activity? Proc. Natl. Acad. Sci. USA. 93:5872–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, R. G., H. G. L. Coster, and J. R. Smith. 1981. The molecular organisation of bimolecular lipid membranes: the dielectric structure of the hydrophilic/hydrophobic interface. Biochim. Biophys. Acta. 643:191–204. [DOI] [PubMed] [Google Scholar]
- Bañó, M. C., L. Braco, and C. Abad. 1991. Conformational transitions of gramicidin A in phospholipid model membranes. A high-performance liquid chromatography assessment. Biochemistry. 30:886–894. [DOI] [PubMed] [Google Scholar]
- Barth, C., and G. Stark. 1991. Radiation inactivation of ion channels formed by gramicidin A. Protection by lipid double bonds and by α-tocopherol. Biochim. Biophys. Acta. 1066:54–58. [DOI] [PubMed] [Google Scholar]
- Becker, M. D., D. V. Greathouse, R. E. Koeppe, and O. S. Andersen. 1991. Amino acid sequence modulation of gramicidin channel function: effects of tryptophan-to-phenylalanine substitutions on the single-channel conductance and duration. Biochemistry. 30:8830–8839. [DOI] [PubMed] [Google Scholar]
- Bevington, P. R. 1969. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill, New York.
- Boggs, J. M. 1987. Lipid intermolecular hydrogen bonding: influence on structural organization and membrane function. Biochim. Biophys. Acta. 906:353–404. [DOI] [PubMed] [Google Scholar]
- Bouchard, M., and M. Auger. 1993. Solvent history dependence of gramicidin-lipid interactions: a Raman and infrared spectroscopic study. Biophys. J. 65:2484–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard, M., J. H. Davis, and M. Auger. 1995. High-speed magic angle spinning solid-state 1H nuclear magnetic resonance study of the conformation of gramicidin A in lipid bilayers. Biophys. J. 69:1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busath, D. D. 1993. The use of physical methods in determining gramicidin channel structure and function. Annu. Rev. Physiol. 55:473–501. [DOI] [PubMed] [Google Scholar]
- Caputo, G. A., and E. London. 2003. Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic α-helix locations within membranes. Biochemistry. 42:3265–3274. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A. 1992. Membrane penetration depth analysis using fluorescence quenching: a critical review. In Biomembranes Structure & Function: The State of the Art. B. P. Gaber, and K. R. K. Easwaran, editors. Adenine Press, Schenectady, New York. 153–163.
- Chattopadhyay, A. 2003. Exploring membrane organization and dynamics by the wavelength-selective fluorescence approach. Chem. Phys. Lipids. 122:3–17. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A., and E. London. 1987. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 26:39–45. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A., and S. Mukherjee. 1993. Fluorophore environments in membrane-bound probes: a red edge excitation shift study. Biochemistry. 32:3804–3811. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A., and S. Mukherjee. 1999a. Depth-dependent solvent relaxation in membranes: wavelength-selective fluorescence as a membrane dipstick. Langmuir. 15:2142–2148. [Google Scholar]
- Chattopadhyay, A., and S. Mukherjee. 1999b. Red edge excitation shift of a deeply embedded membrane probe: implications in water penetration in the bilayer. J. Phys. Chem. B. 103:8180–8185. [Google Scholar]
- Chattopadhyay, A., S. Mukherjee, and H. Raghuraman. 2002. Reverse micellar organization and dynamics: a wavelength-selective fluorescence approach. J. Phys. Chem. B. 106:13002–13009. [Google Scholar]
- Chattopadhyay, A., S. Mukherjee, R. Rukmini, S. S. Rawat, and S. Sudha. 1997. Ionization, partitioning, and dynamics of tryptophan octyl ester: implications for membrane-bound tryptophan residues. Biophys. J. 73:839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay, A., S. S. Rawat, D. A. Kelkar, S. Ray, and A. Chakrabarti. 2003. Organization and dynamics of tryptophan residues in erythroid spectrin: novel structural features of denatured spectrin revealed by the wavelength-selective fluorescence approach. Protein Sci. 12:2389–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G. C., and J. T. Yang. 1977. Two-point calibration of circular dichrometer with d-10-camphorsulphonic acid. Anal. Lett. 10:1195–1207. [Google Scholar]
- Cornell, B. A., F. Separovic, A. J. Baldassi, and R. Smith. 1988. Conformation and orientation of gramicidin A in oriented phospholipid bilayers measured by solid state carbon-13 NMR. Biophys. J. 53:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell, B. A., F. Separovic, D. E. Thomas, A. R. Atkins, and R. Smith. 1989. Effect of acyl chain length on the structure and motion of gramicidin A in lipid bilayers. Biochim. Biophys. Acta. 985:229–232. [DOI] [PubMed] [Google Scholar]
- Cooper, E. C., and L. Y. Jan. 1999. Ion channel genes and human neurological disease: recent progress, prospects and challenges. Proc. Natl. Acad. Sci. USA. 96:4759–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten, M., F. Xu, and T. A. Cross. 1997. Protein stability and conformational rearrangements in lipid bilayers: linear gramicidin, a model system. Biophys. J. 73:614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, K. J., C. Ho, J. V. Lombardi, and C. D. Stubbs. 1992. Gramicidin conformational studies with mixed-chain unsaturated phospholipid bilayer systems. Biochemistry. 31:1112–1118. [DOI] [PubMed] [Google Scholar]
- Daumas, P., F. Heitz, L. Ranjalahy-Rasoloarijao, and R. Lazaro. 1989. Gramicidin A analogs: influence of the substitution of the tryptophans by naphthylalanines. Biochimie. 71:77–81. [DOI] [PubMed] [Google Scholar]
- Demchenko, A. P. 1988. Site-selective excitation: a new dimension in protein and membrane spectroscopy. Trends Biochem. Sci. 13:374–377. [DOI] [PubMed] [Google Scholar]
- Demchenko, A. P. 2002. The red-edge effects: 30 years of exploration. Luminescence. 17:19–42. [DOI] [PubMed] [Google Scholar]
- Dittmer, J. C., and R. L. Lester. 1964. A simple, specific spray for the detection of phospholipids on thin-layer chromatograms. J. Lipid Res. 5:126–127. [PubMed] [Google Scholar]
- Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Eftink, M. R. 1991a. Fluorescence quenching reactions: probing biological macromolecular structure. In Biophysical and Biochemical Aspects of Fluorescence Spectroscopy. T. G. Dewey, editor. Plenum Press, New York. 1–41.
- Eftink, M. R. 1991b. Fluorescence quenching: theory and applications. In Topics in Fluorescence Spectroscopy. Vol. 2: Principles. J. R. Lakowicz, editor. Plenum Press, New York. 53–126.
- Eftink, M. R. 1991c. Fluorescence techniques for studying protein structure. In Methods of Biochemical Analysis, Vol. 35. C. H. Suelter, editor. John Wiley, New York. 127–205. [DOI] [PubMed]
- Fonseca, V., P. Daumas, L. Ranjalahy-Rasoloarijao, F. Heitz, R. Lazaro, Y. Trudelle, and O. S. Andersen. 1992. Gramicidin channels that have no tryptophan residues. Biochemistry. 31:5340–5350. [DOI] [PubMed] [Google Scholar]
- Galbraith, T. P., and B. A. Wallace. 1998. Phospholipid chain length alters the equilibrium between pore and channel forms of gramicidin. Faraday Discuss. 111:159–164. [DOI] [PubMed] [Google Scholar]
- Ghosh, A. K., R. Rukmini, and A. Chattopadhyay. 1997. Modulation of tryptophan environment in membrane-bound melittin by negatively charged phospholipids: implications in membrane organization and function. Biochemistry. 36:14291–14305. [DOI] [PubMed] [Google Scholar]
- Grinvald, A., and I. Z. Steinberg. 1974. On the analysis of fluorescence decay kinetics by the method of least-squares. Anal. Biochem. 59:583–598. [DOI] [PubMed] [Google Scholar]
- Guha, S., S. S. Rawat, A. Chattopadhyay, and B. Bhattacharyya. 1996. Tubulin conformation and dynamics: a red edge excitation shift study. Biochemistry. 35:13426–13433. [DOI] [PubMed] [Google Scholar]
- Ho, C., and C. D. Stubbs. 1992. Hydration at the membrane protein-lipid interface. Biophys. J. 63:897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, W., K.-C. Lee, and T. A. Cross. 1993. Tryptophans in membrane proteins: indole ring orientations and functional implications in the gramicidin channel. Biochemistry. 32:7035–7047. [DOI] [PubMed] [Google Scholar]
- Ippolito, J. A., R. S. Alexander, and D. W. Christianson. 1990. Hydrogen bond stereochemistry in protein structure and function. J. Mol. Biol. 215:457–471. [DOI] [PubMed] [Google Scholar]
- Kelkar, D. A., A. Ghosh, and A. Chattopadhyay. 2003. Modulation of fluorophore environment in host membranes of varying charge. J. Fluoresc. 13:459–466. [Google Scholar]
- Ketchem, R. R., W. Hu, and T. A. Cross. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 261:1457–1460. [DOI] [PubMed] [Google Scholar]
- Killian, J. A. 1992. Gramicidin and gramicidin-lipid interactions. Biochim. Biophys. Acta. 1113:391–425. [DOI] [PubMed] [Google Scholar]
- Killian, J. A., K. U. Prasad, D. Hains, and D. W. Urry. 1988. The membrane as an environment of minimal interconversion. A circular dichroism study on the solvent dependence of the conformational behavior of gramicidin in diacylphosphatidylcholine model membranes. Biochemistry. 27:4848–4855. [DOI] [PubMed] [Google Scholar]
- Kirby, E. P., and R. F. Steiner. 1970. The influence of solvent and temperature upon the fluorescence of indole derivatives. J. Phys. Chem. 74:4480–4490. [Google Scholar]
- Koeppe, R. E., and O. S. Andersen. 1996. Engineering the gramicidin channel. Annu. Rev. Biophys. Biomol. Struct. 25:231–258. [DOI] [PubMed] [Google Scholar]
- Kouyama, T., T. Nishikawa, T. Tokuhisa, and H. Okumura. 2004. Crystal structure of the L intermediate of bacteriorhodopsin: evidence for vertical translocation of a water molecule during the proton pumping cycle. J. Mol. Biol. 335:531–546. [DOI] [PubMed] [Google Scholar]
- Kovacs, F., J. Quine, and T. A. Cross. 1999. Validation of the single-stranded channel conformation of gramicidin A by solid-state NMR. Proc. Natl. Acad. Sci. USA. 96:7910–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer, J. M. H., M. W. J. Esker, C. Pathmamanoharan, and P. H. Wiersema. 1977. Vesicles of variable diameter prepared by a modified injection method. Biochemistry. 16:3932–3935. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer-Plenum Press, New York.
- Lampert, R. A., L. A. Chewter, D. Phillips, D. V. O'Connor, A. J. Roberts, and S. R. Meech. 1983. Standards for nanosecond fluorescence decay time measurements. Anal. Chem. 55:68–73. [Google Scholar]
- Langs, D. A. 1988. Three-dimensional structure at 0.86 Å of the uncomplexed form of the transmembrane ion channel peptide gramicidin A. Science. 241:188–191. [DOI] [PubMed] [Google Scholar]
- Langs, D. A., G. D. Smith, C. Courseille, G. Précigoux, and M. Hospital. 1991. Monoclinic uncomplexed double-stranded, antiparallel, left-handed β5.6-helix (↑↓β5.6) structure of gramicidin A: alternate patterns of helical association and deformation. Proc. Natl. Acad. Sci. U.S.A. 88:5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoGrasso, P. V., F. Moll, and T. A. Cross. 1988. Solvent history dependence of gramicidin A conformations in hydrated lipid bilayers. Biophys. J. 54:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London, E., and A. S. Ladokhin. 2002. Measuring the depth of amino acid residues in membrane-inserted peptides by fluorescence quenching. In Current Topics in Membranes. Vol. 52. D. Benos and S. Simon, editors. Elsevier, San Diego, CA. 89–115.
- Martinac, B., and O. P. Hamill. 2002. Gramicidin A channels switch between stretch activation and stretch inactivation depending on bilayer thickness. Proc. Natl. Acad. Sci. USA. 99:4308–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClare, C. W. F. 1971. An accurate and convenient organic phosphorus assay. Anal. Biochem. 39:527–530. [DOI] [PubMed] [Google Scholar]
- Mentré, P. 2001. Water in the cell. Cell. Mol. Biol. 47:709–970. [PubMed] [Google Scholar]
- Moro, F., F. M. Goñi, and M. A. Urbaneja. 1993. Fluorescence quenching at interfaces and the permeation of acrylamide and iodide across phospholipid bilayers. FEBS Lett. 330:129–132. [DOI] [PubMed] [Google Scholar]
- Mukherjee, S., and A. Chattopadhyay. 1994. Motionally restricted tryptophan environments at the peptide-lipid interface of gramicidin channels. Biochemistry. 33:5089–5097. [DOI] [PubMed] [Google Scholar]
- Mukherjee, S., and A. Chattopadhyay. 1995. Wavelength-selective fluorescence as a novel tool to study organization and dynamics in complex biological systems. J. Fluoresc. 5:237–246. [DOI] [PubMed] [Google Scholar]
- Mukherjee, S., H. Raghuraman, S. Dasgupta, and A. Chattopadhyay. 2004. Organization and dynamics of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: a fluorescence approach. Chem. Phys. Lipids. 127:91–101. [DOI] [PubMed] [Google Scholar]
- O'Connell, A. M., R. E. Koeppe, and O. S. Andersen. 1990. Kinetics of gramicidin channel formation in lipid bilayers: transmembrane monomer association. Science. 250:1256–1259. [DOI] [PubMed] [Google Scholar]
- O'Connor, D. V., and D. Phillips. 1984. Time-Correlated Single Photon Counting. Academic Press, London. 180–189.
- Perochon, E., A. Lopez, and J. F. Tocanne. 1992. Polarity of lipid bilayers: a fluorescence investigation. Biochemistry. 31:7672–7682. [DOI] [PubMed] [Google Scholar]
- Prasad, K. U., T. L. Trapane, D. Busath, G. Szabo, and D. W. Urry. 1983. Synthesis and characterization of (1–13C) Phe9 gramicidin A. Effects of side chain variations. Int. J. Pept. Protein Res. 22:341–347. [DOI] [PubMed] [Google Scholar]
- Prendergast, F. G. 1991. Time-resolved fluorescence techniques: methods and applications in biology. Curr. Opin. Struct. Biol. 1:1054–1059. [Google Scholar]
- Raghuraman, H., and A. Chattopadhyay. 2003. Organization and dynamics of melittin in environments of graded hydration: a fluorescence approach. Langmuir. 19:10332–10341. [Google Scholar]
- Raghuraman, H., D. A. Kelkar, and A. Chattopadhyay. 2003. Novel insights into membrane protein structure and dynamics utilizing the wavelength-selective fluorescence approach. Proc. Ind. Nat. Sci. Acad. A 69:25–35. [Google Scholar]
- Raghuraman, H., and A. Chattopadhyay. 2004. Effect of micellar charge on the conformation and dynamics of melittin. Eur. Biophys. J. Apr 8 2004 [Epub ahead of print]. PMID: 15071759. [DOI] [PubMed]
- Raghuraman, H., S. K. Pradhan, and A. Chattopadhyay. 2004. Effect of urea on the organization and dynamics of Triton X-100 micelles: a fluorescence approach. J. Phys. Chem. B. 108:2489–2496. [Google Scholar]
- Raja, S. M., S. S. Rawat, A. Chattopadhyay, and A. K. Lala. 1999. Localization and environment of tryptophans in soluble and membrane-bound states of a pore-forming toxin from Staphylococcus aureus. Biophys. J. 76:1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, G. N., and R. Chandrasekaran. 1972. Conformation of peptide chains containing both L- and D-residues: part I—helical structures with alternating L- and D-residues with special reference to the LD-ribbon and the LD-helices. Ind. J. Biochem. Biophys. 9:1–11. [PubMed] [Google Scholar]
- Rawat, S. S., and A. Chattopadhyay. 1999. Structural transition in the micellar assembly: a fluorescence study. J. Fluoresc. 9:233–244. [Google Scholar]
- Rawat, S. S., S. Mukherjee, and A. Chattopadhyay. 1997. Micellar organization and dynamics: a wavelength-selective fluorescence approach. J. Phys. Chem. B. 101:1922–1929. [Google Scholar]
- Rees, D. C., G. Chang, and R. H. Spencer. 2000. Crystallographic analyses of ion channels: lessons and challenges. J. Biol. Chem. 275:713–716. [DOI] [PubMed] [Google Scholar]
- Reithmeier, R. A. F. 1995. Characterization and modeling of membrane proteins using sequence analysis. Curr. Opin. Struct. Biol. 5:491–500. [DOI] [PubMed] [Google Scholar]
- Salom, D., E. Pérez-Payá, J. Pascal, and C. Abad. 1998. Environment- and sequence-dependent modulation of the double-stranded to single-stranded conformational transition of gramicidin A in membranes. Biochemistry. 37:14279–14291. [DOI] [PubMed] [Google Scholar]
- Sarges, R., and B. Witkop. 1965. The structure of valine- and isoleucine-gramicidin A. J. Am. Chem. Soc. 87:2011–2020. [DOI] [PubMed] [Google Scholar]
- Schibli, D. J., R. F. Epand, H. J. Vogel, and R. M. Epand. 2002. Tryptophan-rich antimicrobial peptides: comparative properties and membrane interactions. Biochem. Cell Biol. 80:667–677. [DOI] [PubMed] [Google Scholar]
- Seelig, J. 1977. Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys. 10:353–418. [DOI] [PubMed] [Google Scholar]
- Separovic, F., J. Ashida, T. Woolf, R. Smith, and T. Terao. 1999. Determination of chemical shielding tensor of an indole carbon and application to tryptophan orientation of a membrane peptide. Chem. Phys. Lett. 303:493–498. [Google Scholar]
- Slater, S. J., C. Ho, F. J. Taddeo, M. B. Kelly, and C. D. Stubbs. 1993. Contribution of hydrogen bonding to lipid-lipid interactions in membranes and the role of lipid order: effects of cholesterol, increased phospholipid unsaturation, and ethanol. Biochemistry. 32:3714–3721. [DOI] [PubMed] [Google Scholar]
- Smith, R., D. E. Thomas, A. R. Atkins, F. Separovic, and B. A. Cornell. 1990. Solid-state 13C-NMR studies of the effects of sodium ions on the gramicidin A ion channel. Biochim. Biophys. Acta. 1026:161–166. [DOI] [PubMed] [Google Scholar]
- Spande, T. F., and B. Witkop. 1967. Reactivity toward N-bromosuccinimide as a criterion for buried and exposed tryptophan residues in proteins. Methods Enzymol. 11:528–532. [Google Scholar]
- Stutts, M. J., C. M. Canessa, J. C. Olsen, M. Hamrick, J. A. Cohn, B. C. Rossier, and R. C. Boucher. 1995. CFTR as a cAMP-dependent regulator of sodium channels. Science. 269:847–850. [DOI] [PubMed] [Google Scholar]
- Sychev, S. V., L. I. Barsukov, and V. T. Ivanov. 1993. The double ππ5.6 helix of gramicidin A predominates in unsaturated lipid membranes. Eur. Biophys. J. 22:279–288. [DOI] [PubMed] [Google Scholar]
- Ulmschneider, M. B., and M. S. P. Sansom. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta. 1512:1–14. [DOI] [PubMed] [Google Scholar]
- Urry, D. W. 1971. The gramicidin A transmembrane channel: a proposed π(L,D) helix. Proc. Natl. Acad. Sci. USA. 68:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urry, D. W., M. C. Goodall, J. D. Glickson, and D. F. Mayers. 1971. The gramicidin A transmembrane channel: characteristics of head-to-head dimerized π (L,D) helices. Proc. Natl. Acad. Sci. USA. 68:1907–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch, W. R., E. T. Fossel, and E. R. Blout. 1974. The conformation of gramicidin A. Biochemistry. 13:5249–5256. [DOI] [PubMed] [Google Scholar]
- Veatch, W. R., R. Mathies, M. Eisenberg, and L. Stryer. 1975. Simultaneous fluorescence and conductance studies of planar bilayer membranes containing a highly active and fluorescent analog of gramicidin A. J. Mol. Biol. 99:75–92. [DOI] [PubMed] [Google Scholar]
- Verza, G., and L. Bakás. 2000. Location of tryptophan residues in free and membrane bound Escherichia coli α-hemolysin and their role on the lytic membrane properties. Biochim. Biophys. Acta. 1464:27–34. [DOI] [PubMed] [Google Scholar]
- Wallace, B. A. 2000. Common structural features in gramicidin and other ion channels. Bioessays. 22:227–234. [DOI] [PubMed] [Google Scholar]
- Wallace, B. A., and K. Ravikumar. 1988. The gramicidin pore: crystal structure of a cesium complex. Science. 241:182–187. [DOI] [PubMed] [Google Scholar]
- Weber, G. 1960. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J. 75:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, S. H., and W. C. Wimley. 1994. Peptides in lipid bilayers: structural and thermodynamic basis for partitioning and folding. Curr. Opin. Struct. Biol. 4:79–86. [Google Scholar]
- Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- Woolley, G. A., and B. A. Wallace. 1994. Membrane protein structure: lessons from gramicidin. In Membrane Protein Structure: Experimental Approaches. S.H. White, editor. Oxford University Press, New York. 314–334.
- Yau, W.-M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- Yeagle, P. 1987. The Membranes of Cells. Academic Press, Orlando, FL. 89–90.
- Zein, M., and R. Winter. 2000. Effect of temperature, pressure and lipid acyl chain length on the structure and phase behaviour of phospholipid-gramicidin bilayers. Phys. Chem. Chem. Phys. 2:4545–4551. [Google Scholar]
- Zhang, Z., S. M. Pascal, and T. A. Cross. 1992. A conformational rearrangement in gramicidin A: from a double-stranded left-handed to a single-stranded right-handed helix. Biochemistry. 31:8822–8828. [DOI] [PubMed] [Google Scholar]