

Abstract
The ClC family of anion channels mediates the efficient, selective permeation of Cl− across the biological membranes of living cells under the driving force of an electrochemical gradient. In some eukaryotes, these channels are known to exhibit a unique gating mechanism, which appears to be triggered by the permeant Cl− anion. We infer details of this gating mechanism by studying the free energetics of Cl− occupancy in the pore of a prokaryotic ClC homolog. These free energetics were gleaned from 30 ns of molecular dynamics simulation on an ∼133,000-atom system consisting of a hydrated membrane embedded StClC transporter. The binding sites for Cl− in the transporter were determined for the cases where the putative gating residue, Glu148, was protonated and unprotonated. When the glutamate gate is protonated, Cl− favorably occupies an exterior site, Sext, to form a queue of anions in the pore. However, when the glutamate gate is unprotonated, Cl− cannot occupy this site nor, consequently, pass through the pore. An additional, previously undetected, site was found in the pore near the outer membrane that exists regardless of the protonation state of Glu148. Although this suggests that, for the prokaryotic homolog, protonation of Glu148 may be the first step in transporting Cl− at the expense of H+ transport in the opposite direction, an evolutionary argument might suggest that Cl− opens the ClC gate in eukaryotic channels by inducing the conserved glutamate's protonation. During an additional 20 ns free dynamics simulation, the newly discovered outermost site, Sout, and the innermost site, Sint, were seen to allow spontaneous exchange of Cl− ions with the bulk electrolyte while under depolarization conditions.
INTRODUCTION
ClC Cl− channels have served a purpose that has survived much evolution, and therefore, are expressed in a wide range of organisms, from bacteria (Booth et al., 2003) to mammals. In eukaryotes, these channels play a crucial role in the maintenance of osmotic equilibrium to control cell volume, mediate bulk flow associated with transepithelial absorption and secretion, maintain intracellular pH, and influence the electrostatic potential difference across cellular membranes (Estévez and Jentsch, 2002; Foskett, 1998; Maduke et al., 2000). In prokaryotes, ClC acts as a Cl−/H+ antiporter (Accardi et al., 2004; Accardi and Miller, 2004) and plays an important physiological role in bacterial resistance to extreme acidity (Iyer et al., 2002). Nine different types of ClC channels are known to exist in humans, and hereditary defects in the channels are known to cause many diseases, including dominant and recessive myotonia, Bartter's syndrome, and Dent's disease (Pusch et al., 2002).
Although other ion channels share the ability to selectively conduct anions across membranes, ClC is unique in the manner in which its conduction is turned on or off in a process called “fast gating”. Electrophysiological studies have shown that, during fast gating, ClC functions as a dimer, with each pore-forming monomer opening and closing independently in a “double-barrel” fashion (Ludewig et al., 1996; Miller and White, 1984). The probability, Popen, of forming an open monomer, or protochannel, in this process, is voltage dependent and is modulated by extracellular pH (pHO) (Chen and Chen, 2001; Hanke and Miller, 1983; Rychkov et al., 1996, 1997), extracellular Cl− ([Cl−]O) (Chen and Chen, 2001; Chen and Miller, 1996; Pusch et al., 1995; Rychkov et al., 1996), and intracellular Cl− ([Cl−]I) (Chen and Chen, 2003; Chen et al., 2003; Chen and Miller, 1996). At physiological pH, it appears that fast gating is activated by the permeant Cl− anion, rendering the process of conduction and gating intimately coupled (Dutzler et al., 2003; Pusch et al., 1995). This is because Popen is increased as [Cl−]O increases at a given membrane voltage. In addition, it was observed that [Cl−]O increases the depolarization-favored opening rate (Chen and Chen, 2001). On the other hand, lowering pHO or raising [Cl−]I have a different effect on gating. An increase in [H+]O enhances the opening rate mostly at hyperpolarized voltages, while the elevation of [Cl−]I decreases the closing rate of fast gating (Chen and Chen, 2001; Chen et al., 2003).
The high-resolution x-ray structures from Salmonella (StClC) and Escherichia coli (EcClC) (Dutzler et al., 2002, 2003) have confirmed a homodimeric structure of ClC and have provided clues to the fast gating mechanism even though it is likely that in both of these organisms, the protein acts as a Cl−/H+ antiporter (Accardi and Miller, 2004). Structural models of ClC show that each protopore provides an hourglass-shaped passageway for Cl− composed of an antiparallel arrangement of α-helices. The more recently determined structures show that at least two Cl− binding sites exist in the wild-type EcClC selectivity filter. One site is a central site (Scen) where Cl− is coordinated by main-chain amide nitrogen atoms at the end of a helix and by the highly conserved residues Ser107 and Tyr445. The other site (Sint) is located more toward the cytoplasmic end of the transporter, at the interface where aqueous solution from the intracellular side of the pore meets with the selectivity filter. Here, Cl− is coordinated by amide nitrogen atoms from the main chain of the protein.
Just above Scen, (∼4 Å) there exists a negatively charged residue, Glu148, which protrudes into the pore near the interface where aqueous solution from the extracellular vestibule of the transporter meets with the selectivity filter. Mutation of this residue to neutral Ala or Gln has allowed for the refinement of x-ray structures that show Cl− occupying the site where the carboxylate of Glu148 would have been in the wild-type structure, implying that displacement of the Glu148 residue in wild-type ClC by the permeant Cl− anion might serve as the fast gating mechanism (Dutzler et al., 2003). There is also a natural type of “mutation” the wild-type transporter may undergo at lower pH—protonation (Chen and Chen, 2001; Dutzler et al., 2003; Hanke and Miller, 1983; Rychkov et al., 1996). Lowering pHO induces the protonation of a site on the extracellular side of the protochannel, which, in turn, increases Popen (Chen and Chen, 2001; Rychkov et al., 1996). In light of structural observations, the interpretation of this low pH effect has been that when the carboxylate group of Glu148 is protonated, the gate is prone to being open, just as in ClC mutants where the glutamate is replaced with another amino acid (Dutzler et al., 2003).
The structural models of ClC have also afforded the utilization of computational techniques with the aim of gaining knowledge of the molecular details of the conduction mechanism (Cohen and Schulten, 2004; Corry et al., 2004; Miloshevsky and Jordan, 2004; Yin et al., 2004). Despite such studies, however, many striking questions still remain to be answered. If, under physiological conditions, Cl− passes through the pore, how does it overcome the repulsive Coulomb force to approach the negatively charged Glu148 carboxylate to compete for the Sext site? Can Cl− really displace the glutamate gate such that all three known sites, Sint, Scen, and Sext, are occupied simultaneously? Are there additional sites in the pore, and what role could they play? If opening the gate consists of displacing the negatively charged Glu148 with Cl−, and closing it consists of the reverse process (displacing Cl− bound in Sext with the glutamate side chain) why does the modulation of gating in eukaryotes depend on [Cl−]O in such a different way than on [Cl−]I (Chen et al., 2003)? Finally, is it possible that the mechanism of prokaryotic Cl− transport via ClC is related to that of eukaryotic Cl− conduction?
METHODS
All of these questions involve knowledge of the molecular details of the gating mechanism, and might be answered by utilizing molecular dynamics (MD) simulation techniques. Toward this end, we have performed extensive MD simulations of a hydrated dipalmitoylphosphatidylcholine (DPPC) membrane-embedded StClC transporter bathed in NaCl electrolyte at equilibrium in the absence of a transmembrane electrochemical gradient. The transporter was simulated in two protonated states: one where Glu148 was protonated (neutral) and another where it was unprotonated (negatively charged).
We can expect to learn a good deal about the properties of the protopore from such simulations—the position of water molecules and their interaction with Cl− and the behavior of ions in the electrolyte solution of either side of the membrane. However, we cannot expect to observe permeation events in the absence of an electrochemical gradient. In addition, the rarity of a typical permeation event (Accardi et al., 2004) in this prokaryotic transporter would not allow for a robust statistical analysis of the free energetics of Cl− occupancy at the available sites within the pore. Thus, we utilized umbrella sampling simulation techniques to determine the potential of mean force (PMF) (Torrie and Valleau, 1977), or relative free-energy profile (with respect to the bulk electrolyte), as a function of the transport axis (z) for bringing a Cl− ion to various positions in the protopore at various Cl− occupancies. Such a free-energy profile (or hypersurface) is the determining property of the behavior of ions inside channels (Roux, 1999). The total combined time of our equilibrium and umbrella sampling MD simulations was 30 ns, and the PMFs of our work were constructed by analyzing nearly 3 × 106 configurations from the system trajectories. Finally, we performed an additional 20 ns free MD simulation on the system with a protonated Glu148 in which a membrane depolarization was established via applying an external electric field (0.05 V/nm).
Molecular dynamics simulation
Molecular dynamics simulations were run at the Academic Computing Center of the University of North Carolina at Chapel Hill, using the GROMACS package (Berendsen et al., 1995; Lindahl et al., 2001). All simulations used a time step of 4 fs and configurations from the trajectories were saved for analysis every 1 ps. The force-field parameters for lipids were based on the work of Berger (Berger et al., 1997) and those of the protein and ions were taken from the GROMACS force field. The LINCS algorithm was used to constrain all bonds in the system (Hess et al., 1997). Periodic boundary conditions were applied in all three dimensions. Long-range electrostatics were handled using the PME algorithm (Essmann et al., 1995). The temperature in all simulations was maintained at 325 K using the Nose-Hoover scheme with an oscillatory relaxation period of 1.0 ps. The pressure was maintained at 0 atm using the Parrinello-Rahman pressure coupling scheme (Nose and Klein, 1983; Parrinello and Rahman, 1981) with a barostat time constant of 2.0 ps. The analysis of subsequent results was performed using a combination of GROMACS analysis utilities and our own code. Pore radius profiles were calculated with the HOLE algorithm (Smart et al., 1996). Molecular graphics were rendered using VMD (Humphrey et al., 1996) and Raster3D (Merrit and Bacon, 1997).
All simulations in our work stemmed from the same initial configuration. A large hydrated pure DPPC bilayer was initially constructed by replicating a smaller equilibrated bilayer consisting of 64 lipids and 3864 SPC water molecules (obtained from the website of Dr. Peter Tieleman at the University of Calgary, Calgary, Alberta, Canada at http://moose.bio.ucalgary.ca/Downloads/) three times in both the x- and y-dimensions. All lipids escaping the bilayer after 200 ps of simulation were removed from the system, and additional lipids were removed to create a bilayer with an equal number of lipids on each monolayer leaflet.
The initial StClC protein structure (Protein Data Bank code 1KPL) was taken from the work of Dutzler et al. (2002). All incomplete residues in the structure were completed, and the protonation state of each residue was as suggested in previous work (Dutzler et al., 2002). The missing residues on the termini of the crystal structure were omitted in the simulation model, because they are deemed to reside in the cytoplasmic solution and should have little effect on ion behavior inside the pore. The N-terminal of each monomer was capped with a neutral acetyl group, and the C-termini were capped with N-methyl groups, leaving a charge of +15e for the dimer. The Cl− ion occupying the Scen site in each protopore was preserved in the structure. Water molecules were inserted into the pores of each transporter using the program, DOWSER (Zhang and Hermans, 1996). After several passes with the program for each monomer, 55 and 57 water molecules were placed in energetically favorable positions within monomers A and B, respectively. The hydrated dimer was then placed in the center of the bilayer with the transport axis along the bilayer normal (the z axis), and the overlapping water and lipid molecules from the hydrated DPPC bilayer configuration were removed. Water molecules in the surrounding aqueous bath of the biomembrane structure were randomly replaced by 73 Cl− and 60 Na+ ions. The resulting structure was energy minimized using the method of steepest descent while constraining the coordinates of the protein and its internally bound ions to their crystallographic positions.
This configuration consisted of 1 StClC dimer, 75 Cl− ions, 60 Na+ ions, 403 DPPC molecules, and 33,135 water molecules (133,203 atoms). The system was equilibrated under constant NPT conditions for 4.8 ns while restraining the positions of all protein atoms and the Cl− ion in the Scen site within each monomer. During this time the area of the system converged to ∼157.1 nm2. The Glu148 residue of each monomer was then protonated and two Na+ ions were removed to preserve the electroneutrality of the system. Four Cl− ions from the bulk electrolyte were then placed in the interior of the dimer such that the Sext and Sint sites of each monomer were occupied in addition to the Scen site. Their precise positions were determined by optimally structurally aligning the backbone of the recently determined EcClC E148A mutant structure (PDB code 1OTT) to the backbone of the protein structure in our simulated system using a least-squares method. The resulting structure was then energy minimized using the method of steepest descent while constraining the six interior bound ions within the pore to their known binding sites. After a 1-ns equilibration, we performed a 12-ns simulation of the resulting system while restraining the six interior bound ions to their sites in the z-dimension only, allowing them to move freely in the xy-plane. We refer to this simulated system as the “protonated system”. The center of mass (in the z-dimension) for the ions was seen to converge after the first 6 ns of the trajectory, and the protein root-mean-square distance deviation (RMSD) was seen to converge after the first 4 ns of the trajectory (see Fig. 1). Thus, the final 6 ns of the trajectory were used in the analysis of ion densities in the bulk electrolyte solution. After equilibration of the ions, the average, bulk electrolyte concentration was measured to be 78 ± 2 mM.
FIGURE 1.

(Top panel) Time series showing the evolution of the centers of mass for each type of ion in our system for the extracellular (z > 6 nm) and intracellular (z < 6 nm) side of the membrane. Stability in the evolution indicates a converged equilibrium ion distribution for the last 6 ns of the simulation. (Bottom panel) Time series showing the RMSD obtained after performing an optimal least-squares alignment of all protein configurations in the trajectory to the initial protein structure.
The transporter was also simulated with unprotonated Glu148 residues by removing the appropriate proton for each monomer in the resulting structure of the previous simulation and replacing the two Na+ ions in the bulk. After a 1-ns equilibration, we performed a 4-ns simulation of the resulting system while restraining the six interior ions to their “supposed” binding sites (again, in the z-dimension only). We refer to this simulated system as the “unprotonated system”. At this point, we note that according to the postulate of previous studies (Cohen and Schulten, 2004; Dutzler et al., 2002, 2003), the negatively charged carboxylate of Glu148 should remain in an extended conformation, replaced by the negative Cl− as it is restrained in the z-dimension to its known Sext binding site as seen in E148A and E148Q mutant structures. Instead, however, we find that after only a few picoseconds of simulation, the Glu148 carboxylate forms a very robust hydrogen bond to its own amide nitrogen, repelling the Cl− ion and providing a very unstable configuration for the ion. This provides for a protein configuration consistent with the crystal structure of the wild-type, unprotonated EcClC transporter (Dutzler et al., 2003) with two internally bound ions (in the Sint and Scen sites). It also provides evidence that Cl− cannot compete with the negative Glu148 carboxylate for the acceptance of the Glu148 amide nitrogen's hydrogen bond, and thus cannot occupy Sext while the glutamate is in this unprotonated state as was previously supposed (Dutzler et al., 2003). The exchange of ions between the interior and exterior of the transporter was also observed for protonated StClC under the influence of an applied external electric field. The final configuration of the protonated system was taken as the initial configuration and the simulation box was extended in the z-dimension (to 40 nm) to implement slab geometric boundary conditions for the membrane-protein system (Bostick and Berkowitz, 2003; Yeh and Berkowitz, 1999). This choice of boundary conditions is necessary to separate the intracellular and extracellular solutions. Otherwise, purely three-dimensional boundary conditions would provide a situation where ions could simply cross the periodic boundary between simulation cells to relieve the electrochemical gradient supplied by the external electric field (effectively, a short circuit for ion conduction). An external electric field of 0.05 V/nm was applied in the z-dimension (along the transport axis; normal to the bilayer) to simulate a reasonable condition of membrane depolarization. A total of 20 ns was simulated for this system under constant NVT conditions.
Potential of mean force calculations
All PMFs in our work were calculated via umbrella sampling simulations (Torrie and Valleau, 1977) using the final configuration of the unprotonated system trajectory for ClC in the unprotonated state and the final configuration of the protonated system trajectory for ClC in the protonated state. Each PMF profile was constructed from a series of umbrella sampling windows. The probability distribution as a function of the Cl− ion position along the transport axis (z) was generated by combining the statistics from each series of simulations using the weighted histogram analysis method (Kumar et al., 1995; Roux, 1995). Each individual umbrella simulation consisted of 20 ps of equilibration, followed by 100 ps of production MD over which statistics of Cl− occupancy along z were recorded. In each simulation the Cl− ion was allowed to move freely in the plane perpendicular to the transport axis.
The PMF profiles presented in Figs. 3 and 4 both represent cross sections through hypersurfaces of a multiion PMF. This notion is necessary for the interpretation of our displayed data, because the ClC protopore is known to accommodate multiple ions. Hence, the profile on the extracellular side of Scen in Fig. 3 A can more generally be labeled, 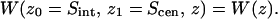 Likewise, the profile on the intracellular side of Scen can be labeled,
Likewise, the profile on the intracellular side of Scen can be labeled, 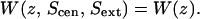 The profile of Fig. 3 B is
The profile of Fig. 3 B is 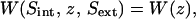 those of Fig. 4 A are
those of Fig. 4 A are 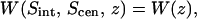 and that of Fig. 4 B is
and that of Fig. 4 B is 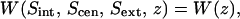 a hypersurface of a four-body PMF. For each of these PMFs, the degrees of freedom that were held constant for sampling (i.e., at the binding site positions, Sint, Scen, or Sext) were held with a harmonic potential of stiffness 5 × 104 kJ/mol/nm2 (effectively constrained) in the z-dimension with respect to the center of mass of the ClC transporter, whereas the single degree of freedom (hypersurface) for which the probability of occupancy was sampled was held with an umbrella potential of stiffness, 3 × 103 kJ/mol/nm2. This allowed us to sample the conditional probability that Cl− occupies the position, z, along the transport axis (given that all other Cl− ions occupying the protopore are located in their respective binding sites) using windows of Δz = 0.05 nm. The position of the umbrella-restrained ion was sampled every MD time step (every 4 fs). The PMFs in Fig. 3 A required 25 and 45 umbrella sampling window simulations for the interior and exterior profiles, respectively, whereas the central PMF profile in Fig. 3 B required nine windows. The PMF in Fig. 4, A and B, corresponding to bringing the ion in from the bulk to Sout, within the unprotonated and protonated transporter required 32 and 29 windows, respectively, making the total simulation time for this study 50 ns. The probability that Cl− occupies the position, z, outside the pore in the bulk electrolyte, was taken from the analysis of the last 6 ns of the 12-ns protonated system trajectory, and from the analysis of the 4-ns unprotonated system trajectory. Because the trajectory in each of these simulations was saved every 1 ps, a total of ∼3 × 106 configurations were analyzed in the construction of the PMFs of this work.
a hypersurface of a four-body PMF. For each of these PMFs, the degrees of freedom that were held constant for sampling (i.e., at the binding site positions, Sint, Scen, or Sext) were held with a harmonic potential of stiffness 5 × 104 kJ/mol/nm2 (effectively constrained) in the z-dimension with respect to the center of mass of the ClC transporter, whereas the single degree of freedom (hypersurface) for which the probability of occupancy was sampled was held with an umbrella potential of stiffness, 3 × 103 kJ/mol/nm2. This allowed us to sample the conditional probability that Cl− occupies the position, z, along the transport axis (given that all other Cl− ions occupying the protopore are located in their respective binding sites) using windows of Δz = 0.05 nm. The position of the umbrella-restrained ion was sampled every MD time step (every 4 fs). The PMFs in Fig. 3 A required 25 and 45 umbrella sampling window simulations for the interior and exterior profiles, respectively, whereas the central PMF profile in Fig. 3 B required nine windows. The PMF in Fig. 4, A and B, corresponding to bringing the ion in from the bulk to Sout, within the unprotonated and protonated transporter required 32 and 29 windows, respectively, making the total simulation time for this study 50 ns. The probability that Cl− occupies the position, z, outside the pore in the bulk electrolyte, was taken from the analysis of the last 6 ns of the 12-ns protonated system trajectory, and from the analysis of the 4-ns unprotonated system trajectory. Because the trajectory in each of these simulations was saved every 1 ps, a total of ∼3 × 106 configurations were analyzed in the construction of the PMFs of this work.
FIGURE 3.

Conditional PMF profiles for Cl− inside the protonated ClC transporter. The z axis is chosen in the same manner as in Fig. 1. The black dashed lines delimit the pore boundaries. (A) Profiles for bringing Cl− from bulk to Sint (to the left of the occupied Scen site marked with a blue stripe at 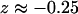 ) and to Sext (to the right of Scen). Because both profiles are given with respect to the bulk electrolyte, we can draw them on the same plot and compare them. The panels a–d are color coded with the features marked on the plot, and show the structural events correlated with minima in the free-energy profiles. Ions are shown as green spheres, and hydrogen bonds are shown as black dotted lines. (B) PMF of Cl− between the occupied Sext and Sint sites (Scen is marked by the dotted line), and the structure (right panel) of Scen.
) and to Sext (to the right of Scen). Because both profiles are given with respect to the bulk electrolyte, we can draw them on the same plot and compare them. The panels a–d are color coded with the features marked on the plot, and show the structural events correlated with minima in the free-energy profiles. Ions are shown as green spheres, and hydrogen bonds are shown as black dotted lines. (B) PMF of Cl− between the occupied Sext and Sint sites (Scen is marked by the dotted line), and the structure (right panel) of Scen.
FIGURE 4.

Conditional PMFs for bringing Cl− from the bulk electrolyte on the outside of the membrane to the pore interior. The z axis is chosen in the same manner as in Fig. 1, and the structural rendering conventions in the panels on the right are adopted from Fig. 2. (A) The profile for the unprotonated transporter is compared to that of the protonated transporter in the case where Sint and Scen are occupied. The inset shows the same profile on the large scale. Positions of the sites, Sext (which only exists for the protonated pore) and Sout, are marked with labeled dotted lines. The Sout site is very close to the third minimum observed in the extracellular profile for the protonated protopore. The panel on the right shows the coordination of the ion leading to the favorable Sout site in the unprotonated pore. A hydrogen-bonded network involving water, Glu148 (hydrogen bonded to its own amide nitrogen), and ions forms a wire of interactions. (B) The profile for the protonated transporter in the case where the three other sites are occupied. A dashed black line marks the outer pore boundary. The coordination leading to favorable position, Sout, is shown in the panel on the right. Again, a hydrogen-bonded network forms a wire in the pore, but in this case, involving only ions and water.
The PMF in units of kBT, where kB is Boltzmann's constant and T is the absolute temperature, was obtained from these measurements of probability, P(z), by using the Boltzmann relation, 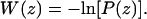 The PMF profiles from the umbrella sampling simulations were matched with the free MD trajectories by shifting them along the W axis. We took the reference state
The PMF profiles from the umbrella sampling simulations were matched with the free MD trajectories by shifting them along the W axis. We took the reference state 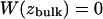 to be in the bulk electrolyte. After matching the PMFs, the profiles were smoothed using (10 iterations of) a three-point window scheme.
to be in the bulk electrolyte. After matching the PMFs, the profiles were smoothed using (10 iterations of) a three-point window scheme.
RESULTS
Fig. 2 B shows the density distributions of various species in the system as a function of the transport (z) axis. The distributions represent an average over the lateral (xy) dimensions of the system, perpendicular to the bilayer normal. Thus, the figure displays projections of the three-dimensional ion density onto the one-dimensional z axis. The vertical dashed black lines in both panels of the figure delimit the beginning and end of the pore. There are a few attributes of the ion densities (bottom panel) that are worthy of comments. First, the fact that the maximum in the Na+ ion density along z is near the transporter's boundaries (dashed black lines) reflects not that Na+ enters the ClC pore, but that, upon binding to the membrane surface, it penetrates more deeply into the polar headgroup region of the bilayer. On the other hand, the Cl− density maximum reflects that it binds the membrane surface at a position further from the bilayer center. This behavior is consistent with previous studies of NaCl electrolyte near a zwitterionic bilayer surface (Böckmann et al., 2003; Pandit et al., 2003a,b). Finally, the extreme dipole moment of the protein along the z axis (2770 ± 70 D) is reflected in the equilibrium ion distribution. As a result of this dipole, the maximum in the Na+ density is seen to be larger on the extracellular membrane surface (at z > 0) than on the intracellular surface (at z < 0). The reverse is true for Cl−.
FIGURE 2.

The simulated StClC system structure. The bilayer normal or transport axis is taken to be in the z-dimension with 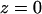 nm at the position of the ClC backbone center of mass. The DPPC headgroups (orange) are shown in a space-filling representation, and the monomers of each homodimer are shown in different shades of blue (dark blue and cyan). Axes are given as a reference. (A) Top-down view of the system from the extracellular side of the membrane (top). The lipid tails are shown as green lines. Nine periodic images of the central simulation cell are shown (top left) and the central cell (top right) shows the protein α-helices as cylinders. A side view of the system (bottom) shows how ClC sits in the membrane. Cl− ions are shown as green spheres, Na+ ions are shown as blue spheres, and water is shown as red dots. (B) Densities of various species in the system are shown in the top panel. All error bars represent the standard error resulting from splitting the trajectory into 1-ns segments. We take the two peaks in the lipid phosphorus (PC Phosphorus) density (at ∼±1.9 nm) to delineate the beginning and end of the transporter along its axis (dotted lines mark the maxima of these peaks). (C) Pore radius as a function of z generated by the HOLE (Smart et al., 1996) algorithm. The protonated ClC pore radius is shown in black, and the unprotonated one in red. The error bars were determined by sampling the pore radius every 250 ps. The location of each known binding site is shown in the inset along with the profile extending the entire pore length. The range of values that the Sext site can take along z are delimited by the blue dashes, and were taken to be the standard deviation in the Cl− density for the ion at this site.
nm at the position of the ClC backbone center of mass. The DPPC headgroups (orange) are shown in a space-filling representation, and the monomers of each homodimer are shown in different shades of blue (dark blue and cyan). Axes are given as a reference. (A) Top-down view of the system from the extracellular side of the membrane (top). The lipid tails are shown as green lines. Nine periodic images of the central simulation cell are shown (top left) and the central cell (top right) shows the protein α-helices as cylinders. A side view of the system (bottom) shows how ClC sits in the membrane. Cl− ions are shown as green spheres, Na+ ions are shown as blue spheres, and water is shown as red dots. (B) Densities of various species in the system are shown in the top panel. All error bars represent the standard error resulting from splitting the trajectory into 1-ns segments. We take the two peaks in the lipid phosphorus (PC Phosphorus) density (at ∼±1.9 nm) to delineate the beginning and end of the transporter along its axis (dotted lines mark the maxima of these peaks). (C) Pore radius as a function of z generated by the HOLE (Smart et al., 1996) algorithm. The protonated ClC pore radius is shown in black, and the unprotonated one in red. The error bars were determined by sampling the pore radius every 250 ps. The location of each known binding site is shown in the inset along with the profile extending the entire pore length. The range of values that the Sext site can take along z are delimited by the blue dashes, and were taken to be the standard deviation in the Cl− density for the ion at this site.
Investigation of the pore size during the simulations of the protonated and unprotonated transporters reveals that, within statistical fluctuations, the protopore in both systems is mostly the same width all along the transport axis (Fig. 2 C). However, there is one significant difference between the two pores at Sext, directly at the site of the titrated Glu148. The size of the pore at this site is nearly two times smaller in the unprotonated transporter (∼0.8 Å) than in the protonated one (∼1.4 Å). The analysis presented in Fig. 2 C probes only the steric aspect of site availability. A Pauling radius of 1.81 Å (Hille, 1992) would make it difficult for Cl− to travel from the outside of the membrane to this site without local steric or electrostatic changes in the protein. Presumably the impetus for such changes must come from Cl−, itself, because it is known to trigger the gating mechanism. We aim to test the recent hypothesis that this rearrangement is due to the displacement of the Glu148 carboxylate (Dutzler et al., 2003).
Fig. 3 shows the calculated PMF for the three internal ions in the pore of the protonated system (see Methods). The free-energy profile shows that protonation of Glu148 allows Cl− to easily penetrate the pore (with a barrier of ∼2.6 kBT) from the extracellular electrolyte and bind favorably to the Sext site 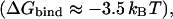 even while the other known binding sites, Sint and Scen, are occupied (Fig. 3 A). This result is in agreement with electrostatic studies, which suggest that neutralization of the glutamate might allow Cl− to pass through the pore by creating an electrostatic trap (Miloshevsky and Jordan, 2004).
even while the other known binding sites, Sint and Scen, are occupied (Fig. 3 A). This result is in agreement with electrostatic studies, which suggest that neutralization of the glutamate might allow Cl− to pass through the pore by creating an electrostatic trap (Miloshevsky and Jordan, 2004).
Interestingly, two substates are resolved in the PMF corresponding to this site. One of these sites (Fig. 3 A (c)) corresponds to Cl− occupying exactly the same position as the negative carboxylate of Glu148 in the unprotonated structure (Dutzler et al., 2002). Here, it successfully gains the hydrogen bond of the glutamate amide nitrogen and is also strongly coordinated by the (now) polar side chain of glutamatic acid in addition to the polar and positively charged groups of Arg147. In this substate it is somewhat more sequestered from water in the extracellular vestibule than in the other (Fig. 3 A (d)) where it loses the amide hydrogen bond donated by Glu148 to gain hydration, but is still coordinated by its hydroxyl group on an extended side chain. There is a third minimum in this profile closer to z ≈ 0.8 nm (z = 0 nm corresponds to the center of mass of the protein backbone), hinting at the possibility of a fourth binding site near the outside of the pore.
The PMF corresponding to bringing Cl− from the intracellular bulk electrolyte to the interior vestibule while Sext and Scen are both occupied reveals an interior binding site very similar to that observed in recent work (Fig. 3 A (b)) (Dutzler et al., 2003). Here, Cl− is coordinated by the backbone amide nitrogens of Gly105 and Gly106, and occasionally by Ser107. The binding free energy for this site 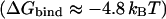 is not much different from the second feature of the profile (Fig. 3 A (a)) closer to the inside of the membrane. The barrier between these two features is low (∼1 kBT) thus making Sint a very weak site. Fig. 3 A (a) shows a broad, favorable region of the pore where Cl− is mostly solvated by water in the large vestibule at the intracellular mouth. At the most favorable point in this region, Cl− interacts with the charged and polar groups of Arg98 and Gln281.
is not much different from the second feature of the profile (Fig. 3 A (a)) closer to the inside of the membrane. The barrier between these two features is low (∼1 kBT) thus making Sint a very weak site. Fig. 3 A (a) shows a broad, favorable region of the pore where Cl− is mostly solvated by water in the large vestibule at the intracellular mouth. At the most favorable point in this region, Cl− interacts with the charged and polar groups of Arg98 and Gln281.
The Scen site in Fig. 3 B, representing the most selective part of the pore, is exactly as seen in the work of Dutzler et al. (2003). Although the binding energy with respect to the bulk (or the other sites) cannot be defined using our PMF methodology (see Methods), we identify its location at the minimum of the parabolic free-energy profile. Cl− at this site is seen to be pinned between its neighboring occupied sites and is coordinated by the polar hydrogens of Ser107, Tyr445, and backbone amide nitrogens. A hydrogen-bonded wire of water and Cl− occasionally develops (Fig. 3 B, right), which presumably would aid in mutual destabilization of the ions in their sites to expedite permeation events.
Thus far, we have only considered how Cl− can occupy a protonated transporter. Strikingly, when Glu148 is unprotonated, Cl− is so repelled that it cannot approach the glutamate occupied Sext site from the outside of the membrane, let alone occupy it (Fig. 4 A). This result implies that the site may be occupied by Cl− if and only if Glu148 is protonated. Perhaps this is why Sext has not been observed in crystallographic structures of wild-type, unprotonated ClC transporters (Dutzler et al., 2003). However, the PMF does show one site, Sout, which is favorable for Cl− at z ≈ 0.86 nm. At this site, Cl− is bound 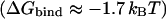 to the positively charged Arg147 side chain, the polar Gly316 side chain, and occasionally to the polar Gln61 side chain.
to the positively charged Arg147 side chain, the polar Gly316 side chain, and occasionally to the polar Gln61 side chain.
A corresponding site also exists when Glu148 is protonated. Fig. 4 B shows the free-energy profile corresponding to bringing Cl− from the outside of the membrane to the interior of the protonated monomer given that the Sext, Scen, and Sint sites are occupied. The site exists for the protonated pore in the range z ≈ 1.0 – 1.2 nm when the other three sites are occupied (Fig. 4 B and Fig. 5). At this site, the ion is coordinated by the polar hydrogens of Gln61, Asn318, and occasionally Gly316. Again, water would seem to play a role here as it forms a network connecting the anion at Sout with the one at Sext (Fig. 4 B). Also, there appears to be a significant rearrangement of the Arg147 side chain when comparing the state represented in Fig. 4 B with that in Fig. 4 A. It seems that the charged side chain prefers to coordinate Cl− at Sext when Glu148 is protonated and displaced (see Figs. 4 B (right), 3 A (c), and 3 A (d)), but prefers to coordinate Cl− at Sout when the Glu148 side chain is unprotonated and occupies Sext.
FIGURE 5.

Schematic drawing of the available sites for Cl− in the StClC pore at a given Glu148 protonation state. The drawing convention has been adopted from Dutzler et al. (2003). Sites for Cl− ions are shown as patterned spheres and hydrogen bonds are shown as dashed lines.
These results allow us to arrive at a picture for site availability within the pore given the protonation state of Glu148. Fig. 5 summarizes this availability. The unprotonated transporter allows Cl− to bind at three possible sites within the pore, Sint, Scen, and Sout, while Sext is occluded by the negatively charged side chain of the glutamate gate. The protonated transporter allows Cl− to bind at all four sites (Sint, Scen, Sext, and Sout). Note that there is a difference in the way Arg147 coordinates Cl− at a given protonation state. When the glutamate gate is unprotonated, the Arg147 side chain can coordinate Cl− at Sout (see Fig. 5 (closed)), whereas when the glutamate is protonated, the Arg147 side chain can coordinate Cl− at Sext (see Fig. 5 (open)). Regardless of this change in coordination of Cl− at Sout the affinity of this site for Cl− is basically the same (see Fig. 4 A) whether or not Glu148 is protonated. Fig. 6 shows all four sites within the global context of a protonated monomer of the ClC transporter. Fig. 6 B illustrates how water can occupy the space between anions in the pore. This presumably would be important, not only for dislodging the anions and solvating them to facilitate their flux through the pore, but also for facilitating the flux of protons in the opposite direction.
FIGURE 6.

A single protonated protopore with four Cl− ions in their equilibrium positions as discussed in the text. (A) The two symmetric halves of the monomer are shown in transparent blue and silver, respectively. Helices are rendered as cylinders. The ions are shown as green spheres. (B) One symmetric half of the transporter is rendered in silver with a smooth molecular surface representation. The conserved residues of the selectivity filter are colored in blue (Dutzler et al., 2002). The water (shown as lines) fills the outer and inner vestibules of the protopore. The vestibules are seen to meet with the ions (green spheres) at the Sout (top-most ion) and Sint (bottom-most ion) sites.
Upon free MD simulation of the protonated system under depolarized conditions (see Methods) for 20 ns, we observe the spontaneous exchange of Cl− ions between Sout and the exterior bulk electrolyte, and Sint and the interior bulk electrolyte during the timescale of the simulation (Fig. 7), although we do not observe permeation. The central two sites, Scen and Sext, enjoy a much higher occupancy while the pore is in this protonated state.
FIGURE 7.

Time series of various Cl− ion positions along the transport axis (ordinate, z axis) during the simulation of the protonated StClC transporter under depolarization conditions with slab-periodic boundary conditions. The dashed lines delimit the pore boundaries. The regions along z for each of the four sites outlined in our work are outlined by the long open rectangles within the pore region. Each region is labeled at the right. Sout and Sint spontaneously exchange Cl− with the bulk electrolyte during the simulation whereas Sext and Scen enjoy a much higher occupancy.
DISCUSSION
The idea that Cl− displaces the negative carboxylate of the Glu148 gate as it approaches from the extracellular vestibule (Cl−/COO− competition) upon membrane depolarization offers a very intuitive way to conceptualize how ClC might be gated by the permeant anion. However, this idea poses a problem when attempting to understand the mechanism of action in muscle-type eukaryotic ClC channels. The mechanism does not explain why the modulation of gating by [Cl−]O is different from that of [Cl−]I (Chen et al., 2003). The idea of Cl−/COO− competition for Sext also conflicts with our results. Our work indicates that the three sites, Sint, Scen, and Sext as seen in the crystallographically determined ClC mutants (Dutzler et al., 2003) can be occupied in the pore when Glu148 is protonated (Fig. 3). We also show that a fourth site, Sout, exists near the outside of the membrane whether or not the glutamate is protonated (see Figs. 4 and 5). Fig. 4 B indicates that Sout can be occupied even when the other three sites are also occupied. On the other hand, when Glu148 is not protonated, we see that Cl− is so repelled that it cannot approach the glutamate occupied Sext site from the outside of the membrane (Figs. 4 A and 5).
The protein in our simulation, StClC, is likely a prokaryotic Cl−/H+ antiporter just like its neighbor, EcClC (Accardi and Miller, 2004). Even though its eukaryotic homologs, the muscle-type ClC channels ClC-0 and ClC-1, are very closely related, it is possible that their mechanism of Cl− conduction is very different from the StClC mechanism of Cl− transport. However, the fact that gating is modulated by both pH and [Cl−] in the muscle-type ClC channels and in their neighbors, the prokaryotic Cl−/H+ antiporters, would argue that this group of proteins have a related mechanism of action. Nonetheless, one must take care when using conclusions drawn from studies of the prokaryotic transporters to make postulations about the mechanisms of the eukaryotic channels.
The PMF calculations, combined with the structural observations of our simulations on StClC allow for a novel interpretation of well-known experimental and structural findings in muscle-type ClC channels. It has been observed experimentally that lowering pHO increases the opening rate when the membrane is more hyperpolarized. Thus, it has been suggested that internal Cl− may interact with external H+ to modulate hyperpolarization-activated gating in ClC (Chen and Chen, 2001). On the other hand, external Cl− increases the depolarization-favored opening rate (Chen and Chen, 2001). These observations imply two opening processes with opposite voltage dependence. In light of our findings, it would be reasonable to suggest that both of these processes hinge around the unifying idea that, in order for Cl− to occupy Sext, and ultimately, pass through the channel, Glu148 must become protonated.
Electrophysiological studies on ClC mutants support this conclusion. It was observed that mutating Glu166 in ClC-0 (the analogous residue to Glu148 in StClC) to either Ala, Gln, or Val causes the gate to stay open (Dutzler et al., 2003) as if the mutation abolishes the fast gating mechanism. Mutation of Glu232 of ClC-1 (the analogous residue to Glu148 in StClC) has been shown to almost totally lack gating (Estévez et al., 2003). Many other ClC channels, including ClC-K, ClC-4, and ClC-5, display either minimal or abolished gating upon mutation of the putative glutamate gate (Estévez et al., 2003). Mutating Glu148 to Ala in the prokaryotic EcClC transporter was shown to transform it into a nearly ideal Cl− channel by eliminating the pH dependence of the Cl− and H+ currents (Accardi et al., 2004). Could the mechanism in prokaryotic ClC that leads to the “antiportation” of Cl− and H+ be related to the fast gating mechanism of the eukaryotic channels? Mutational studies considered along with our own results suggest that a relationship exists, and that it should involve protonation of the glutamate gate. Other residues within the protopore may become protonated/deprotonated while moving Cl− in the prokaryotic transporters. However, the fact that the E148A mutant of EcClC displays diffusive, selective Cl− flux implies a possibility that Glu148 is the only residue that changes its protonation state (Accardi et al., 2004).
Experimental studies have linked gating to a titratable site (Hanke and Miller, 1983) in ClC-0 (Chen and Chen, 2001) and ClC-1 (Rychkov et al., 1997) channels. Although the glutamic acid side chain is normally thought to have a pKa closer to 4.3, we suspect that the presence of negative charge (such as Cl−) in its vicinity can influence the local electric field Glu148 experiences and shift its pKa to values closer to the physiological range than previously thought (Dutzler et al., 2003). Recent electrostatic calculations on the ClC structure support this. Yin et al. have shown that Cl− ions at Scen and at an electrostatically favorable site analogous to Sout can shift the apparent pKa of the Glu148 side chain more than two pH units to ∼7.5 (Yin et al., 2004). Indeed, introducing more negative charge to the pore, either by increasing [Cl−]I or by mutating pore residues appropriately, has been shown to stabilize the open state of the fast gate (Chen et al., 2003). With our own result in hand, suggesting that Cl− cannot pass the glutamate gate unless it is protonated, one can build a causal explanation for such electrostatic control of the gating: the fast gating of eukaryotic ClC channels is a process based upon Cl−-induced protonation. This conclusion also accounts for the coupling between gating and Cl− permeation (Chen and Chen, 2003; Chen and Miller, 1996; Pusch et al., 1995).
Experimental work has long supported the idea that a binding site accessible only to the outside of the membrane controls Cl− permeation at depolarized membrane voltages. This site is linked with protonation of a site within the channel, and must be occupied by Cl− for opening to occur at physiological pHO. Previous work suggests that H+ affects fast gating by modulating the binding of Cl− to this outer site in ClC-1 (Rychkov et al., 1997, 1996). Studies on ClC-0, however, do not demonstrate this H+ modulation effect (Chen and Chen, 2001). Our results for StClC indicate that although the coordinating residues for Cl− change at the Sout site as a function of the protonation of Glu148, (see Fig. 4, A and B, and illustrations in Fig. 5) the site, itself, still exists in either protonation state (as shown in Fig. 4 A). Thus, Cl− binding to Sout in StClC is much like that described for the outer site of ClC-0, because the binding is not modulated by H+. On the other hand, it would seem that Cl− binding to the external site, Sext, in StClC is much like that described for the outer site of ClC-1, because it depends crucially on the protonation of Glu148.
Phenomenological models for eukaryotic channel gating that best explain electrophysiological data follow a scheme in which the Cl− is bound to an outer site on the closed channel. The opening process lumps together the inward movement of Cl− with the opening of the gate (Chen and Chen, 2001). Because the Sext site appears to be more closely linked with closure of the gate by internal Cl− (Chen et al., 2003), the Sout site that we identify in our work presents a prime suspect for the outer site that controls gating. Of the residues that participate in this binding site, Arg147 of StClC (Lys165 in ClC-0 or Lys231 in ClC-1; Dutzler et al., 2002) plays the most likely role by luring Cl− with its positive charge. The fact that we observe the Arg147 side chain to coordinate Cl− at Sext when the glutamate gate is protonated suggests that the other polar residues of the Sout site are also important, and serve to shape the site and coordinate the anion (see Fig. 5).
With all this in mind, let us address the “gating” of the transporter in our simulation. The gating process for the StClC transporter upon membrane depolarization must occur as depicted in Fig. 8 A. The start of this sequence (step 1), is described as the closed state by Dutzler et al. (2003). Here, Sext is blocked by Glu148. Cl− may then bind to Sout (step 2). With a depolarized membrane, H+ must be driven from the intracellular solution to the pore interior where it can protonate Glu148, and the opening process (step 3) lumps together the inward movement of Cl− (to Sext) with the opening of the gate (Chen and Chen, 2001) along with the protonation of Glu148. We do not want to make assumptions about where the proton comes from, but we are willing to entertain the possibility that this transporter works by creating a locally concentrated solution of hydrochloric acid. A continuous supply of Cl− from the extracellular bath will supply Sout with anions, feeding them to Sext. It is possible that Sint and Scen are occupied during this process. Indeed, electrostatic studies might support this (Yin et al., 2004). However, it would seem to be less important than occupancy of Sout and Sext for keeping the glutamate gate protonated and open.
FIGURE 8.

Outline of the StClC gating/transport process under (A) depolarized and (B) hyperpolarized membrane conditions. The four available sites are represented as large open circles. The smaller open circles represent Cl− as it occupies one of the available sites. Glu148 is shown occupying the Sext site when it is unprotonated. In this state, its negative charge is represented by a smaller open circle with a minus sign inside it. In the protonated state (E148-H), the side chain is represented by a smaller open circle without a minus sign inside it.
Hyperpolarization of the membrane drives protons from the extracellular solution into the pore near Glu148 (steps 1 and 2 of Fig. 8 B), and drives Cl− to fill all of the interior sites up to Scen. Again, Glu148 becomes protonated and Sext becomes occupied (step 3). The anions move from the inside of the membrane to the outside, all the while occupying Sext, keeping the gate open.
A similar set of mechanisms might explain eukaryotic channel fast gating. Because these ClC proteins are channels, and not Cl−/H+ antiporters, protons should come from the extracellular solution upon depolarization. This might explain why the rate of fast gate opening is increased when external concentrations of chloride and protons are increased and why there is a difference in the gating dependence on voltage. It also might explain the so-called “foot-in-the-door” effect observed for this channel—the tendency of the gate not to close easily after having been opened and occupied at a key site within the channel (Chen et al., 2003). This effect is seen at high [Cl−]I and hyperpolarized membrane voltages—a situation that can easily supply anions to Sext from the inside of the membrane. Once Sext has continual access to anions, it must inevitably remain continually protonated (and, thus, opened).
Allegorically, if Cl− is the man standing at the site in front of the gate, then the proton is his key for passage into the site, Sext, inside the gate. The depolarization-favored and hyperpolarization-favored mechanisms of the eukaryotic muscle-type ClC channel appear to be two ways in which the man might find his key. Both ways share one common feature: for Cl− to occupy Sext, and ultimately, pass through the ClC protopore, the glutamate gate must become protonated, regardless of the membrane voltage.
Acknowledgments
We thank University of North Carolina-Chapel Hill Academic Computing Services for computational services. We also thank Gennady Miloshevsky for friendly advice and discussion.
This work was supported by the National Science Foundation under grant MCB0315502 and also in part by the Molecular and Cellular Biophysics Program at the University of North Carolina at Chapel Hill under the U.S. Public Health Service training grant T32 GM08570.
References
- Accardi, A., L. Kolmakova-Partensky, C. Williams, and C. Miller. 2004. Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J. Gen. Physiol. 123:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of CLC Cl− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- Berger, O., O. Edholm, and F. Jahnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckmann, R. A., A. Hac, T. Heimberg, and H. Grubmüller. 2003. Effect of sodium chloride on a lipid bilayer. Biophys. J. 85:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth, I. R., M. D. Edwards, and S. Miller. 2003. Bacterial ion channels. Biochemistry. 42:10045–10053. [DOI] [PubMed] [Google Scholar]
- Bostick, D., and M. L. Berkowitz. 2003. The implementation of slab geometry for membrane-channel molecular dynamics simulations. Biophys. J. 85:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.-F., and T.-Y. Chen. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.-F., and T.-Y. Chen. 2003. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 122:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.-Y., M.-F. Chen, and C.-W. Lin. 2003. Electrostatic control and chloride regulation of the fast gating of ClC-0 chloride channels. J. Gen. Physiol. 122:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.-Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, J., and K. Schulten. 2004. Mechanism of anionic conduction across ClC. Biophys. J. 86:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry, B., M. O'Mara, and S.-H. Chung. 2004. Conduction mechanisms of chloride ions in ClC-type channels. Biophys. J. 86:846–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chalt, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Estévez, R., and T. J. Jentsch. 2002. CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 12:531–539. [DOI] [PubMed] [Google Scholar]
- Estévez, R., B. Schroeder, A. Accardi, T. J. Jentsch, and M. Pusch. 2003. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 38:47–59. [DOI] [PubMed] [Google Scholar]
- Foskett, K. J. 1998. ClC and CFTR Chloride channel gating. Annu. Rev. Physiol. 60:689–717. [DOI] [PubMed] [Google Scholar]
- Hanke, W., and C. Miller. 1983. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82:25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaige. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- Hille, B. 1992. Ionic Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Iyer, R., T. M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- Kumar, S., J. M. Rosenberg, D. Bouzida, R. H. Swendsen, and P. A. Kollman. 1995. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 16:1339–1350. [Google Scholar]
- Lindahl, E., B. Hess, and D. van der Spoel. 2001. Gromacs 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- Ludewig, U., M. Pusch, and T. J. Jentsch. 1996. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 383:340–343. [DOI] [PubMed] [Google Scholar]
- Maduke, M., C. Miller, and J. A. Mindell. 2000. A decade of CLC chloride channels: structure, mechanism, and many unsettled questions. Annu. Rev. Biophys. Biomol. Struct. 29:411–438. [DOI] [PubMed] [Google Scholar]
- Merrit, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505–524. [DOI] [PubMed] [Google Scholar]
- Miller, C., and M. M. White. 1984. Dimeric structure of single chloride channels from Torpedo electroplax. Proc. Natl. Acad. Sci. USA. 81:2772–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miloshevsky, G. V., and P. C. Jordan. 2004. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: electrostatic effects of charged residues. Biophys. J. 86:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose, S., and M. L. Klein. 1983. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50:1055–1076. [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003a. Mixed bilayer containing dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylserine: lipid complexation, ion binding, and electrostatics. Biophys. J. 85:3120–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003b. Molecular dynamics simulation of a dipalmitoylphosphatidylcholine bilayer with NaCl. Biophys. J. 84:3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello, M., and A. Rahman. 1981. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52:7182–7190. [Google Scholar]
- Pusch, M., A. Accardi, A. Liantonio, P. Guida, S. Traverso, D. C. Camerino, and F. Conti. 2002. Mechanisms of block of muscle type CLC chloride channels (Review). Mol. Membr. Biol. 19:285–292. [DOI] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage dependent chloride channel ClC-0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Roux, B. 1995. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 91:275–282. [Google Scholar]
- Roux, B. 1999. Statistical mechanical equilibrium theory of selective ion channels. Biophys. J. 77:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov, G. Y., D. S. J. Astill, B. Bennetts, B. P. Hughes, A. H. Bretag, and M. L. Roberts. 1997. pH-dependent interactions of Cd2+ and a carboxylate blocker with the rat ClC-1 chloride channel and its R30E mutant in the Sf-9 insect cell line. J. Physiol. 501:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov, G. Y., M. Pusch, D. S. J. Astill, M. L. Roberts, T. J. Jentsch, and A. H. Bretag. 1996. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. 497:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart, O. S., J. G. Neduvelil, X. Wang, B. A. Wallace, and M. S. P. Sansom. 1996. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14:354–360. [DOI] [PubMed] [Google Scholar]
- Torrie, G. M., and J. P. Valleau. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. Journal of Computational Physics. 23:187–199. [Google Scholar]
- Yeh, I., and M. L. Berkowitz. 1999. Ewald summation for systems with slab geometry. J. Chem. Phys. 111:3155–3162. [Google Scholar]
- Yin, J., Z. Kuang, U. Mahankali, and T. L. Beck. 2004. Ion transit pathways and gating in ClC chloride channels. http://www.arxiv.cornell.edu/abs/physics/0401022. [DOI] [PubMed]
- Zhang, L., and J. Hermans. 1996. Hydrophilicity of cavities in proteins. Proteins. 24:433–438. [DOI] [PubMed] [Google Scholar]