

Abstract
Photoactive yellow protein is the protein responsible for initiating the “blue-light vision” of Halorhodospira halophila. The dynamical processes responsible for triggering the photoactive yellow protein photocycle have been disentangled with the use of a novel application of dispersed ultrafast pump-dump-probe spectroscopy, where the photocycle can be started and interrupted with appropriately tuned and timed laser pulses. This “incoherent” manipulation of the photocycle allows for the detailed spectroscopic investigation of the underlying photocycle dynamics and the construction of a fully self-consistent dynamical model. This model requires three kinetically distinct excited-state intermediates, two (ground-state) photocycle intermediates, I0 and pR, and a ground-state intermediate through which the protein, after unsuccessful attempts at initiating the photocycle, returns to the equilibrium ground state. Also observed is a previously unknown two-photon ionization channel that generates a radical and an ejected electron into the protein environment. This second excitation pathway evolves simultaneously with the pathway containing the one-photon photocycle intermediates.
INTRODUCTION
Photoreceptor proteins activate the signaling pathways that transmit information regarding ambient light conditions in living organisms. The resulting responses include important biological functions such as human eyesight, directed growth of plants, and bacterial phototaxis (Hellingwerf, 2000). Often, these photoreceptor responses are triggered by small light-induced structural changes of intrinsically bound chromophores within these proteins. The process of eliciting biological responses from these structural changes results from a complex relationship between the chromophore and the surrounding protein environment, including the formation and breaking of hydrogen bonds, stabilizing charge redistribution, and the manipulation of the potential energy surfaces responsible for structure transitions. The photoactive yellow protein (PYP) is an excellent model system for studying this relationship because of its well-studied photophysical properties, including both static and time-resolved IR (Brudler et al., 2001; Groot et al., 2003; Xie et al., 1996, 2001), Raman (Unno et al., 2002, 2003; Zhou et al., 2001), and visible dynamics (Baltuška et al., 1997; Changenet et al., 1998; Devanathan et al., 1999; Premvardhan et al., 2003). Moreover, the available static (Genick et al., 1998) and transient x-ray structures (Genick et al., 1997; Perman et al., 1998) combined with recent theoretical studies (Groenhof et al., 2004, 2002a,b; Ko et al., 2003; Molina and Merchan, 2001) further aids in exploring the PYP system. However, it is still unknown which microscopic properties determine the dynamics and the resulting quantum yield of the photocycle. With the aid of ultrafast dispersed pump-dump-probe (PDP) spectroscopy, where the photocycle can be initiated and halted with appropriately tuned and timed laser pulses, we have explored the initial photoinduced steps of the excitation dynamics of PYP.
PYP is a small 125-amino-acid protein found in the purple sulfur bacterium Halorhodospira halophila that initiates the pathway responsible for negative phototaxis of the organism upon exposure to blue light (Sprenger et al., 1993). This “blue-light vision” arises from the excitation of a p-coumaric acid (pCA) chromophore that is covalently bound to a unique cysteine (Cys-69) residue of the protein (Baca et al., 1994; van Beeumen et al., 1993). Since the discovery of PYP in the mid-1980s (Meyer et al., 1987), much effort has been spent in characterizing its photoinduced photocycle dynamics, which can be decomposed into several important basic steps, including the isomerization (Genick et al., 1997; Groot et al., 2003) and protonation (Xie et al., 1996) of the pCA chromophore, the partial unfolding of the protein (Rubinstenn et al., 1998), and their subsequent recovery reactions (van Brederode et al., 1996). For the wild-type PYP system, the ensuing photocycle includes dynamics extending from tens of femtoseconds to hundreds of milliseconds (Cusanovich and Meyer, 2003; Hellingwerf et al., 2003; Hoff et al., 1994; Meyer et al., 1987). In particular, dispersed ultrafast pump-probe (PP) measurements have proven insightful for exploring the initial ultrafast dynamics of this photocycle (Baltuška et al., 1997; Devanathan et al., 1999; Gensch et al., 2002; Imamoto et al., 2001; Ujj et al., 1998). A simplified scheme of the room temperature photocycle of PYP would read as pG* → I0 → pR → pB → pG, where the pG* state is the initially photoexcited PYP system and pB is a partially unfolded intermediate(s) with a protonated chromophore (Cusanovich and Meyer, 2003; Hellingwerf et al., 2003). Several nomenclatures have been proposed to discuss the photoproduct state in the PYP photocycle; the rationale for selecting the above nomenclature is presented elsewhere (Hellingwerf et al., 2003). The time window used for the ultrafast measurements limits the study of dynamics to only to the rise of the pR state, and different experimental setups/techniques are required to investigate the longer times dynamics of the photocycle (Devanathan et al., 1998; Hoff et al., 1994).
Although a powerful technique, PP signals are not definitive in determining the events of the initiation of the PYP photocycle because of overlapping spectral and temporal properties of the transient species. For example, the near-complete overlap of the stimulated emission band with the absorption of the first photoproduct, I0, in the photocycle precludes directly correlating the quenching timescales of the excited state with the timescale of photocycle initiation (details below). Consequently, the excited-state population that is responsible for the observed fluorescence may not necessarily be the same population that is responsible for initiating the photocycle. Addressing this question is especially important for exploring systems that exhibit dual fluorescence like many charge transfer chromophores (Lapouyade et al., 1992); recently dual fluorescence (multiple bands) has been observed in a model PYP chromophore in the gas phase (Ryan et al., 2002), which were ascribed to the cis and trans conformers. Furthermore, a strong charge transfer aspect to the photoexcitation of the PYP protein (Premvardhan et al., 2003) and its free chromophore in solution (Premvardhan et al., 2004) has been observed in electro-optical Stark measurements. In the time-domain, different kinetic schemes have been proposed for modeling the initial dynamics of the PYP photocycle, ranging from evolution of the transient states in series (Devanathan et al., 2001), in parallel (Gensch et al., 2002), or admixtures of the two (Imamoto et al., 2001; Unno et al., 2002). With the aid of multiple pulse transient absorption techniques, we present a self-consistent model describing the first five nanoseconds of the PYP photocycle dynamics after absorption of a 395-nm photon.
The PDP technique (also referred to as stimulated emission pumping) has been used successfully to study ground-state properties in gas phase experiments (Ishikawa et al., 1997; Pique et al., 1988) and has recently been employed in the study of several biological systems (Changenet-Barret et al., 2000; Gai et al., 1997; Larsen et al., 2003a, 2004; Ruhman et al., 2002). We extend this technique to include the collection of dispersed signals, i.e., the simultaneous collection of both wavelength and time-resolved data. The generated multidimensional data were analyzed using a detailed global analysis methodology (Hoff et al., 1994; Holzwarth, 1996; Krueger et al., 2001; van Stokkum et al., 2004). This combination has been shown to be a powerful tool for disentangling the underlying dynamics of complicated systems (Larsen et al., 2003a), including the quenching dynamics of the isolated PYP chromophore (Larsen et al., 2004) and green fluorescent protein (Vengris et al., 2004) chromophore in solution and the excited-state dynamics of carotenoids (Larsen et al., 2003a). The power of PDP spectroscopy lies in its ability to effectively “control” reactions as they evolve, by manipulating the ensuing transient species with applied laser pulses (e.g., the application of a laser pulse with carefully selected spectral and temporal properties can transfer part of the excited-state population to the ground electronic state). Thus, the PDP technique can be viewed as the “incoherent control” counterpart to more complex “coherent control” techniques (Rice and Zhao, 2000) and the resulting PDP spectroscopic signals can be explained solely in terms of manipulating electronic state populations. This simplification allows for straightforward interpretations of the collected multidimensional data.
This article describes our exploration of the initiation dynamics of PYP with this dispersed PDP technique and supplemented with excitation-wavelength-dependent and power-dependent PP signals. We identify and characterize an interfering multiphoton ionization pathway that evolves in parallel with the single photon photocycle and results from exciting the PYP protein with high intensity ultrafast excitation 395-nm light. From this data, we propose a kinetic model that describes not only the successful, but also the failed attempts in the initiation of the PYP photocycle.
EXPERIMENTAL
The PYP sample was produced and isolated as a hexahistidine-tagged apo-protein in Escherichia coli (Kort et al., 1996) and was reconstituted with the 1,1-carbonyldiimidazole derivative of the p-coumaric acid chromophore (Sigma, St. Louis, MO) (Hendriks et al., 2002). The protein samples were purified with a Ni2+ column and were used without removal of the hexahistidine tag in 10 mM Tris/HCl, pH 8. The absorption spectra of the prepared PYP samples (Fig. 1) were measured before and after data collection and no significant change was observed.
FIGURE 1.

Normalized static absorption (solid line) and fluorescence (dashed line) spectra of PYP overlapping the spectra of the 395-nm pump (shaded) and the 505-nm dump (light shaded) laser pulses. The arrow at 470 nm indicates the wavelength used for red-edge excitation of the sample in the excitation-wavelength-dependent experiment.
The used multipulse experimental system is described in detail elsewhere (Larsen et al., 2004) and is briefly outlined below. An amplified ultrafast Ti:Sapphire laser system (Mira Seed, Coherent, Santa Clara, CA; and Alpha 1000, BMI, Paris, France) operating at 1 kHz produces short duration (∼40 fs full width at half-maximum) laser pulses at 790 nm. These amplified pulses are split into three parts. The first portion is frequency-doubled in a 0.5-mm β-barium borate crystal, producing 395 nm light, which is used as an excitation source. A second portion is used to generate a white-light continuum by focusing the ultrafast 800-nm light into a slowly translating 2-mm-thick CaF2 plate. The generated white light extends from ∼320 nm to the near-infrared and is used as a broadband probe beam. The remaining portion of the 790-nm pulses is used to operate a homebuilt, 395-nm pumped non-co-linear optical parametric amplifier (NOPA) that is tuned to generate dump pulses at 505 nm. For measuring the excitation-wavelength-dependent PP data, the NOPA was tuned to 470 nm instead of 505 nm, and the data was collected with the same experimental setup.
Setting the time delay between the pump and the probe was achieved by varying the distance traversed by the probe beam with a motorized mechanical stage. Both the pump and the dump beam are focused in the sample (spot size ∼300 μm) with separate 20-cm focal length quartz lenses; the probe light is focused (spot size ∼80 μm) with off-axis (90°) parabolic mirrors to reduce dispersion. All three laser beams are focused within the same plane with a ∼1.5° angle between adjacent beams. After the sample, the probe light was dispersed onto a double-diode array detector with an imaging spectrograph (Oriel, Mountain View, CA). The diode signals were then digitized and collected with an attached computer for further analysis. The timing accuracy in the dispersion-corrected data is limited to <50 fs and the pump and dump pulse intensities were 120 nJ and 150 nJ, respectively. The instrument response function is 120 fs (full width at half-maximum), the wavelength resolution is 1 nm and the polarizations of the two laser beams are parallel to each other, whereas the white light probe is set at magic angle (54.7°) to eliminate effects of reorientation dynamics. The PYP sample (OD ∼0.7 at the absorption maximum) was pumped through a rapidly translating 1-mm quartz flow cell (rapid translation ensured that the sample was renewed between the consecutive laser pulses, and the flowing allowed us to avoid the accumulation of any potential photoproducts).
Traditional pump-probe (PP) techniques involve the impinging of two short pulses (pump and probe) onto the sample, producing one-dimensional data. When the probe is a broad-band pulse and is spectrally dispersed onto a multichannel detector, two-dimensional information is collected. Furthermore, the introduction of an additional dump laser pulse between the pump and the probe pulses generates three-dimensional signals (two time delays and one wavelength); measuring and presenting the data in different ways can thus exploit this multidimensionality. The most intuitive way to collect such three-dimensional data is in the form of a “kinetic trace,” where both the first and second laser pulses are set at fixed times and the signals are collected by varying the probe pulse delay, as in PP experiments. An alternative procedure for collecting and viewing the PDP signals is the “action trace,” which is collected by delaying the second pulse with fixed pump and probe pulse times. By varying the second pulse delay (with a fixed probe time), the asymptotic dump-induced effects, including dump efficiencies, photoproduct yields, and selective depletion on the specific PP spectrum are directly monitored. Whereas kinetic traces directly monitor the dump-induced dynamics, action traces measure the asymptotic dump-induced effect at a specific probe time and describe the magnitude of the change in the ΔOD as a function of the timing of the dump pulse with respect to the pump pulse. As discussed below, action traces are useful for identifying the pathway(s) of connectivity between transient states in PP spectra.
Previously, multicompartmental global analysis has proved successful in interpreting dispersed PP studies (Hoff et al., 1994; Holzwarth, 1996; Krueger et al., 2001). Here, we expanded this methodology to include the additional coupling between transient states induced by the externally applied dump pulse (van Stokkum et al., 2004; Vengris et al., 2004a). The construction of a connectivity scheme, k, is central to the type of global analysis used here. The connectivity scheme describes the interactions between the observed time-resolved states, nj(t), in addition to the coupling due to the external electric fields of the pump and dump laser pulses. Assuming first-order (rate) kinetics, a set of differential equations can then be constructed and numerically solved that describes the time evolution of the constituent transient states,
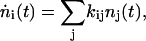 |
(1) |
where the matrix elements of the connectivity scheme, kij, is the rate constant that connects the ith state with the jth state. For the PDP signals, these states are further coupled by the electric field of both the pump and dump pulses (van Stokkum et al., 2004; Vengris et al., 2004a). Spectral information is then introduced by adding an explicit wavelength dependence to the obtained solution, resulting in wavelength-dependent pre-factors before the transient state concentration components,
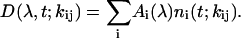 |
(2) |
For each transient state, a wavelength-dependent factor, Ai(λ), is obtained, which is called the species-associated difference spectrum (SADS, Hoff et al., 1994; Holzwarth, 1996). The “true” transient spectra, the species-associated spectra (SAS), can then be determined by subtracting a common bleach spectrum from the corresponding SADS. The SAS, in combination with the connectivity scheme and the corresponding decay times, provide a straightforward approach for interpreting the complex and unwieldy dispersed PP and PDP data collected in our experiment. The SAS and the timescales that relate the growth and decay kinetics associated with these individual states are estimated from the data by means of nonlinear regression (Hoff et al., 1994; Holzwarth, 1996). Further information about the details behind using global analysis to describe measured PDP signals can be found elsewhere (van Stokkum et al., 2004). It should be emphasized that the combination of fitting both the collected PP and PDP data, encompassing over 100,000 data points (covering time range from tens of femtoseconds to 5 ns and wavelength range from 320 nm to 650 nm), and the intuition provided by the added dump pulses, allows for an overdetermined extraction of the timescales and SAS values of the multiple transient states.
RESULTS
In Fig. 1, the PYP absorption spectrum is shown overlapping the pump and dump laser spectra. The pump pulse (395 nm) excites the ground-state PYP population on the blue edge of the absorption spectrum, whereas the dump pulse (505 nm) de-excites the excited-state population near the peak of the stimulated emission band. The dump wavelength was selected to overlap well with the stimulated emission band and poorly with the ground-state absorption spectrum; otherwise the dump pulse will not only remove population from the excited state, but will also initiate a photocycle from the remaining ground-state population. This allows for a clean interpretation of the collected data; if the dump pulse were resonant with an excited state absorption (ESA) band also, then competing signals would also be observed (Gai et al., 1997; Larsen et al., 2003a). However, in PYP, no clearly resolvable ESA band was observed to overlap with the applied 505-nm dump pulse in previous PP measurements (Baltuška et al., 1997; Changenet et al., 1998; Devanathan et al., 1999; Premvardhan et al., 2003).
As the PDP technique extends on the PP results, a brief discussion of the PP signals for PYP is needed for interpreting the PDP signals. Five representative PP transient spectra are shown in Fig. 2, spanning the probe time from 180 fs to 4 ns. The collected spectra and dynamics of the PYP system are similar to the data measured in previous dispersed PP measurements (Baltuška et al., 1997; Devanathan et al., 1999; Imamoto et al., 2001; Ujj et al., 1998), with the main difference that the probe window in the data presented in Fig. 2 extends into the UV below 395 nm. All transient spectra show a ground-state bleach peaking at the maximum of the absorption spectrum (446 nm). The 180-fs and 2-ps spectrum also exhibit a broad negative band at 520 nm and a pronounced positive band at 370 nm, which are ascribed to stimulated emission (SE) and excited-state absorption (ESA), respectively. In contrast, the 35-ps, 500-ps, and 4-ns spectra do not exhibit clear SE bands, but instead show positive product state absorption bands peaking at 500 nm and 470 nm, respectively, which are ascribed to the product state absorptions of the initial red-shifted intermediates in the PYP photocycle, I0 and pR (Cusanovich and Meyer, 2003; Hellingwerf et al., 2003). The 35-ps, 500-ps, and 4-ns PP spectra also exhibit a noticeable positive sharply peaked band at 360 nm that has not been previously ascribed to either the I0 or pR product state band. Since ultrafast fluorescence measurements show that no excited-state population exists at this time (Changenet et al., 1998; Chosrowjan et al., 1997; Vengris et al., 2004b), this sharply featured band is not ascribed to the similar UV ESA band observed in the 180-fs and 2-ps spectra. Furthermore, a weak broad absorption is observed in the 4-ns spectrum at ∼600 nm. A similar band in the same spectral region was observed in isolated model PYP chromophores in solution (Larsen et al., 2003b, 2004) and was ascribed to radical absorption bands generated from a multiphoton ionization process.
FIGURE 2.

Representative dispersion-corrected transient PP spectra. The region around the pump excitation wavelength (395 nm) is corrupted due to scatter from the pump pulse.
Shown in Fig. 3 are representative kinetic traces of only two PDP data sets dumped at 500 fs (open circles) and 2 ps (solid circles) and are overlapping the PP signals (open triangles). The dump pulse demotes part of the excited-state population back to the ground state, which is noticeable as a rapid loss of SE (550 nm) and ESA (375 nm) bands along with a concomitant recovery in the bleach region (445 nm). However, the relative magnitude of the bleach recovery at 445 nm (∼25%) is not comparable with the depletion of the stimulated emission and ESA (∼50%), suggesting the involvement of an intermediate state that temporarily stores ground-state population before refilling the bleach on a longer timescale. This ground-state intermediate (GSI) is more clearly observed as a dump-induced absorption in the 490-nm trace, where a local maximum in both the PP and the PDP signals is observed. Although observed in the PP data, the magnitude of the induced absorption in this spectral region is transiently enhanced by the application of the dump pulse. A similar ground-state intermediate was also observed in the PYP chromophore in solution, but exhibited a faster relaxation lifetime (Larsen et al., 2004).
FIGURE 3.

Selected PP (open triangles) and PDP traces dumped at 500 fs (open circles) and 2 ps (solid circles). Symbols are the experimental data and the solid lines are the results of the global fits to these data. Note that the time axis is linear up to 5 ps, and then logarithmic to 1 ns.
In the UV spectral region, a clear difference of dynamics is observed between the 350-nm and 375-nm PDP kinetic traces (Fig. 3). Whereas the dump induces a loss in both signals, which are positive during the first 10-ps, the 375-nm trace eventually decays to a negative value and the 350-nm traces stays positive for the duration of the experiment. Moreover, the nature and amplitude of the dump-induced changes in these UV traces differ, which has been characterized extensively for the isolated PYP chromophore in solution (Larsen et al., 2004). This difference can be better observed in the measured action trace signals 500 ps in Fig. 4. Here, the relative dump-induced change to the PP signals, also referred to as the double-difference OD signal, ΔΔODrel = (PDP-PP)/PP, is plotted versus the dump delay time. Even though a measurable PP signal is observed across the complete wavelength region at this probe time, the dump-induced changes are spectrally limited. The ΔΔODrel signals measured in the I0 (solid square) and bleach (open squares) bands show a clear dump-induced depletion of the respective PP signals, resulting from the reduced photoproduct yield by shifting excited-state population back into the ground state before photocycle initiation. In strong contrast, the UV radical band (open circles) and the ejected electron band (solid circles) are completely unaffected by the 505-nm dump pulse. Overlapping the action trace is the simulated time-dependent excited-state population estimated from the global analysis (dashed curve), showing that the observed time-dependent yield does not follow the observed fluorescence decay dynamics exactly. The non-zero action traces clearly highlight that the photocycle proceeds through the part of the potential energy surface that is accessible with the 505-nm dump pulse. In sharp contrast, the species responsible for the UV and red product bands do not.
FIGURE 4.

Action trace of PYP, measured with probe time at 500 ps. Plotted is the ΔΔODrel signal (PDP-PP)/PP versus the dump time. The open squares are the dump-induced depletion of the bleach (446 nm) and the solid squares are the induced-depletion of the I0 photoproduct absorption (510 nm). The solid and open circles are the depletion of the radical (370 nm) and the ejected electron (620 nm), respectively. The amplitude of the PP signals were 13 mOD (radical), 30 mOD (bleach), 12 mOD (I0), and 4 mOD (electron). Overlapping the bleach and I0 signals is the modeled excited-state population with the parameters from Table 1 (dashed line).
To explore if the additional signals observed in the action trace data originated from the same resonantly-enhanced ionization mechanism found for the PYP chromophore in solution (Larsen et al., 2003b, 2004), we supplemented the PDP data with excitation-power dependence and excitation-wavelength dependence data on selected time-gated PP spectra. Using the same multipulse experimental setup described above and by tuning the NOPA to 470-nm instead of 505-nm, the PDP setup can be used to measured two PP data sets with different excitation wavelengths in a near-simultaneous manner (alternating every ∼6 ms with mechanical choppers). In Fig. 5, the 2-ps and 200-ps PP spectra collected after excitation at the high- and low-energy sides of the absorption band (Fig. 1), with 395-nm (solid curve) and 470-nm (dashed curve) pulses respectively are contrasted. This real-time collection of two different excitation-wavelength PP signals guarantees identical experimental conditions for both data sets, rejects potential errors due to sample degradation between sample runs, and cleanly resolves any potential differences in the PP signals resulting from different excitation wavelengths (Larsen et al., 2003a). Qualitatively similar spectroscopic signals are observed for both excitation wavelengths, albeit with clearly resolvable differences. The 2-ps PP spectra (Fig. 5 A) probe the PYP system when the pCA chromophore is still electronically excited, hence a negative SE at 500 nm is observed. However, this SE band is absent in the 200-ps spectra (Fig. 5 B), the I0 photoproduct is observed instead in the same spectral region. The 395-nm pumped signals were scaled to achieve similar amplitudes for the SE and I0 product bands for each spectrum, respectively. These data suggest that the transient spectra measured after 395-nm excitation can be decomposed into a sum of two components: 1), one that is essentially identical to the spectrum obtained with 470-nm excitation and 2), an additional spectrum that is ascribed to photoproducts which are generated from a secondary photoexcitation process initiated only after 395-nm excitation (see Fig. 5, insets). Based on the similarity between the spectrum of this additional pathway and the terminal spectrum observed in the PP measurements of the PYP chromophore in solution (Larsen et al., 2003a, 2004), we ascribe this additional pathway to a multiphoton ionization process that evolves in parallel with the photocycle dynamics after excitation with 395-nm ultrafast laser light. A similar excitation-wavelength dependence of the PP dynamics has been previously addressed where a complex wavelength-dependent excited-state model was proposed to address the observed differences in the signals (Devanathan et al., 1999; Gensch et al., 2002).
FIGURE 5.

Excitation-wavelength dependence on the 2-ps (A) and 200-ps (B) PP spectra. The solid and dashed curves are the PP spectra generated after either 395-nm or 470-nm excitation, respectively.
We explored the pump power dependence of the 200-ps spectrum after excitation with 395-nm light, to further explore if the ionization mechanism is responsible for the additional pathway observed in the PYP protein. Fig. 6 shows the measured transient spectrum generated from varying the pump power at this probe time. Increasing the pump intensity clearly amplifies the amplitudes in all observed bands. However, as Fig. 6 B highlights, the observed power dependence for individual wavelengths in the spectrum is complex and cannot be simply classified as either linear or quadratic. These complex power-dependent traces result partially from overlapping spectral features of the species generated from the different pathways suggested by the wavelength-dependent data in Fig. 5 and partially from the complicated underlying mechanisms involved in their generation. The composition and characterization of these data into contributions from the two pathways are achieved with the aid of global analysis (see below).
FIGURE 6.

Power dependence of the PP data with 395-nm excitation. (A) Power dependence of the 200-ps PP spectrum. The region around the pump excitation wavelength is corrupted due to scatter from the pump pulse. The arrows indicates the direction of the signal change with increasing laser power. (B) Power dependence of representative wavelengths (as labeled). (Inset) (Negative) dependence of the bleach because of its greater range.
DISCUSSION
A central goal in measuring the dispersed PDP data is exploring how the PYP photocycle evolves after exciting the embedded pCA chromophore. The first question we posed was whether the PYP photocycle evolves via the same part of the excited-state potential energy surface that is also responsible for the observed SE bands in PP measurements (Baltuška et al., 1997; Devanathan et al., 1999; Imamoto et al., 2001) and the fluorescence in time-resolved emission studies (Changenet et al., 1998; Chosrowjan et al., 1997; Vengris et al., 2004b). The action trace data in Fig. 4 show that this is indeed the case and moreover, a secondary pathway initiated by the 395-nm excitation pulse can be clearly identified that does not evolve via the same point in the potential energy surface. The first subsection of the discussion characterizes this secondary pathway, whereas the second subsection elaborates on the details extracted from the PDP data on the photoproduct pathway.
Ionization pathway
The PDP action trace signals, the wavelength-dependent, and the power-dependent PP data, show the existence of a secondary pathway that co-exists with the PYP photocycle dynamics when excited with 395-nm laser pulses. Moreover, this pathway is not initiated when PYP is excited with 470-nm ultrafast light (Fig. 5). We ascribe this pathway to an interfering ionization reaction, which results in the generation of a pCA radical and an ejected electron into the protein pocket. Similar two-photon ionization has been observed in the PP studies of isolated PYP chromophore analogs in solution (Changenet-Barret et al., 2001; Larsen et al., 2004), where two photons from the ultrafast 395-nm excitation pulse doubly excite the PYP chromophore: the first photon excites the ground state into the excited S1 state, whereas the second photon excites the S1 into an Sn state via an ESA band in the near UV that overlaps the excitation wavelength (Fig. 5). This double excitation imparts enough energy, ∼50,000 cm−1, to exceed the ionization potential of the chromophore.
To further characterize if this mechanism is applicable, the power-dependent data shown in Fig. 6 was globally analyzed and then interpreted within a resonantly-enhanced ionization model. In this analysis, the measured power-dependent 200-ps spectra is decomposed into a sum of two spectra (Fig. 7 A) with intensity-dependent amplitudes (Fig. 7 B). It must be emphasized that no power dependence was explicitly or implicitly introduced into this decomposition. The resulting spectra extracted from this decomposition agree with the wavelength-dependent PP data in Fig. 5, where the pure photocycle spectrum constituting only the I0 photoproduct and corresponding bleach bands (solid curve) is significantly different from the spectrum associated with ionization (dashed curve). The other spectrum also exhibits not only a bleach component but a positive band in the UV and a broad featureless electron spectrum extending to the red beyond our spectral window. This estimated spectrum is near-identical with the ionization spectrum predicted from the wavelength-dependent PP measurements (Fig. 5, inset), and can be observed in the PP data of Fig. 2. Moreover, this spectrum is strikingly similar to the terminal spectrum measured after photoionization of the isolated PYP chromophore in solution (Larsen et al., 2003b, 2004). The portion of this extracted ionization spectrum that is attributed to the detached electron is also qualitatively similar to the spectrum of the solvated electron in water, with the exception that its gently increasing slope is noticeably weaker than that observed in aqueous solution (Jou and Freeman, 1979).
FIGURE 7.

Analysis of the power-dependent PP spectra at 200 ps. (A) Estimated spectra associated with the photocycle (solid curve) and ionization (dashed curve) pathways. (B) Power-dependent concentration of the two pathways overlapping the predicted curves for the proposed model in the inset. (C) Abridged multistate photocycle model to explain observed power dependence. Fitting parameters: {k01S, k10S}= 44,000 cm2/mmol and {k12S, k21S}= 45,000 cm2/mmol. The used ke = 1/100 fs and kp = 1/2 ps time constants are relatively insensitive in modeling a single PP spectrum and are mean values from the full analysis shown in Table 1.
The global analysis simplifies the complex power dependence observed in Fig. 6 B into the power-dependent concentrations displayed in Fig. 7 B. The 200-ps probe time was selected so that all excited-state contributions have completely decayed (Fig. 2), and much of the complexity of the PYP photocycle is not required in modeling the power-dependence of a single PP spectrum (see below). We interpret these concentration curves with a simplified model (Fig. 7 C) with five linearly coupled components. Similar multistate models have been proposed to interpret other multipulse experiments including PDP (Gai et al., 1997) and PrPP (Logunov et al., 2001) signals in bacteriorhodopsin and the power-dependence of the activation of the green fluorescence protein (Vengris et al., 2004a) and PYP chromophores (Larsen et al., 2004). In this model, one 395-nm photon from the pump pulse excites the PYP system into the first excited electronic state, E1; however, since a strong ESA overlaps the 395-nm excitation wavelength (Figs. 2 and 5), an additional photon can also be absorbed from the pump pulse, which promotes the already excited PYP system into a highly excited electronic state, called E2 in the model. From this high energy electronic state, the 50,000 cm−1 of imparted excitation energy is enough to overcome the ionization threshold of the chromophore and detach the electron from the protein pocket, leaving a radical behind. The dynamics of the population of the different states are modeled with the following system of differential equations,
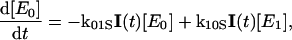 |
(3a) |
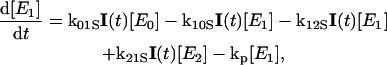 |
(3b) |
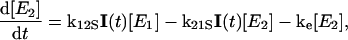 |
(3c) |
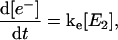 |
(3d) |
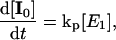 |
(3e) |
where E0, E1, and E2 are the ground, first, and second electronic excited states, respectively; e− is the solvated electron/radical concentration; and I0 is the photocycle population (the complete dynamics of which are excluded in this simplified model). The electronic states are coupled via external electric fields (e.g., using pump or dump laser pulses) with extinction coefficients for absorption and stimulated emission transitions, kij, from the ith to jth electronic state. Spontaneous relaxation and internal conversion pathways are excluded from this analysis because probing the spectrum at a single probe time is insensitive to these pathways; a complete model base on the full PDP signal is presented below. The I0 photoproduct and electron/radical species are generated from the E1 and E2 electronic states, respectively. The rate of photoproduct and radical/electron formation are kp and ke, respectively; the Gaussian intensity profile (number of photons in pump pulse) of the excitation pulse is I(t).
The above differential equations were solved numerically with a Runge-Kutta algorithm (MATLAB). The predicted power dependence from these equations, with the parameters in the caption of Fig. 7, satisfactorily describes the observed power dependence of both the radical/electron and I0 photoproduct spectra. At lower pulse intensities, the power dependence of the photocycle population increases linearly, whereas the ionization population behaves more like the quadratic dependence expected for a true two-photon transition and a similar power dependence was observed in the ionization of small molecules in solution (Lachish et al., 1975). Once the E1 population is appreciable (∼70 μJ) these trends deviate, where the photocycle population is effectively “stolen” by the ionization pathway. At this point, the photocycle power dependence starts to saturate, whereas the ionization evolves into a more linear dependence. This continues until the photocycle population starts to decrease and the ionization steals more of the excited-state population. Consequently, a steady-state E1 population cannot be maintained at high pump intensities with 395-nm ultrafast laser light and this depletion may explain the lack of generating an appreciable photoproduct in early time-resolved x-ray measurements on PYP with ∼395-nm excitation, whereas successful measurements were eventually performed on the far red edge of the absorption spectrum (∼490 nm) (Genick et al., 1997; Perman et al., 1998; Ren et al., 2001).
Even though the ionization pathway is a two-photon process, the observed pseudolinear power dependence originates from the fact that the E2 ← E1 transition is as strongly coupled to the excitation pulse as the E1 ← E0 transition. Since the ionization is resonantly enhanced with two sequential single-photon transitions, both with appreciable extinction coefficients, this ionization pathway cannot be reduced by decreasing the excitation power (using 395-nm ultrafast excitation light), without decreasing the signal/noise ratio of the single-photon photocycle signals to an unacceptable level. An alternative and more useful way to suppress the ionization pathway would be to excite the system at a wavelength that has good overlap with the ground-state absorption and poor overlap with the ESA. Since ultrafast laser pulses ∼400-nm are relatively easy to generate with widely available Ti:Sapphire laser systems, many experimental ultrafast studies potentially include contributions from this parallel ionization pathway, which may complicate interpretations.
Just how radicals and ejected electrons behave (and recombine) in a protein environment is poorly understood. Presumably, the ejected electron does not escape from the vicinity of the chromophore-binding pocket and may transiently combine with a low-redox potential residue or alternatively be stabilized by nearby amino-acid residues (similar to solvation). An analogous electron transfer mechanism has been ascribed to the observed decarboxylation of a glutamic acid residue in the green fluorescent protein that results in the generation of free CO2 molecules (van Thor et al., 2002). Regardless of the specific dynamics of the potential ionization pathway, transient PP studies are sensitive to not only the inherent one-photon dynamics, but also to potential two-photon radical/electron dynamics (e.g., recombination, scavenging, and radical protonation) when excited with high energy pulses.
Kinetic models
The kinetic traces of Fig. 3 highlight the ability of the PDP technique to interrupt the reaction at specific and controlled times and then observe the resulting dynamics on both the excited and ground electronic states, which contributes to a greater understanding of the underlying processes in the PYP protein. Due to the complex multidimensional nature of the dispersed PDP data, we find that global analysis (simultaneously fitting the PP and PDP signals) is crucial in interpreting our data, particularly since the photoproduct absorption bands (Io and pR) overlap both spectrally and temporally with the excited- and ground-state intermediate bands. The modeling of the PP signals alone does not provide a clear separation of decay timescales and spectral signatures of the ground transient states; to accomplish this, the simultaneous fitting of both PP and PDP data is required. The information yielded by PDP experiments provides valuable insight in the ground-state dynamics and reveals the origin of different overlapping bands. When the dump pulse de-excites the excited PYP protein, the SE is depleted and concomitantly, the nonequilibrium ground-state absorption at 490 nm increases (Fig. 3). This provides a qualitative explanation to the differences in the PDP kinetics between the two induced absorption bands peaking at ∼360 nm and 470 nm. Whereas the UV band is a true ESA (along with the radical band underneath it) and disappears upon dumping, the 490-nm band is due to the absorption of unrelaxed GSI, which increases upon dumping. The dynamics of the GSI is sensitive to when the excited-state population was dumped; in the early dump times, a single exponential decay of the GSI into the bleach is observed, whereas, when dumped at later times, a bi-exponential relaxation is observed (extracted from global analysis).
The kinetic model underlying the global analysis (k in Eq. 1) must account for both one-photon dynamics with evolution along both excited and ground electronic states and the competing two-photon ionization mechanism. This ionization process is introduced as an additional pathway that is initiated within the excitation pulse and evolves in parallel with the single-photon dynamics. Even though subpicosecond spectral dynamics from photoinduced ejected electrons in solution have been observed previously (Migus et al., 1987; Vilchiz et al., 2001), the identification and characterization of such spectral evolution was not possible in our data and was excluded in the modeling. This simplification allows us to disentangle the interfering two-photon ionization process from the single-photon dynamics.
On the basis of the fitting from the PP and PDP data, we construct two kinetic models to describe the observed dynamics that underlies the global analysis. These two models, contrasted in Fig. 8, both produce similar quality fits to the measured PDP data, within the corresponding signal/noise level. The obtained results are self-consistent with the simultaneously evolving pathways suggested from the excitation-wavelength and power-dependent PP signals (Figs. 5 and 6). The nonexponential dynamics observed in the kinetic traces of Fig. 3 emphasizes the complex photodynamics observed in PYP; similar multiexponential excited-state lifetimes have been previously observed (Baltuška et al., 1997; Changenet et al., 1998; Devanathan et al., 1999; Mataga et al., 2003; Vengris et al., 2004b). A long-standing problem in interpreting measured multiexponential relaxation decays from an ensemble of chromophores (or protein-chromophore complexes, in this case) is whether the observed multiexponential behavior results from inherent homogeneous excited-state dynamics of the chromophore, e.g., evolution over a complex excited-state potential energy surface, or alternatively if it results from an inhomogeneous ground-state population. The inhomogeneous model postulates that the equilibrated ground-state population consists of several subensembles (e.g., slightly different chromophore conformations or nearby amino-acid orientations), with each exhibiting a different excited-state lifetime. The excitation pulse would consequently excite a portion of each subensemble and thus the measured signals would exhibit nonexponential decays.
FIGURE 8.

Connectivity schemes compatible with the data and used in the global analysis: (A) inhomogeneous model and (B) homogeneous model. Dynamical states are separated into four classes: excited state (red), ground state (blue), photocycle products (green), and two-photon ionization dynamics (black). ESI1, ESI2, and ESI3 refer to the excited-state lifetimes #1, #2, and #3, respectively. pG is the equilibrated ground-state species, and GSI is the ground-state intermediate. Thick solid arrows represent the initial excitation process from the laser pulse and thin solid arrows dynamics represent the “natural” PP population dynamics. The dashed arrows represent the population transfer dynamics that may be enhanced with the dump pulse.
In Fig. 8, both the homogeneous and inhomogeneous kinetic models describe the observed PDP and PP dynamics as an evolution between discrete interconnected transient states, which can be separated into four categories: 1), excited-state dynamics (red boxes); 2), ground-state dynamics (blue boxes); 3), photocycle dynamics (green boxes); and 4), an ionization channel (black boxes). The estimated SAS from this global analysis (Fig. 9), representing the spectra of these underlying transient states, in combination with their corresponding decay times (Table 1), provide a straightforward and intuitive basis to interpret the multidimensional PDP data. Although the connectivity schemes underlying each model differ, the resulting SAS and timescales obtained from the global analysis of the data with each model do not differ appreciably and can be represented with the same set of spectra. This similarity further complicates determining which model is most appropriate for describing the underlying PYP photodynamics, even when aided with dispersed multidimensional PDP data.
FIGURE 9.

Species-associated spectra (SAS) estimated from the global analysis of the PP and PDP traces. The red curve is the SAS associated with the ESI1, ESI2, and ESI3 states. The dashed blue curve is the ground-state intermediate, and the solid green and dashed green curves are the I0 and pR photoproducts, respectively. The solid blue curve is the bleach common to all transient ground states. The black curve is the pCA radical spectrum. These estimated spectra were also supported with the global fit of polarization-dependent PP data, which is published elsewhere (van Stokkum et al., 2004).
TABLE 1.
Global analysis parameters
| Excited-state intermediates
|
Ground-state intermediates
|
Photocycle states
|
|||||
|---|---|---|---|---|---|---|---|
| State | ESI1 | ESI2 | ESI3 | GSI1 | GSI2* | I0 | pR |
| Relative ratio | 50% | 40% | 10% | — | — | — | — |
| Lifetime | 600 ± 100 fs | 2.8 ± 0.3 ps | 40 ± 10 ps | 3.6 (1.2)* ± 0.5 ps | 4.0 ± 0.5 ps | 1.5 ± 0.5 ns | ∞† |
| Inhomogeneous branching yields (%)‡ | 50 (GSB) | 80 (GSI) | 99 (GSI) | — | — | 50 (pR) | — |
| 10 (GSI) | 20 (Io) | 1 (Io) | 50 (GSB) ± 10% | ||||
| 40 (Io) | |||||||
| Homogeneous branching yields (%)‡ | 25 (GSB) | 60 (GSI) | 95 (GSI) | 100 (GSB) | 100 (into GSB) | 50 (pR) | — |
| 6 (GSI) | 30 (Io) | 5 (Io) | 100* (GSI2) | 50 (GSB) ± 10% | |||
| 22 (Io) | 10 (ESI3) | ||||||
| 47 (ESI2) | |||||||
Decay lifetimes and branching yields of transient states extracted from the global analysis of the measured data. Relative errors of the branching yields are estimated at <10%. Key: GSB, ground-state bleach; GSI, ground-state intermediate; ESI, excited-state intermediate.
A bi-exponential ground-state decay is observed only in 1 ps and later dump data.
Infinite with respect to the timescale of the experiment (∼5 ns).
Relative branching yields of the specific state into other states in parentheses.
The spectral and temporal properties of the SE and ESA bands directly probe the excited-state dynamics. We observed three timescales with 600-fs, 2.8-ps, and 40-ps lifetimes and ascribed each decay time with a specific excited-state intermediate (ESI)—ESI1, ESI2, and ESI3, respectively. Similar timescales were observed in other excited-state quenching measurements (Changenet et al., 1998; Mataga et al., 2000; Vengris et al., 2004b). No clear time-dependent spectral shifting is observed in the emission bands of these ESI states and we constrain the fit, making all three ESI states have the same spectral features (Fig. 9, red curve). Similar nonshifting multiexponential quenching dynamics have been observed in ultrafast fluorescence upconversion measurements, however, with a small narrowing of the fluorescence on an ∼100-fs timescale (Mataga et al., 2003; Vengris et al., 2004b). This fast spectroscopic change was not resolvable in the PP and PDP data presented here, due to the overlapping bleach and electron bands. Multiexponential decay kinetics are common in systems that are typically inhomogeneous, like polymers and proteins, hence these three excited-state intermediates are not different excited states (i.e., extracted from solving the Schrödinger equation), but are used to refer to the different kinetics involved.
The kinetic models in Fig. 8 differ primarily in the respective connectivity for the excited-state evolution and which state(s) is initially excited by the applied laser pulse (thick black lines). Whereas the inhomogeneous model ascribes the multiexponential excited-state behavior to a superposition of multiple subpopulations with differing decay times, the homogeneous model ascribes the multiexponential decay to evolution along the excited-state potential energy surface. Since the excited-state lifetimes measured in previous PP and time-resolved fluorescence signals of isolated PYP chromophores in solution exhibit predominantly single-exponential decays (Changenet-Barret et al., 2002, 2001; Larsen et al., 2003b; Vengris et al., 2004b), the observed multiexponential dynamics of PYP is a direct consequence of the protein environment. This is suggestive (at least partially) that an inhomogeneous model is involved in the protein. Furthermore, high-level ab initio calculations of the potential energy surfaces for the PYP chromophore in vacuum versus the protein environment suggest that the nearby amino acids manipulate the nature of the excited-state evolution and isomerization yield (Groenhof et al., 2004). In particular, the presence of the arginine 52-amino-acid residue, which is located just above the PYP chromophore in the protein pocket (Genick et al., 1998), has been identified as the major factor in modifying the potential energy surface (Groenhof et al., 2002a,b). Solution-phase NMR measurements and computational molecular dynamics simulations have identified that this residue can adopt two conformations in the ground state of the protein: either parallel or perpendicular with respect to the embedded PYP chromophore (Dux et al., 1998; Groenhof et al., 2002b). Consequently, the inhomogeneity in the chromophore pocket, in addition to chromophore conformational disorder, may result in pronounced multiexponential quenching dynamics. The inhomogeneous model is further supported by the fact that the absorption spectrum of PYP reconstituted with the isomerization-locked chromophore is narrower than the absorption spectrum of wild-type PYP (Cordfunke et al., 1998; Larsen et al., 2004). The inhomogeneity in the twisting degree of freedom of the native chromophore may also explain (at least partially) the observed broadening of the measured PYP absorption spectrum. Also, fluorescence excitation spectra on the low-temperature PYP samples suggest such an inhomogeneous model may be involved (Hoff et al., 1992).
The PDP data favor the inhomogeneous over the homogeneous model since each ESI does not have the identical yield in initiating the PYP photocycle (Table 1). The PDP technique can directly probe the reaction yield of each ESI by dumping the ESI at different times and then probing the magnitude of the change of the I0 photoproduct absorption (Fig. 4). This is similar to the previous use of the PDP techniques to characterize the (non)changing emitting dipole moment in the bacteriorhodopsin photoreceptor (Ruhman et al., 2002); however, because the I0 spectrum strongly overlaps with the SE band, this observation is not clear from investigating the signals directly. However, the action trace signals in Fig. 4 in combination with the global analysis of the kinetic trace PDP signals, support the observation that ESI1 has the greatest reaction yield (∼40%), followed by ESI2 (20%) and then ESI3 with a near-negligible yield (∼1%) in initiating the photocycle. Whereas both the inhomogeneous and homogeneous kinetic models account for this observation (Table 1), their respective interpretations differ.
A homogenous model would require that the excited-state population would have a time-dependent yield and evolve across a convoluted potential energy surface with quenching pathway(s) that compete with photocycle generation; however, such a model is not supported by recent ab initio calculations (Groenhof et al., 2004). The inhomogeneous model, in contrast, is simpler to interpret where each ESI and their respective probability of initiating the photocycle is directly connected within the context of an inhomogeneous ground-state distribution. Assuming the photoproduct yield is then dependent on the protein-chromophore conformation, or orientations of the residues surrounding the chromophore, the specific subensembles that are less likely to initiate the photocycle will result in the longer excited-state lifetime than the ones that initiate the cycle promptly. The observed long-living 40-ps excited-state lifetime component that was also observed in time-resolved fluorescence measurements (Changenet et al., 1998; Vengris et al., 2004), which exhibits a near-zero photoproduct yield (Table 1), would consequentially result from a very unfavorable initial conformation of the protein-chromophore system and the protein pocket would act essentially as a viscous medium to hinder quenching. Recent mixed quantum-classical molecular dynamics simulations on the complete PYP protein emphasize a similar dependence of the dynamics and the context of the amino acids surrounding the chromophore, specifically the Arg-52 residue (Groenhof et al., 2004, 2002b).
The evolution of the photoproducts from I0 to pR (Fig. 9, solid and dashed green curves, respectively) occurs on a 1.5-ns timescale, and is in agreement with previous studies (Imamoto et al., 2001; Ujj et al., 1998). The estimated yield from I0 to generate pR appears not to be unity, but is ∼50 ± 10%. The origin of this loss is unknown and can be partially observed in the recovery dynamics in the bleach (445 nm) signal in Fig. 3; however, part of this rise is also ascribed to the rise of the blue-shifted pR spectrum, hence knowledge of the underlying spectra is required to extract these overlapping dynamics. We model this lost population as a pathway through the GSI. However, this is not well resolved since a small population is generated by the slow nanosecond evolution and a different ground-state recovery pathway, separated from the GSI, may be involved. A more descriptive investigation of these dynamics, including comparing global analysis results of these data with differing yields can be found elsewhere (van Stokkum et al., 2004). It should be noted that recent time-resolved IR measurements do not suggest that this I0 → pR loss is as significant as our visible PDP results suggest here (Groot et al., 2003) and further studies are planned to explore this issue. An intermediate photoproduct state, 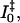 has been proposed to exist between the I0 and pR photocycle states (Devanathan et al., 1999; Ujj et al., 1998; Zhou et al., 2001). However, we cannot resolve an intermediate state on these timescales in either our PP and PDP data, and consequently do not introduce this state into our model. Alternatively, we suggest that the unaccounted long-living 40 ps and its corresponding SE band state lifetime may partially explain the previous observation of
has been proposed to exist between the I0 and pR photocycle states (Devanathan et al., 1999; Ujj et al., 1998; Zhou et al., 2001). However, we cannot resolve an intermediate state on these timescales in either our PP and PDP data, and consequently do not introduce this state into our model. Alternatively, we suggest that the unaccounted long-living 40 ps and its corresponding SE band state lifetime may partially explain the previous observation of 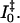
With the PDP spectroscopy, we identify a GSI through which failed attempts at initiating the photocycle evolve. For the isolated PYP chromophore in solution, a similar GSI was observed with near-identical spectral properties to the GSI observed in the PYP protein, although it decayed on a threefold-faster timescale (Table 1; see also Larsen et al., 2004). The GSI identified in isolated chromophores was ascribed to the ground-state recovery of twisted chromophores back into their equilibrated geometry after the internal conversion of excited twisted molecules. Other mechanisms, including vibrational cooling and ground-state solvation, were excluded as main contributors to this observation and similar arguments are applicable for the PYP protein case. Both the excited- and ground-state solvation dynamics for chromophores embedded in proteins have been shown to evolve quickly (∼200 fs), which is considerably faster than the 4-ps decay observed for the GSI here. The PDP traces in Fig. 3 show that the dumping of the excited state increases the population of the GSI band at 490 nm, even though ∼75% of the initial excitation energy is removed by the stimulated emission photons (395-nm pump versus 505-nm dump). This would result in less vibrational energy introduced into the nascent ground-state species and hence reduced GSI amplitudes for the PDP than the PP signals. However, both data sets can be simultaneously fit with the model using the same spectra for the GSI, irrespective of the mechanism of generation (i.e., natural quenching or dumping with a laser pulse) suggesting that the GSI is not a vibrational hot state of the original structurally equilibrated ground state.
The nascent ground-state population generated by the dump pulse is produced directly into the GSI state (blue curve, Fig. 9), which subsequently evolves into the equilibrated ground state, refilling the bleach, on a 3.5-ps timescale when dumped early at 500 fs, and with 1-ps and 4-ps timescales when dumped at later times. The structural basis of this intermediate is not known and may originate from a competing structural rearrangement motion (e.g., rotation around one of the σ-bonds, instead of the double bond) or from a local minimum in the ground-state potential energy surface of the isomerization coordinate. However, our results show clearly that this intermediate evolves directly into the equilibrated ground state, and does not evolve into the photoproducts; hence population of this intermediate state results from unsuccessful attempts at initiating the photocycle.
It should be noted that even though the global analysis that was used to analyze the PP and PDP data was able to estimate the radical spectrum (Fig. 9) from the overlapping photocycle signals, the separation was not as successful for the ejected electron. Our analysis was not able to separate the dynamics of the electron in the red from the I0→ pR evolution. It appears that the electron spectrum does decay, whereas no evolution was observed for the radical, suggesting a secondary recombination reaction is occurring before the final recovery between electron and radical.
CONCLUSIONS
We have shown that the application of appropriately tuned (395 nm and 505 nm) and timed laser pulses can manipulate the PYP reaction by “short-circuiting” the photoinduced dynamics and help interpret the underlying dynamics of overlapping spectra. The observed photo-dynamics of PYP, after excitation with 395-nm ultrafast laser pulses, can be separated into a one-photon photocycle initiation plus a stepwise sequential two-photon ionization process, which both result in spectrally and temporally overlapping signals. The photocycle yield is a time-dependent property that decreases as the excited state evolves. Failed attempts at initiating the photocycle evolve through a ground-state intermediate that decays to the ground state on a timescale longer than the principle excited-state lifetimes. The power-dependent PP signals show that the ionization pathway results from exciting the PYP complex with an ultrafast laser wavelength that overlaps with the ESA. Decreasing the pump intensity does decrease this contribution to the signal; however, because this reaction proceeds via a resonantly enhanced mechanism, a power intensity cannot be identified that simultaneously removes 100% of the ionization pathway and maintains an acceptable signal/noise level.
The multiexponential kinetics prompts an important question: Is the observed multiexponential quenching the result of inherent chromophore isomerization dynamics in initiating the photocycle (i.e., a sequential connectivity model) or the consequence of structural inhomogeneity that leads to a distribution of excited-state lifetimes (i.e., a parallel connectivity model)? Although both the inhomogeneous and homogeneous models equally fit the measured PP data, our PDP results favor the inhomogeneous model because of the observed time-dependent photocycle initiation yields. An alternative dynamical experiment can be suggested where a second laser pulse with a wavelength centered at the ground-state intermediate band (∼485 nm) can re-excite unsuccessful photocycle initiation attempts and thus investigate whether the failed photocycle initiation sample can initiate the photocycle if given a “second chance,” or must always fail, due to the nature of the specific conformation. Experiments are currently in progress to explore this aspect of incoherent control in the PYP photocycle.
Acknowledgments
This research was supported by the Netherlands Organization for Scientific Research via the Dutch Foundation for Earth and Life Sciences. D.S.L. is grateful to the Human Frontier Science Program Organization for providing financial support with a long-term fellowship. M.V. received financial support from the Stichting voor Fundamenteel Onderzoek der Materie, Netherlands.
Delmar S. Larsen's present address is Dept. of Chemistry, University of Southern California, Los Angeles, CA 90089.
Abbreviations used: PYP, photoactive yellow protein; pCA, p-coumaric acid; PP, pump-probe; PDP, pump-dump probe; ΔΔOD, double-difference optical density.
References
- Baca, M., G. E. Borgstahl, M. Boissinot, P. M. Burke, D. R. Williams, K. A. Slater, and E. D. Getzoff. 1994. Complete chemical structure of photoactive yellow protein: novel thioester-linked 4-hydroxycinnamyl chromophore and photocycle chemistry. Biochemistry. 33:14369–14377. [DOI] [PubMed] [Google Scholar]
- Baltuška, A., I. H. M. van Stokkum, A. Kroon, R. Monshouwer, K. J. Hellingwerf, and R. van Grondelle. 1997. The primary events in the photoactivation of yellow protein. Chem. Phys. Lett. 270:263–266. [Google Scholar]
- Brudler, R., R. Rammelsberg, T. T. Woo, E. D. Getzoff, and K. Gerwert. 2001. Structure of the I1 early intermediate of photoactive yellow protein by FTIR spectroscopy. Nat. Struct. Biol. 8:265–270. [DOI] [PubMed] [Google Scholar]
- Changenet, P., H. Zhang, M. J. van der Meer, K. J. Hellingwerf, and M. Glasbeek. 1998. Subpicosecond fluorescence upconversion measurements of primary events in yellow proteins. Chem. Phys. Lett. 282:276–282. [Google Scholar]
- Changenet-Barret, P., C. Choma, E. Gooding, W. DeGrado, and R. M. Hochstrasser. 2000. Ultrafast dielectric response of proteins from dynamics stokes shifting of coumarin in calmodulin. J. Phys. Chem. B. 104:9322–9329. [Google Scholar]
- Changenet-Barret, P., A. Espagne, N. Katsonis, S. Charier, J.-B. Baudin, L. Jullien, P. Plaza, and M. M. Martin. 2002. Excited-state relaxation dynamics of a PYP chromophore model in solution: influence of the thioester group. Chem. Phys. Lett. 365:285–291. [Google Scholar]
- Changenet-Barret, P., P. Plaza, and M. M. Martin. 2001. Primary events in the photoactive yellow protein chromophore in solution. Chem. Phys. Lett. 336:439–444. [Google Scholar]
- Chosrowjan, H., N. Mataga, N. Nakashima, Y. Imamoto, and F. Tokunaga. 1997. Femtosecond-picosecond fluorescence studies on excited state dynamics of photoactive yellow protein from Ectothiorhodospira halophila. Chem. Phys. Lett. 270:267–272. [Google Scholar]
- Cordfunke, R., R. Kort, A. Pierik, B. Gobets, G. J. Koomen, J. W. Verhoeven, and K. J. Hellingwerf. 1998. Trans/cis (Z/E) photoisomerization of the chromophore of photoactive yellow protein is not a prerequisite for the initiation of the photocycle of this photoreceptor protein. Proc. Natl. Acad. Sci. USA. 95:7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich, M. A., and T. E. Meyer. 2003. Photoactive yellow protein: a prototypic PAS domain sensory protein and development of a common signaling mechanism. Biochemistry. 42:4759–4770. [DOI] [PubMed] [Google Scholar]
- Devanathan, S., U. K. Genick, I. L. Canestrelli, T. E. Meyer, M. A. Cusanovich, E. D. Getzoff, and G. Tollin. 1998. New insights into the photocycle of Ectothiorhodospira halophila photoactive yellow protein: photorecovery of the long-lived photobleached intermediate in the Met100Ala mutant. Biochemistry. 37:11563–11568. [DOI] [PubMed] [Google Scholar]
- Devanathan, S., S. Lin, M. A. Cusanovich, N. Woodbury, and G. Tollin. 2001. Early photocycle kinetic behavior of the E46A and the Y42F mutants of photoactive yellow protein. Femtosec. Spectrosc. Biophys. J. 81:2314–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanathan, S., A. Pacheco, L. Ujj, M. Cusanovich, G. Tollin, S. Lin, and N. Woodbury. 1999. Femtosecond spectroscopic observations of initial intermediates in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys. J. 77:1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dux, P., G. Rubinstenn, G. W. Vuister, R. Boelens, F. A. Mulder, K. Hard, W. D. Hoff, A. R. Kroon, W. Crielaard, K. J. Hellingwerf, and R. Kaptein. 1998. Solution structure and backbone dynamics of the photoactive yellow protein. Biochemistry. 37:12689–12699. [DOI] [PubMed] [Google Scholar]
- Gai, F., J. C. McDonald, and P. Anfinrud. 1997. Pump-dump-probe spectroscopy of bacteriorhodopsin: evidence for a near-IR excited state absorbance. J. Am. Chem. Soc. 119:6201–6202. [Google Scholar]
- Genick, U. K., G. E. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Srajer, T. Y. Teng, W. Schildkamp, D. E. McRee, K. Moffat, and E. D. Getzoff. 1997. Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science. 275:1471–1475. [DOI] [PubMed] [Google Scholar]
- Genick, U. K., S. M. Soltis, P. Kuhn, I. L. Canestrelli, and E. D. Getzoff. 1998. Structure at 0.85Å resolution of an early protein photocycle intermediate. Nature. 392:206–209. [DOI] [PubMed] [Google Scholar]
- Gensch, T., C. C. Gradinaru, I. H. M. van Stokkum, J. Hendriks, K. Hellingwerf, and R. van Grondelle. 2002. The primary photoreaction of photoactive yellow protein (PYP): anisotropy changes and excitation wavelength dependence. Chem. Phys. Lett. 356:347–354. [Google Scholar]
- Groenhof, G., M. Bouxin-Cademartory, B. Hess, S. de Visser, H. Brendendsen, M. Olivucci, A. E. Mark, and M. A. Robb. 2004. Why photon absorption triggers isomerization of the chromophore in photoactive yellow protein. J. Am. Chem. Soc. 126:4228–4233. [DOI] [PubMed] [Google Scholar]
- Groenhof, G., M. F. Lensink, H. J. C. Berendsen, and A. E. Mark. 2002a. Signal transduction in the photoactive yellow protein. II. Proton transfer initiates conformational changes. Proteins Struct. Funct. Gen. 48:212–219. [DOI] [PubMed] [Google Scholar]
- Groenhof, G., M. F. Lensink, H. J. C. Berendsen, J. G. Snijders, and A. E. Mark. 2002b. Signal transduction in the photoactive yellow protein. I. Photon absorption and the isomerization of the chromophore. Proteins Struct. Funct. Gen. 48:202–211. [DOI] [PubMed] [Google Scholar]
- Groot, M. L., L. van Wilderen, D. S. Larsen, M. A. van der Horst, I. H. M. van Stokkum, K. J. Hellingwerf, and R. van Grondelle. 2003. Initial steps of signal generation in photoactive yellow protein revealed with femtosecond mid-infrared spectroscopy. Biochemistry. 42:10054–10059. [DOI] [PubMed] [Google Scholar]
- Hellingwerf, K. J. 2000. Key issues in the photochemistry and signalling-state formation of photosensor proteins. J. Photochem. Photobiol. B Biol. 54:94–102. [DOI] [PubMed] [Google Scholar]
- Hellingwerf, K. J., J. Hendriks, and T. Gensch. 2003. Photoactive yellow protein, a new type of photoreceptor protein: will this “yellow lab” bring us where we want to go? J. Phys. Chem. A. 107:1082–1094. [Google Scholar]
- Hendriks, J., T. Gensch, L. Hviid, M. A. van der Horst, K. J. Hellingwerf, and J. J. van Thor. 2002. Transient exposure of hydrophobic surface in the photoactive yellow protein monitored with Nile Red. Biophys. J. 82:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, W. D., S. L. S. Kwa, R. van Grondelle, and K. J. Hellingwerf. 1992. Low temperature absorbance and fluorescence spectroscopy of the photoactive yellow protein from Ectothiorhodospira halophila. Photochem. Photobiol. 56:529–539. [Google Scholar]
- Hoff, W. D., I. H. M. van Stokkum, H. J. van Ramesdonk, M. E. van Brederode, A. M. Brouwer, J. C. Fitch, T. E. Meyer, R. van Grondelle, and K. J. Hellingwerf. 1994. Measurement and global analysis of the absorbance changes in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys. J. 67:1691–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzwarth, A. R. 1996. Data analysis in time-resolved measurements. In Biophysical Techniques in Photosynthesis. J. Amesz and A. J. Hoff, editors. Kluwer, Dordrecht, The Netherlands.
- Imamoto, Y., M. Kataoka, F. Tokunaga, T. Asahi, and H. Masuhara. 2001. Primary photoreaction of photoactive yellow protein studied by subpicosecond-nanosecond spectroscopy. Biochemistry. 40:6047–6052. [DOI] [PubMed] [Google Scholar]
- Ishikawa, H., C. Nagao, N. Mikami, and R. Field. 1997. Observation of the “isomerization states” of HCP by stimulated emission pumping spectroscopy: comparison between theory and experiment. J. Chem. Phys. 106:2980–2983. [Google Scholar]
- Jou, F.-Y., and G. R. Freeman. 1979. Temperature and isotope effects on the shape of the optical absorption spectrum of solvated electrons in water. J. Phys. Chem. 83:2383–2387. [Google Scholar]
- Ko, C., B. Levine, A. Toniolo, L. Monohar, S. Olsen, H.-J. Werner, and T. J. Martínez. 2003. Ab initio excited-state dynamics of the photoactive yellow protein chromophore. J. Am. Chem. Soc. 125:12710–12711. [DOI] [PubMed] [Google Scholar]
- Kort, R., W. D. Hoff, M. Van West, A. R. Kroon, S. M. Hoffer, K. H. Vlieg, W. Crielaand, J. J. Van Beeumen, and K. J. Hellingwerf. 1996. The xanthopsins: a new family of eubacterial blue-light photoreceptors. EMBO J. 15:3209–3218. [PMC free article] [PubMed] [Google Scholar]
- Krueger, B. P., S. S. Lampoura, I. H. M. van Stokkum, E. Papagiannakis, J. M. Salverda, C. C. Gradinaru, D. Rutkauskas, R. G. Hiller, and R. van Grondelle. 2001. Energy transfer in the peridinin chlorophyll-a protein of Amphidinium carterae studied by polarized transient absorption and target analysis. Biophys. J. 80:2843–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachish, U., A. Shafferman, and G. Stein. 1975. Intensity dependence in laser flash photolysis experiments: hydrated electron formation form ferrocyanide, tyrosine, and tryptophan. J. Chem. Phys. 64:4205–4211. [Google Scholar]
- Lapouyade, R., K. Czeschka, W. Majenz, W. Rettig, E. Gilabert, and C. Rulliere. 1992. Photophysics of donor-acceptor substituted stilbenes—a time-resolved fluorescence study using selectively bridged dimethylamino cyanomodel compounds. J. Phys. Chem. 96:9643–9650. [Google Scholar]
- Larsen, D. S., E. Papagiannakis, I. H. M. van Stokkum, M. Vengris, J. T. M. Kennis, and R. van Grondelle. 2003a. Excited state dynamics of beta-carotene explored with dispersed multi-pulse transient absorption. Chem. Phys. Lett. 381:733–742. [Google Scholar]
- Larsen, D. S., M. Vengris, I. H. M. van Stokkum, M. van der Horst, R. Cordfunke, K. J. Hellingwerf, and R. van Grondelle. 2003b. Initial photoinduced dynamics of the photoactive yellow protein chromophore in solution. Chem. Phys. Lett. 369:563–569. [Google Scholar]
- Larsen, D. S., M. Vengris, I. H. M. van Stokkum, M. van der Horst, F. L. de Weerd, K. J. Hellingwerf, and R. van Grondelle. 2004. Photoisomerization and photoionization of the photoactive yellow protein chromophore in solution. Biophys. J. 86:2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logunov, S. L., V. V. Volkov, M. Braun, and M. A. El-Sayed. 2001. The relaxation dynamics of the excited electronic states of retinal in bacteriorhodopsin by two-pump probe femtosecond studies. Proc. Natl. Acad. Sci. USA. 98:8475–8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataga, N., H. Chosrowjan, Y. Shibata, Y. Imamoto, F. Tokunaga, and F. Tanaka. 2000. Femtosecond fluorescence studies on ultrafast reaction dynamics of photoactive proteins. J. Luminesc. 98–89:821–823. [Google Scholar]
- Mataga, N., H. Chosrowjan, S. Taniguchi, N. Hamada, F. Tokunaga, Y. Imamoto, and M. Kataoka. 2003. Ultrafast photoreactions in protein nanospaces as revealed by femtosecond fluorescence dynamics measurements on photoactive yellow protein and related systems. Phys. Chem. Chem. Phys. 5:2454–2460. [Google Scholar]
- Meyer, T. E., E. Yakali, M. A. Cusanovich, and G. Tollin. 1987. Properties of a water-soluble, yellow protein isolated from a halophilic phototrophic bacterium that has photochemical activity analogous to sensory rhodopsin. Biochemistry. 26:418–423. [DOI] [PubMed] [Google Scholar]
- Migus, A., Y. Gauduel, L. J. Martin, and A. Antonetti. 1987. Excess electrons in liquid water: first evidence of a prehydrated state with femtosecond lifetime. Phys. Rev. Lett. 58:1559–1562. [DOI] [PubMed] [Google Scholar]
- Molina, V., and M. Merchan. 2001. On the absorbance changes in the photocycle of the photoactive yellow protein: a quantum-chemical analysis. Proc. Natl. Acad. Sci. USA. 98:4299–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perman, B., V. Srajer, Z. Ren, T. Teng, C. Pradervand, T. Ursby, D. Bourgeois, F. Schotte, M. Wulff, R. Kort, K. J. Hellingwerf, and K. Moffat. 1998. Energy transduction on the nanosecond time scale: early structural events in a xanthopsin photocycle. Science. 279:1946–1950. [DOI] [PubMed] [Google Scholar]
- Pique, J., Y. Engel, R. Levine, Y. Chen, R. Field, and J. Kinsery. 1988. Broad spectral features in the stimulated-emission pumping spectrum of acetylene. J. Chem. Phys. 88:5972–5974. [Google Scholar]
- Premvardhan, L. L., F. Buda, M. A. van der Horst, D. Luehrs, K. J. Hellingwerf, and R. van Grondelle. 2004. The impact of photon absorption on the electronic properties of p-coumaric acid derivatives of the photoactive yellow protein chromophore. J. Phys. Chem. B. 108:5138–5148. [Google Scholar]
- Premvardhan, L. L., M. A. van der Horst, K. J. Hellingwerf, and R. van Grondelle. 2003. Stark spectroscopy on photoactive yellow protein, E46Q, and a nonisomerizing derivative, probes photoinduced charge motion. Biophys. J. 84:3226–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Z., B. Perman, V. Srajer, T.-V. Teng, C. Pradervand, D. Bourgeois, F. Schotte, T. Ursby, R. Kort, M. Wulff, and K. Moffat. 2001. A molecular movie at 1.8Å resolution displays the photocycle of photoactive yellow protein, a eubacterial blue-light receptor, from nanoseconds to seconds. Biochemistry. 40:13788–13801. [DOI] [PubMed] [Google Scholar]
- Rice, S. A., and M. Zhao. 2000. Optical Control of Molecular Dynamics. Wiley Interscience, New York.
- Rubinstenn, G., G. W. Vuister, F. A. Mulder, P. E. Dux, R. Boelens, K. J. Hellingwerf, and R. Kaptein. 1998. Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat. Struct. Biol. 5:568–570. [DOI] [PubMed] [Google Scholar]
- Ruhman, S., B. Hou, N. Freidman, M. Ottolenghi, and M. Sheves. 2002. Following evolution of bacteriorhodopsin in its reactive excited state via stimulated emission pumping. J. Am. Chem. Soc. 124:8854–8858. [DOI] [PubMed] [Google Scholar]
- Ryan, W., D. J. Gordon, and D. H. Levy. 2002. Gas phase photochemistry of the photoactive yellow protein chromophore trans-p-coumaric acid. J. Am. Chem. Soc. 124:6194–6201. [DOI] [PubMed] [Google Scholar]
- Sprenger, W. W., W. D. Hoff, J. P. Armitage, and K. J. Hellingwerf. 1993. The eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J. Bacteriol. 175:3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujj, L., S. Devanathan, T. E. Meyer, M. A. Cusanovich, G. Tollin, and G. H. Atkinson. 1998. New photocycle intermediates in the photoactive yellow protein from Ectothiorhodospira halophila: picosecond transient absorption spectroscopy. Biophys. J. 75:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno, M., M. Kumauchi, J. Sasaki, F. Tokunaga, and S. Yamauchi. 2002. Resonance Raman spectroscopy and quantum chemical calculations reveal structural changes in the active site of photoactive yellow protein. Biochemistry. 41:5668–5674. [DOI] [PubMed] [Google Scholar]
- Unno, M., M. Kumauchi, J. Sasaki, F. Tokunaga, and S. Yamauchi. 2003. Assignment of resonance Raman spectrum of photoactive yellow protein in its long-lived blue-shifted intermediate. J. Phys. Chem. B. 107:2837–2845. [Google Scholar]
- van Beeumen, J. J., B. V. Devreese, S. M. van Bun, W. D. Hoff, K. J. Hellingwerf, T. E. Meyer, D. E. McRee, and M. A. Cusanovich. 1993. Primary structure of a photoactive yellow protein from the phototrophic bacterium Ectothiorhodospira halophila, with evidence for the mass and the binding site of the chromophore. Protein Sci. 2:1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Brederode, M. E., W. D. Hoff, I. H. M. van Stokkum, M. L. Groot, and K. J. Hellingwerf. 1996. Protein folding thermodynamics applied to the photocycle of the photoactive yellow protein. Biophys. J. 71:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Stokkum, I. H. M., D. S. Larsen, and R. van Grondelle. 2004. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta. 1657:82–104. [DOI] [PubMed] [Google Scholar]
- van Thor, J. J., T. Gensch, K. J. Hellingwerf, and L. N. Johnson. 2002. Phototransformation of green fluorescent protein with UV and visible light leads to decarboxylation of glutamate 222. Nat. Struct. Biol. 9:37–41. [DOI] [PubMed] [Google Scholar]
- Vengris, M., M. van der Horst, G. Zgrablić, I. H. M. van Stokkum, S. Haacke, M. Chergui, K. J. Hellingwerf, R. van Grondelle, and D. S. Larsen. 2004b. Contrasting the excited-state dynamics of the photoactive yellow protein chromophore: protein vs. solvent environments. Biophys. J. 87:1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengris, M., I. H. M. van Stokkum, X. He, A. Bell, P. J. Tonge, R. van Grondelle, and D. S. Larsen. 2004a. Ultrafast excited and ground-state dynamics of the green fluorescent protein chromophore in solution. J. Phys. Chem. B. 108:4587–4598. [Google Scholar]
- Vilchiz, V. H., J. A. Kloepfer, A. C. Germaine, V. A. Lenchenkov, and S. E. Bradforth. 2001. Map for the relaxation dynamics of hot photoelectrons injected into liquid water via anion threshold photodetachment and above threshold solvent ionization. J. Phys. Chem. A. 105:1711–1723. [Google Scholar]
- Xie, A., W. D. Hoff, A. R. Kroon, and K. J. Hellingwerf. 1996. Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein. Biochemistry. 35:14671–14678. [DOI] [PubMed] [Google Scholar]
- Xie, A., L. Kelemen, J. Hendriks, B. J. White, K. J. Hellingwerf, and W. D. Hoff. 2001. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 40:1510–1517. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., L. Ujj, T. E. Meyer, M. A. Cusanovich, and G. H. Atkinson. 2001. Photocycle dynamics and vibrational spectroscopy of the E46Q mutant of photoactive yellow protein. J. Phys. Chem. A. 105:5719–5726. [Google Scholar]