

Abstract
An agonist-initiated Ca2+ signaling model for calmodulin (CaM) coupled to the phosphorylation of myosin light chains was created using a computer-assisted simulation environment. Calmodulin buffering was introduced as a module for directing sequestered CaM to myosin light chain kinase (MLCK) through Ca2+-dependent release from a buffering protein. Using differing simulation conditions, it was discovered that CaM buffering allowed transient production of more Ca2+-CaM-MLCK complex, resulting in elevated myosin light chain phosphorylation compared to nonbuffered control. Second messenger signaling also impacts myosin light chain phosphorylation through the regulation of myosin light chain phosphatase (MLCP). A model for MLCP regulation via its regulatory MYPT1 subunit and interaction of the CPI-17 inhibitor protein was assembled that incorporated several protein kinase subsystems including Rho-kinase, protein kinase C (PKC), and constitutive MYPT1 phosphorylation activities. The effects of the different routes of MLCP regulation depend upon the relative concentrations of MLCP compared to CPI-17, and the specific activities of protein kinases such as Rho and PKC. Phosphorylated CPI-17 (CPI-17P) was found to dynamically control activity during agonist stimulation, with the assumption that inhibition by CPI-17P (resulting from PKC activation) is faster than agonist-induced phosphorylation of MYPT1. Simulation results are in accord with literature measurements of MLCP and CPI-17 phosphorylation states during agonist stimulation, validating the predictive capabilities of the system.
INTRODUCTION
The construction of models that can be used for simulation of biological systems provides a means to formulate new hypotheses about the system and then test these hypotheses experimentally. However, the complexity of biological systems and the differences in experimental starting points make it difficult to establish a standard formula for modeling a given system. Although several mathematical suites and simulation environments are available, few provide a simple interface to mathematical algorithms and differential equation solvers that are necessary to provide understandable graphical and numerical output for the cell biologist or biochemist. To this end, the Virtual Cell provides a common, user-operable interface for designing and launching biochemical simulations (Loew and Schaff, 2001; Slepchenko et al., 2003). Models of Ca2+ mobilization in neurons (Slepchenko et al., 2002; Fink et al., 2000) and smooth muscle cells (Fink et al., 1999) provide the framework for building more extensive models capable of simulating the entire pathway from agonist to cellular endpoint. One of the ubiquitous cellular signaling pathways involves mobilization of Ca2+ and activation of calmodulin (CaM) targeted enzymes. In a previous report, a model of agonist-mediated Ca2+ mobilization coupled to Ca2+-CaM signaling in smooth muscle cells was created and validated with respect to agonist sensitivity and Ca2+-mediated CaM activation of a downstream protein kinase (Lukas, 2004). With this model, it is now possible to explore additional features of the Ca2+ signaling pathway including the questions of calmodulin buffering and details of the regulation of a biochemical system. The CaM-myosin light chain kinase (MLCK) system and its associated regulatory elements are the focus of this report. The strategies used for integrating multiple kinase and phosphatase regulatory systems are outlined, as well as the concept of specific CaM buffering, a process for delivering CaM to a subset of Ca2+-dependent processes.
Using simulation data to predict CaM-buffer properties, myosin Ic was implicated as a CaM buffering protein associated with the smooth muscle cytoskeleton. One of the three CaM-binding IQ domain motifs of Myosin Ic was identified as a CaM buffering domain based upon the measured CaM-binding properties of a synthetic peptide analog. Simulation data suggest that the Ca2+-independent binding and release of CaM by myosin Ic delivers CaM to the filament-bound MLCK, thus providing a conduit for activation of myosin-based contraction. Details of the regulation of myosin phosphatase activity were also elucidated by simulation data. An estimate of the level of phosphorylated MYPT1 subunit was obtained by running simulations with various levels of prephosphorylated subunit and allowing additional phosphorylation during a typical Ca2+ transient. Similarly, the dynamic role of CPI-17 activated by phosphorylation in modulating myosin light chain phosphatase (MLCP) activity was also elaborated by comparing simulation output to existing experimental results.
METHODS
Model building strategy
The modules built for regulation of the phosphorylation state of smooth muscle myosin are shown in Fig. 1. Agonist-induced Ca2+ generation and sequestration were done as described (Lukas, 2004). In the prototype system, the MLCK and MLCP concentrations are equal (10 μM), and the levels of other regulatory molecules estimated from the literature (Table 1) and optimized by simulation as needed. As outlined below, each step of the process was compared by simulation to any existing data, and the model parameters further refined to achieve a close fit. The system was also found to be scalable to other types of cells that contain different amounts of MLCK, MLCP, and calmodulin.
FIGURE 1.

Downstream signaling elements in the cytoplasmic compartment. Shown are the details of the MLC phosphorylation/dephosphorylation module. The simulation system contains dynamic regulation of MLCK activity through Ca2+-CaM and MLCP activity through phosphorylation by Rho kinase and inhibition by phosphorylated CPI-17. CPI-17 is phosphorylated by PKC that is activated by DAG produced by activated phospholipase C. RhoA that is generated by G-protein exchange with receptor activated G proteins activates Rho kinase, and cG kinase is activated by cGMP.
TABLE 1.
Ca2+, CaM, MLCK, PKC, and MLCP kinetic parameters
| Reaction | Kd (μM) | kforward M−1 s−1 | References |
|---|---|---|---|
| Ca2+-CaM + MLCK ↔ Ca2+-CaM-MLCK | 0.0011 | 28 | (Johnson et al., 1996; Kasturi et al., 1993) |
| Ca2+-CaM + MLCK-P ↔ Ca2+-CaM-MLCK-P | 0.023 | 8 | (Johnson et al., 1996; Kasturi et al., 1993) |
| CaM + BufferP ↔ CaM-BufferP | 5 | 5 | See text |
| CaM-BufferP + 2Ca2+ ↔ Ca2+-CaM + BufferP | 2.5 | 75 | See text |
| MLCP + CPI-17P ↔ MLCP-CPI-17P | 0.005 | 5 | (Feng et al., 1999b), see text |
| Enzyme kinetics | Km μM | kcat s−1 | |
| Rho Kinase + MYPT → MYPT-P (MLCP-P) | 2.5 | 2.5 | (Feng et al., 1999b) |
| RhoA-Rho Kinase + MYPT → MYPT-P (MLCP-P) | 2.5 | 1.5 | (Feng et al., 1999b) |
| PKC + CPI-17 → CPI-17P | 1.0 | 4.0 | (Senba et al., 1999), see text |
| CPI-17P (dephosphorylation) → CPI-17 | 1.0 | 1.5 | See text |
| MYPT-P + (cG kinase-activated) → MYPT | 1.0 | 1.0 | See text |
Values were optimized by comparison to experimental data.
All simulations presented in this work used the Runge-Kutta fourth-order solver with a fixed time step of 0.1 ms. Ordinary differential equations (ODE) were generated by the Virtual Cell suite (www.nrcam.uchc.edu). The biomodel system (including the ODEs) can be exported to other simulation programs by way of the export functions on the XML part of the Virtual Cell model page. Thus, the simulation suite is designed to be portable and available to a wide variety of researchers. The described Virtual Cell biomodel can be found in the public directory Ca release-coupled myosin light chain (MLC).
Calmodulin buffering
The total buffer concentration was 15 μM and a Kd of 10 μM was assumed. Thus, the model was built around a single class of CaM-buffering proteins that could sequester CaM proximal to other CaM targets (such as MLCK) and then liberate CaM during a Ca2+ flux. Thus, CaM delivery to MLCK was derived using Eqs. 1–3:
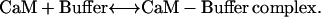 |
(1) |
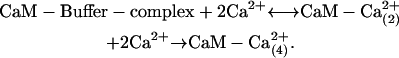 |
(2) |
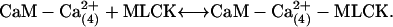 |
(3) |
The scheme assumes that 2 mol of Ca2+ are needed to dissociate CaM from the buffer complex and that this transient CaM with two bound Ca2+ rapidly acquires two more Ca2+ and then binds to and activates MLCK. The kinetic constants for these steps were further refined by simulations (see Results).
Regulation of myosin light chain phosphatase
The Km and specific activity values for the constitutive and RhoA stimulated activity (Table 1) were used to parameterize MYTP1 phosphorylation. Stimulated MYTP1 phosphorylation was coupled to the production of RhoA. Activated RhoA (GTP-bound) is generated by exchange from G11/12 species, coupled to a subpopulation of agonist receptors (Somlyo and Somlyo, 2000). This process is catalyzed by RhoA exchange factors (RhoGEFS) (Blomquist et al., 2000), but reaction kinetics or binding parameters are not available for these species. Therefore, RhoA generation was set up in a hypothetical manner by exchanging a portion of the activated (GTP-bound) Gq to RhoA-GTP. RhoA-GTP was allowed to inactivate through hydrolysis of its bound GTP at a basal rate of 0.1/s. The second regulatory mechanism for MLCP involves inhibition through the small protein inhibitor CPI-17. CPI-17 phosphorylated at Thr-38 binds to MLCP with a Kd of 1–5 nM and inhibits activity (Hayashi et al., 2001; Kitazawa et al., 2000). Experimental data suggest that diacylglycerol (DAG)-activated PKC (α and γ) phosphorylate CPI-17 in smooth muscle cells (Kitazawa et al., 2003), tissues (Woodsome et al., 2001; Ogut and Brozovich, 2000), and in some nonmuscle cells such as platelets (Watanabe et al., 2001). Thus, CPI-17 phosphorylation was coupled to DAG production and PKC activation using enzyme explicit Michaelis parameters and a PKC concentration of 1 μM (see Table 1). In the current model, the concentration of MLCP was set equal to MLCK (10 μM) and CPI-17 was set at 6 μM near the upper limit estimated in smooth muscle cells (Woodsome et al., 2001). The dephosphorylation of the CPI-17P-MLCP complex and CPI-17P were formulated by irreversible Michaelis-Menton kinetics with a kcat of 1/s and Km of 1 μM.
Countering the downregulation of MLCP, static levels of cGMP were coupled to MLCP activation through a direct or indirect reversal of the inhibitory Rho kinase-mediated phosphorylation (Surks et al., 1999; Sandu et al., 2001). Although cG kinase (the target of cGMP) phosphorylates MYPT1, it apparently does not alter MLCP activity (Nakamura et al., 1999). The parameters for cG kinase-mediated reversal of MLCP inhibition were based upon cGMP binding and activation of cG kinase with an EC50 for cGMP of 0.57 μM (Smith et al., 2000). The cG-kinase concentration was fixed at 0.1 μM (Francis et al., 1988) and the concentration of cGMP was fixed at the estimated cGMP concentration (0.1 μM) in smooth muscle (Smith et al., 2000; Francis et al., 1988). The phosphatase that is most effective in dephosphorylating MYPT1 is PPA2c (Takizawa et al., 2002b); however, a direct link between cG kinase and this phosphatase has not been established. Thus, cG kinase-induced dephosphorylation of MYPT1 was done in a hypothetical fashion “catalyzed” by cG kinase.
Binding of CaM to IQ-domain peptides
Dansyl bovine brain calmodulin (Dansyl-CaM) was prepared as described (Vorherr et al., 1990). The stoichiometry was 0.9–1.2 mol/mol and the fluorescent protein used at a concentration of 200 nM. Synthetic peptides based upon myosin Ic IQ domains, IQ1 = RKHSIATFLQARWRGYHQRQKFL and IQ2 = HMKHSAVEIQSWWRGTIGRRKAA, were prepared by standard solid-phase methods using Fmoc-protected amino acids as described previously (Lukas et al., 1999). Peptides were purified by reverse-phase high-performance liquid chromatography on a Phenomenex C5 column. Purified peptides gave the expected MH+ ions (IQ1 = 2928.6, calc = 2928.4; IQ2 = 2706.1, calc = 2706.2) in matrix-assisted laser desorption ionization time-of-flight mass spectra.
Peptide binding was done in 25 mM Tris, 1 mM EDTA, 150 mM NaCl, pH 7.5). Solutions of Dansyl-CaM in the binding buffer were placed into a 96-well microtiter plate (Falcon). Peptide was then added to the desired final concentration from a concentrated stock solution in water. Total volume remained constant at 110 μL. Fluorescence was read in a Victor 2 spectrometer using 335-nm excitation and 535-nm emission filters. The data were corrected for background fluorescence and expressed as the ratio F/Fo, where Fo is the fluorescence of Dansyl-CaM in the absence of peptide and F is the measured fluorescence in the presence of increasing concentration of peptide. Data were fit to Eq. 4 to obtain the binding constant, K:
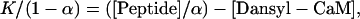 |
(4) |
where α = F − Fo/F∞ − Fo and F∞ is the calculated endpoint fluorescence value. The binding constant K equals the reciprocal slope of a plot of 1/1 − α versus [Peptide]/α (Vorherr et al., 1990).
RESULTS
The details of the regulatory model for myosin light chain phosphorylation are illustrated in Fig. 1. The importance of each of the subsystems involved in the regulation of the maximal amount of Ca2+ -CaM-MLCK complex will be evaluated using simulations based upon the model and compared to existing results.
Properties of CaM buffering proteins
The level of free CaM in the cytoplasm under resting conditions has been estimated at 0.25–1 μM using various techniques (Zimmermann et al., 1995; Tran et al., 2003). The total amount of CaM, however, is on the order of 20–50 μM (Zimmermann et al., 1995; Tran et al., 2003). Thus, without release from CaM-buffering proteins, the activation of CaM targets through increases in Ca2+ is apparently limited by the free CaM concentration. However, certain CaM targets that bind CaM (at low Ca2+) provide a source of CaM that is released to activate targets during a Ca2+ flux. Although some CaM remains associated with isolated actomyosin cytoskeletons (Wilson et al., 2002), it is not clear how much CaM is actually bound to MLCK compared to other CaM binding proteins. In the formulation of the model for smooth muscle, 10 μM MLCK and 1 μM free CaM at equilibrium provided 0.36 μM CaM associated with MLCK under resting conditions. Under these conditions, simulations predicted that the activated MLCK would yield only ∼28% of the peak phosphorylated MLC (0.6 mol/mol) observed in agonist-stimulated smooth muscle cells (Wilson et al., 2002; Taylor and Stull, 1988), suggesting that MLCK alone does not appear to sequester enough CaM at resting Ca2+ to provide a physiological level of MLC phosphorylation in smooth muscle. The concentration of a hypothetical CaM-buffer protein was adjusted to buffer 3 μM CaM, similar to that estimated in smooth muscle cytoskeleton (Wilson et al., 2002).
Fig. 2 A illustrates the effect of changing the Ca2+ dissociation constant of a CaM-buffer protein complex over the 1–20 μM range assuming that the buffer protein dissociates CaM upon binding Ca2+. Compared with the MLC phosphorylation activity obtained in the absence of buffering, it appears that in this system, the Ca2+ dissociation constant for a CaM-buffer protein should be less than 5 μM to enhance phosphorylation more than twofold. Surveying the known CaM-binding proteins associated with the cytoskeleton, a plausible CaM buffering protein is myosin Ic (Coluccio, 1997). Myosin Ic contains three potential CaM-binding IQ motifs that occur in tandem (Coluccio, 1997; Gillespie and Cyr, 2002). Experiments done in vitro have demonstrated that 1 mol/mol of CaM is released by myosin Ic at elevated Ca2+ concentrations (Collins et al., 1990), supporting its potential role as a CaM buffer. Recent studies of synthetic peptide analogs of the IQ domains of myosin Ic indicated that two IQ domain peptide analogs fit the requirement of being able to bind CaM in the absence of Ca2+ and weakly in the presence of Ca2+ (Gillespie and Cyr, 2002). Using the published data on CaM binding to the peptide analogs and assuming that binding of at least two calcium ions causes CaM to dissociate from the complex (Zhu et al., 1998), the calculated Ca2+ sensitivity of the dissociation of CaM from these IQ domains is in the range of 2–4 μM, comparable with the values obtained from the simulations. As shown in Fig. 2 B, the difference in the peak of MLC phosphorylation is at least threefold greater with CaM buffering than in its absence and the level of activated Ca2+-CaM-MLCK complex increases proportionately (Fig. 2 B, inset) Thus, based upon these results, myosin Ic is predicted to be a CaM buffer for MLCK activation in smooth muscle.
FIGURE 2.

Predicted effects of calmodulin buffering on peak MLC phosphorylation and Ca2+-CaM-MLCK complex formation. (A) Effect of changing the Ca2+ dissociation constant for the CaM-buffer protein complex on the peak of MLC phosphorylation. (B) Profile of MLC phosphorylation (MLC_P) and Ca2+-CaM-MLCK complex formation (inset) in the presence (solid line) and absence (dotted line) of CaM buffering. Simulations were run at a fixed ligand (100-nM) concentration.
To determine which IQ domain peptide analog of myosin Ic was a more representative CaM-buffering domain required reinvestigation of their CaM binding properties. Fig. 3 shows that one peptide analog (IQ1) binds well to Dansyl-CaM (Kd = 2 μM) in the presence of 1 mM Ca2+ chelator, whereas the other peptide analog from the adjacent IQ domain (IQ2) of myosin Ic has weaker affinity for CaM (Kd > 20 μM). The IQ1 peptide (up to 40 μM) does not change the fluorescence of Dansyl-CaM in the presence of 1 mM Ca2+, suggesting that it only weakly interacts with the Ca2+-bound form of CaM (data not shown).
FIGURE 3.

Binding of myosin Ic IQ-domain peptide analogs to CaM. Dansyl-bovine brain calmodulin (200 nM) was titrated with peptide IQ1, RKHSIATFLQARWRGYHQRQKFL (squares) or IQ2, HMKHSAVEIQSWWRGTIGRRKAA (circles) in 50 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 7.5. Error bars are from duplicates run in the titration assay. Fluorescence emission was measured at 535 nm with excitation at 335 nm (10-nm band pass) using a Victor 2 96-well plate reading fluorometer. Data are corrected for background fluorescence in the absence of Dansyl-CaM.
Simulations forecast the effects of changing MLCP regulatory elements
Inhibition of MLCP is mediated through phosphorylation of one or more sites in the MYPT regulatory subunit (Thr 695 and/or Thr-850) (Feng et al., 1999a; Kawano et al., 1999)-human MYPT numbering. Several kinases phosphorylate MYPT in vitro, including Rho kinase (Feng et al., 1999a; Kawano et al., 1999), p21-activated kinase (PAK) (Takizawa et al., 2002a), integrin-linked kinase (Kiss et al., 2002), and Raf-1 (Broustas et al., 2002). Phosphorylation at Thr-695 of MYPT decreases MLCP activity (Borman et al., 2002) and sensitizes muscle tissue to contract at lower Ca2+ (Somlyo and Somlyo, 2000). Studies with phosphorylation site-directed antibodies indicate that MYPT1 phosphorylation state increases 1.5- to 4-fold upon agonist treatment (Watanabe et al., 2001; Seko et al., 2003). Thus, a portion of MYPT1 is phosphorylated in resting cells, but the ratio of phosphorylated to unphosphorylated MYPT1 in resting tissues is not known; therefore, this initial value was estimated by the simulation process. Applying the known parameters for enzymatic activities (Table 1), simulation data indicates that the level of MLCP activity limits the maximal MLC phosphorylation realized by an agonist-stimulated Ca2+ transient. As illustrated in Fig. 4 A, the initial level of phosphorylated MYPT subunit of MLCP was varied. At the peak, nearly all of the MLC is phosphorylated when the initial stoichiometry of MYPT1 subunit phosphorylation is >0.5. This level of activity is inconsistent with experiments in smooth muscle tissues and cells where MLC phosphorylation peaks at ∼60% of total (Taylor and Stull, 1988). Thus, the initial level of phosphorylated MLCP was set at 0.25 (25%) allowing for a further increase in phosphorylation state during a Ca2+ transient, consistent with previous experimental results (Kitazawa et al., 2003; Watanabe et al., 2001).
FIGURE 4.

Predictions of effects of myosin phosphatase regulatory elements on MLC phosphorylation (MLC_P). (A) Simulation of MLC phosphorylation as a function of the percent of MYTP prephosphorylated in a model cell stimulated with 100 nM G protein-coupled receptor ligand. MLC_P production increases with MYPT phosphorylation at 0 (thinnest line), 25%, 50%, and 75% (thickest line) prephosphorylated enzyme. (B) Simulation of MLC_P production in the presence and absence of CPI-17P inhibitor generation, and stimulated MYPT phosphorylation: control (solid line), no CPI-17 phosphorylation (dashed line), no additional MYTP1 phosphorylation (dotted line), and no additional MYPT phosphorylation or CPI-17P (dot-dash line).
Depending upon the cell or tissue, one or both mechanisms of MLCP regulation (phosphorylation and inhibition by CPI-17) dynamically decrease MLCP activity (Kitazawa et al., 2003; Watanabe et al., 2001). How these parameters vary with agonist concentration under various conditions is not clear because the endpoint measurement reflects the convergence of multiple enzymatic processes that occur in different time frames. The model was tested in simulations representing various conditions and the effects of inhibiting CPI-17 phosphorylation, stimulated MLCP phosphorylation (via Rho kinase), or both (Fig. 4 B). Inhibition of CPI-17 or MLCP phosphorylation (enhancing MLCP activity) decreases the maximal MLC phosphorylation by >50%. The shape of the MLC phosphorylation profile is multiphasic, with both CPI-17 and MLCP phosphorylation contributing to the overall response. The contribution of each toward modulating MLCP activity is dependent upon the agonist concentration. CPI-17 phosphorylation parallels that of MLC phosphorylation at most ligand concentrations (Fig. 5). The simulated results also predict that RhoA kinase-stimulated MLCP phosphorylation is slow even at high agonist concentrations. At low agonist concentrations, the time to MLC phosphorylation peak is slower (∼15 s) compared to higher agonist concentrations (6–8 s) but the peak Ca2+-CaM-MLCK complex occurs between 3 and 6 s (Fig. 5). Thus, within this time window the level of MLC phosphorylation achieved is limited because the MLCP has not yet been inhibited by CPI-17P. This is seen as an inflection on the MLC profile at 3 s (Fig. 5). At the lowest ligand concentration (1 nM), MLC phosphorylation parallels CPI-17P-mediated inhibition, resulting in a delayed peak response. These data suggest that agonist-induced MLC phosphorylation state in some stimulated cells or tissues may exhibit a rapid peak of MLC phosphorylation followed by a slow decay phase.
FIGURE 5.

Ligand dependence of MLCP regulatory activities. Illustrated are examples of simulated output of MLC_P production (solid lines), MYPT phosphorylation (dashed lines), and CPI-17P-MLCP complex (dotted lines) at (A) 1 nM, (B) 10 nM, and (C) 100 nM ligand.
The model also incorporates static regulation of MLCP activity by the action of cyclic GMP (cGMP). This cyclic nucleotide has been long recognized to be a relaxing agent for smooth muscle cells, but how its main effector cGMP-dependent protein kinase (cG kinase) achieves this activity is still being investigated (Lincoln et al., 2001). Two parallel mechanisms exist for relaxation by cGMP. The first involves the down-regulation of Ca2+ channels (both plasma membrane and intracellular), whereas the other involves direct effects on MLCP or pathways that lead to MLCP down-regulation (Cornwell and Lincoln, 1989; Lincoln et al., 2001). To test whether the modeled elements provide some level of cGMP control, simulations were performed at different cGMP concentrations ranging from 0.03 to 3 μM. First, the cGMP concentration was changed in the absence of ligand and the system allowed to equilibrate (∼200 s). This resulted in a new level of prephosphorylated MYTP1 subunit that was entered as the new initial state. For example, at 3 μM cGMP, the amount of phosphorylated MYTP1 decreased to 3% of the total. When the cGMP was lowered to 0.03 μM, the level of prephosphorylated MYTP1 increased to 47%. As shown earlier in Fig. 4, changing the basal level of MYPT1 phosphorylation dramatically affects the endpoint of MLC phosphorylation. Thus, consistent with experimental data (Lincoln et al., 2001), elevated cGMP results in a desensitized state, whereas decreased cGMP results in sensitization.
Secondary regulation of MLCK activity
The current model does not include dynamic regulation of MLCK activity through phosphorylation. It is known, however, that MLCK activity may be modulated (in vitro) through phosphorylation by kinases such as CaM-dependent kinase II (CaMKII) (Stull et al., 1993; Ikebe and Reardon, 1990), mitogen-activated protein kinase (ERK-1) (Morrison et al., 1996; Klemke et al., 1997), and PAK (Goeckeler et al., 2000; Sanders et al., 1999). A 10–50% increase in MLCK specific activity upon phosphorylation by ERK-1 in vitro has been reported (Klemke et al., 1997; Morrison et al., 1996), whereas phosphorylation by CaMKII (Ikebe and Reardon, 1990) or PAK-2 (Goeckeler et al., 2000; Sanders et al., 1999) decreases the Ca2+-CaM sensitivity of the kinase complex with little change to its specific activity (Ikebe and Reardon, 1990). Kinetic analysis indicates that phosphorylated MLCK binds more slowly to Ca2+-CaM and dissociates more rapidly when Ca2+ is removed (Table 3) (Johnson et al., 1996; Kasturi et al., 1993). Simulation parameters can be adjusted to such changes in activation, but, unlike the regulatory subunit of MLCP (MYPT), there is little data on the extent of MLCK phosphorylation at specific sites in agonist-stimulated cells (Stull et al., 1990). However, correlation of the desensitization of MLCK activity by CaMKII phosphorylation of sites near the CaM regulatory domain has been shown in cultured smooth muscle cells and tissues (Tansey et al., 1992, 1994; Stull et al., 1990). Fig. 6 provides examples of the predicted effects of complete phosphorylation (100%) of MLCK by CaMKII or PAK, phosphorylation by ERK, and the combined effect of both. In the case of CaMKII, reduced Ca2+/CaM sensitivity decreases peak MLC phosphorylation by at least 50%. As expected, ERK-1 phosphorylation of MLCK potentiates MLC phosphorylation but fails to overcome the inhibitory effects of phosphorylation by CaMKII. It should be pointed out, however, that these predictions are at the extremes because MLCK is rapidly dephosphorylated in vitro by the PP1 and PP2A phosphatases (Nomura et al., 1992). Thus, the timing and duration of MLCK phosphorylation with respect to other control elements will determine its relevance in agonist-stimulated contractile events.
FIGURE 6.

Predicted effects of MLCK prephosphorylation on MLC_P production in agonist-stimulated cells. Models were modified to contain phosphorylated MLCK with enhanced specific activity (ERK phosphorylation) or decreased Ca2+-CaM sensitivity (CaMKII phosphorylation). Simulations were done at 100 nM ligand (10× Kd) with no MLCK phosphorylation (solid line), stoichiometric ERK phosphorylation (dashed line), stoichiometric CaMKII phosphorylation (dotted lines), and a combination of both (dot-dash lines).
DISCUSSION
In this article, a comprehensive model of Ca2+ signaling through CaM and target proteins was tested in different applications to biological problems on the downstream side of the pathway. The first problem is that of calmodulin buffering. Most cellular CaM is bound to buffering molecules, some of which must be located proximal to other CaM targets that are activated by Ca2+. To evaluate the CaM binding and Ca2+-release properties of the buffering molecules, the simulation process proved useful in providing a plausible set of parameters for which a candidate protein, myosin Ic, was identified and a putative buffering domain characterized. Based upon the simulation results and estimates of myosin Ic concentration in smooth muscle cells (Wilson et al., 2002), less than 15% of the total CaM is sequestered by myosin Ic, suggesting additional CaM buffering proteins are present. There are two modes of CaM-releasing activity of CaM buffering proteins. The difference in affinity between Ca2+-dependent and Ca2+-independent binding of CaM, as in myosin Ic, allows buffering, but this property is not common to all IQ motif-containing proteins (Bahler and Rhoads, 2002). In addition, phosphorylation of some of the neuron-associated IQ-motif proteins, such as neurogranin and neuromodulin, antagonizes CaM binding and releases CaM to bind to other targets (Slemmon et al., 1996, 2000). CaM buffering proteins with similar properties in nonneuronal tissues remain to be identified. It is anticipated that some of these will contain IQ domains similar in properties but not necessarily primary structure to the myosin Ic domains. Indeed, a recent report by Tran et al. (2003) demonstrates that the CaM-binding domain of endothelial cell nitric-oxide synthase serves as an internal CaM buffer that sequesters a significant part (up to 25%) of the total cellular CaM in certain endothelial cell lines.
The next problem investigated using the model and predictive simulations was the complex regulation of myosin phosphatase activity. This process involves multiple protein phosphorylation events. In the control of MLCP phosphatase activity, modeling efforts were focused on Rho kinase and the CPI-17 inhibitor protein. Because of the ability of the selective Rho kinase inhibitor, Y27632, to block Ca2+ sensitization of smooth muscle cells (Fu et al., 1998) and tissue (Seko et al., 2003), this is probably the most ubiquitous pathway for MLCP regulation. However, the relatively low activity of RhoA-activated kinase suggests that constitutive MLCP phosphorylation by Rho kinase and other known MYPT kinases such as ZIP kinase and integrin-linked kinase may contribute toward the initial set point of cellular MLCP activity (unphosphorylated + phosphorylated enzyme). Using simulations, this set point was found to be ∼25% phosphorylated MLCP.
In contrast to Rho kinase, the results from simulations predict that CPI-17 plays a key role in the dynamic control (i.e., during a Ca2+ transient) of MLCP activity. Activation of PKC and phosphorylation of CPI-17 occurs within the same time window as MLCK activation, whereas Rho kinase activation is slower and serves to prolong MLC phosphorylation (Figs. 4 and 5). Although CPI-17 expression was thought to be restricted to smooth muscle, recent work has shown that this protein is expressed in some nonmuscle cells such as platelets (Watanabe et al., 2001). Using selective PKC and Rho-kinase inhibitors, the contribution of CPI-17 and MYPT phosphorylation to MLC phosphorylation state has been addressed. In platelets (Watanabe et al., 2001) or smooth muscle cells (Kitazawa et al., 2003) inhibition of either pathway reduced the MLC phosphorylation peak ∼50%, consistent with the current results predicted by the model and simulation (Fig. 4 B). In different types of smooth muscle cells (e.g., artery versus portal vein), similar experiments also suggested that CPI-17 phosphorylation induced by agonist stimulation may be more important for Ca2+ sensitization in cells that express high levels of CPI-17 compared to MLCP (Woodsome et al., 2001; Kitazawa et al., 1999).
The last problem investigated the role of phosphorylation of MLCK by other protein kinases. Stimulatory phosphorylation of MLCK by MAP kinase (ERK-1) could be important in certain nonmuscle cell types (Klemke et al., 1997), but the model and simulation data indicate that phosphorylation of MLCK by ERK-1 may not reverse the down-regulation of Ca2+ signaling caused by stoichiometric CaMKII or PAK-1 phosphorylation. Thus, in the current model for a smooth muscle cell, the simulation results are consistent with experimental data in smooth muscle tissues where the inhibition of MAP kinases did not influence contractility (Gorenne et al., 1998).
Shortcomings of simulation data
The primary limitation of the model and simulation results is the lack of quantification of MLCP expression and various regulatory domain (MYPT) isoforms present. Some existing data suggest that regulation of MLCP will differ depending upon the composition of the complex. For example, adult chicken gizzard does not contain the MYPT isoform that has a cG kinase-binding domain and is resistant to relaxation by cGMP (Ogut and Brozovich, 2000). In contrast, aorta contains mostly the MYPT isoform that binds cG kinase and is very sensitive to relaxation by cGMP (Ogut and Brozovich, 2000). In some tissues, increases in cGMP levels also regulate MLCP activity through the phosphorylation and inhibition of RhoA, thus pushing the basal level of MYPT phosphorylation downward and decreasing Ca2+ sensitivity (Murthy et al., 2003; Begum et al., 2000). Drugs such as sildenafil citrate (Viagra) that inhibit cGMP phosphodiesterases work through this pathway in vivo (Turko et al., 1999). Thus, MYPT isoform expression data as well as quantitative determination of the amounts of phosphorylated MYPT and various phosphorylated MLCK species present in a given tissue will allow more accurate modeling of contractility in specific smooth muscle tissues.
Acknowledgments
The Virtual Cell Portal is provided by the National Resource for Cell Analysis and Modeling supported by National Institutes of Health grant 5P41RR013186.
References
- Bahler, M., and A. Rhoads. 2002. Calmodulin signaling via the IQ motif. FEBS Lett. 513:107–113. [DOI] [PubMed] [Google Scholar]
- Begum, N., N. Duddy, O. Sandu, J. Reinzie, and L. Ragolia. 2000. Regulation of myosin-bound protein phosphatase by insulin in vascular smooth muscle cells: evaluation of the role of Rho kinase and phosphatidylinositol-3-kinase-dependent signaling pathways. Mol. Endocrinol. 14:1365–1376. [DOI] [PubMed] [Google Scholar]
- Blomquist, A., G. Schworer, H. Schablowski, A. Psoma, M. Lehnen, K. H. Jakobs, and U. Rumenapp. 2000. Identification and characterization of a novel Rho-specific guanine nucleotide exchange factor. Biochem. J. 352:319–325. [PMC free article] [PubMed] [Google Scholar]
- Borman, M. A., J. A. MacDonald, A. Murany, D. J. Hartshorne, and T. A. J. Haystead. 2002. Smooth muscle myosin phosphatase-associated kinase induces Ca2+ sensitization via myosin phosphatase inhibition. J. Biol. Chem. 277:23441–23446. [DOI] [PubMed] [Google Scholar]
- Broustas, C. G., N. Grammatikakis, M. Eto, P. Dent, D. L. Brautigan, and U. Kasid. 2002. Phosphorylation of the myosin-binding subunit of myosin phosphatase by Raf-1 and inhibition of phosphatase activity. J. Biol. Chem. 277:3053–3059. [DOI] [PubMed] [Google Scholar]
- Collins, K., J. R. Sellers, and P. Matsudaira. 1990. Calmodulin dissociation regulates brush border myosin I (110-kD-calmodulin) mechanochemical activity in vitro. J. Cell Biol. 110:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio, L. M. 1997. Myosin I. Am. J. Physiol. 273:C347–C359. [DOI] [PubMed] [Google Scholar]
- Cornwell, T. L., and T. M. Lincoln. 1989. Regulation of intracellular Ca2+ levels in cultured vascular smooth muscle cells. Reduction of Ca2+ by atriopeptin and 8-bromo-cyclic GMP is mediated by cyclic GMP-dependent protein kinase. J. Biol. Chem. 264:1146–1155. [PubMed] [Google Scholar]
- Feng, J., M. Ito, K. Ichikawa, N. Isaka, M. Nishikawa, D. J. Hartshorne, and T. Nakano. 1999a. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J. Biol. Chem. 274:37385–37390. [DOI] [PubMed] [Google Scholar]
- Feng, J., M. Ito, Y. Kureishi, K. Ichikawa, M. Amano, N. Isaka, K. Okawa, A. Iwamatsu, K. Kaibuchi, D. J. Hartshorne, and T. Nakano. 1999b. Rho-associated kinase of chicken gizzard smooth muscle. J. Biol. Chem. 274:3744–3752. [DOI] [PubMed] [Google Scholar]
- Fink, C. C., B. Slepchenko, and L. M. Loew. 1999. Determination of time-dependent inositol-1,4,5-trisphosphate concentrations during calcium release in a smooth muscle cell. Biophys. J. 77:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink, C. C., B. M. Slepchenko, I. I. Moraru, J. Watras, J. C. Schaff, and L. M. Loew. 2000. An image-based model of calcium waves in differentiated neuroblastoma cells. Biophys. J. 79:163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S. H., B. D. Noblett, B. W. Todd, J. N. Wells, and J. D. Corbin. 1988. Relaxation of vascular and tracheal smooth muscle by cyclic nucleotide analogs that preferentially activate purified cGMP-dependent protein kinase. Mol. Pharmacol. 34:506–517. [PubMed] [Google Scholar]
- Fu, X., M. C. Gong, T. Jia, A. V. Somlyo, and A. P. Somlyo. 1998. The effects of the Rho-kinase inhibitor Y-27632 on arachidonic acid-, GTPgammaS-, and phorbol ester-induced Ca2+-sensitization of smooth muscle. FEBS Lett. 440:183–187. [DOI] [PubMed] [Google Scholar]
- Gillespie, P. G., and J. L. Cyr. 2002. Calmodulin binding to recombinant myosin-1c and myosin-1c IQ peptides. BMC Biochem. 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeler, Z. M., R. A. Masaracchia, Q. Zeng, T.-L. Chew, P. A. Gallagher, and R. B. Wysolmerski. 2000. Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. J. Biol. Chem. 275:18366–18374. [DOI] [PubMed] [Google Scholar]
- Gorenne, I., X. Su, and R. S. Moreland. 1998. Inhibition of p42 and p44 MAP kinase does not alter smooth muscle contraction in swine carotid artery. Am. J. Physiol. 275:H131–H138. [DOI] [PubMed] [Google Scholar]
- Hayashi, Y., S. Senba, M. Yazawa, D. L. Brautigan, and M. Eto. 2001. Defining the structural determinants and a potential mechanism for inhibition of myosin phosphatase by the protein kinase C-potentiated inhibitor protein of 17 kDa. J. Biol. Chem. 276:39858–39863. [DOI] [PubMed] [Google Scholar]
- Ikebe, M., and S. Reardon. 1990. Phosphorylation of smooth muscle myosin light chain kinase by smooth muscle Ca2+/calmodulin-dependent multifunctional protein kinase. J. Biol. Chem. 265:8975–8978. [PubMed] [Google Scholar]
- Johnson, J. D., C. Snyder, M. P. Walsh, and M. Flynn. 1996. Effects of myosin light chain kinase and peptides on Ca2+ exchange with the N- and C-terminal Ca2+ binding sites of calmodulin. J. Biol. Chem. 271:761–767. [DOI] [PubMed] [Google Scholar]
- Kasturi, R., C. Vasulka, and J. D. Johnson. 1993. Ca2+, caldesmon, and myosin light chain kinase exchange with calmodulin. J. Biol. Chem. 268:7958–7964. [PubMed] [Google Scholar]
- Kawano, Y., Y. Fukata, N. Oshiro, M. Amano, T. Nakamura, M. Ito, F. Matsumura, M. Inagaki, and K. Kaibuchi. 1999. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 147:1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, E., A. Murany, C. Csortos, P. Gergely, M. Ito, D. J. Hartshorne, and F. Erdodi. 2002. Integrin-linked kinase phosphorylates the myosin phosphatase target subunit at the inhibitory site in platelet cytoskeleton. Biochem. J. 365:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa, T., M. Eto, T. P. Woodsome, and D. L. Brautigan. 2000. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J. Biol. Chem. 275:9897–9900. [DOI] [PubMed] [Google Scholar]
- Kitazawa, T., M. Eto, T. P. Woodsome, and M. Khalequzzaman. 2003. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J. Physiol. 546:879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa, T., N. Takizawa, M. Ikebe, and M. Eto. 1999. Reconstitution of protein kinase C-induced contractile Ca2+ sensitization in triton X-100-demembranated rabbit arterial smooth muscle. J. Physiol. 520:139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke, R. L., S. Cai, A. L. Giannini, P. J. Gallagher, P. de Lanerolle, and D. A. Cheresh. 1997. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln, T. M., N. Dey, and H. Sellak. 2001. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J. Appl. Physiol. 91:1421–1430. [DOI] [PubMed] [Google Scholar]
- Loew, L. M., and J. C. Schaff. 2001. The Virtual Cell: a software environment for computational cell biology. Trends Biotechnol. 19:401–406. [DOI] [PubMed] [Google Scholar]
- Lukas, T. J. 2004. A signal transduction pathway prototype I: from agonist to cellular endpoint. Biophys. J. 87:1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas, T. J., S. Mirzoeva, U. Slomczynska, and D. M. Watterson. 1999. Identification of novel classes of protein kinase inhibitors using combinatorial peptide chemistry based on functional genomics knowledge. J. Med. Chem. 42:910–919. [DOI] [PubMed] [Google Scholar]
- Morrison, D. L., J. S. Sanghera, J. Stewart, C. Sutherland, M. P. Walsh, and S. L. Pelech. 1996. Phosphorylation and activation of smooth muscle myosin light chain kinase by MAP kinase and cyclin-dependent kinase-1. Biochem. Cell Biol. 74:549–557. [DOI] [PubMed] [Google Scholar]
- Murthy, K. S., H. Zhou, J. R. Grider, and G. M. Makhlouf. 2003. Inhibition of sustained smooth muscle contraction by PKA and PKG preferentially mediated by phosphorylation of RhoA. Am. J. Physiol. Gastrointest. Liver Physiol. 284:G1006–G1016. [DOI] [PubMed] [Google Scholar]
- Nakamura, M., K. Ichikawa, M. Ito, B. Yamamori, T. Okinada, N. Isaka, Y. Yoshida, S. Fujita, and T. Nakano. 1999. Effects of the phosphorylation of myosin phosphatase by cyclic GMP-dependent protein kinase. Cell. Signal. 11:671–676. [DOI] [PubMed] [Google Scholar]
- Nomura, M., J. T. Stull, K. E. Kamm, and M. C. Mumby. 1992. Site-specific dephosphorylation of smooth muscle myosin light chain kinase by protein phosphatases 1 and 2A. Biochemistry. 31:11915–11920. [DOI] [PubMed] [Google Scholar]
- Ogut, O., and F. V. Brozovich. 2000. Determinants of the contractile properties in the embryonic chicken gizzard and aorta. Am. J. Physiol. 279:C1722–C1732. [DOI] [PubMed] [Google Scholar]
- Sanders, L. C., F. Matsumura, G. M. Bokoch, and P. de Lanerolle. 1999. Inhibition of myosin light chain kinase by p21-activated kinase. Science. 283:2083–2085. [DOI] [PubMed] [Google Scholar]
- Sandu, O. A., M. Ito, and N. Begum. 2001. Selected contribution: insulin utilizes NO/cGMP pathway to activate myosin phosphatase via Rho inhibition in vascular smooth muscle. J. Appl. Physiol. 91:1475–1482. [DOI] [PubMed] [Google Scholar]
- Seko, T., M. Ito, Y. Kureishi, R. Okamoto, N. Moriki, K. Onishi, N. Isaka, D. J. Hartshorne, and T. Nakano. 2003. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 92:411–418. [DOI] [PubMed] [Google Scholar]
- Senba, S., M. Eto, and M. Yazawa. 1999. Identification of trimeric myosin phosphatase (PP1M) as a target for a novel PKC-potentiated protein phosphatase-1 inhibitory protein (CPI17) in porcine aorta smooth muscle. J. Biochem. (Tokyo). 125:354–362. [DOI] [PubMed] [Google Scholar]
- Slemmon, J. R., B. Feng, and J. A. Erhardt. 2000. Small proteins that modulate calmodulin-dependent signal transduction: effects of PEP-19, neuromodulin, and neurogranin on enzyme activation and cellular homeostasis. Mol. Neurobiol. 22:99–113. [DOI] [PubMed] [Google Scholar]
- Slemmon, J. R., J. I. Morgan, S. M. Fullerton, W. Danho, B. S. Hilbush, and T. M. Wengenack. 1996. Camstatins are peptide antagonists of calmodulin based upon a conserved structural motif in PEP-19, neurogranin, and neuromodulin. J. Biol. Chem. 271:15911–15917. [DOI] [PubMed] [Google Scholar]
- Slepchenko, B. M., J. C. Schaff, J. H. Carson, and L. M. Loew. 2002. Computational cell biology: spatiotemporal simulation of cellular events. Annu. Rev. Biophys. Biomol. Struct. 31:423–441. [DOI] [PubMed] [Google Scholar]
- Slepchenko, B. M., J. C. Schaff, I. Macara, and L. M. Loew. 2003. Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 13:570–576. [DOI] [PubMed] [Google Scholar]
- Smith, J. A., R. B. Reed, S. H. Francis, K. Grimes, and J. D. Corbin. 2000. Distinguishing the roles of the two different cGMP-binding sites for modulating phosphorylation of exogenous substrate (heterophosphorylation) and autophosphorylation of cGMP-dependent protein kinase. J. Biol. Chem. 275:154–158. [DOI] [PubMed] [Google Scholar]
- Somlyo, A. P., and A. V. Somlyo. 2000. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol. 522:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull, J. T., L. C. Hsu, M. G. Tansey, and K. E. Kamm. 1990. Myosin light chain kinase phosphorylation in tracheal smooth muscle. J. Biol. Chem. 265:16683–16690. [PubMed] [Google Scholar]
- Stull, J. T., M. G. Tansey, D. C. Tang, R. A. Word, and K. E. Kamm. 1993. Phosphorylation of myosin light chain kinase: a cellular mechanism for Ca2+ desensitization. Mol. Cell. Biochem. 127–128:229–237. [DOI] [PubMed] [Google Scholar]
- Surks, H. K., N. Mochizuki, Y. Kasai, S. P. Georgescu, K. M. Tang, M. Ito, T. M. Lincoln, and M. E. Mendelsohn. 1999. Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase 1α. Science. 286:1583–1587. [DOI] [PubMed] [Google Scholar]
- Takizawa, N., Y. Koga, and M. Ikebe. 2002a. Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem. Biophys. Res. Commun. 297:773–778. [DOI] [PubMed] [Google Scholar]
- Takizawa, N., N. Niiro, and M. Ikebe. 2002b. Dephosphorylation of the two regulatory components of myosin phosphatase, MBS and CPI17. FEBS Lett. 515:127–132. [DOI] [PubMed] [Google Scholar]
- Tansey, M. G., K. Luby-Phelps, K. E. Kamm, and J. T. Stull. 1994. Ca(2+)-dependent phosphorylation of myosin light chain kinase decreases the Ca2+ sensitivity of light chain phosphorylation within smooth muscle cells. J. Biol. Chem. 269:9912–9920. [PubMed] [Google Scholar]
- Tansey, M. G., R. A. Word, H. Hidaka, H. A. Singer, C. M. Schworer, K. E. Kamm, and J. T. Stull. 1992. Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent protein kinase II in smooth muscle cells. J. Biol. Chem. 267:12511–12516. [PubMed] [Google Scholar]
- Taylor, D. A., and J. T. Stull. 1988. Calcium dependence of myosin light chain phosphorylation in smooth muscle cells. J. Biol. Chem. 263:14456–14462. [PubMed] [Google Scholar]
- Tran, Q. K., D. J. Black, and A. Persechini. 2003. Intracellular coupling via limiting calmodulin. J. Biol. Chem. 278:24247–24250. [DOI] [PubMed] [Google Scholar]
- Turko, I. V., S. A. Ballard, S. H. Francis, and J. D. Corbin. 1999. Inhibition of cyclic GMP-binding cyclic GMP-specific phosphodiesterase (Type 5) by sildenafil and related compounds. Mol. Pharmacol. 56:124–130. [DOI] [PubMed] [Google Scholar]
- Vorherr, T., P. James, J. Krebs, A. Enyedi, D. J. McCormick, J. T. Penniston, and E. Carafoli. 1990. Interaction of calmodulin with the calmodulin binding domain of the plasma membrane Ca2+ pump. Biochemistry. 29:355–365. [DOI] [PubMed] [Google Scholar]
- Watanabe, Y., M. Ito, Y. Kataoka, H. Wada, M. Koyama, J. Feng, H. Shiku, and M. Nishikawa. 2001. Protein kinase C-catalyzed phosphorylation of an inhibitory phosphoprotein of myosin phosphatase is involved in human platelet secretion. Blood. 97:3798–3805. [DOI] [PubMed] [Google Scholar]
- Wilson, D. P., C. Sutherland, and M. P. Walsh. 2002. Ca2+ activation of smooth muscle contraction: evidence for the involvement of calmodulin that is bound to the triton insoluble fraction even in the absence of Ca2+. J. Biol. Chem. 277:2186–2192. [DOI] [PubMed] [Google Scholar]
- Woodsome, T. P., M. Eto, A. Everett, D. L. Brautigan, and T. Kitazawa. 2001. Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J. Physiol. 535:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, T., K. Beckingham, and M. Ikebe. 1998. High affinity Ca2+ binding sites of calmodulin are critical for the regulation of myosin Ibeta motor function. J. Biol. Chem. 273:20481–20486. [DOI] [PubMed] [Google Scholar]
- Zimmermann, B., A. V. Somlyo, G. C. Ellis-Davies, J. H. Kaplan, and A. P. Somlyo. 1995. Kinetics of prephosphorylation reactions and myosin light chain phosphorylation in smooth muscle. Flash photolysis studies with caged calcium and caged ATP. J. Biol. Chem. 270:23966–23974. [DOI] [PubMed] [Google Scholar]