

Abstract
The complex between calmodulin and the calmodulin-binding portion of smMLCKp has been studied. Electrostatic interactions have been anticipated to be important in this system where a strongly negative protein binds a peptide with high positive charge. Electrostatic interactions were probed by varying the pH in the range from 4 to 11 and by charge deletions in CaM and smMLCKp. The change in net charge of CaM from ∼−5 at pH 4.5 to −15 at pH 7.5 leaves the binding constant virtually unchanged. The affinity was also unaffected by mutations in CaM and charge substitutions in the peptide. The insensitivity of the binding constant to pH may seem surprising, but it is a consequence of the high charge on both protein and peptide. At low pH it is further attenuated by a charge regulation mechanism. That is, the protein releases a number of protons when binding the positively charged peptide. We speculate that the role of electrostatic interactions is to discriminate against unbound proteins rather than to increase the affinity for any particular target protein.
INTRODUCTION
Electrostatic interactions are one major determinant of the stability and function of charged macromolecules in aqueous solutions. This is true for technical formulations, naturally occurring suspensions, as well as in biological systems. Electrostatic interactions modulate the binding of metal ions, protons, small charged substrates, and other macromolecules to proteins (Getzoff et al., 1983; Kesvatera et al., 1994; Linse et al., 1991). Given the large variation in protein net charge and charge distribution, the electrostatic contribution to a protein-protein interaction will vary considerably from case to case and it is important to understand the basis for this variation. One type of protein-protein interaction for which a significant electrostatic contribution to the binding free energy has been suggested is CaM-target recognition (Ikura et al., 1992; Meador et al., 1992). In this system, the strongly negatively charged calmodulin commonly binds to an amphiphatic α-helical segment that is positively charged and overlaps with an auto-inhibitory domain of a target enzyme (Andersson and Malencik, 1986; Crivici and Ikura, 1995).
Calmodulin is a ubiquitous eukaryotic Ca2+ sensor that transfers Ca2+ signals to an impressively large number of proteins (Zhu et al., 2001) including protein kinases, ion channels (Gu and Cooper, 1999; Lee et al., 1999) and IP3 receptors (Missiaen et al., 1999). The molecular basis for the high affinity for all these different proteins in combination with the discrimination against the nonrecognized proteins is an intriguing issue. CaM has a dumbbell shape with the two domains separated by a central helix (Chattopadhyaya et al., 1992). In solution, this helix is disrupted in the center with considerable mobility around residues 78–81, forming a flexible tether that allows the two domains to adjust their relative orientation and come together in an optimal fashion for the binding of target proteins (Chou et al., 2001; Persechini and Kretsinger, 1988). The calmodulin binding regions of most target enzymes contain a considerable fraction of basic and hydrophobic residues. These segments can be produced as linear peptides that bind to calmodulin in an α-helical form with maintained affinity. Peptides derived from myosin light chain kinases from both smooth and skeletal muscle (smMLCK and skMLCK, respectively) are bound in a channel between the two domains of calmodulin (Ikura et al., 1992; Meador et al., 1992), sometimes referred to as the wrap-around mode of binding (Fig. 1). The two conserved hydrophobic residues of each MLCK-peptide (Trp and Leu in smMLCKp, Trp and Phe in skMLCKp) are found in hydrophobic pockets in the channel (Crivici and Ikura, 1995). It has been convincingly shown that a peptide derived from skMLCK induces the same structure in CaM as the full protein (Kranz et al., 2002). In recent years, it has become clear that the wrap-around mode of binding the MLCK peptides is only one of many possible ways in which CaM interacts with its targets (Hoeflich and Ikura, 2002).
FIGURE 1.

Structure of calmodulin in complex with smMLCKp. Calmodulin has two globular domains with two Ca2+ binding EF-hands each. The position of substituted residues in CaM are shown in red and the charged residues in the peptide in blue. The figure was prepared using the program MOLMOL (Koradi et al., 1996) from the x-ray coordinates (Meador et al., 1992).
Significant electrostatic contributions to the binding of targets to calmodulin have been suggested. However, the electrostatic component is poorly understood as experimental data are scarce and seemingly contradictory. Data on CaM binding of MLCK peptides suggest that the enthalpy of binding does not change between 0 and 100 mM NaCl (Wintrode and Privalov, 1997), that any one residue can be replaced by alanine with a resulting gain in affinity (Montigiani et al., 1996), and that in the absence of Ca2+, the affinity for the peptide is reduced at increased concentrations of salt (Tsvetkov et al., 1999).
To investigate the role of electrostatic interactions in target recognition by calmodulin, we have performed a combined theoretical and experimental study of the binding of smMLCKp to calmodulin. The charges of both the protein and peptide were modified by site-specific substitutions or by varying the pH. The affinity between protein and peptide was assessed through fluorescence measurements using a tryptophan side chain in the peptide as a reporter. Monte Carlo simulations of the binding process were performed in a semigrand canonical ensemble allowing the protein to adjust its charge according to solution condition. That is, salt concentration, protein concentration, and solution pH, as well as the binding process itself, were allowed to affect the titration status of the protein. The study was conducted at low ionic strength to avoid screening by added salt and to maintain a maximum of electrostatic interactions.
EXPERIMENTAL PROCEDURES
Chemicals
Unless otherwise stated, all solvents and reagents were purchased from commercial suppliers and used without further purification. N-terminally Fmoc-protected amino acids alanine (Fmoc-Ala·H2O), glutamine (Fmoc-Gln(Trt)), glutamic acid (Fmoc-Glu(OBut)·H2O), glycine (Fmoc-Gly), leucine (Fmoc-Leu), phenylalanine (Fmoc-Phe), arginine (Fmoc-Arg(Pbf), lysine (Fmoc-Lys(Boc), tryptophan (Fmoc-Trp(Boc), threonine (Fmoc-Thr(But), serine (Fmoc-Ser(But), histidine (Fmoc-His(Trt), isoleucine (Fmoc-Ile), and valine (Fmoc-Val) were purchased from Advanced ChemTech (Louisville, KY). The coupling reagent O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), and PAL peptide support resin (0.311 mmol/g) were purchased from PerSeptive Biosystems (Framingham, MA). Anisole, 1,2-ethanedithiol, piperidine, and thioanisole were purchased from Aldrich Chemical (St. Louis, MO). Acetic anhydride, acetonitrile, CH2Cl2, N,N-DMF, N-methylmorpholine, and TFA were purchased from Fisher Scientific (Loughborough, UK). Diethyl ether was purchased from VWR Scientific (West Chester, PA). All solvents used for high-performance liquid chromatography (HPLC) were of HPLC grade.
Protein preparation
Vertebrate CaM was expressed from a modified Pet-vector containing a synthetic gene with codons optimized for production in Escherichia coli (Waltersson et al., 1993) and purified as previously described (Waltersson et al., 1993). The CaM mutants E11Q, E83Q, E84Q, and D78N were produced from this vector using QuikChange (Stratagene, La Jolla, CA), and the forward primers GAA GAG CAG ATT GCA CAG TTC AAA GAG GCT TTT TCT CTG for E11Q, GAT ACA GAT AGC GAA CAA GAA ATT CGT GAA GCG TTC CGT GTG for E83Q, GAT ACA GAT AGC GAA GAA CAA ATT CGT GAA GCG TTC CGT GTG for E84Q, and ATG GCG CGC AAA ATG AAA AAT ACA GAT AGC GAA GAA for D87N, plus their complementary reverse primers, were used to construct the mutants. Recombinant protein was produced in E. coli and purified using the same protocol as for wild-type (wt) CaM except that the charge difference led to pooling of slightly different ion exchange fractions. Purity was confirmed by agarose gel electrophoresis, SDS-polyacrylamide gel electrophoresis, and 1H NMR. The concentration of Ca2+-loaded CaM was determined by absorbance at 280 nm using an extinction coefficient of 3200 M−1cm−1 (Klee 1977) for all mutants and confirmed by amino acid analysis after acid hydrolysis.
Peptide synthesis and purification
Peptide synthesis was carried out by standard solid phase methodology employing a Rainin Instrument (Woburn, MA) PS3 peptide synthesizer. The sequence of the wt smMLCK peptide is ARRKWQKTGHAVRAIGRLSS, which corresponds to residues 796–815 of the full-length protein. The three peptide variants contain the following single substitutions: K7Q, K7G, and K7E. A fourth variant, named or referred to as K4QK7QR17Q, contains three substitutions: K4Q + K7Q + R17Q. In the synthesis, Fmoc chemistry was used with DMF as the solvent. All peptides were made on the Wang resin, except smMLCKp-am and smMLCKp-am-ac that was made on the PAL resin to furnish a C-terminal carboxamide. The coupling steps were 45-min long employing HBTU as the coupling reagent. The following amino acids were double-coupled: Arg, Lys, Ala, Ile, Ser, and, in some cases, Gln. The N-terminus of smMLCKp-ac was acetylated with 0.5 M acetic anhydride and pyridine in DMF for 20 min. The N-terminus of each mutant was not acetylated, but kept as the free amine. The cleavage of the peptide from the resin was achieved with TFA/thioanisole/1,2-ethanedithiol/anisole (9.0:0.5:0.3:0.2) for 2 h. After evaporation of the solvent with a stream of N2, the crude peptide was precipitated with ice-cold diethyl ether, collected by filtration, dissolved in water and acetonitrile, and lyophilized. The peptides were purified by reversed phase high-performance liquid chromatography on a C4 column using a Rainin Instrument Dynamax Solvent Delivery System, equipped with a Rainin Dynamax Absorbance Detector Model UV-1, or on a C8 column using a Rainin HPXL dual solvent delivery system, equipped with a Rainin Dynamax detector model UV-D II. Analytical HPLC was carried out using a Hewlett-Packard (Palo Alto, CA) Series 1100 QuatPump equipped with a Hewlett-Packard Series 1100 Diode-Array Detector. The solvent system consisted of 0.1% TFA in H2O (A) and 0.1% TFA in 90% acetonitrile/H2O (B). Unless otherwise indicated, samples were detected at 220 nm and were prepared at 1 mg/mL for analytical runs or 10 mg/mL for preparatory runs. Volume flow rates were 1 mL/min for analytical runs or 10 mL/min for preparatory runs. The following gradients were used: smMLCKp 15–35% B, 40 min; smMLCKp-am-ac 15–30% B, 30 min; smMLCKp-am 15–30% B, 30 min; smMLCKp-am 15–30% B, 30 min; smMLCKp-ac 15–30% B, 30 min; K7Q 13–28% B, 30 min; K7E 10–30% B, 40 min; and K7G 15–30% B, 30 min.
The purity of the peptides was estimated from analytical HPLC on a reversed phase C18 column and was found to be >95% for all peptides. Electrospray ionization mass spectrometry was performed by SynPep (Dublin, CA) to verify peptide masses. Mass of peptides (expected mass, in daltons): smMLCKp 2278 (2278.6), smMLCKp-am-ac 2319 (2319.6), 7KE 2279 (2279.6), K4QK7QR17Q 2250 (2250.5), smMLCKp-ac 2319 (2320), K7G 2207 (2207.5), smMLCKp-am 2277 (2278), and K7Q 2278 (2278.6).
The peptide concentration was determined spectrophotometrically at 280 nm using an extinction coefficient of 5500 M−1cm−1 (Pace et al., 1995) for all peptides and confirmed by amino acid analysis after acid hydrolysis.
Fluorescence spectroscopy
Binding constants were measured in 5 mM buffer with 1 mM CaCl2 at pH ranging from 4 to 11. No salt was added. The peptide concentration was between 0.3 and 2 μM, and calmodulin aliquots were added from a concentrated stock solution. Fluorescence emission spectra were recorded on a PerkinElmer (Foster City, CA) Luminescence Spectrometer LS 50 B connected to a Julabo F25 thermostatic water bath set at 25°C. Emission spectra were recorded between 310 and 400 nm using an excitation wavelength of 295 nm. Data for fluorescence titrations were obtained by excitation at 295 nm and emission at 335 nm. Excitation and emission slits were set to 3–5 nm and 5–10 nm, respectively. Each titration point was obtained by integration of the signal over 30 s after 1–1.5 min of equilibration. Alternately, the measured intensity after each titration was determined by averaging the intensity at the chosen wavelength from 10 scans. The obtained binding constants are averages of at least two independent measurements. The accuracy of the binding constant depends on the strength of the binding and peptide concentration used. Here, the error in binding constants is <±0.2 log units. In some titrations the first few data points produce a slightly sigmoidal shape at the start of the binding curve. Test experiments in the presence of 100 mM NaCl yield the same binding constant values as reported in several other studies (Afshar et al., 1994; Cox et al., 1985). Concentrations of protein and peptide were always kept at low enough concentrations to yield a significant fraction of unbound molecules during titrations to allow a good estimate of the binding constant. Representative titration curves can be seen in Fig. 2.
FIGURE 2.

Fluorescence titrations of peptides binding to CaM. Experimental data for smMLCKp at pH 4.9 (▪), pH 10.0 (○), and K4QK7QR17Q at pH 7.5 (X). The titration data for smMLCKp binding at pH 10.0 include more points not shown here. The solid line represents the fitted curves using Eq. 2.
Data analysis
Data were analyzed according to a 1:1 binding model,
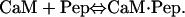 |
The concentration of free peptide after each addition can be calculated from:
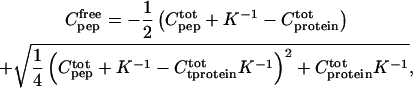 |
(1) |
where 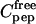 is the free peptide concentration,
is the free peptide concentration, 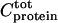 the total CaM concentration,
the total CaM concentration, 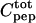 the total peptide concentration, and K the stoichiometric binding constant. The total intensity at each titration point, Icalc, is a combination of the intensity of free peptide, Ifree, and the intensity of the CaM-peptide complex, Ibound, weighted by their respective concentrations according to
the total peptide concentration, and K the stoichiometric binding constant. The total intensity at each titration point, Icalc, is a combination of the intensity of free peptide, Ifree, and the intensity of the CaM-peptide complex, Ibound, weighted by their respective concentrations according to
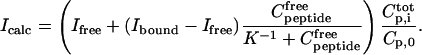 |
(2) |
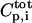 is the total peptide concentration after each addition and the ratio
is the total peptide concentration after each addition and the ratio 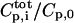 takes care of dilution effects. The binding curves are fitted directly to the experimental quantity using least-square fitting with Caligator software (Andre and Linse, 2002).All parameters were allowed to adjust in the fit (
takes care of dilution effects. The binding curves are fitted directly to the experimental quantity using least-square fitting with Caligator software (Andre and Linse, 2002).All parameters were allowed to adjust in the fit (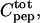 K, Ifree, Icalc).
K, Ifree, Icalc).
pH profile
In the study of the pH profile, the pH range from 4 to 11 was covered by the following buffers: sodium acetate, MES, bis-Tris, Tris, bis-Tris propane, tricine, and CAPS. Below pH 5, 3–5 mM Ca2+ was necessary to saturate CaM. Overlapping buffering areas were used to test for the absence of significant specific buffer effect on the binding. The pH readings taken before and after titration agreed within ±0.1 units.
COMPUTATIONAL DETAILS
The binding constant
The thermodynamic binding constant, KTH, for a process where a protein (P) binds a ligand (L) and forms a complex (PL) can be formally written as,
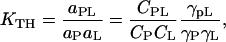 |
(3) |
where the a's, C's, and γ's are activities, concentrations, and activity factors for the molecules indicated by subscripts, respectively. In Eq. 3 we have also made use of the relation a = γC. The first ratio on the right-hand side of Eq. 3
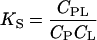 |
(4) |
is the stoichiometric binding constant, which is the quantity measured in the experiments.
Thus, since KTH is a true constant, any measured change in KS reflects a change in the activity factors. The activity factor is related to the excess chemical potential,
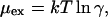 |
(5) |
which is the quantity obtained from the Monte Carlo simulations.
We will restrict ourselves to measured changes in the stoichiometric binding constant. Hence, from Eqs. 3 to 5 it follows that the ratio of two stoichiometric binding constants is given by
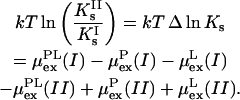 |
(6) |
The notation I and II could, for example, correspond to the binding at two different pH. Equation 6 can be made more compact by defining the excess chemical potential of the bound ligand as,
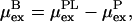 |
(7) |
which is directly accessible in the simulations and hence the shift in the binding constant becomes
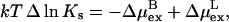 |
(8) |
with 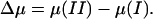 The excess chemical potential in Eq. 8 is averaged over all protonation states of the protein.
The excess chemical potential in Eq. 8 is averaged over all protonation states of the protein.
So far no approximations have been introduced and the above equations are formally exact. In the following, the focus will be on electrostatic interactions only and we will assume that the change in binding constant is solely due to electrostatics. Note that this does not mean that structural changes or molecular details of solvation upon binding are neglected, but only that they are assumed to be the same irrespective of salt concentration, pH, etc., which is a much weaker condition.
The dielectric continuum model
The aim of this study is to investigate the importance of electrostatic interactions when a small peptide binds to calmodulin. It therefore seems natural to use a dielectric continuum model for the description of the protein solution. Thus, the atomic details of the solvent (water) is assumed to be of secondary importance and the water is instead described as a structureless continuum characterized only by its bulk dielectric permittivity, ɛr = 78.3, at room temperature. However, the protein atoms and the salt particles are treated explicitly as independent particles. Negatively charged amino acids, Glu, Asp, and the C-terminus, are given a charge of −e divided equally between the two carboxylic oxygens. A positive unit charge is assigned to the appropriate nitrogen atoms of basic amino acid residues including Lys, Arg, His, and the N-terminus. The remaining protein atoms are treated as hard spheres with a radius of 2 Å—the same hard core radius is assigned to charged protein atoms and any added positive and negative salt ions. With this model, the protein has a nonuniform charge distribution and the detailed form of the protein is taken into account. The protein coordinates are taken from an x-ray determination of the complex between calmodulin containing four calcium atoms and smMCLKp (Meador et al., 1992). The dielectric properties of the protein itself are essentially unknown and previous experience with calmodulin (Svensson et al., 1993) as well as other calcium binding proteins (Juffer and Vogel, 2000; Kesvatera et al., 1994; Svensson et al., 1993) has convincingly shown that the assumption of a high dielectric response from the protein gives the best agreement with experiments. Also in other proteins, it has been advocated that the assumption of a high dielectric response from the protein gives better agreement when comparing theoretical and experimental apparent acid constants of charged amino acids (Antosiewicz et al., 1996; Juffer and Vogel, 2000; Kesvatera et al., 2001). Two-dielectric models have shown reasonable agreement if a high protein dielectric constant is used (Antosiewicz et al., 1996; Juffer and Vogel, 2000). Most charged side chains are also directly solvent exposed in proteins. The dielectric response of the protein interior has been discussed by Warshel and co-workers in several publications (Lee et al., 1992; Muegge et al., 1998; Sham et al., 1997, 1998; Warshel et al., 1984). One of their conclusions is that the protein relaxation leads to a significant reduction of charge-charge interactions and is a major component in an effective dielectric constant. This means that the effective dielectric response in the protein is much higher than in a pure hydrocarbon phase. Thus, in the calculations presented here, we have assumed a uniform dielectric constant throughout the solution and equal to the value for pure water. The interaction energy between any two particles can be formally described by
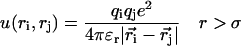 |
(9) |
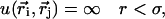 |
where e and ɛ0 are the elementary charge and electric permittivity for vacuum, respectively. Hence the total energy is a sum over all charged particles
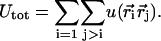 |
(10) |
Monte Carlo simulations
The electrostatic interactions described above define the Hamiltonian, which forms the basis for a Monte Carlo simulation of calmodulin in a salt solution. We use the standard Metropolis algorithm (Metropolis et al., 1953) and the protein atoms are kept fixed at the experimental x-ray coordinates, whereas counter ions and salt particles are subject to moves in the Monte Carlo (MC) algorithm. By using MC simulations to calculate the free energy of binding, we avoid the approximations inherent in the Poisson-Boltzmann approach commonly used in similar studies. In addition to the interactions described in the previous section, we have also introduced a confining sphere for the protein and the ions and whose radius defines the protein concentration. The ionization status of acidic and basic amino acid residues is in principle unknown and varies with pH, salt concentration, and protein concentration, as well as the binding of any ligand, be it calcium ions or a target peptide. This property has to be taken into account in the simulations by extending the canonical Metropolis algorithm to a semicanonical approach. Thus, the MC procedure consists of two types of moves: i), random displacement of mobile salt particles, and ii), random change of the ionization status of titrating residues mentioned in the previous section. The acceptance of the second type of move is controlled by a change in electrostatic interactions plus the cost for ionizing/neutralizing the randomly chosen amino acid. The appropriate Boltzmann factor reads,
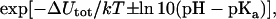 |
(11) |
where pH is the chosen pH and pKa is the acid constant for the particular amino acid.
The second term in the exponential can be either positive or negative, depending on whether the group is ionized or neutralized. After completion of this semicanonical MC scheme, one obtains the average charge on each titrating residue and hence the proper net charge of the protein. Note that this procedure mimics the experimental situation, in which a proton released from the protein is absorbed by buffer maintaining a constant pH. In a few simulations, we have suppressed the titration and instead used fixed charges appropriate for that particular pH. The results from these simulations give an indication of the importance of charge regulation upon peptide binding.
The free energy of binding for the peptide has been obtained from the MC simulations using a modified Widom insertion technique (Svensson and Woodward, 1988; Widom, 1963). Both the excess chemical potential of the bound and free peptide are obtained from the same simulation. In the first case, the peptide is inserted in the binding site, whereas in the latter the peptide is inserted at random in the MC sphere. The excess chemical potential is then obtained as a canonical average,
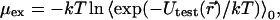 |
(12) |
where Utest(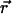 ) is the interaction energy between a peptide inserted at position
) is the interaction energy between a peptide inserted at position 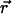 and all other particles. The brackets denote an ensemble average over the unperturbed system.
and all other particles. The brackets denote an ensemble average over the unperturbed system.
In other words, the Widom method is nonperturbative and does not affect the Markov chain underlying the Metropolis algorithm, and hence μex for several peptides of varying charge and size can be obtained in a single MC simulation. The accuracy of the Widom method goes down with increasing peptide charge and/or size. With 100,000 passes and an equal number of insertions, one obtains an estimated error in μex for an octavalent peptide of a few tenths of a kT. The computed average charge on a titrating residue, on the other hand, is obtained with three significant digits.
RESULTS
Design of calmodulin charge mutants
The binding of smMLCKp by CaM has been suggested to depend on ion pairing interactions because five negative charges are found close to the smMLCK peptide (Meador et al., 1992). Specifically, Glu-7, Glu-11, Glu-84, and Glu-114 in CaM are found <4 Å away from the nearest charged residue in the peptide. Charge deletions in CaM were made in positions that are in close contact (Glu-11, Glu-84) with the peptide, but also at positions further away from the peptide (Asp-78, Glu-84) in the complex (Fig. 1). All mutants were expressed with high yield comparable to wild-type CaM.
Design of smMLCK peptide analogs
The wt smMLCK peptide has a formal net charge of +7. Lys-7 found in the center of the peptide was varied with Gln, Gly, or Glu (K7Q, K7G, and K7E). In K7Q and K7G, the net charge is reduced by one unit (to +6), and in K7E by two units (to +5). The nonelectrostatic effect of substitution was tested by using the small nonpolar Gly. To allow for a large charge substitution effect, a triple mutant was constructed, K4QK7QR17Q (+4). Ionic charges in the peptide were further varied through amidation of carboxy terminus and acetylation of the amino terminus of the peptide with wt sequence to obtain smMLCKp-am (+8), smMLCKp-ac (+6) and smMLCKp-am-ac (+7).
Simulation of calmodulin net charge
CaM with four calcium ions bound, all acidic groups deprotonated, and all basic groups protonated results in a net charge of −15. From simulations performed at pH 7 and low salt and protein concentration, the net charge is found to be −14.6. The average charge of acidic residues is in the range from −1 to −0.9, and His-107 has an average charge of 0.54. The pH value has a significant effect on the net charge, and as seen in Fig. 3 the isoelectric point for CaM is around pH 4, in good agreement with experiment (Klee and Vanaman, 1982). The exact value depends on both salt and protein concentration. Some glutamates and aspartates have substantially up-shifted apparent pKa values. For example, the pKa value of Glu-83 is up-shifted by ∼1 pK unit at low salt and protein concentration. His-107 shows a similar behavior.
FIGURE 3.

Simulated net charge of CaM as a function of pH: Cp = 0.1 and Cs = 1.1 mM.
Binding experiments at different pH values
The pH dependence of the CaM-binding constant of wt smMLCKp was studied in the range from pH 4 to 11 (Fig. 4). In the experiments, the affinity was found to increase linearly with pH, but with a slope of only ∼0.1 log K units per pH unit. The total difference in affinity between pH 4.5 and 11 is ∼1 order of magnitude. Similarly weak pH dependence is observed for the wt peptide with protected end groups (smMLCKp-am-ac). A dramatic decrease in affinity is observed at pH below 4.5. The capability of CaM to bind calcium goes down at low pH, and it is well-known that CaM must be fully calcium saturated to bind smMLCKp with high affinity (Martin et al., 2000). We have, however, ensured that the CaCl2 concentration used in the experiments is sufficient to maintain a calcium-saturated protein even at pH 4.
FIGURE 4.

Experimental pH dependence of peptide binding of log K as a function of pH for smMLCKp (▪) and smMLCKp-am-ac (⋄) binding to CaM.
Simulation at different pH values
To understand the observed pH dependence of complex formation between CaM and peptides, we carried out a simulation study of binding positively charged peptides to CaM. It is straightforward to perform simulations both at a preset pH with titrating residues and at a fixed charge distribution. In the former case, the protein charges will fluctuate and the protein can adjust its charge upon binding the positively charged peptide. Charges of individual residues are shown in Fig. 5, indicating that CaM releases protons upon binding of smMLCKp, in particular at pH values where amino acids in the protein can easily titrate. That is, the charge response is much larger at pH 5 than at pH 7. At pH 5, the largest response is shown by Glu-11 and Glu-84, which both reduce their net charge from −0.5 to close to −1.0. The accumulated charge change over all ionizable residues results in a reduction of the CaM net charge of ∼−3.5 units. At pH 7, the largest change upon peptide binding is demonstrated by His-107, which reduces its charge from 0.46 to 0.30. The glutamates and aspartates now show a smaller response, and the cumulative effect of their partial titration means that the net charge by CaM is changed by only −1.0 unit upon binding of the peptide. The smMCLK peptide contains one histidine, which is close to neutral in the free peptide, but which increases its net charge to 0.53 when the peptide binds to CaM. These results were obtained by performing three separate simulations: one for CaM using the x-ray coordinates of Ca2+-loaded CaM (Protein Data Bank access code 1CLL, ref 12), one for smMCLKp, and one for the CaM-peptide complex. In the two latter simulations, the x-ray coordinates for the CaM-smMCLKp complex were used (1CDL.pdb, (Meador et al., 1992)). The simulated binding constant shifts shown in Fig. 6 confirm the experimental results with a nearly constant peptide binding over a large pH interval. However, at sufficiently low pH where the calmodulin net charge is close to zero, or even positive, the binding constant shows a dramatic drop.
FIGURE 5.

Simulated change in net charge of titratable acidic amino acids in CaM upon binding of the smMCLK peptide at two different pH values, pH 7 (•) and pH 5 (□). Cs = 1.1 and Cp = 0.1 mM. Glutamic acids 7, 11, and 84 show the largest response at pH 4.
FIGURE 6.

Influence of peptide charge on CaM binding at different pH values. Simulated curves representing the behavior of different peptides K4QK7QR17Q (solid line and filled circles), K7G (dashed line and filled squares) and smMLCK-am-ac (dot-dashed line and solid triangles). For each peptide, the log K shift (Δlog K) is calculated relative pH 7; Cs = 1.1 and Cp = 0.1 mM. Fat curves without symbols are obtained with a titratable protein, whereas curves with symbols are obtained with a fixed charge distribution on all amino acid residues. Note that the absolute value of log K has no meaning; only differences between the simulated numbers are of interest.
Calmodulin and peptides with charge substitutions
The affinity of peptides for CaM is summarized in Table 1. All charge substitutions variants show binding constants for CaM that are within 0.1 log units from that of the wt peptide. The measured binding constants of the peptides are hence similar within the error limits, which is a surprising result bearing in mind that the charge has changed up to three units. Somewhat larger effects are observed when the charges of the end groups are modified. The peptide with amidated C-terminus binds CaM 0.6 log K units weaker than does unblocked wt, although the modification leads to increased positive charge of the peptide. In principle, it is possible that nonelectrostatic effects counteract the charge substitution effects. However, there is no significant difference in affinity between the peptide variants K7Q and K7G that have the same charge deletion but otherwise very different side-chain perturbations.
TABLE 1.
Experimental binding constant logarithm values for binding of smMLCK peptide analogs to wt CaM in 5 mM Tris at pH 7.5 and 25°C
| Peptide Label | Sequence | Charge | Log K |
|---|---|---|---|
| smMLCKp-am | +ARRKWQKTGHAVRAIGRLSS−NH2 | +8 | 6.9 |
| smMLCKp | +ARRKWQKTGHAVRAIGRLSS− | +7 | 7.5 |
| smMLCKp-am-ac | Ac-ARRKWQKTGHAVRAIGRLSS−NH2 | +7 | 7.6 |
| smMLCKp-ac | Ac-ARRKWQKTGHAVRAIGRLSS− | +6 | 7.3 |
| K7Q | +ARRKWQQTGHAVRAIGRLSS− | +6 | 7.5 |
| K7G | +ARRKWQGTGHAVRAIGRLSS− | +6 | 7.5 |
| K7E | +ARRKWQETGHAVRAIGRLSS− | +5 | 7.4 |
| K4QK7QR17Q | +ARRQWQQTGHAVRAIGQLSS− | +4 | 7.6 |
The affinities of wt smMLCKp were measured for wt CaM as well as the E11Q, E83Q, E84Q, and D78N mutants (see Table 2). The binding constants were all within the error limit of the measurement. Neither the total charge of the peptide nor neutralization of selected negative charges in CaM affects the affinity for smMLCK peptide at low ionic strength. Thus, the binding is not dependent on specific ion pairs.
TABLE 2.
Experimental binding constant logarithm values for binding of smMLCK-am-ac peptide to wt CaM and its mutant forms with negative charge deletions in 5 mM Tris at pH 7.5 and 25°C
| Protein | Log K |
|---|---|
| wt CaM | 7.6 |
| E11Q | 7.5 |
| D78N | 7.5 |
| E83Q | 7.4 |
| E84Q | 7.4 |
DISCUSSION
Electrostatic interactions have been suggested to play an important role in target recognition by CaM (Cox et al., 1985; Crivici and Ikura, 1995; Ikura et al., 1992; Meador et al., 1992). This view has emerged from the x-ray structures of complexes showing oppositely charged residues in close proximity as well as from opposite net charges of CaM and target recognition sequences (Meador et al., 1992). However, there is no convincing experimental proof to these assumptions.
Lack of charge sensitivity
The experimentally measured affinities between CaM and peptides are seemingly independent of electrostatic interactions. Neither changes in the net charge nor elimination of specific charges affects the affinity. The experimental results are highly surprising because of the large and opposite net charges of smMLCKp and CaM, and the ionic contacts observed in the structure of the complex. The binding displays insensitivity toward charge changes but this does not mean that the electrostatic binding free energy is small. It is possible that the insensitivity of peptide charge can be explained by conformational degrees of freedom. For example, there should be a cost of bringing the charges together in the α-helical peptide from the unfolded state of the peptide. This cost will increase with the charge of the peptide and can be a factor that reduces the affinity for more highly charged peptides. Molecular dynamics simulations indicate that the cost of forming a helix increases by ∼1 kJ/mol when the charge of the smMLCK peptide is increased by one unit (data not shown). Our data rely on the assumption that structural changes in CaM or in the peptide upon binding are independent of pH, salt concentration, protein mutations, and peptide charge. This is probably a valid approximation except in the last case, hence a comparison between experiment and simulation is not relevant when making charge modifications in the peptide.
All seven basic residues in the smMLCK peptide have been suggested to form salt bridges to acidic groups in CaM (Crivici and Ikura, 1995). If specific interactions like ion pairing were important, removal of these interactions by mutation should have a large effect on the affinity. However, neither of the mutations E11Q or E84Q affects the affinity. It can therefore be concluded that ion pairing with E11 and E84 is not crucial for the affinity of the peptide. The entropic cost of bringing two charges in close proximity may overcome the electrostatic attraction. In addition, the charge density in the CaM-smMLCKp is so high that the interaction between two close oppositely charged residues could not be regarded as specific. Removal of distant charges, as in E83Q and D87N, also has no effect on the binding.
The insensitivity to charge perturbations is a general phenomenon that will occur in any system where highly and oppositely charged molecules interact.
The binding of highly charged ligands
Changes in pH were utilized to alter the charge of CaM more drastically. When pH is varied from 4.5 to 11, the net charge of the protein changes from ∼−3 to −17, yet the peptide affinity changes only marginally. The same result is found in the simulations. It is straightforward to reduce pH even further in the simulations. and for pH < 4, a significant decrease in the binding is found. At these pH values, the net charge of CaM is slightly positive. The insensitivity of peptide binding to the net charge of CaM at pH > 4 is unexpected, but it can be explained as a consequence of the strong electrostatic interactions.
One way to understand these findings is by considering a simpler binding model. For example, let us take a spherical aggregate of radius R and charge Qa, which is contained in a sphere of radius Rc. A charged ligand, Ql binds to the surface of the aggregate. The excess chemical potential for the peptide in the bound site is
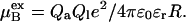 |
(13) |
The corresponding quantity for the free peptide can be approximated with
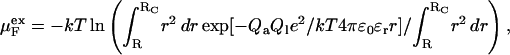 |
(14) |
where k and T are the Boltzmann constant and temperature, respectively. The integration is over the whole cell, but the integrand in the numerator is a rapidly decaying function and the result will, if the interaction is strong, be dominated by integrand values where 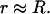 This has the consequence that the difference in excess chemical potential between the bound and free peptide becomes approximately constant independent of aggregate charge (Fig. 7).
This has the consequence that the difference in excess chemical potential between the bound and free peptide becomes approximately constant independent of aggregate charge (Fig. 7).
FIGURE 7.

Binding constant shifts for the simple model described in Eq. 14 as a function of aggregate charge. The curves correspond to different ligand charges: thick solid line, Ql = 7; dashed line, Ql = 5; dot-dashed line, Ql = 3; and thin solid line, Ql = 1. R = 10 Å and RC = 150 Å.
The protein releases a number of protons when binding the positively charged peptide, because peptide binding changes the electrostatic environment of acidic groups and their pKa values are shifted downward. Fig. 5 shows that CaM releases 3–4 protons when it binds the peptide at pH 5. Simulations show that the average charge of acidic residues in CaM at pH 7 is in the range from −1 to −0.9 and His-107 has an average charge of 0.54. Thus, the histidine and some glutamates and aspartates have substantially up-shifted pKa values. For example, the pKa value of Glu-83 is up-shifted by ∼1 pK unit and His-107 has a pKa of ∼7. This charge regulation mechanism contributes to make the binding constant less sensitive to changes of the protein net charge. For CaM it is seen at low pH, where the simulations with a titrating protein maintain its strong peptide binding to lower pH values than the CaM model with fixed charges (see Fig. 6). The magnitude of the charge regulation is related to the protein capacitance, which happens to be large for CaM around pH 4 (Lund and Jönsson, unpublished). Thus, the charge response by titratable groups is a mechanism for a protein to extend the pH range of high-affinity binding of highly charged ligands. The peptide can in principle also contribute to charge regulation, but its capacitance is close to zero around pH 4. To reduce computation time in this study, the charge regulation by peptide was ignored and the peptide was assigned fixed charges on its titratable groups.
Specificity for bound targets or discrimination against nonbound ones?
The balance of forces involved in the target recognition by CaM is an interesting issue due to the diversity in CaM-peptide complex structures. There is little sequence identity among the more than 100 proteins that bind to CaM, and it is unclear how CaM can have target specificity under these circumstances. It is possible that the core requirement for CaM binding is only the presence of basic terminal sequence with high α-helical propensity and a fair amount of hydrophobic residues. This is supported by results obtained using designed synthetic peptides (Cox et al., 1985). CaM is able to bind to a peptide from δ-hemolysin, which has zero net charge (Cox et al., 1985) but no peptide with negative net charge has been reported to bind CaM. Our data suggest that the charge of the peptide itself does not increase the specificity of the binding. Why are then all known CaM-binding regions positively charged? Maybe the question should be turned around from finding the molecular basis for target recognition to the molecular basis for discrimination against unbound proteins.
A net negative charge is important for calmodulin to function as a Ca2+ sensor, as it ensures a high on-rate for Ca2+ (Martin et al., 1990). The recognition sequences of target enzymes may need to be charged to avoid aggregation due to its amphiphatic character. For this purpose, the peptide could be negative or positive; however, a negative peptide would be repelled by CaM. Therefore, the sequence needs to be positive. The negative charge of CaM prevents it from binding to anything. Most cytosolic proteins are negatively charged, as is DNA, and effectively repel CaM. Discrimination against nonwanted targets through repulsive electrostatic interactions seems to be more fruitful than a strong optimization of target binding. Hence, CaM can bind to a large number of different targets with apparent specificity, although the protein is actually not strictly optimized for binding to any one of them. Indeed, Shifman and Mayo (2002) showed that increased specificity toward one target led to decreased affinity toward others.
It may be a general scenario that electrostatic interactions in protein-protein complexes are utilized to avoid unwanted partners through repulsive forces rather than to attract particular targets. Avoiding electrostatic repulsion seems to be a more fruitful regulatory mechanism than employing attractive electrostatic interactions, due to counteracting factors from the entropic costs of fixation, and from desolvation. There is an analogy with protein folding in which electrostatic repulsion to avoid misfolding should be more profitable than guidance to the correct fold through electrostatic attraction.
CONCLUSIONS
The binding of highly positively charged peptides to calmodulin is surprisingly insensitive to the net charges of both peptide and protein. This means that the difference in excess chemical potential of bound and free peptide is approximately constant and independent of the details of protein-peptide interaction as long as it is strong. This insensitivity is further emphasized by the charge response of titrating acidic groups. That is, the net charge of both calmodulin and the target peptide changes in the binding process. We speculate that in target recognition the main function of the high negative charge on calmodulin is to avoid unwanted complexation, rather than to enhance the interaction with target peptides.
Acknowledgments
This work was supported by Swedish Natural Science Foundation (I.A., B.J., and S.L.), and by the Estonian Science Foundation (T.K.) K.S.Å. was supported by NFS Career Grant No. 9996074.
Abbreviations used: smMLCKp, a synthetic peptide corresponding to residues 796–815 in chicken gizzard in smooth muscle myosin light chain kinase in this work numbered 1–20; smMLCK, smooth muscle myosin light chain kinase; skMLCK, skeletal muscle myosin light chain kinase; skMLCKp, a synthetic peptide corresponding to residues 494–513 in rabbit skeletal muscle myosin light chain kinase; CaM, calmodulin; DMF, N, N-dimethyl formamide; MES, 2-morpholinoethanesulfonic acid; CAPS, 3-(cyclohexylamino)-1-propanesulfonic acid; TFA, trifluoroacetic acid.
References
- Afshar, M., L. S. Caves, L. Guimard, R. E. Hubbard, B. Calas, G. Grassy, and J. Haiech. 1994. Investigating the high affinity and low sequence specificity of calmodulin binding to its targets. J. Mol. Biol. 244:554–571. [DOI] [PubMed] [Google Scholar]
- Andersson, S. R., and D. A. Malencik. 1986. Peptides recognizing calmodulin. In Calcium and Cell Function, Vol. 6. W. Y. Cheung, editor. Academic Press, New York. 1–42.
- Andre, I., and S. Linse. 2002. Measurement of Ca2+-binding constants of proteins and presentation of the Caligator software. Anal. Biochem. 305:195–205. [DOI] [PubMed] [Google Scholar]
- Antosiewicz, J., J. A. McCammon, and M. K. Gilson. 1996. The determinants of pKas in proteins. Biochemistry. 35:7819–7833. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya, R., W. E. Meador, A. R. Means, and F. A. Quiocho. 1992. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 228:1177–1192. [DOI] [PubMed] [Google Scholar]
- Chou, J. J., S. Li, C. B. Klee, and A. Bax. 2001. Solution structure of Ca(2+)-calmodulin reveals flexible hand-like properties of its domains. Nat. Struct. Biol. 8:990–997. [DOI] [PubMed] [Google Scholar]
- Cox, J. A., M. Comte, J. E. Fitton, and W. F. DeGrado. 1985. The interaction of calmodulin with amphiphilic peptides. J. Biol. Chem. 260:2527–2534. [PubMed] [Google Scholar]
- Crivici, A., and M. Ikura. 1995. Molecular and structural basis of target recognition by calmodulin. Annu. Rev. Biophys. Biomol. Struct. 24:85–116. [DOI] [PubMed] [Google Scholar]
- Getzoff, E. D., J. A. Tainer, P. K. Weiner, P. A. Kollman, J. S. Richardson, and D. C. Richardson. 1983. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 306:287–290. [DOI] [PubMed] [Google Scholar]
- Gu, C., and D. M. Cooper. 1999. Calmodulin-binding sites on adenylyl cyclase type VIII. J. Biol. Chem. 274:8012–8021. [DOI] [PubMed] [Google Scholar]
- Hoeflich, K. P., and M. Ikura. 2002. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 108:739–742. [DOI] [PubMed] [Google Scholar]
- Ikura, M., G. M. Clore, A. M. Gronenborn, G. Zhu, C. B. Klee, and A. Bax. 1992. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 256:632–638. [DOI] [PubMed] [Google Scholar]
- Juffer, A. H., and H. J. Vogel. 2000. pK(a) calculations of calbindin D(9k): effects of Ca(2+) binding, protein dielectric constant, and ionic strength. Proteins. 41:554–567. [PubMed] [Google Scholar]
- Kesvatera, T., B. Jonsson, E. Thulin, and S. Linse. 1994. Binding of Ca2+ to calbindin D9k: structural stability and function at high salt concentration. Biochemistry. 33:14170–14176. [DOI] [PubMed] [Google Scholar]
- Kesvatera, T., B. Jonsson, E. Thulin, and S. Linse. 2001. Focusing of the electrostatic potential at EF-hands of calbindin D(9k): titration of acidic residues. Proteins. 45:129–135. [DOI] [PubMed] [Google Scholar]
- Klee, C. B. 1977. Conformational transition accompanying the binding of Ca2+ to the protein activator of 3′,5′-cyclic adenosine monophosphate phosphodiesterase. Biochemistry. 16:1017–1024. [DOI] [PubMed] [Google Scholar]
- Klee, C. B., and T. C. Vanaman. 1982. Calmodulin. Adv Protein Chem. 35:213–321. [DOI] [PubMed] [Google Scholar]
- Koradi, R., M. Billeter, and K. Wuthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- Kranz, J. K., E. K. Lee, A. C. Nairn, and A. J. Wand. 2002. A direct test of the reductionist approach to structural studies of calmodulin activity: relevance of peptide models of target proteins. J. Biol. Chem. 277:16351–16354. [DOI] [PubMed] [Google Scholar]
- Lee, A., S. T. Wong, D. Gallagher, B. Li, D. R. Storm, T. Scheuer, and W. A. Catterall. 1999. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 399:155–159. [DOI] [PubMed] [Google Scholar]
- Lee, F. S., Z. T. Chu, M. B. Bolger, and A. Warshel. 1992. Calculations of antibody-antigen interactions: microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. 5:215–228. [DOI] [PubMed] [Google Scholar]
- Linse, S., C. Johansson, P. Brodin, T. Grundstrom, T. Drakenberg, and S. Forsen. 1991. Electrostatic contributions to the binding of Ca2+ in calbindin D9k. Biochemistry. 30:154–162. [DOI] [PubMed] [Google Scholar]
- Martin, S. R., S. Linse, C. Johansson, P. M. Bayley, and S. Forsen. 1990. Protein surface charges and Ca2+ binding to individual sites in calbindin D9k: stopped-flow studies. Biochemistry. 29:4188–4193. [DOI] [PubMed] [Google Scholar]
- Martin, S. R., L. Masino, and P. M. Bayley. 2000. Enhancement by Mg2+ of domain specificity in Ca2+-dependent interactions of calmodulin with target sequences. Protein Sci. 9:2477–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador, W. E., A. R. Means, and F. A. Quiocho. 1992. Target enzyme recognition by calmodulin: 2.4 Å structure of a calmodulin-peptide complex. Science. 257:1251–1255. [DOI] [PubMed] [Google Scholar]
- Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092. [Google Scholar]
- Missiaen, L., J. B. Parys, A. F. Weidema, H. Sipma, S. Vanlingen, P. De Smet, G. Callewaert, and H. De Smedt. 1999. The bell-shaped Ca2+ dependence of the inositol 1,4, 5-trisphosphate-induced Ca2+ release is modulated by Ca2+/calmodulin. J. Biol. Chem. 274:13748–13751. [DOI] [PubMed] [Google Scholar]
- Montigiani, S., G. Neri, P. Neri, and D. Neri. 1996. Alanine substitutions in calmodulin-binding peptides result in unexpected affinity enhancement. J. Mol. Biol. 258:6–13. [DOI] [PubMed] [Google Scholar]
- Muegge, I., T. Schweins, and A. Warshel. 1998. Electrostatic contributions to protein-protein binding affinities: application to Rap/Raf interaction. Proteins. 30:407–423. [DOI] [PubMed] [Google Scholar]
- Pace, C. N., F. Vajdos, L. Fee, G. Grimsley, and T. Gray. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4:2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persechini, A., and R. H. Kretsinger. 1988. The central helix of calmodulin functions as a flexible tether. J. Biol. Chem. 263:12175–12178. [PubMed] [Google Scholar]
- Sham, Y. Y., Z. T. Chu, and A. Warshel. 1997. Consistent calculations of pK(a)'s of ionizable residues in proteins: semi-microscopic and microscopic approaches. J. Phys. Chem. B. 101:4458–4472. [Google Scholar]
- Sham, Y. Y., I. Muegge, and A. Warshel. 1998. The effect of protein relaxation on charge-charge interactions and dielectric constants of proteins. Biophys. J. 74:1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman, J. M., and S. L. Mayo. 2002. Modulating calmodulin binding specificity through computational protein design. J. Mol. Biol. 323:417–423. [DOI] [PubMed] [Google Scholar]
- Svensson, B., B. Jonsson, E. Thulin, and C. E. Woodward. 1993. Binding of Ca2+ to calmodulin and its tryptic fragments: theory and experiment. Biochemistry. 32:2828–2834. [DOI] [PubMed] [Google Scholar]
- Svensson, B. R., and C. E. Woodward. 1988. Widom method for uniform and non-uniform electrolyte-solutions. Mol. Phys. 64:247–259. [Google Scholar]
- Tsvetkov, P. O., I. I. Protasevich, R. Gilli, D. Lafitte, V. M. Lobachov, J. Haiech, C. Briand, and A. A. Makarov. 1999. Apocalmodulin binds to the myosin light chain kinase calmodulin target site. J. Biol. Chem. 274:18161–18164. [DOI] [PubMed] [Google Scholar]
- Waltersson, Y., S. Linse, P. Brodin, and T. Grundstrom. 1993. Mutational effects on the cooperativity of Ca2+ binding in calmodulin. Biochemistry. 32:7866–7871. [DOI] [PubMed] [Google Scholar]
- Warshel, A., S. T. Russell, and A. K. Churg. 1984. Macroscopic models for studies of electrostatic interactions in proteins: limitations and applicability. Proc. Natl. Acad. Sci. USA. 81:4785–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom, B. 1963. Some topics in theory of fluids. J. Chem. Phys. 39:2808–2812. [Google Scholar]
- Wintrode, P. L., and P. L. Privalov. 1997. Energetics of target peptide recognition by calmodulin: a calorimetric study. J. Mol. Biol. 266:1050–1062. [DOI] [PubMed] [Google Scholar]
- Zhu, H., M. Bilgin, R. Bangham, D. Hall, A. Casamayor, P. Bertone, N. Lan, R. Jansen, S. Bidlingmaier, T. Houfek, T. Mitchell, P. Miller, R. A. Dean, M. Gerstein, and M. Snyder. 2001. Global analysis of protein activities using proteome chips. Science. 293:2101–2105. [DOI] [PubMed] [Google Scholar]