

Abstract
Protein stability plays an extremely important role not only in its biological function but also in medical science and protein engineering. Osmolytes provide a general method to protect proteins from the unfolding and aggregation induced by extreme environmental stress. In this study, the effect of glycerol on protection of the model enzyme creatine kinase (CK) against heat stress was investigated by a combination of spectroscopic method and thermodynamic analysis. Glycerol could prevent CK from thermal inactivation and aggregation in a concentration-dependent manner. The spectroscopic measurements suggested that the protective effect of glycerol was a result of enhancing the structural stability of native CK. A further thermodynamic analysis using the activated-complex theory suggested that the effect of glycerol on preventing CK against aggregation was consistent with those previously established mechanisms in reversible systems. The osmophobic effect of glycerol, which preferentially raised the free energy of the activated complex, shifted the equilibrium between the native state and the activated complex in favor of the native state. A comparison of the inactivation rate and the denaturation rate suggested that the protection of enzyme activity by glycerol should be attributed to the enhancement of the structural stability of the whole protein rather than the flexible active site.
INTRODUCTION
Protein stability plays an extremely important role not only in its biological function but also in protein engineering, including protein design, refolding, storage, and transfer (Chi et al., 2003). When compared to the physiological conditions, isolated proteins are usually subjected to severe environmental conditions which might lead to inactivation, denaturation, and aggregation. Particularly, irreversible protein aggregation is a common feature in protein engineering, and much effort has been devoted to investigating the mechanism and driving force of protein aggregation (Chi et al., 2003; Dong et al., 1995; Yan et al., 2004), the protection of proteins from aggregation (Chi et al., 2003 and references therein; Meng et al., 2001), and refolding proteins from aggregates (Meersman and Heremans, 2003) in recent years. Development of techniques to protect proteins, especially enzymes, against denaturing stresses such as extreme temperature is always one of the major topics of protein science and engineering.
Many organisms including plants, animals, and microorganisms that live in extreme environments have had to adapt to the environmental stresses by evolving means to protect themselves. Besides the complex regulation of specific transcriptional control, a very common mechanism that these organisms evolve in response to environmental stress involves accumulation of intracellular low-molecular-weight organic compounds, known as osmolytes (Yancey et al., 1982; Somero, 1986; Bolen and Baskakov, 2001). These naturally occurring compounds include specific carbohydrates, methylamines, amino acids, and their derivatives (Yancey et al., 1982). Previous studies have shown that these osmolytes might have the ability not only to protect the intracellular proteins of certain organisms, but also to provide any protein with general protection against denaturation stress (Yancey et al., 1982). The protective mechanism of osmolytes has traditionally been attributed to “preferential hydration” of the protein (Gekko and Timasheff, 1981a,b; Priev et al., 1996; Timasheff, 2002), which suggested that the exclusion of the cosolvent molecules from the protein surface led to a minimization of the protein surface without changing its conformation. From the results of hydrogen/deuterium (H/D) exchange studies, Bolen and his coauthors (Bolen, 2001; Bolen and Baskakov, 2001) further suggested a mechanism of “solvophobic thermodynamic force,” which indicated that the unfavorable interaction between the osmolytes and the peptide backbone raised the free energy of the denatured state and as a result, protected the protein by shifting the equilibrium in favor of the native state. However, the evidence of this mechanism was based mostly on the H/D exchange and reversible folding studies. In this study, the investigation focused on the effect of glycerol, which has long been used to protect the enzyme activity and native structure of proteins against various types of denaturation (Rariy and Klibanov, 1997; Timasheff, 1998; Meng et al., 2001), on the thermodynamics of irreversible thermal denaturation of rabbit creatine kinase.
Creatine kinase (CK; ATP:creatine n-phosphoryltransferase, EC 2.7.3.2), which catalyzes the reversible transfer of the phosphoryl group from MgATP to creatine, plays an important role in cellular energy metabolism in vertebrates (Wallimann et al., 1992). The folding and unfolding problems of CK have been investigated with various stresses, such as alkaline pH (Yang et al., 1997), urea (Zhou and Tsou, 1986), guanidine hydrochloride (Yao et al., 1982), SDS (Wang et al., 1995), and temperature (Lyubarev et al., 1999). Moreover, the effects of various ions, small organic compounds, and chaperones on the CK refolding were thoroughly studied by us previously (for example, Yang et al., 1997; Meng et al., 2001; Ou et al., 2001). Thus the two-state irreversible thermal transition of CK was taken as a model system in this study to investigate the stabilizing effect of glycerol on proteins using the thermodynamic method.
MATERIALS AND METHODS
Sample preparation and activity measurement
ATP, creatine, and acrylamide were purchased from Sigma Chemical (St. Louis, MO). All other reagents were local products of analytical grade. Purification of rabbit muscle CK was the same as that described previously (Yao et al., 1982). The purity of the enzyme was checked by electrophoresis. The enzyme concentration was determined by measuring the absorbance at 280 nm with 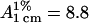 (Yao et al., 1982). CK activity was assayed using the pH-calorimetry method (Yao et al., 1982) at 25°C and the reaction system contained 24 mM creatine, 4 mM ATP, 5 mM Mg2+, 0.01% Thymol Blue (w/v), and 5 mM glycine-NaOH buffer (pH 9.0). All activity measurements were carried out with a Perkins-Elmer Lambda Bio U/V spectrophotometer (Wellesley, MA). Samples for spectroscopic and calorimetric experiments were prepared by using a Tris-HCl buffer, pH 8.05.
(Yao et al., 1982). CK activity was assayed using the pH-calorimetry method (Yao et al., 1982) at 25°C and the reaction system contained 24 mM creatine, 4 mM ATP, 5 mM Mg2+, 0.01% Thymol Blue (w/v), and 5 mM glycine-NaOH buffer (pH 9.0). All activity measurements were carried out with a Perkins-Elmer Lambda Bio U/V spectrophotometer (Wellesley, MA). Samples for spectroscopic and calorimetric experiments were prepared by using a Tris-HCl buffer, pH 8.05.
Spectroscopic measurements
For all spectroscopic measurements except for thermal aggregation studies, all samples were prepared by heating the enzyme solutions at 50°C for 30 min and then quickly cooling to room temperature before measurements. The intrinsic fluorescence emission spectra were measured with an Hitachi 850 spectrofluorometer (Tokyo, Japan) using 1-cm-pathlength cuvettes with an excitation wavelength of 295 nm. Circular dichroism (CD) spectra were recorded on a Jasco J-715 spectrophotometer (Jasco Research, Tokyo, Japan) over a wavelength range of 200–250 nm with a 2-mm-pathlength cell. Each spectrum was the result of eight scans obtained by collecting data with a resolution of 0.5 nm, and an integration time of 0.5 s. The aggregation of CK at 50°C was monitored by measuring the turbidity at 400 nm with a Perkins-Elmer Lambda Bio U/V spectrophotometer using a final protein concentration of 1 mg/ml. Infrared (IR) spectra were measured with a Perkin-Elmer Spectrum 2000 spectrometer equipped with a dTGS detector (Yan et al., 2003). IR samples were prepared by dissolving 50 mg protein in 1 ml D2O with a final pD of 8.45 (uncorrected value). The samples were stored overnight and a sample of ∼30 μl was placed between a pair of CaF2 windows separated by a 50-μm Teflon spacer. IR spectra were collected continuously at 50°C every 5 min. All spectroscopic experiments were repeated at least twice to ensure the reproducibility of the data.
Calorimetric measurements
Calorimetric measurements were taken using a Setaram Micro DSC III (Caluire, France) differential scanning calorimeter (DSC) with a 0.8-ml cell. The DSC curves were obtained using a scanning rate of 1 K/min from 20 to 95°C. Protein concentration was 0.8–1.2 mg/ml. Reversibility of the thermal transition was examined by reheating of the sample after cooling from the first scan. The chemical baseline was subtracted using the procedure of Takahashi and Sturtevant (1981).
The DSC curves were analyzed using the two-state irreversible model (Sanchez-Ruiz et al., 1988; Freire et al., 1990; Sanchez-Ruiz, 1995; Kurganov et al., 1997) of
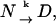 |
(1) |
where N is the native protein, D is the denatured state, and k is the first-order rate constant.
Kinetic behavior of the system following this model is described by the differential equation
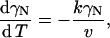 |
(2) |
where γN is the mole fraction of the native state, T is the absolute temperature, and v is the scanning rate. The rate constant can be determined by the Arrhenius equation
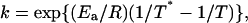 |
(3) |
where Ea is the energy of activation, R is the gas constant, and T* is the absolute temperature at which k = 1 min−1. The rate constant can also be determined by the equation of the activated-complex theory (Glasstone et al., 1941; Johnson et al., 1954; Miles, 1993; Miles et al., 1995; Jensen et al., 1997; Lyubarev and Kurganov, 2000)
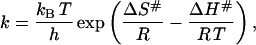 |
(4) |
where kB is Boltzmann's constant, h is Planck's constant, ΔH# and ΔS# are the enthalpy and entropy of activation, respectively.
The excess heat capacity (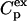 ) is determined by the equation
) is determined by the equation
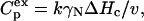 |
(5) |
where ΔHc is calorimetric enthalpy (enthalpy of denaturation).
The following equation was also used in the model
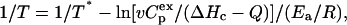 |
(6) |
where Q is the heat absorbed during heating of the protein to the temperature T, and
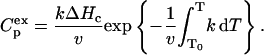 |
(7) |
Eq. 6 was used for plotting the linear anamorphoses of DSC curves, as well as for estimating the initial parameters during the fitting procedure. Fitting of Eq. 7 to the experimental data was carried out using an original program for IBM-compatible computers (Kurganov et al., 1997). Fitting of the system of Eqs. 2 and 5 to the experimental data was carried out using the commercial software, “Scientist” (MicroMath Scientific Software, Salt Lake City, UT). Both experimental and theoretical curves were built using the interval between the points of 0.01–0.02 K.
RESULTS
Effect of glycerol on CK thermal aggregation at 50°C
Our previous study suggested that the thermal denaturation of CK was an irreversible two-state transition with a Tm value of ∼56°C (Lyubarev et al., 1999). To further characterize the behavior of the protein heated at a relatively high temperature, the aggregation of CK at 50°C was investigated first in this study. The conformational changes of CK upon heat stress were monitored by FTIR spectra, and the results are presented in Fig. 1 A. No significant difference was found between the spectrum recorded at 30°C (data not shown) and the first spectrum recorded after 5 min at 50°C, which suggested that most of the protein molecules favored the native state at 50°C. However, by heating the protein at this temperature, the intensity of the IR bands from native structures (1629, 1639, 1651, and 1660 cm−1) gradually decreased, whereas two new bands, a strong band at ∼1614 cm−1 and a weak band at ∼1682 cm−1, appeared in the IR spectra (Fig. 1 A). These two new bands have been previously assigned to the characteristic bands of intermolecular β-sheet structures in aggregates (Dong et al., 1995). The extent of aggregation and the effect of glycerol were investigated by monitoring the turbidity at 400 nm, and the results are presented in Fig. 1 B. The time before the appearance of aggregation was delayed by addition of glycerol and the aggregation was totally inhibited when the concentration of glycerol reached 25%. It should be noted that the turbidity of the protein heated at 50°C in 5% glycerol was slightly higher than that in Tris-HCl buffer. This phenomenon was observed in all repeated experiments and might be caused by the fast deposition of the aggregates in Tris-HCl buffer, whereas the deposition was slowed by the existence of glycerol due to increased viscosity.
FIGURE 1.

Second derivative FTIR spectra in the amide I region of CK in D2O (pD 8.45, uncorrected value) incubated at 50°C (A) and kinetic course of the aggregation of CK in Tris-HCl buffer (pH 8.0) incubated at 50°C in various concentrations of glycerol monitored by turbidity at 400 nm (B). The arrows in A indicate the direction of the intensity changes along with incubation time, and the wavenumbers indicated are 1682, 1666, 1660, 1651, 1639, 1629, and 1614 cm−1, respectively.
Effect of glycerol on CK thermal inactivation
The enzyme activity of CK in the presence of glycerol with a concentration ranging from 0% to 30% was carried out by the pH-calorimetry method (Yao et al., 1982) after heating at 50°C for a given time ranging from 0 to 60 min. The kinetic data shown in Fig. 2 indicated that thermal inactivation of CK was a one-stage process and glycerol can slow down the inactivation rate (k). The enzyme lost ∼90% of its activity after being heated at 50°C for 30 min, although only ∼50% was lost in the presence of >10% glycerol. The decrease in the k-value exhibited a glycerol concentration-dependent manner, which suggested that the effect of glycerol was more likely to reduce the rate of conformational changes by enhancing the structural stability of the native enzyme. This result was quite consistent with previously proposed mechanisms of osmolytes (Bolen and Baskakov, 2001; Chi et al., 2003; Timasheff, 2002). Thus the protective effect of glycerol on CK thermal inactivation was a result of maintaining the native conformation of the enzyme. A comparison of Figs. 1 B and 2 indicated that not all of the gradual loss of activity induced by heat could be attributed to irreversible protein aggregation. After heating at 50°C for 30 min, the turbidity reached ∼30% of its maximum value for the protein in Tris-HCl buffer, whereas the activity was almost totally lost. Thus the effect of glycerol on preventing the protein from off-pathway aggregation was also a result of enhancing the structural stability and decreasing the rate of the formation of nonnative aggregates.
FIGURE 2.

Semilogarithmic plots of the activities of CK incubated in 20 mM Tris-HCl buffer, pH 8.05, with different concentrations of glycerol at 50°C for up to 60 min. Glycerol concentrations were 0% (•), 5% (▵), 10% (*), 20% (○), and 30% (▪). Enzyme activity was determined in aliquots taken at suitable time intervals, and the final enzyme concentration was 4 μM.
To confirm the protective mechanism of glycerol by enhancing structural stability on CK inactivation, fluorescence and CD spectra were used to identify the tertiary and secondary structural changes of CK under extreme thermal conditions. After CK solution was incubated without glycerol at 50°C for 30 min, the maximum emission wavelength of CK shifted from 331.0 to 339.0 nm, whereas in solutions with glycerol concentrations of 5%, 10%, 20%, and 30%, the maximum emission wavelength of CK shifted to 337.0, 336.5, 334.0, and 332.0 nm, respectively (Fig. 3 A). These data indicated that the presence of glycerol could help the enzyme prevent against heat-induced unfolding in the experimental concentration range. Similar to the result from intrinsic fluorescence spectra, the data from CD spectra (Fig. 3 B) further indicated that the presence of glycerol could efficiently prevent the change of the native secondary structure induced by heat stress. All the spectral data confirmed that the protection of glycerol in thermal inactivation of CK was a result of the enhancement of its structural stability.
FIGURE 3.

The intrinsic fluorescence spectra (A) and circular dichroism spectra (B) of CK after 30 min incubation at 50°C in the presence of glycerol. The fluorescence and CD spectra of native CK in 20 mM Tris-HCl buffer (dashed lines) were measured at 25°C. The CD spectrum of unfolded CK was measured using a sample completely unfolded by 4 M guanidine hydrochloride.
Qualifying the energetic characteristics of the contribution of glycerol to CK thermal stability
The results from the activity and spectroscopic measurements above suggested that the glycerol's effect on CK thermal stability behaved in a concentration-dependent manner. Meanwhile, no aggregation was found for the protein in 30% glycerol solution after incubation at 50°C for 2 h (data not shown). To further characterize the effect of glycerol on the irreversible thermodynamic process, the behavior of the protein in various concentrations of glycerol was studied by DSC. The heat flow versus temperature profile of CK samples contained an asymmetric peak at the temperatures ranging from 50°C to 60°C (Fig. 4). Further increase in temperature brought out a small knob which was associated with the aggregation of CK (data not shown, also see Lyubarev et al., 1999). When the samples of the enzyme were heated up to 65°C and immediately cooled at a rate of 1 K/min, the reheating scan demonstrated no thermal effect in the area of the main peak. These results were quite consistent with our previous study (Lyubarev et al., 1999), and indicated that the thermal denaturation of CK samples was calorimetrically irreversible. The excess heat capacity versus temperature profiles (Fig. 4) indicated that the maximum point of the denaturation peak shifted toward higher temperatures as the glycerol concentration increased. Our previous study (Lyubarev et al., 1999) suggested that thermal denaturation of CK followed a two-state irreversible model. One of the validity criteria of this model (Kurganov et al., 1997) is the linearity of the plot in the coordinates 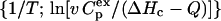 (Eq. 6). As presented in Fig. 5, the plots were linear for all samples and the existence of glycerol did not affect the irreversible two-state thermal transition of the protein. Thus this model was also used here to obtain the quantitative thermodynamic description of the effect of glycerol on CK thermal denaturation.
(Eq. 6). As presented in Fig. 5, the plots were linear for all samples and the existence of glycerol did not affect the irreversible two-state thermal transition of the protein. Thus this model was also used here to obtain the quantitative thermodynamic description of the effect of glycerol on CK thermal denaturation.
FIGURE 4.

Temperature dependence of the excess heat capacity of CK in the presence of glycerol with concentrations of 0% (•) 5% (▵), 10% (*), 20% (○), and 30% (▪). The enzyme concentration was 9.3 μM and the temperature scanning rate was 1 K/min.
FIGURE 5.

The linear anamorphosis (Kurganov et al., 1997; Lyubarev et al., 1999) of the DSC data in Fig. 4. The symbols used are the same as those used in Fig. 4. All the points with Q/ΔHc values between 0.05 and 0.95 were used.
The parameters of the two-state irreversible model were estimated by fitting the experimental data to Eq. 7 and to the system described by Eq. 2 and Eq. 5. The values of k were determined by Eqs. 3 and 4. The resultant parameters (Table 1), ΔH# and ΔS# were calculated from the absolute rate theory described by Eq. 4, whereas T* was obtained from the Arrhenius equation (Eq. 3). The ΔHc values obtained from Eq. 3 and Eq. 4 were the same. The values of the parameter Ea in the Arrhenius equation (Eq. 3) were closely related to the parameter ΔH#, with the only difference being the value of RT (∼2.7 kJ/mol), and were not presented here. As shown in Table 1, all four parameters increased as glycerol concentration increased. The values of ΔH# and ΔS# were found to increase simultaneously, which was due to the compensatory effect. The increase of the value of T* corresponded to the high-temperature shift of the maximum point of the denaturation peak on the DSC curve (Fig. 4).
TABLE 1.
Kinetic parameters estimated for thermal denaturation of CK in solutions with various concentrations of glycerol
| Glycerol (v/v) | ΔH# (kJ mol−1)* | ΔS# (kJ mol−1 K−1)* | ΔHc (kJ mol−1)† | T* (K)‡ | r§ |
|---|---|---|---|---|---|
| 0% | 429 ± 7¶ | 1.016 ± 0.02 | 1016 ± 40 | 331.3 ± 0.1 | 0.9989 |
| 5% | 446 ± 8 | 1.063 ± 0.02 | 1075 ± 50 | 331.8 ± 0.2 | 0.9986 |
| 10% | 450 ± 7 | 1.072 ± 0.02 | 1106 ± 40 | 332.9 ± 0.1 | 0.9990 |
| 20% | 473 ± 5 | 1.137 ± 0.02 | 1270 ± 30 | 333.8 ± 0.1 | 0.9994 |
| 30% | 478 ± 8 | 1.148 ± 0.02 | 1346 ± 60 | 334.6 ± 0.1 | 0.9988 |
ΔH# and ΔS# are the enthalpy and entropy of activation, and were estimated using Eq. 4.
ΔHc is the heat absorbed through the denaturation process (calorimetric enthalpy) and was estimated using Eq. 3 and Eq. 4.
T* is the absolute temperature at which k = 1 min−1 and was estimated using Eq. 3.
The effect of glycerol on the thermal stability of proteins has been studied by some researchers using both thermal inactivation and calorimetric methods. They found that the presence of glycerol could shift the maximum temperature of the DSC peak or the temperature at which the enzyme lost 50% of its activity to higher temperatures (Back et al., 1979; Gekko and Timasheff, 1981a,b; Gekko, 1982; Hottiger et al., 1994; Wu et al., 1995; Saburova et al., 1996). The results here of the CK irreversible thermal denaturation are consistent with those previous works. However, less information about the effect of glycerol on calorimetric enthalpy change (ΔHc) could be obtained. Gekko (1982) indicated that 50% glycerol could increase the value of ΔHc for lysozyme by 44%. In contrast, Saburova et al. (1996) were unable to find the difference in the value of ΔHc within the accuracy of the analysis (20%) for lactate dehydrogenase in the absence or the presence of glycerol (6–56%). Our results (Table 1) indicated that the presence of 30% glycerol could increase the value of ΔHc for CK by ∼30%.
DISCUSSION
The results in this article indicated that the rate of CK thermal inactivation was significantly slowed in the presence of glycerol and this effect was concentration-dependent. The results of spectroscopic measurements confirmed that during the thermal inactivation, glycerol could protect both the tertiary and secondary structure of CK from heat-induced unfolding and thus keep the protein from further aggregation.
The well-established thermodynamic theories (Gekko and Timasheff, 1981a,b; Priev et al., 1996; Timasheff, 1998 and 2002; Bolen and Baskakov, 2001) are more applicable to reversible denaturation when equilibrium is attained between the native (N) and denatured (D) states of the protein molecule than to the irreversible process. However, the denaturation of many proteins has been found to proceed irreversibly. Moreover, the protection of proteins against aggregation has become a problematic issue in biotechnology and pharmacology (Chi et al., 2003; Stefani and Dobson, 2003). The need for a thermodynamic description of the osmolyte effect on irreversible denaturation is urgent. In this study, the activated-complex theory (Glasstone et al., 1941; Johnson et al., 1954), which could accurately describe protein irreversible denaturation in terms of thermodynamic quantities, was used to explain how glycerol protects CK against heat-induced inactivation and aggregation. The transition from reactants into products has an intermediate state, which has been called the activated complex. Thus the transformation of reactants into products is connected with the energetic barrier, and the activated complex (transition state) corresponds to the maximum of the dependence of the potential energy on the reaction coordinate. The main postulate of the activated-complex theory is the existence of equilibrium between reactants and the activated complex. Within this theoretical framework, the kinetic scheme of irreversible denaturation of the protein has the form
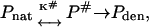 |
(8) |
where Pnat and Pden are the native and denatured states of the protein and P# is the activated complex. The equilibrium constant K# is described as
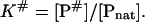 |
(9) |
The free energy of activation ΔG# is connected with the equilibrium constant K# by the relationship
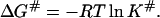 |
(10) |
In accordance with the activated-complex theory, the rate constant of denaturation k is calculated by the formula
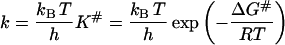 |
(11) |
(also see Eq. 4). As can be seen from this equation, any factor that raises the free energy of activation ΔG# will reduce the rate of the reaction.
The thermodynamic cycle shown in Scheme 1 above allows the influence of osmolytes on the free energy of activation ΔG# for protein denaturation to be characterized. Pnat,w and Pnat,osm are the native states of the protein in water and in the solution with osmolytes at a given concentration, whereas  and
and  are the activated complex in water and in the solution with osmolytes, respectively. In this scheme, ΔGP,osm and
are the activated complex in water and in the solution with osmolytes, respectively. In this scheme, ΔGP,osm and 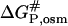 are the changes of free energy for transfer of the native protein and activated complex from water to the solution with osmolytes, whereas
are the changes of free energy for transfer of the native protein and activated complex from water to the solution with osmolytes, whereas 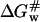 and
and 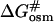 are the free energies of activation for protein denaturation in water and in the solution with osmolytes. It has been found that osmolytes exhibit a stabilizing effect through preferential exclusion from the immediate vicinity of both the native and unfolded states of the protein (Gekko and Timasheff, 1981a and 1981b; Priev et al., 1996; Timasheff, 1998 and 2002). Preferential exclusion between a small organic solute and protein implies net unfavorable (solvophobic) interactions between the protein and osmolyte, and as a result implies an increase in the free energy of the protein species (Bolen, 2001; Bolen and Baskakov, 2001). The solvent exposure of the activated complex P# is much larger than the native state, and thus the activated complex is more solvophobic than the native state, i.e., the change in free energy
are the free energies of activation for protein denaturation in water and in the solution with osmolytes. It has been found that osmolytes exhibit a stabilizing effect through preferential exclusion from the immediate vicinity of both the native and unfolded states of the protein (Gekko and Timasheff, 1981a and 1981b; Priev et al., 1996; Timasheff, 1998 and 2002). Preferential exclusion between a small organic solute and protein implies net unfavorable (solvophobic) interactions between the protein and osmolyte, and as a result implies an increase in the free energy of the protein species (Bolen, 2001; Bolen and Baskakov, 2001). The solvent exposure of the activated complex P# is much larger than the native state, and thus the activated complex is more solvophobic than the native state, i.e., the change in free energy 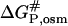 is greater than
is greater than 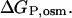
SCHEME 1.

The following relationship holds true for the changes in free energy in the thermodynamic cycle (Scheme 1),
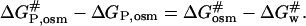 |
(12) |
This relationship suggests that 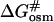 (the free energy of activation in an osmolyte solution) is more positive than
(the free energy of activation in an osmolyte solution) is more positive than 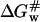 (the free energy of activation in water), and consequently, the rate of irreversible denaturation of the protein in the solution with osmolytes is lower than that in water. Alternatively, the protein is more stable in the solution with osmolytes than in water because osmolytes raise the free energy of the activated complex far more than the native state. Several works have addressed the effect of glycerol on the activation parameters of irreversible protein denaturation. Simonova et al. (1995) found that the activation energy for the inactivation of lactate dehydrogenase increased as the glycerol concentration increased. However, Jensen et al. (1997) found that the ΔH# and ΔS# values for inactivation of malate dehydrogenase in 6.1 M glycerol were less than those in water, and TΔS# value decreased much more than the ΔH# value did. The results in the present work were similar to those of Simonova et al. (1995). The ΔH# values for CK denaturation increased as the glycerol concentration increased (Table 1), and the increase of ΔH# was greater than that of TΔS#. Thus
(the free energy of activation in water), and consequently, the rate of irreversible denaturation of the protein in the solution with osmolytes is lower than that in water. Alternatively, the protein is more stable in the solution with osmolytes than in water because osmolytes raise the free energy of the activated complex far more than the native state. Several works have addressed the effect of glycerol on the activation parameters of irreversible protein denaturation. Simonova et al. (1995) found that the activation energy for the inactivation of lactate dehydrogenase increased as the glycerol concentration increased. However, Jensen et al. (1997) found that the ΔH# and ΔS# values for inactivation of malate dehydrogenase in 6.1 M glycerol were less than those in water, and TΔS# value decreased much more than the ΔH# value did. The results in the present work were similar to those of Simonova et al. (1995). The ΔH# values for CK denaturation increased as the glycerol concentration increased (Table 1), and the increase of ΔH# was greater than that of TΔS#. Thus 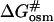 was larger than
was larger than 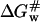 at the temperatures under investigation.
at the temperatures under investigation.
It is noteworthy that the loss of enzyme activity was somehow faster than protein aggregation (Figs. 1 B and 2). The existence of 30% glycerol could successfully inhibit CK from aggregation (Fig. 1 B) and maintain the tertiary and secondary structures of the protein that were almost the same as the native state (Fig. 3). However, loss of enzyme activity could still be observed (Fig. 2). Moreover, no significant difference was found in CK activity without heat treatment between the samples with and without glycerol at concentrations ranging from 5% to 20%, whereas a slight decrease (<10%) was found for the sample in 30% glycerol (data not shown). Previous H/D exchange studies indicated that the exchange rate of the slow-exchanging amide protons was more affected by the addition of osmolytes than the exchange rate of fast-exchanging protons (Wang et al., 1995; Qu and Bolen, 2003). It is imaginable that the active site, which has been proposed to be more flexible than the molecule as a whole (Zhou and Tsou, 1986; Tsou, 1998), was less protected than the central structure of the enzyme and thus the inactivation rate constants were larger than the denaturation rate constants even though glycerol was present. This lower degree of protection might also be the reason why osmolytes have no effect on protein function. Assuming that the unfolding of the region of the active site (AS) obeys the activated-complex theory,
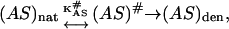 |
(13) |
where (AS)nat and (AS)unf are the native and denatured states of the region of the active site and (AS)# is the corresponding activated complex. A higher rate of the loss in the enzymatic activity of CK suggests that the free energy of activation 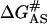 is less positive than that for unfolding of the whole protein molecule ΔG#. Thus it could be concluded that the protective effect of glycerol on CK activity could be due to the enhancement of the structural stability of the whole molecule, but not of the stability of the active site. This explanation was also applicable to general proteins, in which the functional region, which is usually composed of the relative flexible region of the whole molecule, might be less affected by osmolytes.
is less positive than that for unfolding of the whole protein molecule ΔG#. Thus it could be concluded that the protective effect of glycerol on CK activity could be due to the enhancement of the structural stability of the whole molecule, but not of the stability of the active site. This explanation was also applicable to general proteins, in which the functional region, which is usually composed of the relative flexible region of the whole molecule, might be less affected by osmolytes.
In conclusion, the effects of glycerol on the protection of CK from irreversible thermal aggregation that we have shown here, and which were analyzed according to the activated-complex theory, are totally consistent with those well-established mechanisms of osmophobic effect on reversible unfolding (Timasheff, 1998, 2002; Bolen, 2001). Since the effect of osmolytes is general to any protein and to any stress (Yancey et al., 1982), the result here provides the key to understanding the mechanism involved in the protective effect of osmolytes on irreversible inactivation and aggregation of enzymes. Moreover, the effect of osmolytes on the structural stability of the whole protein rather than on the active site was found to be more responsible to the protective effect on enzyme activity. This result suggested that less protection of the functional loops of the activated site might not be responsible for the maintaining of protein function by osmolytes. It also suggested that although the extraordinary ability of glycerol, as well as other osmolytes, provided a general method to protect proteins from aggregation and unfolding induced by extreme environmental stress, the limitations indicated above should also be taken into account when storing enzymes for use in the biotechnology industry.
Acknowledgments
The authors thank Prof. Su-Qin Sun and Mrs. Xiao-Lan Ding (Tsinghua University) for their expert technical assistance. The authors also thank the anonymous reviewer for the helpful suggestions.
This research was supported by the National Key Basic Research Specific Fund, People's Republic of China (No. G 1999075607), the Basic Research Funds (No. JC2002047 and JC2003061), the 985 Fund and Tsinghua Laboratory Fund (THSJZ) from Tsinghua University, People's Republic of China, and funds from the State Key Laboratory of Biomembranes, People's Republic of China. This research was also supported by the Russian Foundation for Basic Research (grants 02-04-48704 and 02-04-49099), the Program “Molecular and Cellular Biology” of the Russian Academy of Sciences, the Program for the Support of the Leading Schools in Russia (grant 813.2003.4), and the international association for the promotion of cooperation with scientists from the new independent states of the former Soviet Union (INTAS) (grant 03-51-4813).
Fan-Guo Meng and Yuan-Kai Hong contributed equally to this work.
Fan-Guo Meng's present address is Albert Einstein College of Medicine, Dept. of Molecular Pharmacology, Bronx, NY, 10461.
Yuan-Kai Hong's present address is Dept. of Biophysics, School of Basic Medical Science, Peking University, Beijing 100083, China.
References
- Back, J. F., D. Oakenfull, and M. B. Smith. 1979. Increased thermal stability of proteins in the presence of sugars and polyols. Biochemistry. 18:5191–5196. [DOI] [PubMed] [Google Scholar]
- Bolen, D. W. 2001. Protein stabilization by naturally occurring osmolytes. Methods Mol. Biol. 168:17–36. [DOI] [PubMed] [Google Scholar]
- Bolen, D. W., and I. V. Baskakov. 2001. The osmophobic effect: natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310:955–963. [DOI] [PubMed] [Google Scholar]
- Chi, E. Y., S. Krishnan, T. W. Randolph, and J. F. Carperter. 2003. Physical stability of proteins in aqueous solutions: mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 20:1325–1336. [DOI] [PubMed] [Google Scholar]
- Dong, A., S. J. Prestrelski, S. D. Allison, and J. F. Carpenter. 1995. Infrared spectroscopic studies of lyophilization- and temperature-induced protein aggregation. J. Pharm. Sci. 84:415424. [DOI] [PubMed] [Google Scholar]
- Freire, E., W. W. van Osdol, O. L. Mayorga, and J. M. Sanchez-Ruiz. 1990. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Annu. Rev. Biophys. Biophys. Chem. 19:159–188. [DOI] [PubMed] [Google Scholar]
- Gekko, K., and S. N. Timasheff. 1981a. Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry. 20:4667–4676. [DOI] [PubMed] [Google Scholar]
- Gekko, K., and S. N. Timasheff. 1981b. Thermodynamic and kinetic examination of protein stabilization by glycerol. Biochemistry. 20:4677–4686. [DOI] [PubMed] [Google Scholar]
- Gekko, K. 1982. Calorimetric study on thermal denaturation of lysozyme in polyol-water mixtures. J. Biochem. 91:1197–1204. [DOI] [PubMed] [Google Scholar]
- Glasstone, S., K. J. Laidler, and H. Eyring. 1941. The Theory of Rate Processes: The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena. McGraw-Hill, New York.
- Hottiger, T., C. De Virgilio, M. N. Hall, T. Boller, and A. Wiemken. 1994. The role of trehalose synthesis for the acquisition of thermotolerance in yeast. II. Physiological concentration of trehalose increase the thermal stability of proteins in vitro. Eur. J. Biochem. 219:187–193. [DOI] [PubMed] [Google Scholar]
- Jensen, W. A., J. M. Armstrong, J. De Giorgio, and M. T. W. Hearn. 1997. Thermodynamic analysis of the stabilization of pig heart mitochondrial malate dehydrogenase and maize leaf phosphoenolpyruvate carboxylase by different salts, amino acids and polyols. Biochim. Biophys. Acta. 1338:186–198. [DOI] [PubMed] [Google Scholar]
- Johnson, F. H., H. Eyring, and M. J. Polisar. 1954. The Kinetic Basis of Molecular Biology. Wiley and Sons, New York.
- Kurganov, B. I., A. E. Lyubarev, J. M. Sanchez-Ruiz, and V. L. Shnyrov. 1997. Analysis of differential scanning calorimetry data for proteins. Criteria of validity of one-step mechanism of irreversible protein denaturation. Biophys. Chem. 69:125–135. [DOI] [PubMed] [Google Scholar]
- Lyubarev, A. E., and B. I. Kurganov. 2000. Analysis of DSC data relating to proteins undergoing irreversible thermal denaturation. J. Therm. Anal. Cal. 62:51–62. [Google Scholar]
- Lyubarev, A. E., B. I. Kurganov, V. N. Orlov, and H. M. Zhou. 1999. Two-state irreversible thermal denaturation of muscle creatine kinase. Biophys. Chem. 79:199–204. [DOI] [PubMed] [Google Scholar]
- Meersman, F., and K. Heremans. 2003. Temperature-induced dissociation of protein aggregates: accessing the denatured state. Biochemistry. 42:14234–14241. [DOI] [PubMed] [Google Scholar]
- Meng, F. G., Y. D. Park, and H. M. Zhou. 2001. Role of proline, glycerol, and heparin as protein folding aids during refolding of rabbit muscle creatine kinase. Int. J. Biochem. Cell. B. 33:701–709. [DOI] [PubMed] [Google Scholar]
- Miles, C. A. 1993. Kinetics of collagen denaturation in mammalian lens capsules studied by differential scanning calorimetry. Int. J. Biol. Macromol. 15:265–271. [DOI] [PubMed] [Google Scholar]
- Miles, C. A., T. V. Burjanadze, and A. J. Bailey. 1995. The kinetics of the thermal denaturation of collagen in unrestrained rat tail tendon determined by differential scanning calorimetry. J. Mol. Biol. 245:437–446. [DOI] [PubMed] [Google Scholar]
- Ou, W. B., W. Luo, Y. D. Park, and H. M. Zhou. 2001. Chaperone-like activity of peptidyl-prolyl cis-trans isomerase during creatine kinase refolding. Protein Sci. 10:2346–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priev, A., A. Almagor, S. Yedgar, and B. Gavish. 1996. Glycerol decreases the volume and compressibility of protein interior. Biochemistry. 35:2061–2066. [DOI] [PubMed] [Google Scholar]
- Qu, Y., and D. W. Bolen. 2003. Hydrogen exchange kinetics of RNase A and the urea:TMAO paradigm. Biochemistry. 42:5837–5849. [DOI] [PubMed] [Google Scholar]
- Rariy, R. V., and A. M. Klibanov. 1997. Correct protein folding in glycerol. Proc. Natl. Acad. Sci. USA. 94:13520–13523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saburova, E. A., N. N. Khechinashvili, and L. I. Elfimova. 1996. Polyhydric alcohol-protein interactions. Microcalorimetry of lactate dehydrogenase denaturation in water-glycerol solutions. Mol. Biol. 30:733–738. [PubMed] [Google Scholar]
- Sanchez-Ruiz, J. M. 1995. Differential scanning calorimetry of proteins. Subcell. Biochem. 24:133–176. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ruiz, J. M., J. L. Lopez-Lacomba, M. Cortijo, and P. L. Mateo. 1988. Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry. 27:1648–1652. [DOI] [PubMed] [Google Scholar]
- Simonova N. B., E. A. Saburova, G. Shirshikova, L. I. Elfimova, N. A. Pronina, T. N. Falkovich, and V. E. Semenenko. 1995. The effect of glycerol on thermal stability of lactate dehydrogenase from halophilic alga Dunaliella salina and glicophilic Chlamydomonas reinhardtii. Dokl. Akad. Nauk. 345:549–554 (in Russian). [Google Scholar]
- Somero, G. N. 1986. Protons, osmolytes, and fitness of internal milieu for protein function. Am. J. Physiol. 251:197–213. [DOI] [PubMed] [Google Scholar]
- Stefani, M., and C. M. Dobson. 2003. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81:678–699. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., and J. M. Sturtevant. 1981. Thermal denaturation of Streptomyces subtilisin inhibitor, subtilisin BPN′, and the inhibitor-subtilisin complex. Biochemistry. 20:6185–6190. [DOI] [PubMed] [Google Scholar]
- Timasheff, S. N. 1998. Control of protein stability and reactions by weakly interacting cosolvents: the simplicity of the complicated. Adv. Protein Chem. 51:355–432. [DOI] [PubMed] [Google Scholar]
- Timasheff, S. N. 2002. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry. 41:13473–13482. [DOI] [PubMed] [Google Scholar]
- Tsou, C. L. 1998. Active site flexibility in enzyme catalysis. Ann. N. Y. Acad. Sci. 864:1–8. [DOI] [PubMed] [Google Scholar]
- Wallimann, T., M. Wyss, D. Brdiczka, K. Nicolay, and H. M. Eppenberger. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the “phosphocreatine circuit” for cellular energy homeostasis. Biochem. J. 281:21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A. J., A. D. Robertson, and D. W. Bolen. 1995. Effects of a naturally occurring compatible osmolyte on the internal dynamics of ribonuclease A. Biochemistry. 34:15096–15104. [DOI] [PubMed] [Google Scholar]
- Wang, Z. F., M. Q. Huang, X. M. Zou, and H. M. Zhou. 1995. Unfolding, conformational change of active sites and inactivation of creatine kinase in SDS solutions. Biochim. Biophys. Acta. 1251:109–114. [DOI] [PubMed] [Google Scholar]
- Wu, J., D. G. Long, and R. M. Weis. 1995. Reversible dissociation and unfolding of the Escherichia coli aspartate receptor cytoplasmic fragment. Biochemistry. 34:3056–3065. [DOI] [PubMed] [Google Scholar]
- Yan, Y. B., Q. Wang, H. W. He, X. Y. Hu, R. Q. Zhang, and H. M. Zhou. 2003. Two-dimensional infrared correlation spectroscopy study of the heat-induced unfolding and aggregation process of myoglobin. Biophys. J. 85:1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Y. B., Q. Wang, H. W. He, and H. M. Zhou. 2004. Protein thermal aggregation involves distinct regions: sequential events in the heat-induced unfolding and aggregation of hemoglobin. Biophys. J. 86:1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey, P. H., M. E. Clark, S. C. Hand, R. D. Bowlus, and G. N. Somero. 1982. Living with water stress: evolution of osmolyte systems. Science. 217:1214–1222. [DOI] [PubMed] [Google Scholar]
- Yang, H. P., H. N. Zhong, S. Li, and H. M. Zhou. 1997. Salt-induced folding of alkaline denatured creatine kinase under high pH conditions. Biochem. Mol. Biol. Int. 41:257–267. [DOI] [PubMed] [Google Scholar]
- Yao, Q. Z., L. X. Hou, H. M. Zhou, and C. L. Tsou. 1982. Conformational changes of creatine kinase during guanidine denaturation. Sci. Sin. B. 25:1186–1193. [PubMed] [Google Scholar]
- Zhou, H. M., and C. L. Tsou. 1986. Comparison of activity and conformation changes during refolding of urea-denatured creatine kinase. Biochim. Biophys. Acta. 869:69–74. [DOI] [PubMed] [Google Scholar]