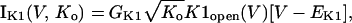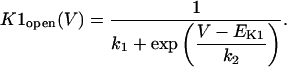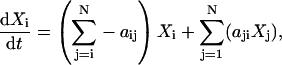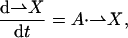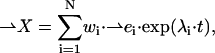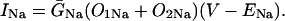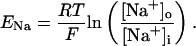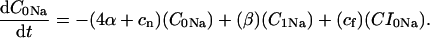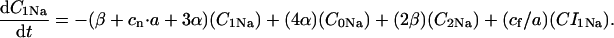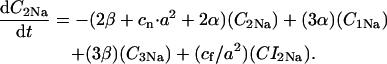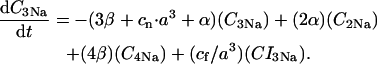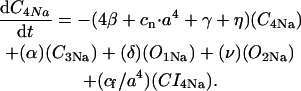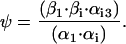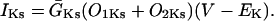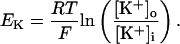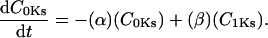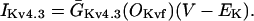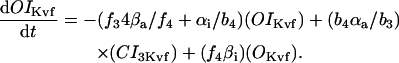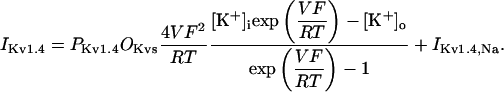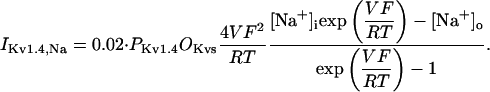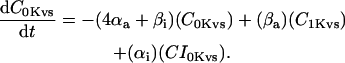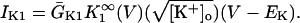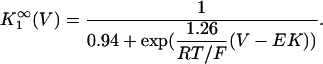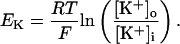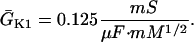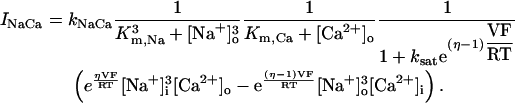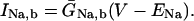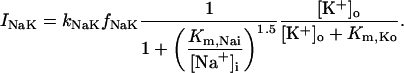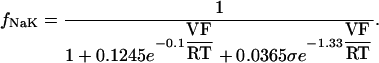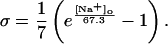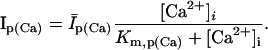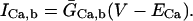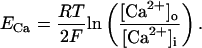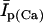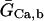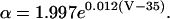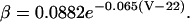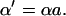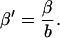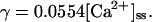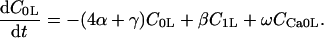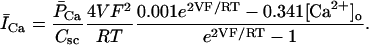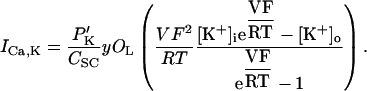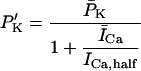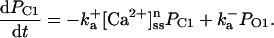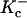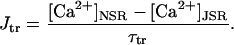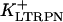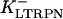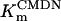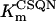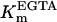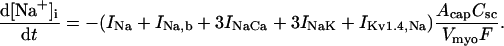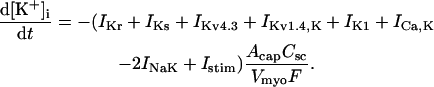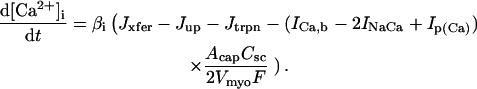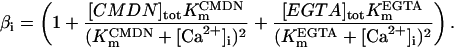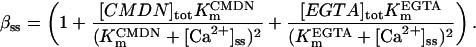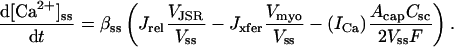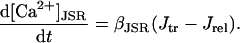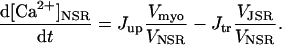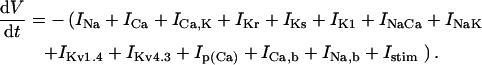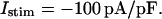An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
1The Center for Cardiovascular Bioinformatics and Modeling and the Whitaker Biomedical Engineering Institute, The Johns Hopkins University School of Medicine and Whiting School of Engineering, Baltimore, Maryland
1The Center for Cardiovascular Bioinformatics and Modeling and the Whitaker Biomedical Engineering Institute, The Johns Hopkins University School of Medicine and Whiting School of Engineering, Baltimore, Maryland
1The Center for Cardiovascular Bioinformatics and Modeling and the Whitaker Biomedical Engineering Institute, The Johns Hopkins University School of Medicine and Whiting School of Engineering, Baltimore, Maryland
1The Center for Cardiovascular Bioinformatics and Modeling and the Whitaker Biomedical Engineering Institute, The Johns Hopkins University School of Medicine and Whiting School of Engineering, Baltimore, Maryland
Address reprint requests to Raimond L. Winslow, PhD, Rm. 201B, Clark Hall, The Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21093. Tel.: 410-516-5417; Fax: 410-516-5294; E-mail: rwinslow@bme.jhu.edu.
A computational model of the human left-ventricular epicardial myocyte is presented. Models of each of the major ionic currents present in these cells are formulated and validated using experimental data obtained from studies of recombinant human ion channels and/or whole-cell recording from single myocytes isolated from human left-ventricular subepicardium. Continuous-time Markov chain models for the gating of the fast Na+ current, transient outward current, rapid component of the delayed rectifier current, and the L-type calcium current are modified to represent human data at physiological temperature. A new model for the gating of the slow component of the delayed rectifier current is formulated and validated against experimental data. Properties of calcium handling and exchanger currents are altered to appropriately represent the dynamics of intracellular ion concentrations. The model is able to both reproduce and predict a wide range of behaviors observed experimentally including action potential morphology, ionic currents, intracellular calcium transients, frequency dependence of action-potential duration, Ca2+-frequency relations, and extrasystolic restitution/post-extrasystolic potentiation. The model therefore serves as a useful tool for investigating mechanisms of arrhythmia and consequences of drug-channel interactions in the human left-ventricular myocyte.
INTRODUCTION
Computational models of the mammalian ventricular myocyte have become important tools for understanding the biophysical basis of the ventricular action potential (AP), for relating changes in gene/protein expression and effects of gene mutations to alterations of AP and calcium (Ca2+) transient morphology, and for investigating mechanisms of arrhythmia (Clancy and Rudy, 1999; Luo and Rudy, 1991, 1994; Noble et al., 1998; Pandit et al., 2001; Winslow et al., 1999). These models describe properties of voltage-gated ionic currents, membrane pumps and exchangers, intracellular Ca2+ cycling, Ca2+-induced Ca2+-release, and processes regulating intracellular sodium (Na+) and potassium (K+) concentrations. Models of ventricular myocytes isolated from guinea pig (Luo and Rudy, 1994; Noble et al., 1998), rat (Pandit et al., 2001), canine (Winslow et al., 1999), and human (Priebe and Beuckelmann, 1998) heart have been developed.
The Priebe-Beuckelmann model and simplifications thereof (Bernus et al., 2002) are the first human ventricular myocyte models developed and were derived via parameter variation from the Luo-Rudy phase II ventricular myocyte model. Ten Tusscher et al. (2003) subsequently presented a human myocyte model in which properties of membrane currents are based on more recent whole-cell current data measured in isolated human ventricular myocytes. Each of these models is able to reconstruct qualitative features of AP and Ca2+ transient morphology; however, they make limited use of what is now substantial experimental data on properties of both wild-type and recombinant channels, whole-cell currents, excitation-contraction coupling processes, and processes regulating intracellular Na+ concentration (Nai) in the human ventricular myocyte. The model described here is based on these more extensive experimental data.
The resulting myocyte model exhibits long-term stability of intracellular ion concentrations over a range of pacing frequencies and is able to reproduce and predict diverse behaviors measured experimentally in isolated human ventricular myocytes including: 1), properties of voltage-dependent ionic currents; 2), AP morphology and rate-dependent changes of AP duration (APD); 3), Ca2+-transient morphology and Ca2+-frequency relations; and 4), properties of extrasystolic restitution and post-extrasystolic potentiation.
METHODS
Model development
The human myocyte model consists of a system of ordinary differential-algebraic equations describing the time-rate of change of channel-state occupancy probabilities and intracellular concentrations of Na+, K+, and Ca2+, whole-cell current through ion channels, membrane pumps and exchangers, and the dynamics of intracellular Ca2+ cycling processes. Complete model equations and parameters are given in the Appendix.
Fast inward Na+ current (INa)
INa is modeled based on the formulation of Irvine et al. (1999). This model is constrained by single channel and whole-cell measurements of recombinant cardiac hH1 channels, and is able to reproduce diverse experimental data including ionic currents, gating currents, tail currents, steady-state inactivation, recovery from inactivation, and single channel open time distribution. Model state transition rates are explicit functions of enthalpy, entropy, voltage, and temperature. The original model behavior has been verified over the temperature range of 10°-23°C, but does not extrapolate directly to body temperature. Model parameters are therefore adjusted in this work to match Na+ current dynamics at physiologic temperature (Nagatomo et al., 1998; Wang et al., 2000). Analytical methods used to extrapolate INa to 33°C are given in the Appendix. The INa density is also adjusted to reproduce measurements of AP amplitude (Li et al., 1998; Nabauer et al., 1996) and phase 0 upstroke velocity (Vmax) measured experimentally in human ventricular subepicardium and midmyocardium (Drouin et al., 1995; Pereon et al., 2000). Model AP amplitude is 126 mV at 1 Hz pacing, which is in the midrange of experimental measurements (100 mV, Li et al., 1999; 132 mV, Li et al., 1998; and 135 mV, Nabauer et al., 1996). Model Vmax is 350 V/s (experimental measurements range from 228 ± 20 V/s, Drouin et al., 1995, to 446 ± 46 V/s, Pereon et al., 2000).
Simulated behavior of INa is compared with experimental data from recombinant human hH1 channels in Fig. 1. Data on activation, inactivation, and recovery from inactivation largely fall within the experimental error bars. Simulated steady-state activation and inactivation functions are in agreement with experimental data, having a half-maximal activation voltage of −39.6 mV and slope factor 7.2 (compared with measurements of −39.6 ± 2.2 mV and 7.1 ± 0.5; Nagatomo et al., 1998), and half-maximal inactivation voltage of −82.4 mV and slope factor 5.8 (compared with measurements of −85.8 ± 2.7 and 5.8 ± 0.1; Nagatomo et al., 1998).
Comparison of simulated (solid line) versus experimentally measured properties, from Nagatomo et al. (1998) and Wang et al. (2000), of INa (▵) at 33°C. (A) Model voltage-clamp current responses compared with experimental data (▵) over the range −50 mV to +30 mV. Activation and inactivation kinetics were studied using a voltage-clamp protocol with a holding potential of -150 mV and steps of 24-mS duration to various depolarizing potentials at 33°C. (B) Comparison of time to peak current (ordinate, ms) as a function of test potential (abscissa, mV) for model versus experiments; see Fig 3, B–E, in Nagatomo et al. (1998). (C) The decay phase of the current traces at potentials from −50 mV to +30 mV was fit with a sum of two weighted exponentials. C shows the fast time constant of inactivation (ordinate, ms) plotted against test potential (abscissa, mV). (D) Slow time constant of inactivation (ordinate, ms) plotted against test potential (abscissa, mV) from fitting procedure described in C. (E) Fractional amplitude of the fast component of inactivation (ordinate) plotted against test potential (abscissa, mV) from fitting procedure described in C. (F) Time course of recovery from inactivation expressed as fraction of maximum current (ordinate) versus recovery interval (abscissa, ms). Data obtained using a two-pulse protocol with a variable interpulse recovery interval (abscissa). Current in second pulse was normalized to current in first pulse and plotted as the ordinate. Experimental data provided for comparison from Fig. 4 in Wang et al. (2000).
Delayed rectifier K+ currents (IKr and IKs)
In human left ventricle, the delayed rectifier current (IK) consists of two components-a rapidly activating and inactivating component (IKr) and a slowly activating component (IKs) (Iost et al., 1998; Li et al., 1996). A model for human IKr was developed recently by Mazhari et al. (2001) based on recordings from recombinant co-assembled human KCNH2/KCNE2 channels. This model has been shown to reproduce a broad range of IKr current characteristics and was incorporated without change into the human whole-cell model. IKr conductance was set to 0.0186 mS/μF to match recordings in isolated human ventricular myocytes (Magyar et al., 2000).
Recent studies of recombinant co-assembled human KCNQ1/KCNE1 channels, which likely encode IKs, have revealed details on its gating structure (Pusch, 1998; Pusch et al., 2000, 2001, 1998; Tristani-Firouzi and Sanguinetti, 1998). Based on this evidence, a Markov model for the gating of IKs channels is used. This model incorporates four sequential states (C1-C2-O3–O4), two of which are nonconducting (C1 and C2), followed by two open, conducting states (O3 and O4). Two open states are included on the basis of experiments in co-assembled KCNQ1/KCNE1 channels: 1), Pusch et al. (2001) deduced two conducting states with differential sensitivity to intracellular Na+ block; and 2), Pusch et al. (2001) recorded a sigmoidal tail current decay characteristic of two open states in the presence of extracellular rubidium (which interferes with and slows channel deactivation). Initially, four closed states were included in the model to reflect the tetrameric structure of the channel. However, to simplify the model, the rapid equilibrium approximation (Hille, 1972) was used to lump the three rapid closed-state transitions into a single activation step (C1-C2), followed by gating into conducting states (C2 to O3 and O3 to O4). This resulted in no significant change in the ability of the model to fit experimental data.
Although studies have reported difficulty in isolating IKs in human myocardium (Veldkamp, 1998), recent experiments have established the presence of this current in human left (Virág et al., 2001) and right (Li et al., 1996) ventricle. The data of Virág et al. (2001) are used to constrain activation and deactivation properties of the channel model. Constraining data include tail current magnitudes, responses to an envelope of tails protocol, activation time constants, and deactivation time constants. The whole-cell conductance of IKs was set to 0.0035 mS/μF to match tail current magnitudes measured in human ventricle (Magyar et al., 2000; Virág et al., 2001). Comparison of model IKs behavior with experimental data is provided in Fig. 2.
Comparison of simulated versus measured (Virág et al., 2001) IKs properties. (A) Simulated IKs in response to voltage-clamp steps from a holding potential of −40 mV to potentials from −10 mV to 50 mV. (B) Tail current magnitudes at −40 mV (ordinate, pA/pF) were computed after depolarization to test potentials (abscissa, mV) from −10 mV to 50 mV for 5000 ms. Experimental tail current magnitudes provided for comparison; from Fig. 2 C, in Virág et al. (2001). (C) Envelope of tails protocol. Voltage-clamp step to +50 mV of varying duration was followed by return to holding potential. Resultant tail currents (ordinate, pA/pF) plotted versus duration of voltage-clamp step (abscissa, ms), overlaid on step current. Peak tail currents are denoted by asterisks. (D) Normalized peak tail currents from protocol in C (ordinate), plotted versus voltage clamp duration (abscissa, ms), highlighting time course of activation of tail currents. Both model and experiment show a slowly activating outward current without evidence of inactivation at +50 mV. Experimental data from Fig. 3 in Virág et al. (2001). (E) Time constant of activation calculated from protocol in D (ordinate, ms), computed for step potentials from +10 to +50 mV (abscissa, mV). Data from Fig. 4 in Virág et al. (2001). (F) Kinetics of deactivation. Tail currents were elicited at potentials between −50 mV and 0 mV and the decay phase was fit with a single exponential. Deactivation time constants (ordinate, ms) were calculated and compared with experimental findings as a function of step potential (abscissa, mV). Experimental data from Fig. 4 in Virág et al. (2001).
Transient outward Ca2+-independent K+ current (Ito1)
In human ventricle, Ito1 consists of two components. Ito1,f is likely encoded by KCND3 and exhibits fast recovery from inactivation (Dixon et al., 1996). Ito1,s is likely encoded by KCNA4 and exhibits relatively slow kinetics of recovery from inactivation (Po et al., 1992). Continuous-time Markov chain models for the gating of human KCND3 and KCNA4 channels have been developed by Greenstein et al. (2000) and have been shown to reproduce human Ito1 current properties accurately. These models are incorporated into the human myocyte model. The fast recovery component, which comprises 89% of human Ito1 (Nabauer et al., 1996), was implemented without adjustment. The slowly recovering component was implemented with one modification—changing the time constant of inactivation to 45 ms for potentials between −10 mV and +40 mV (Nabauer et al., 1996). Density of Ito1 was adjusted to a value of 10 pA/pF in response to a voltage-clamp step to +60 mV from a holding potential of −80 mV, corresponding to experimental measurements in human ventricular subepicardium (Konarzewska et al., 1995; Nabauer et al., 1996; Wettwer et al., 1998).
Inward rectifier K+ current (IK1)
In human ventricle, IK1 activates with a half-maximal activation voltage equal to the reversal potential for K+ (Kubo and Murata, 2001), has a density proportional to the square-root of the extracellular K+ concentration (Bailly et al., 1998), and gates sufficiently fast over physiologic potentials for gating kinetics to be considered instantaneous (Koumi et al., 1995). Thus, activation curves for IK1 were modeled as instantaneous functions of voltage, and current density was set to be a function of the square-root of extracellular potassium. The formulation for IK1 thus becomes (complete equations and parameter values given in Appendix)
(1)
(2)
The voltage dependence of channel rectification (K1open) was constrained using recordings from recombinant human KCNJ2 channels in Xenopus oocytes (Kubo and Murata, 2001), and current density (GK1) was constrained using recordings of slope conductance in human left-ventricular subepicardial myocytes at different extracellular K+ concentrations (Bailly et al., 1998). Properties of the model IK1 are compared with experimental data in Fig. 3.
Comparison of simulated (asterisk symbol) versus measured (▵) (Bailly et al., 1998; Kubo and Murata, 2001) IK1 properties. (A) Steady-state current (ordinate, pA/pF) as a function of voltage (abscissa, mV), referred to as an I-V curve, computed by multiplying the activation curve B by whole-cell conductance and electrical driving force. (B) Experimental I-V curve from Fig. 3 in Li et al. (1998). (C) Steady-state open fraction (ordinate) as a function of membrane potential (abscissa, mV) for model IK1 overlaid with experimental measurements obtained from human KCNJ2 channels; from Fig. 2 C in Kubo and Murata (2001). (D) Open channel conductance (ordinate, mS/μF) as a function of extracellular K+ concentration (abscissa, mV). Experimental data from Fig. 2 C in Bailly et al. (1998).
L-type Ca2+ current (ICaL)
The formulation for ICaL was adopted from Winslow et al. (1999). Rate constants were adjusted to represent the kinetics of inactivation and recovery from inactivation measured in human ventricular myocytes (Li et al., 1999; Magyar et al., 2002). Characteristics of steady-state inactivation were also modified to match experimental findings in human ventricle (Li et al., 1999). Whole-cell conductance was set to achieve a peak inward current density of −6 pA/pF in the presence of 10 mM EGTA, which is in the midrange of values measured in human ventricle (Beuckelmann and Erdmann, 1992; Li et al., 1999; Magyar et al., 2002; Mewes and Ravens, 1994). Fig. 4 contrasts model versus experimental responses to voltage-clamp stimuli.
Comparison of model (lines) versus experimental (▵) (Li et al., 1999; Mewes and Ravens, 1994) ICaL properties. Experiments and simulations were performed using 10 mM EGTA. (A) Normalized model peak I-V relation (holding potential −40 mV) contrasted with experimental data (from Fig. 6 B in Li et al., 1999). (B) Inactivation recovery properties assessed using a two pulse protocol with a variable interpulse interval at holding potential of −80 mV. Current in the second pulse is normalized to current in the first pulse (ordinate), and plotted as a function of interpulse interval (abscissa, ms). Experimental data from Fig. 5 B in Li et al. (1999). (C) Currents in response to voltage-clamp stimuli. From a holding potential of −40 mV, EGTA-loaded myocytes were depolarized from −35 mV to +5 mV. (D) Experimental family of voltage-clamp-elicited currents under same protocol as C, from Fig 4 A in Mewes and Ravens (1994).
INaCa and ISERCA
Sodium-calcium exchanger current (INaCa) is modeled after Winslow et al. (1999). INaCa current density in myocytes isolated from human left-ventricular subepicardium has been measured to be 0.54 ± 0.04 pA/pF at −40 mV; extracellular Na+ and Ca2+ concentrations of 0 and 1 mM, respectively; pipette Na+ concentration of 10 mM; and free Ca2+ at 100 nM (Su et al., 1999). Setting the current density scaling factor kNaCa (see Appendix) to a value of 0.44 elicits a 0.54 pA/pF current density under these conditions.
The SR-Ca2+ ATPase current (ISERCA) is constrained using data on the rate of decline of Ca2+ transients in voltage-clamped human ventricular myocytes in physiological and Na+-free solutions (Beuckelmann et al., 1992, 1995). The decay kinetics of the Ca2+ transient are influenced by two major Ca2+ extrusion mechanisms-INaCa and ISERCA (Bers, 2001). ISERCA can be constrained by fixing INaCa density using the values estimated above, and then varying the magnitude of ISERCA to match experimental Ca2+ transient decay rates. The ISERCA scaling factor KSR (see Appendix) was set to a value of 1.2 to reproduce two findings in human ventricular myocytes. A KSR value of 1.2 produces a time to half-decay of the Ca2+ transient (from peak value) of 270 ms in 5 mm Na+ (reported experimentally as 270 ms and 320 ± 68 ms; see Beuckelmann et al., 1992). ISERCA was also estimated using data from experiments in which pipette and extracellular Na+ concentrations were 0 mM (Beuckelmann et al., 1995), thus ablating INaCa. A model KSR value of 1.25, in an INaCa-free simulation, yielded Ca2+ transient decay rates matching that measured experimentally (305 ± 16 ms). Since this independent estimate provided a similar value for KSR, the original value of 1.2 is selected as the value for KSR in the human model.
Intracellular Na+ handling
The INaK model from Winslow et al. (1999) was adjusted to reduce Na+ efflux at fast pacing frequencies (2.5 Hz and above) by reducing dependence on intracellular Na+ concentration (Nai) and increasing voltage dependence of current density (see Appendix). Modified current-voltage relations compare favorably to curves obtained experimentally from guinea pig (human data not available) (Nakao and Gadsby, 1989). INaK and INab current densities were adjusted to achieve a steady-state Nai of 8 mM at a pacing frequency of 0.25 Hz (see Fig. 5 B of Pieske et al., 2002) and to yield changes of steady-state Nai similar to those measured using SBFI fluorescence at pacing frequencies of 0.5, 1.0, and 2.0 Hz in human left-ventricular myocardium (see Fig. 4 in Pieske et al., 2002). The resulting parameter choices yielded a steady-state value for Nai of 8 mM at 0.25 Hz, 9.8 mM at 1 Hz, and 13.5 mM at 2 Hz, values that are in the physiological range for Nai in human myocytes.
AP and major membrane currents at pacing frequencies of 0.5 Hz (thick solid trace), 1 Hz (thin solid trace), and 2 Hz (dashed trace). (A) AP. (B) Experimental APs from Fig. 1 A in Li et al. (1999). (C) IKr; (D) INaCa; (E) IKs; (F) INaCa; (G) Ito1; (H) INab; (I) IK1; (J) ICaL; and (K) INa.
Intracellular Ca2+ handling
The current model adopts a common pool formulation of intracellular Ca2+ cycling similar to that described in Jafri et al. (1998), using continuous-time Markov chain models of sarcolemmal LCCs, RyRs, Ca2+ compartmentalization including a model subspace, and realistic intracellular Ca2+ buffering.
Numerical methods
For each membrane current, fitting of model transition rates was performed in two steps. First, a cost function defined as the squared error between simulated and experimental responses was minimized globally over the parameter space using a simulated annealing algorithm (Corana, 1987). Second, parameters from the simulated annealing algorithm were selected as initial values for a Neldar-Mead simplex search algorithm (MATLAB), which accepts downhill moves only and has been shown to be superior to the annealing algorithm if the initial guess is in the vicinity of the global minimum (Corana, 1987; Goffe, 1994). See Appendix for further details.
The whole-cell model is composed of a system of 67 coupled nonlinear differential equations which are integrated numerically to simulate the AP. Numerical integration was performed using the LSODE software package (Hindmarsh, 1983), with 10−4 relative error tolerance. The cell model was paced using a stimulus current density of −100 pA/pF and duration of 0.5 ms. The pacing stimulus current was assigned K+ as a charge carrier to eliminate drift of intracellular Na+ and K+ concentrations. This drift has been attributed to neglecting charge contributed by the pacing stimulus during long-term stimulation (Hund et al., 2001). This change allows model ion concentrations to reach a long-term (∼103 s) steady state over a broad range of pacing frequencies. Steady-state initial conditions at each pacing frequency are provided in the Appendix. Use of these numerical methods enables simulation of cell electrophysiological responses in real time, despite the high dimensionality of the model.
RESULTS
APs and rate dependence of AP duration
Fig. 5 shows model APs at pacing frequencies of 0.5 Hz (thick solid line), 1 Hz (thin solid line), and 2 Hz (dashed line). Model (Fig. 5) and experimental (see Fig. 1 in Li et al., 1999) frequency dependence of AP shape and duration are in close agreement. Model AP duration to 90% repolarization (APD90) decreases monotonically with increasing pacing frequency (407 ms at 0.5 Hz, 322 ms at 1 Hz, and 202 ms at 2 Hz).
Experimental APs vary in shape and duration depending on solutions used, potential ion chelators, overall integrity of the cell after harvesting, precise depth of the cell within the myocardium, and pacing history. Thus, experimentally reported figures for APD in human ventricular myocytes vary substantially. Overall, the model APD at 1 Hz (322 ms) lies in the midrange of experimentally recorded values (336 ± 16 ms, 271 ± 13 ms, and 355 ± 50 ms at 1 Hz pacing frequency; see Li et al., 1999, 1998, 1996).
The contribution of specific membrane currents to rate-dependent adaptation of APD90 in human ventricular myocytes remains uncertain (Li et al., 1999). On the basis of experiments and simulation studies in human myocytes and in other species, IKs (Stengl et al., 2003; Viswanathan et al., 1999); ICaL (Li et al., 1999); Ito1 (Nabauer et al., 1996); and INaCa and INaK (Faber and Rudy, 2000) may be involved. Fig. 5 shows APs and ionic current densities at three pacing frequencies studied after pacing to long-term steady state. APD90 decreases from 448 ms at 0.5 Hz to 221 ms at 2 Hz. Corresponding values of Nai are 8.5 mM and 13.5 mM, respectively. The most obvious increases in outward current density at 2-Hz pacing are Na+-dependent increases of INaCa (0.5 pA/pF) and INaK (0.3 pA/pF) during the initial phase of the AP plateau. These changes contribute to APD shortening at higher pacing frequencies. Fig. 5 also shows that the late component of inward ICaL is increased at 2 Hz. An increase of pacing frequency increases SR Ca2+ loading, Ca2+ release flux through RyR and thus Ca2+-mediated inactivation of ICaL. However, the reduced plateau potential in response to increased outward INaCa and INaK increases the driving force on Ca2+, leading to a net increase of ICaL. Because of its slow activation properties, the relative contribution of IKs to repolarization is largest at slower pacing frequencies (the IKr/IKs ratio is ∼2.5:1 at 0.5 Hz, and is ∼5:1 at 2 Hz). However, even at these slower pacing frequencies, its contribution to repolarization remains secondary to that of IKr. The model suggests that the remaining membrane currents (Ito1, IK1, INa, and INab) have little or no role in determining rate-dependence of APD90.
Adaptation of APD90 likely occurs as a result of changes in ionic currents due to accumulation of intracellular ions after long-term pacing, as well as short-term changes in current amplitudes due to kinetics of channel gating. The static APD restitution protocol can isolate short-term changes, as intracellular ions do not accumulate significantly over the short extrasystolic interval. In this protocol, after sustained stimulation at a pacing frequency at steady state (S1 stimuli), a single premature extrasystolic stimulus is delivered at a given diastolic interval, termed an “S2 stimulus”. Extrasystolic APD monotonically increases, rapidly approaching control APD within 200 ms. These data are in qualitative agreement with static restitution curves measured in human left ventricle by Morgan et al. (1992; see also Fig. 6, A–B, this article). Examination of membrane currents during the extrasystolic AP (Fig. 6 C, dashed lines) versus priming control AP (solid line) reveals the mechanism for the short-term changes in APD. The smaller extrasystolic Ca2+ transient (Fig. 6 D, dashed line) induces less calcium-dependent inactivation of ICaL, such that plateau ICaL is slightly higher in the extrasystole (Fig. 6 F). However, augmentation of repolarizing IKs due to incomplete deactivation (Fig. 6 E) and early decreases in depolarizing ICaL due to incomplete recovery also contribute to APD shortening.
Static restitution protocol. (A) APD (ordinate, ms) as a function of S1-S2 diastolic interval (abscissa, ms) at three different priming frequencies. (B) Experimental restitution curves recorded by Morgan et al. (1992). (C) APs in response to S1 (solid line) and S2 (dashed line). S1-S2 interval for C-E set at 350 ms, BCL 1000 ms. (D) Intracellular Ca2+ transients (ordinate, nM) during the S1 AP (solid line) and the S2 premature AP (dashed line). (E) IKs (ordinate, pA/pF) during the S1 (solid line) and the S2 APs (dashed line). (F) ICaL (ordinate, pA/pF) during the S1 (solid line) and S2 APs (dashed line).
Ca2+ transients
The model is able to predict intracellular Ca2+ transients with waveform similar to that observed experimentally. Experimental verification of the steady-state Ca2+ transient at 0.25 Hz against the voltage-clamped Ca2+ transients recorded by Beuckelmann et al. (1992) is given in Fig. 7, A–B. For both model and experiment, a Ca2+ transient was elicited with a voltage clamp step to +10 mV from a holding potential of −40 mV. Model and experimental Ca2+ transients compare well, with similar diastolic and systolic amplitudes (68 nM and 865 nM, respectively, versus experimental values of 60 nM and 810 nM) and similar time course of activation (time to peak activation 45 ms) and decay (time to half-decay 270 ms for both model and experiment).
Ca2+ transient and Ca2+-frequency relation. (A) Model Ca2+ transient elicited upon voltage clamp to +10 mV (ordinate, nM). (B) Experimental recording of Ca2+ transient elicited upon voltage clamp to +10 mV, from Beuckelmann et al. (1992). (C) After pacing to long-term steady state at each frequency (abscissa, Hz), percent increase in Ca2+ transient amplitude (systolic minus diastolic) from value at 0.25 Hz are plotted for model (ordinate, asterisks) and experiment (ordinate, symbols with error bars; from Fig. 2 B in Maier et al., 2000).
Ca2+-frequency relations
Experimental studies in nonfailing human myocardium have shown that at physiologic pacing rates (from 0.25 to 3 Hz), intracellular Ca2+ levels and twitch force increase monotonically with pacing frequency in a characteristic relation known as the positive Ca2+/force-frequency relation (Maier et al., 2000; Pieske et al., 1995). The model Ca2+-frequency relation was constructed by pacing the cell at frequencies ranging from 0.25 to 3 Hz. Once a long-term steady state was reached at each pacing frequency, peak cytosolic Ca2+ was computed. The Ca2+ transient amplitude (systolic minus diastolic levels) at each frequency, expressed as percent increase relative to baseline value at 0.25 Hz, is plotted versus pacing frequency in Fig. 7 C. Model peak Ca2+ levels rise from 438 nM at 0.25 Hz to 1301 nM at 3 Hz-a diastolic/peak amplitude increase of 69% at 1 Hz, 91% at 2 Hz, and 113% at 3 Hz. These percent increases are consistent with those measured experimentally in paced human ventricular myocytes, where intracellular Ca2+ (systolic minus diastolic) is reported to increase from the 0.25 Hz basal level by 41 ± 17% at 1 Hz, 86 ± 26% at 2 Hz, and 124 ± 36% at 3 Hz (Pieske et al., 1995). The mechanism for the positive Ca2+-frequency relation in the model is twofold: 1), increased loading of JSR Ca2+ at higher pacing frequencies leads to larger release events and thus larger intracellular Ca2+ transients (see Fig. 7 D); and 2), higher Nai at higher pacing frequencies increase reverse mode INaCa (see above), leading to greater Ca2+ influx during systole.
Extrasystolic restitution and post-extrasystolic potentiation
The model is also able to predict short-term extrasystolic restitution (ESR) and post-extrasystolic potentiation (PESP) relationships measured experimentally. In the ESR/PESP protocol, myocytes are primed at a basal stimulation rate. After steady state has been reached, a premature beat is delivered at a time S1 after the previous stimulation, where S1 is less than the basic cycle length. An additional stimulus is then delivered after a fixed post-extrasystolic period S2. The premature beat is termed the “extrasystole,” and the beat that occurs S2 ms after the extrasystole is termed the “post-extrasystole.” This protocol is repeated for extrasystolic periods of various durations, and twitch force or intracellular calcium is measured for the steady-state beat, the extrasystolic beat, and the post-extrasystolic beat.
Normal human myocardium shows a restitution of the extrasystolic beat-Ca2+ transients gradually grow with increasing S1. The post-extrasystolic beat is potentiated, and produces large calcium transients corresponding to short S1 and smaller transients corresponding to longer S1 (Gwathmey et al., 1990). The human myocyte model is able to predict qualitative features of these changes. Fig. 8 contrasts model response with the data of Wier and Yue (1986) recorded in ferret ventricle.
Extrasystolic restitution/post-extrasystolic potentiation protocol; see text for details. (A) Model Ca2+ transients (ordinate, nM) versus time (abscissa, ms). Thick trace represents beats at priming basic cycle length of 1500 ms. (B) Experiment in ferret ventricle with fluorescence (ordinate, arbitrary units) versus time (abscissa, ms, adapted from Wier and Yue, 1986). L, uncalibrated luminescence signal.
DISCUSSION
A new computational model of the human left-ventricular epicardial myocyte is presented. Models of the voltage-dependent ionic currents INa, IKr, IKs, Ito1, IK1, and ICaL, constrained by data obtained from recombinant human channels and/or myocytes isolated from human ventricular epicardium, are shown to accurately reproduce a broad range of experimental data. INaCa density is constrained using experimental data obtained under conditions where intra- and extracellular Na+ and Ca2+ concentrations and membrane potential are controlled. ISERCA density is constrained using experimental data on Ca2+ transient decay rate under conditions in which INaCa is either fixed or ablated. INaK and INab densities are constrained using data on the rate of increase of Nai as a function of pacing frequency.
A critical test of any model is its ability to predict experimental data not included in the fitting process. Experimental data that are predicted as emergent properties of the human myocyte model include: 1), qualitative and quantitative changes of APD90 as a function of pacing frequency; 2), quantitative reconstruction of the peak amplitude, time to peak, and half-decay time of the intracellular Ca2+ transient at 1-Hz pacing; 3), monotonic increase of the intracellular Ca2+ transient peak amplitude as a function of pacing frequency; and 4), qualitative prediction of extrasystolic restitution and post-extrasystolic potentiation responses.
Model results suggest that changes in INaCa and INaK produced by changes in Nai at different pacing frequencies have significant effect on APD90, with factors contributing to increased Nai producing shortening of the AP. This finding is consistent with recent observations that: 1), increases of Nai produce substantial shortening of APD90 in left-ventricular myocytes isolated from failing human heart through Na+-induced increases of reverse-mode Na+-Ca2+ exchanger current (Weisser-Thomas et al., 2003); and 2), changing Nai from 5 to 15 mM decreases APD90 of normal canine left-ventricular midmyocardial myocytes from an average of 499.6-327.5 mS through a change in electrical driving force that favors outward current during the early plateau phase of the AP (See Fig. 1 of Armoundas et al., 2003). This finding differs from that of previous simulation studies (Faber and Rudy, 2000; Viswanathan et al., 1999) conducted using models of small mammalian species, which suggest that slow deactivation of IKs induces channel accumulation in the open state at higher pacing frequencies, thereby leading to APD shortening. One reason may be that unlike in guinea pig, IKs in canine ventricular myocytes deactivates rapidly, thereby reducing accumulation of IKs channels in the open state at high pacing frequencies. This, along with the small magnitude of IKs relative to that of IKr, may limit the role of IKs in attenuation of APD at high pacing frequencies in canine and other large mammalian species. However, short-term changes in membrane currents, including IKs and ICaL, are important in reducing APD in situations where Nai remains relatively constant, such as in the static restitution protocol. When a single extrasystolic beat is delivered to the model, significant reductions in depolarizing ICaL and a small increase in IKs is observed. These findings are in agreement with recent experiments by Li et al. (1999), who observed incomplete recovery from inactivation of ICaL at 2 Hz in human ventricular myocytes stimulated by a voltage-clamped action potential waveform.
The model predicts that increased JSR Ca2+ load at high pacing frequencies is responsible for the positive Ca2+-frequency relation, a concept well-established experimentally (Maier et al., 2000; Pieske et al., 1999). The increase of JSR Ca2+ load is in response to increased Ca2+ flux through LCCs (the fraction of time these channels are open is increased at higher pacing frequencies). The model predicts an increase in systolic Ca2+ transients even at high pacing rates (>2 Hz), suggesting that in human myocytes, the increase in SR load more than compensates for the negative effects of adaptation of RyR release channels at high stimulation rates. This finding differs from the conclusions of Ten Tusscher et al. (2003) in modeling studies but is consistent with recent recordings of calcium-frequency relations in human ventricle (Maier et al., 2000; Pieske et al., 1995). Model analysis also reveal that increases in reverse-mode INaCa (brought about by accumulation of Nai) contribute significantly, but are not necessary to the development of a positive Ca2+-frequency relationship. In simulations in which Na+ is clamped at a constant value, systolic Ca2+ levels still rise with pacing frequency.
Some controversy exists about the role of INaCa in the ventricular action potential, with some sources indicating that it is entirely or almost entirely an inward current (forward mode) during the action potential (Piacentino et al., 2003; Weber et al., 2002), and others suggesting that it may operate in outward (reverse mode) significantly into the plateau phase (Armoundas et al., 2003; Despa et al., 2002; Pieske et al., 2002; Weisser-Thomas et al., 2003). In the present model, INaCa is outward for a significant duration of the action potential, suggesting that it plays a role in repolarization and may be important in maintaining SR Ca2+ load. To indirectly validate the INaCa balance, the effects of exchange inhibitory peptide (XIP, a blocker of INaCa) were simulated in the model by reducing exchanger density by 50%. Action potential duration is prolonged at intracellular Na+ of 15 mM and to a lesser degree at 10 mM, but is shortened at 5 mM, similar to changes seen experimentally in XIP-loaded canine myocytes (see Fig. 5 B in Armoundas et al., 2003). These data suggest that, in both model and experiment, INaCa contributes a significant positive repolarizing current at Na+ of 10 mM or greater (such that inhibition of the current prolongs action potential duration) whereas at low Na+ of 5 mM the current is predominantly inward. The model thus likely correctly represents the balance of outward versus inward INaCa at physiological Nai levels.
The model described here has several limitations. First, only qualitative experimental data on the rate-dependence of Nai concentration are available at present. In addition, the model is overly simplistic in its description of Na+ handling, since significant Nai gradients may exist due to higher Na+ concentration in a subsarcolemmal “fuzzy space” (Silverman et al., 2003). One recent study by the Bers group (Weber et al., 2003) suggests that the subsarcolemmal Na+ elevation does not significantly augment INaCa in a rabbit model, lending support to a simpler single-compartment model. However, further experimentation in human myocytes is needed to determine the properties of Na+ handling, as large changes may affect several features of the model, including myocyte repolarization and Ca2+ handling secondarily via INaCa.
A second weakness of the model is that it retains the common-pool formulation of CICR (Stern, 1992). Recent experimental data from both isolated myocytes (Linz and Meyer, 1998; Sipido et al., 1995) as well as recombinant channels expressed in HEK293 cells (Peterson et al., 1999, 2000) show that voltage-dependent inactivation of ICaL is slow and incomplete whereas Ca2+-mediated inactivation is strong, dominating the inactivation process. The opposite is true of existing common pool ventricular myocyte models (see Fig. 10 of Greenstein and Winslow, 2002, and Fig. 5 of Winslow et al., 2001). When Ca2+-dependent inactivation of ICaL is made the dominant inactivation process in common pool models (in which Ca2+ release is all or none), ICaL inactivation also exhibits all-or-none behavior. This all or none Ca2+-induced inactivation of ICaL in turn destabilizes the plateau phase of the AP so that APs alternate between those with short (when JSR Ca2+ release is regenerative and strongly inactivates ICaL) and long (in the absence of JSR Ca2+ release) duration. Thus, common pool models require a dominant voltage-dependent inactivation of ICaL to assure AP stability. A recently developed ventricular myocyte model describing local control of JSR Ca2+ release incorporates both the experimentally measured balance between ICaL voltage- and Ca2+-dependent inactivation processes while exhibiting graded JSR Ca2+ release, voltage-dependent excitation-contraction coupling gain, and stable APs (Greenstein and Winslow, 2002). Future work will address the incorporation of the ionic currents described here for the human myocyte model into this local-control model.
Finally, the model presented here does not attempt to reconstruct AP morphology as a function of transmural location. Rather, the focus has been on developing both a reproductive and predictive model of the left-ventricular epicardial myocyte since the majority of experimental data from human ventricular myocytes have been obtained from such cells. Future work will build from this model to address the issue of transmural variation of AP properties.
Acknowledgments
This work is supported by grants from the National Institutes of Health (RO1 HL-61711, RO1 HL-60133, RO1 HL-72488, P50 HL-52307, and NO1 HV-28180), The Falk Medical Trust, The Whitaker Foundation, and IBM Corporation.
APPENDIX
Model fitting process
The differential equations describing the time rate of change of the occupancy probabilities of each state in a Markov process can be written (by the law of mass action) as the sum of the transitions entering the current state minus the sum of transitions leaving the state as
(A1)
where Xi is the ith state in the system, aij is the rate constant for the transition from state i to state j, and N is the number of states in the system.
This can be written in matrix notation as
(A2)
where X is a column vector containing the probability of occupying each state and A is the state transition matrix (Aij being the rate constant for the transition between state i and state j). Under conditions of applied voltage-clamp, A represents a linear time invariant system. The solution of this system is thus a weighted sum of exponentials,
(A3)
where λi is the ith eigenvalue of the matrix A, ei is its corresponding eigenvector, and the weights wi are constants of integration determined by the initial values X(t = 0).
Model fitting was accomplished by minimizing a sum-squared error function, generated by comparing solutions obtained from Eq. A3 to goal data obtained in various voltage-clamp protocols. Data sets utilized for each current are fully described in Methods. Parameters of the model controlling voltage dependence and kinetics of transitions are systematically varied using the simulated annealing algorithm (Corana, 1987). This algorithm randomly searches the parameter space while incrementally decreasing the search radius. Whereas many minimization algorithms accept only downhill moves and tend to converge on local minima, the simulated annealing algorithm accepts uphill moves as well, and thus is more likely to find the global minimum. Uphill moves are accepted based on the Metropolis criterion, a probabilistic function determined from the difference between the new and old errors and the annealing temperature. The annealing temperature controls the rate of convergence by influencing what uphill moves are accepted and by limiting the search radius. To reach a minimum, the annealing temperature is decreased by 5% per 20 N function evaluations, where N is the number of parameters to be determined, as the algorithm converges on a solution. The algorithm is terminated when there is no more than 0.1% change in error since the last temperature reduction.
For fine tuning of the optimal parameter set, the output of the annealing algorithm is fed into a Neldar-Mead simplex search algorithm (in which only downhill moves are accepted). This approach has been shown to be superior in finding the absolute minimum of functions of several variables (Goffe, 1994).
Model equations and parameters
All rate constants are expressed in units of ms−1 unless otherwise noted. Similarly, all concentrations are expressed in mM unless otherwise noted.
Intracellular ion concentrations and membrane potential
(103)
(104)
(105)
(106)
(107)
(108)
(109)
(110)
(111)
(112)
(113)
Reza Mazhari's current address is Artesian Therapeutics, Inc., 20 Firstfield Road, Suite 190, Gaithersburg, MD 20878.
References
Armoundas, A. A., I. A. Hobai, G. F. Tomaselli, R. L. Winslow, and B. O'Rourke. 2003. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 93:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bailly, P., M. Mouchoniere, J. P. Benitah, L. Camilleri, G. Vassort, and P. Lorente. 1998. Extracellular K+ dependence of inward rectification kinetics in human left ventricular cardiomyocytes. Circulation. 98:2753–2759. [DOI] [PubMed] [Google Scholar]
Bernus, O., R. Wilders, C. W. Zemlin, H. Verschelde, and A. V. Panfilov. 2002. A computationally efficient electrophysiological model of human ventricular cells. Am. J. Physiol. Heart Circ. Physiol. 282:H2296–H3308. [DOI] [PubMed] [Google Scholar]
Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers, Dordrecht, The Netherlands.
Beuckelmann, D. J., and E. Erdmann. 1992. Ca2+-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res. Cardiol. 87:S235–S243. [DOI] [PubMed] [Google Scholar]
Beuckelmann, D. J., M. Nabauer, and E. Erdmann. 1992. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 85:1046–1055. [DOI] [PubMed] [Google Scholar]
Beuckelmann, D. J., M. Nabauer, C. Kruger, and E. Erdmann. 1995. Altered diastolic [Ca2+]i handling in human ventricular myocytes from patients with terminal heart failure. Am. Heart J. 129:684–689. [DOI] [PubMed] [Google Scholar]
Clancy, C. E., and Y. Rudy. 1999. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 400:566–569. [DOI] [PubMed] [Google Scholar]
Corana, M. 1987. Minimizing multimodal functions of continuous variables with the simulated annealing algorithm. ACM Trans. Math. Software. 13:262–280. [Google Scholar]
Despa, S., M. A. Islam, C. R. Weber, S. M. Pogwizd, and D. M. Bers. 2002. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 105:2543–2548. [DOI] [PubMed] [Google Scholar]
Dixon, J. E., W. Shi, H. S. Wang, C. McDonald, H. Yu, R. S. Wymore, I. S. Cohen, and D. McKinnon. 1996. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ. Res. 79:659–668. [DOI] [PubMed] [Google Scholar]
Drouin, E., F. Charpentier, C. Gauthier, K. Laurent, and H. Le Marec. 1995. Electrophysiologic characteristics of cells spanning the left ventricular wall of human heart: evidence for presence of M cells. J. Am. Coll. Cardiol. 26:185–192. [DOI] [PubMed] [Google Scholar]
Faber, G. M., and Y. Rudy. 2000. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys. J. 78:2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Goffe, W. 1994. Global optimization of statistical functions with simulated annealing. J. Econometrics. 60:65–100. [Google Scholar]
Greenstein, J. L., and R. L. Winslow. 2002. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys. J. 83:2918–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Greenstein, J. L., R. Wu, S. Po, G. F. Tomaselli, and R. L. Winslow. 2000. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ. Res. 87:1026–1033. [DOI] [PubMed] [Google Scholar]
Gwathmey, J. K., M. T. Slawsky, R. J. Hajjar, G. M. Briggs, and J. P. Morgan. 1990. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J. Clin. Invest. 85:1599–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hille, B. 1972. Ionic Channels in Excitable Membranes. Sinauer Associates, Sunderland, MA.
Hindmarsh, A. C. 1983. ODEPACK, a systematized collection of ODE solvers. In Scientific Computing. R. S. Stepleman, editor. North-Holland, Amsterdam, The Netherlands. 55–64.
Hund, T. J., J. P. Kucera, N. F. Otani, and Y. Rudy. 2001. Ionic charge conservation and long-term steady state in the Luo-Rudy dynamic cell model. Biophys. J. 81:3324–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Iost, N., L. Virág, M. Opincariu, J. Szécsi, A. Varró, and J. G. Papp. 1998. Delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc. Res. 40:508–515. [DOI] [PubMed] [Google Scholar]
Irvine, L. A., M. S. Jafri, and R. L. Winslow. 1999. Cardiac sodium channel Markov model with temperature dependence and recovery from inactivation. Biophys. J. 76:1868–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jafri, M. S., J. J. Rice, and R. L. Winslow. 1998. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 74:1149–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Konarzewska, H., G. A. Peeters, and M. C. Sanguinetti. 1995. Repolarizing K+ currents in nonfailing human hearts. Similarities between right septal subendocardial and left subepicardial ventricular myocytes. Circulation. 92:1179–1187. [DOI] [PubMed] [Google Scholar]
Koumi, S., C. L. Backer, and C. E. Arentzen. 1995. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation. 92:164–174. [DOI] [PubMed] [Google Scholar]
Kubo, Y., and Y. Murata. 2001. Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J. Physiol. 531:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li, G. R., J. Feng, L. Yue, and M. Carrier. 1998. Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am. J. Physiol. 275:H369–H377. [DOI] [PubMed] [Google Scholar]
Li, G. R., J. Feng, L. Yue, M. Carrier, and S. Nattel. 1996. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ. Res. 78:689–696. [DOI] [PubMed] [Google Scholar]
Li, G. R., B. Yang, J. Feng, R. F. Bosch, M. Carrier, and S. Nattel. 1999. Transmembrane ICa contributes to rate-dependent changes of action potentials in human ventricular myocytes. Am. J. Physiol. 276:H98–H106. [DOI] [PubMed] [Google Scholar]
Linz, K. W., and R. Meyer. 1998. Control of L-type calcium current during the action potential of guinea-pig ventricular myocytes. J. Physiol. (Lond). 513:425–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Luo, C. H., and Y. Rudy. 1991. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ. Res. 68:1501–1526. [DOI] [PubMed] [Google Scholar]
Luo, C. H., and Y. Rudy. 1994. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74:1071–1096. [DOI] [PubMed] [Google Scholar]
Magyar, J., N. Iost, Á. Körtvély, T. Bányász, L. Virág, P. Szigligeti, A. Varró, M. Opincariu, J. Szécsi, J. G. Papp, and P. P. Nánási. 2000. Effects of endothelin-1 on calcium and potassium currents in undiseased human ventricular myocytes. Pflugers Archiv. Eur. J. Physiol. 441:144–149. [DOI] [PubMed] [Google Scholar]
Magyar, J., N. Szentandrassy, T. Bányász, L. Fulop, A. Varró, and P. P. Nánási. 2002. Effects of thymol on calcium and potassium currents in canine and human ventricular cardiomyocytes. Br. J. Pharmacol. 136:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Maier, L. S., P. Barckhausen, J. Weisser, I. Aleksic, M. Baryalei, and B. Pieske. 2000. Ca2+ handling in isolated human atrial myocardium. Am. J. Physiol. Heart Circ. Physiol. 279:H952–H958. [DOI] [PubMed] [Google Scholar]
Mazhari, R., J. L. Greenstein, R. L. Winslow, E. Marban, and H. B. Nuss. 2001. Molecular interactions between two long-QT syndrome gene products, HERG and KCNE2, rationalized by in vitro and in silico analysis. Circ. Res. 89:33–38. [DOI] [PubMed] [Google Scholar]
Mewes, T., and U. Ravens. 1994. L-type calcium currents of human myocytes from ventricle of non-failing and failing hearts and from atrium. J. Mol. Cell. Cardiol. 26:1307–1320. [DOI] [PubMed] [Google Scholar]
Morgan, J. M., D. Cunningham, and E. Rowland. 1992. Electrical restitution in the endocardium of the intact human right ventricle. Br. Heart J. 67:42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nabauer, M., D. J. Beuckelmann, P. Uberfuhr, and G. Steinbeck. 1996. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 93:168–177. [DOI] [PubMed] [Google Scholar]
Nagatomo, T., Z. Fan, B. Ye, G. S. Tonkovich, C. T. January, J. W. Kyle, and J. C. Makielski. 1998. Temperature dependence of early and late currents in human cardiac wild-type and long Q-T δKPQ Na+ channels. Am. J. Physiol. 275:H2016–H2024. [DOI] [PubMed] [Google Scholar]
Nakao, M., and D. C. Gadsby. 1989. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. J. Gen. Physiol. 94:539–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Noble, D., A. Varghese, P. Kohl, and P. Noble. 1998. Improved guinea-pig ventricular cell model incorporating a diadic space, IKr and IKs, and length- and tension-dependent processes. Can. J. Cardiol. 14:123–134. [PubMed] [Google Scholar]
Pandit, S. V., R. B. Clark, W. R. Giles, and S. S. Demir. 2001. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophys J. 81:3029–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pereon, Y., S. Demolombe, I. Baro, E. Drouin, F. Charpentier, and D. Escande. 2000. Differential expression of KvLQT1 isoforms across the human ventricular wall. Am. J. Physiol. Heart Circ. Physiol. 278:H1908–H1915. [DOI] [PubMed] [Google Scholar]
Peterson, B. Z., C. D. DeMaria, J. P. Adelman, and D. T. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558. [DOI] [PubMed] [Google Scholar]
Peterson, B. Z., J. S. Lee, J. G. Mulle, Y. Wang, M. de Leon, and D. T. Yue. 2000. Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca2+ channels. Biophys. J. 78:1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Piacentino III, V., C. R. Weber, X. Chen, J. Weisser-Thomas, K. B. Margulies, D. M. Bers, and S. R. Houser. 2003. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92:651–658. [DOI] [PubMed] [Google Scholar]
Pieske, B., B. Kretschmann, M. Meyer, C. Holubarsch, J. Weirich, H. Posival, K. Minami, H. Just, and G. Hasenfuss. 1995. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 92:1169–1178. [DOI] [PubMed] [Google Scholar]
Pieske, B., L. S. Maier, D. M. Bers, and G. Hasenfuss. 1999. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ. Res. 85:38–46. [DOI] [PubMed] [Google Scholar]
Pieske, B., L. S. Maier, V. Piacentino III, J. Weisser, G. Hasenfuss, and S. Houser. 2002. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 106:447–453. [DOI] [PubMed] [Google Scholar]
Po, S., D. J. Snyders, R. Baker, M. M. Tamkun, and P. B. Bennett. 1992. Functional expression of an inactivating potassium channel cloned from human heart. Circ. Res. 71:732–736. [DOI] [PubMed] [Google Scholar]
Priebe, L., and D. J. Beuckelmann. 1998. Simulation study of cellular electric properties in heart failure. Circ. Res. 82:1206–1223. [DOI] [PubMed] [Google Scholar]
Pusch, M. 1998. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with minK. Pflugers Arch. 437:172–174. [DOI] [PubMed] [Google Scholar]
Pusch, M., L. Bertorello, and F. Conti. 2000. Gating and flickery block differentially affected by rubidium in homomeric KCNQ1 and heteromeric KCNQ1/KCNE1 potassium channels. Biophys. J. 78:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pusch, M., L. Ferrera, and T. Friedrich. 2001. Two open states and rate-limiting gating steps revealed by intracellular Na+ block of human KCNQ1 and KCNQ1/KCNE1 K+ channels. J. Physiol. 533:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pusch, M., R. Magrassi, B. Wollnik, and F. Conti. 1998. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Silverman, B. Z., A. Warley, J. I. Miller, A. F. James, and M. J. Shattock. 2003. Is there a transient rise in sub-sarcolemmal Na and activation of Na/K pump current following activation of INa in ventricular myocardium? Cardiovasc. Res. 57:1025–1034. [DOI] [PubMed] [Google Scholar]
Sipido, K. R., G. Callewaert, and E. Carmeliet. 1995. Inhibition and rapid recovery of Ca2+ during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ. Res. 76:102–109. [DOI] [PubMed] [Google Scholar]
Stengl, M., P. G. Volders, M. B. Thomsen, R. L. Spatjens, K. R. Sipido, and M. A. Vos. 2003. Accumulation of IKs in canine ventricular myocytes. J. Physiol. 551:777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Su, Z., J. H. Bridge, K. D. Philipson, K. W. Spitzer, and W. H. Barry. 1999. Quantitation of Na/Ca exchanger function in single ventricular myocytes. J. Mol. Cell. Cardiol. 31:1125–1135. [DOI] [PubMed] [Google Scholar]
Ten Tusscher, K. H., D. Noble, P. J. Noble, and A.V. Panfilov. 2003. A model for human ventricular tissue. Am. J. Physiol. Heart Circ Physiol. 286:H1573–H1589. [DOI] [PubMed] [Google Scholar]
Tristani-Firouzi, M., and M. C. Sanguinetti. 1998. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 510:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Veldkamp, M. W. 1998. Is the slowly activating component of the delayed rectifier current, Iks, absent from undiseased human ventricular myocardium? Cardiovasc. Res. 40:433–435. [DOI] [PubMed] [Google Scholar]
Virág, L., N. Iost, M. Opincariu, J. Szolnoky, J. Szécsi, G. Bogáts, P. Szenohradszky, A. Varró, and J. G. Papp. 2001. The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc. Res. 49:790–797. [DOI] [PubMed] [Google Scholar]
Viswanathan, P. C., R. M. Shaw, and Y. Rudy. 1999. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 99:2466–2474. [DOI] [PubMed] [Google Scholar]
Wang, D. W., N. Makita, A. Kitabatake, J. R. Balser, and A. L. George, Jr. 2000. Enhanced Na+ channel intermediate inactivation in Brugada syndrome. Circ. Res. 87:E37–E43. [DOI] [PubMed] [Google Scholar]
Weber, C. R., K. S. Ginsburg, and D. M. Bers. 2003. Cardiac submembrane [Na+] transients sensed by Na+-Ca2+ exchange current. Circ. Res. 92:950–952. [DOI] [PubMed] [Google Scholar]
Weber, C. R., V. Piacentino III, K. S. Ginsburg, S. R. Houser, and D. M. Bers. 2002. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ. Res. 90:182–189. [DOI] [PubMed] [Google Scholar]
Weisser-Thomas, J., V. Piacentino III, J. P. Gaughan, K. Margulies, and S. R. Houser. 2003. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc. Res. 57:974–985. [DOI] [PubMed] [Google Scholar]
Wettwer, E., H. M. Himmel, G. J. Amos, Q. Li, F. Metzger, and U. Ravens. 1998. Mechanism of block by tedisamil of transient outward current in human ventricular subepicardial myocytes. Br. J. Pharmacol. 125:659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wier, W. G., and D. T. Yue. 1986. Intracellular calcium transients underlying the short-term force-interval relationship in ferret ventricular myocardium. J. Physiol. 376:507–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Winslow, R., J. Greenstein, G. Tomaselli, and B. O'Rourke. 2001. Computational models of the failing myocyte: relating altered gene expression to cellular function. Phil. Trans. Math. Phys. Eng. Sci. 359:1187–1200. [Google Scholar]
Winslow, R. L., J. Rice, S. Jafri, E. Marban, and B. O'Rourke. 1999. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. II. Model studies. Circ. Res. 84:571–586. [DOI] [PubMed] [Google Scholar]