

Abstract
The binding of the restriction endonuclease EcoRI to DNA is exceptionally specific. Even a single basepair change (“star” sequence) from the recognition sequence, GAATTC, decreases the binding free energy of EcoRI to values nearly indistinguishable from nonspecific binding. The difference in the number of waters sequestered by the protein-DNA complexes of the “star” sequences TAATTC and CAATTC and by the specific sequence complex determined from the dependence of binding free energy on water activity is also practically indistinguishable at low osmotic pressures from the 110 water molecules sequestered by nonspecific sequence complexes. Novel measurements of the dissociation rates of noncognate sequence complexes and competition equilibrium show that sequestered water can be removed from “star” sequence complexes by high osmotic pressure, but not from a nonspecific complex. By 5 Osm, the TAATTC “star” sequence complex has lost almost 90 of the ∼110 waters initially present. It is more difficult to remove water from the CAATTC “star” sequence complex. The sequence dependence of water loss correlates with the known sequence dependence of “star” cleavage activity.
INTRODUCTION
It is generally thought that hydration water likely plays an important role in sequence recognition reactions, particularly in specific sequence DNA-protein binding (Hard and Lundback, 1996; Jen-Jacobson, 1997; Schwabe, 1997; Szwajkajzer and Carey, 1997). We have focused on measuring differences in hydration that distinguish specific and nonspecific DNA-protein interactions particularly of the restriction endonuclease EcoRI, an exceptionally stringent sequence specific DNA binding protein. Changes in hydration linked to binding reactions can be assessed using an experimental approach termed the osmotic stress technique (Parsegian et al., 1995, 2000). Differences in the numbers of solute excluding water molecules associated with specific and nonspecific DNA-protein complexes can be measured from the dependence of the relative free energy difference on osmotic pressure (or water chemical potential), using the same thermodynamics that allows differences in proton or salt binding accompanying macromolecular reactions to be probed by the sensitivity of the reaction to pH or salt activity. The osmotic stress technique has been widely used to measure the changes in water binding accompanying the DNA binding of several proteins: Escherichia coli gal (Garner and Rau, 1995), lac (Fried et al., 2002), and tyr (Poon et al., 1997) repressors, E. coli CAP protein (Vossen et al., 1997), Hin recombinase (Robinson and Sligar, 1996), ultrathorax and deformed homeodomains (Li and Matthews, 1997), the restriction endonucleases EcoRI (Robinson and Sligar,1998; Sidorova and Rau, 1996, 2001), BamHI (Lynch and Sligar, 2000), and EcoRV (Wenner and Bloomfield, 1999), HhaI methyl transferase (Swaminathan et al., 2002), Sso7d protein (Lundback et al., 1998), and the TATA binding protein (TBP) (Khrapunov and Brenowitz, 2004; Wu et al., 2001).
We have previously measured differences in water release for the binding of the EcoRI to specific and nonspecific DNA sequences (Sidorova and Rau, 1996). Several very different solutes, ranging from glycine and glycerol to triethylene glycol, were used to vary the water chemical potential. The free energy difference between specific and nonspecific DNA-EcoRI complexes was linearly dependent on the water chemical potential. The observed osmotic dependence indicated that the nonspecific complex retains some 110 waters more than the specific complex with the recognition sequence. All six osmolytes gave the same difference in waters within 15% experimental error. This slight sensitivity of the difference in the number of water molecules retained by specific and nonspecific complexes to the solute identity is much less than expected for a change in exposed surface area (Courtenay et al., 2000; Davis-Searles et al., 1998; Preisler et al., 1995; Timasheff, 1993). This led us to conclude that this water is sequestered in a space that is sterically inaccessible to solutes. Because the x-ray structure of the EcoRI-DNA specific complex (Rosenberg, 1991; Kim et al., 1990) shows that interface of this complex is essentially dry, the 110 waters are likely at the protein-DNA interface of the nonspecific complex. A structure for the nonspecific complex of EcoRI is not available, but x-ray structures for both specific sequence and noncognate sequence complexes of a closely related type II restriction endonuclease, BamHI, have been solved (Newman et al., 1995; Viadiu and Aggarwal, 2000). Unlike the extensive, direct protein-DNA contacts seen in the specific complex structure, the nonspecific complex structure shows a gap between the BamHI and DNA major groove surfaces that is large enough to hold ∼150 waters. The large difference in sequestered water between nonspecific and specific complexes of EcoRI is in marked contrast to other solution components. Although salt and pH strongly affect the binding of EcoRI to the specific recognition sequence, there is practically no difference in their effect on the relative binding to specific and nonspecific sequences (Sidorova and Rau, 2001).
The observed osmotic sensitivity of the specific-nonspecific equilibrium binding constant is also reflected in the dissociation rate constant of the recognition sequence complex (Sidorova and Rau, 2000, 2001). For DNA-binding proteins that can efficiently locate their target sequences by initially binding nonspecifically and then diffusing along the DNA as EcoRI, the dissociation of nonspecifically bound protein is typically the rate-limiting reaction step. We showed that the observed osmotic dependence of the specific complex dissociation rate is dominated by the difference of 110 waters between the specific and nonspecific binding modes of the enzyme. The dissociation of nonspecifically bound protein from the DNA is coupled to an uptake of ∼10–45 additional waters depending on the osmolyte used to set water activity. This dependence on solute nature is typical for a reaction that results in an increased exposed surface area of the complex.
At low pressures, the osmotic stress technique can be used as a convenient tool for probing differences in water release accompanying nonspecific versus specific DNA-protein binding without perturbing the system. A linear dependence of binding free energy on solute osmotic pressure indicates that the difference in retained water between complexes remains constant. In principle, however, any sequestered water can be removed by applying high enough osmotic stress. Essentially, the pressure-volume work, ΠΔV, done in removing water from a complex or in stabilizing an alternate conformation with fewer sequestered waters is balanced by the resulting unfavorable interactions and conformational changes of the complex. A differential loss of water from a noncognate DNA-protein complex would be seen as a nonlinear dependence of the free energy difference between specific and noncognate complexes on osmotic stress at high pressures. Control experiments are necessary to ensure the solute is still acting osmotically at these high osmotic pressures.
We observed previously (Sidorova and Rau, 1999) that at low osmotic pressures an EcoRI complex with a DNA sequence that only differs from the recognition sequence by only a single basepair (a “star” sequence) sequesters the same amount of water as a complex with a nonspecific sequence oligonucleotide. Because the enzyme will cleave “star” sequences with low activity, but not sequences with two or more wrong basepairs (Gardner et al., 1982; Goodman et al., 1977; Hsu and Berg, 1978; Lesser et al., 1990; Malyguine et al., 1980; Polisky et al., 1975; Rosenberg and Greene, 1982; Tikchonenko et al., 1978), “star” sequence complexes cannot be indistinguishable from nonspecific complexes. Additionally, it has been shown that EcoRI distinctly pauses at “star” sites while diffusing along DNA (Jeltsch et al., 1994). We extend our previous work (Sidorova and Rau, 1999) and measure to high stresses (∼6 Osm) the osmotic dependence of the EcoRI dissociation rate from complexes with oligonucleotides containing either the TAATTC or CAATTC “star” sequence, both of which differ from the recognition sequence, GAATTC, by a single first basepair substitution, or an inverted recognition sequence, CTTAAG. We also measure the osmotic dependence of the relative binding free energies from equilibrium competition experiments at lower stresses (<2 Osm). Kinetic measurements allow use of much higher osmotic pressures than is practical for equilibrium studies. We measure the dissociation rate of “star” and inverted sequence complexes from the kinetics of appearance of specific complex after the enzyme is added to a mixture of specific and noncognate sequence DNA. We develop the equations necessary to describe the kinetics.
At low pressures (<1.2 Osm), the osmotic dependence of binding free energies relative to the specific complex indicates that EcoRI complexes with oligonucleotides containing “star” sequences and the inverted sequence all sequester about the same number of waters. The relative binding free energy of the inverted sequence oligonucleotide complex continues to vary linearly with osmotic pressure through 2 Osm. Both “star” sequence complexes, however, begin to show nonlinearity at the higher pressures, consistent with a loss of water from the complex or the stabilization of an alternate, dehydrated complex. The osmotic dependence of the dissociation rate from the inverted sequence complex shows no loss of water through 6 Osm (∼150 atmospheres), the highest osmotic pressure used. Betaine glycine continues to act osmotically on this EcoRI complex even at these high stresses. Much of the water in the TAATTC “star” sequence complex, however, is removed at high osmotic pressures. Somewhat less water, but still a significant amount, is removed from the CAATTC “star” sequence complex. We estimate that a work of ∼3 kcal/mol is needed to remove substantially all the water from the initial fully hydrated TAATTC “star” sequence complex and ∼6 kcal/mol for the CAATTC complex. These energies are consistent with the observed enzymatic “star” activities for these sequences (Lesser et al., 1990).
MATERIALS AND METHODS
Materials
A DNA fragment containing a single EcoRI recognition sequence was isolated from the plasmid derived from pNEB193 (New England Biolabs, Beverly, MA) using standard techniques. The 360-bp fragment was purified from a PvuII (New England Biolabs) digestion of the plasmid. The double-stranded 30-bp-long oligonucleotides used in competition and kinetic experiments were:
Specific sequence GAATTC oligo: ACGACGGCCAGTGAATTCGAGCTCGGTACC
TAATTC sequence oligo: ACGACGGCCAGTTAATTCGAGCTCGGTACC
CAATTC sequence oligo: ACGACGGCCAGTCAATTCGAGCTCGGTACC
CTTAAG sequence oligo: ACGACGGCCAGTCTTAAGGAGCTCGGTACC.
These oligonucleotides only differ in the central six basepairs shown in bold letters. The specific sequence oligonucleotide contains the EcoRI cognate recognition site, GAATTC. The TAATTC and CAATTC oligonucleotides contain different first basepair substitutions of the recognition sequence and are commonly termed “star” sites (Gardner et al., 1982; Goodman et al., 1977; Hsu and Berg, 1978; Lesser et al., 1990; Malyguine et al., 1980; Polisky et al., 1975; Rosenberg and Greene, 1982; Tikchonenko et al., 1978). The CTTAAG oligonucleotide contains an inverted specific sequence or a nonspecific site with all six basepairs wrong. Sequence differences from the recognition sequence in this central six basepair region are underlined. The oligonucleotides shown above and their complements were purchased from Gibco BRL, and dissolved in STE buffer (100 mM NaCl, 10 mM TrisCl (pH 7.5), 1 mM EDTA). Complementary strands were mixed in 1:1 proportion, heated to 92°C, and annealed by slow cooling to 25°C. Small molecular mass impurities were removed using P6 Bio-Spin columns (Bio-Rad Laboratories, Hercules, CA) at room temperature. Double-stranded oligonucleotides were then additionally purified using high-performance liquid chromatography (Waters (Milford, MA) model 2690, Protein Pak Q ion exchange column), ethanol precipitated, and dissolved in TE buffer (10 mM TrisCl (pH 7.5), 1 mM EDTA). The purity of the double-stranded oligonucleotides was confirmed by polyacrylamide gel electrophoresis. The concentrations of the DNA fragment and oligonucleotides were determined spectrophotometrically, using an extinction coefficient of 0.013 (μM basepairs)−1 at 260 nm. Absorption spectra were obtained with a Perkin Elmer (Wellesley, MA) Lambda 800 ultraviolet-visible spectrophotometer.
DNA-binding experiments were performed with highly purified EcoRI restriction endonuclease (kind gift of Dr. L. Jen-Jacobson). Titration of the EcoRI sample with known concentration with specific DNA fragment (1–10-nM concentration range) confirmed that stoichiometry of binding was one EcoRI dimer per 360-bp fragment with a single EcoRI specific recognition sequence (data not shown). Active protein concentrations for the individual binding experiments were determined by direct titration of the EcoRI with the 360-bp DNA fragment under conditions of stoichiometric binding as described previously (Sidorova and Rau, 1996).
Betaine glycine was purchased from United States Biochemical (Cleveland, OH) and used without further purification. Osmolal concentrations of betaine were determined by direct measurement using a vapor pressure osmometer operating at room temperature (Wescor, Logan, UT; model 5520XR). Changes in water chemical potentials are linearly proportional to solute osmolal concentrations, i.e., 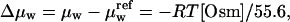 where μw and
where μw and 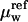 are the water chemical potentials of the solutions with and without added osmolyte. We confirmed previously that betaine glycine acts on EcoRI-DNA binding osmotically up to very high concentrations (at least up to 4 Osm) (Sidorova and Rau, 1996, 1999). The high solubility of betaine glycine coupled with its low viscosity makes it an excellent solute for experiments requiring high osmotic pressures.
are the water chemical potentials of the solutions with and without added osmolyte. We confirmed previously that betaine glycine acts on EcoRI-DNA binding osmotically up to very high concentrations (at least up to 4 Osm) (Sidorova and Rau, 1996, 1999). The high solubility of betaine glycine coupled with its low viscosity makes it an excellent solute for experiments requiring high osmotic pressures.
Equilibrium competition experiments
Specific-nonspecific equilibrium competition experiments were performed as described previously (Sidorova and Rau, 1996). Briefly, mixtures of EcoRI (∼1 nM), the specific site 360-bp fragment (∼2 nM), and the oligonucleotide competitor (between 0 and ∼50 μM in oligonucleotide or 0 and ∼1.5 mM in bp), were incubated at 20°C for 90 min. The loss of specific site binding as the concentration of nonspecific competitor DNA increased was determined by the gel mobility shift assay (Garner and Revzin, 1981; Fried and Crothers, 1981). In the experiments reported here all samples contained 20 mM imidazole (pH 7.5), 2 mM EDTA, 1 mM DTT, 0.1 mg/ml bovine serum albumin, and 2.5% ficoll. The total reaction volume was 30 μl. Because the specific sequence complex dissociation rate is strongly dependent on osmotic pressure (Sidorova and Rau, 2000, 2001), the salt concentration was adjusted for different osmotic stress regimes such that the slowest dissociation half-life time is ∼18 min, to ensure the reaction reaches equilibrium within 90 min. NaCl concentrations were 120 mM and 160 mM in the 0–1 and 1–2 Osm betaine glycine ranges, respectively. We have shown previously (Sidorova and Rau, 2001) that water activity and salt are acting as independent thermodynamic parameters. To prevent redistribution of EcoRI between specific site DNA fragment and competitor oligonucleotide between the time that the sample is loaded onto the gel and when the complex enters the gel, a quench reaction was used that “freezes” the equilibrium fraction of specifically bound EcoRI. After the 90-min incubation, 10 μl of 0.5 μM specific sequence oligonucleotide in 65 mM imidazole (pH 7.0), 10 mM NaCl, and 6.5 Osm betaine glycine was added to the 30-μl reaction volume. The specific sequence oligonucleotide was added to bind EcoRI dissociating from nonspecific complexes to prevent binding to the specific DNA fragment. The combination of increased osmotic pressure and decreased salt and pH lengthens the dissociation half-life time of the specific sequence complex to several hours or more, depending on the initial salt and osmotic pressure conditions. Control experiments with stoichiometrically bound enzyme show no loss of EcoRI from specific site DNA fragment to specific site oligonucleotide under these conditions. In the absence of Mg2+, we observed no measurable cleavage of the DNA.
Dissociation kinetics
To measure the dissociation kinetics of noncognate complexes EcoRI was added to a mixture of the 360-bp specific sequence DNA fragment and nonspecific oligonucleotide. The final concentrations of EcoRI and the specific site 360-bp fragment were 1.5 nM and 3 nM, respectively. The ratio of oligonucleotide and specific sequence fragment concentrations was varied between 50 and 1000. The mixture was incubated for various times, quenched, and the appearance of specific site binding measured using the gel mobility shift assay.
Solution conditions for the kinetic experiments were 20 mM imidazole (pH 7.0), 90 mM NaCl, 2 mM EDTA, 1 mM DTT, and 0.1 mg/ml BSA, in 30 μl volume. Samples were incubated at 20°C. The reaction was quenched by adding a 100-fold excess of specific sequence oligonucleotide and betaine glycine to a final concentration of at least 6 Osm. The samples were sufficiently dense with the added betaine glycine that ficoll was not necessary. The final salt concentration after quenching was 75 mM NaCl. The total reaction volume after quenching was 60 μl.
The ratio of association rates between the specific recognition sequence 360-bp fragment and the specific sequence oligonucleotide was determined from the initial partitioning of EcoRI binding. EcoRI was added to a mixture of the DNA fragment and the specific sequence oligonucleotide and incubated for 10 min at 20°C in the same buffer as used for the dissociation kinetics experiments. The DNA fragment and protein concentrations were ∼3 nM and ∼1.5 nM, respectively. The ratio of oligonucleotide and DNA fragment concentrations was varied between 0.5 to 3 and the osmotic pressure between 1.5 and 5 Osm. After incubation the reaction was quenched by adding an equal volume of 2 molal betaine glycine, 20 mM imidazole (pH 7.0), and 60 mM NaCl. The distribution of the EcoRI between specific fragment and specific oligonucleotide was measured using the gel mobility shift assay. Under these conditions, binding is complete within 5 min incubation and the dissociation half-life time of the specific sequence complex is >6 h even at the lowest osmotic pressure (Sidorova and Rau, 2001). Thus, the distribution of EcoRI between the specific DNA fragment and specific oligonucleotide on the 10-min timescale of the experiment is determined only by association rates.
The coupled reaction scheme we consider is:
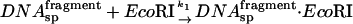 |
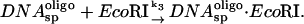 |
If the ratio of specific site concentrations, 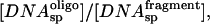 is C and the fraction of specific sequence fragment DNA complex measured after the 10-min incubation is f and f* in the presence and absence of specific sequence oligonucleotide, respectively, then the ratio of association rates, k1/k3, is given by,
is C and the fraction of specific sequence fragment DNA complex measured after the 10-min incubation is f and f* in the presence and absence of specific sequence oligonucleotide, respectively, then the ratio of association rates, k1/k3, is given by,
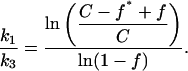 |
(1) |
Gel mobility-shift experiments
Reaction mixtures from equilibrium and kinetic experiments were electrophoresed in a 10% polyacrylamide gel, TAE (45 mM Tris, 22.5 mM acetic acid, 1 mM EDTA, pH 8.3) buffer. Samples were loaded on a gel at 150 V, and gel was run for 40 min at this voltage. The voltage was then reduced to 60 V and the gel was run overnight at 20°C to separate free DNA fragments and EcoRI-bound complexes. EcoRI-specific DNA fragment complexes are remarkably stable in the polyacrylamide gels, no change in fractions of bound fragment was observed between 8- and 16-h runs.
Electrophoretic bands containing free DNA and DNA-protein complex were stained with the fluorescent dye SYBR Green I (Molecular Probes, Eugene, OR). The summed intensities of the two bands are constant within 5% over a wide range of protein binding (from no added protein to 70% of the DNA present as complex), indicating there is no significant difference in staining efficiency of free and EcoRI-bound DNA. The gels were imaged with a luminescent image analyzer LAS-1000 plus (Fuji Film, Valhalla, NY) that includes a 1.3-megapixel cooled charge-coupled device camera, epi-illumination at 470 nm (light-emitting diode), and a dichroic optical filter suitable for SYBR Green I. The LAS-1000 plus was interfaced to a Pentium (Intel, Santa Clara, CA) PC. Band intensities were quantified using ImageGauge (v.3.122) for Windows. The linearity of DNA fluorescent staining over the range of DNA concentrations studied was confirmed using pBR322 DNA fragments generated by MspI digestion.
Equilibrium competition data analysis
As was developed previously (Sidorova and Rau, 1996), the ratio of specific (sp) and nonspecific (nonsp) association binding constants (Ksp/Knonsp) can be determined from the loss of specifically bound complex as the concentration of a nonspecific oligonucleotide competitor DNA is increased. If fb and 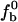 are the fractions of EcoRI-bound specific sequence fragment with and without added oligonucleotide competitor, then under conditions of virtually stoichiometric protein binding and for much weaker nonspecific than specific binding (Knonsp « Ksp) the change in specific sequence binding is given by,
are the fractions of EcoRI-bound specific sequence fragment with and without added oligonucleotide competitor, then under conditions of virtually stoichiometric protein binding and for much weaker nonspecific than specific binding (Knonsp « Ksp) the change in specific sequence binding is given by,
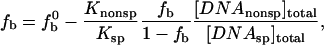 |
(2) |
where [DNAnonsp]total and [DNAsp]total are the molar concentrations of the competing oligonucleotide and the specific sequence fragment, respectively. Relative binding constants, Ksp/Knonsp, were straightforwardly calculated from the linear dependence of fb on 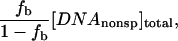 measured at constant specific sequence DNA and protein concentrations. The difference in the numbers of solute excluding waters between specifically and nonspecifically bound protein is calculated from the dependence of Ksp/Knonsp on the solute osmolal concentration (Parsegian et al., 1995, 2000) by
measured at constant specific sequence DNA and protein concentrations. The difference in the numbers of solute excluding waters between specifically and nonspecifically bound protein is calculated from the dependence of Ksp/Knonsp on the solute osmolal concentration (Parsegian et al., 1995, 2000) by
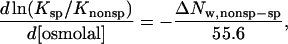 |
(3) |
where ΔNw,nonsp-sp = Nw,sp − Nw,nonsp, the difference in the numbers of waters associated with specific and nonspecific complexes that exclude solute.
THEORY
The dissociation rate from noncognate DNA sequence complexes can be determined by adding EcoRI to a mixture of noncognate oligonucleotide and specific sequence DNA fragment and measuring the time course for specific binding. Under conditions of fast association, the rate of appearance of specific complex depends on three factors: 1), the ratio of specific and nonspecific association rate constants; 2), the ratio of specific sequence fragment and nonspecific oligonucleotide concentrations; and 3), the nonspecific complex dissociation rate.
The coupled reaction scheme we consider is:
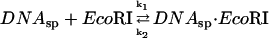 |
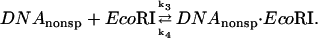 |
The rate constants are DNA length dependent. The dissociation rate constants from specific and nonspecific sites are very different. For proteins as EcoRI that initially bind nonspecifically then diffuse along the DNA in search of recognition sequences (Ehbrecht et al., 1985; Jack et al., 1982; Terry et al., 1985; Wright et al., 1999), association rate constants for specific and nonspecific sites are not expected to differ significantly for DNA of the same length.
We solve the kinetic equations with two assumptions. First, virtually all protein is bound to DNA over the time course of the experiment. For the experimental conditions used here, this means that the specific site concentration should be at least 1 nM. This is also equivalent to stating that the association rates, k1[DNAsp]tot and k3[DNAnonsp]tot, are fast compared to the experimental timescale. Kinetic measurements of protein association in the absence of oligonucleotide show that binding is complete within 5 min (data not shown). Secondly, virtually all protein is bound to the specific site fragment at equilibrium. This is equivalent to stating that the dissociation half-life time from the specific site fragment is much longer than the timescale of the experiment. Our extensive measurements of specific site dissociation rates confirm this (Sidorova and Rau, 2001).
With these assumptions,
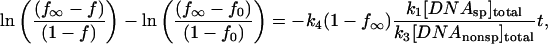 |
(4) |
where f is the fraction of specific site DNA fragment with bound protein at time t, f0 = f at t = 0, and f∞ = f at equilibrium. Under the conditions of the experiment, f∞, can be measured from protein binding in the absence of oligonucleotide and f0 can be calculated from the transcendental equation,
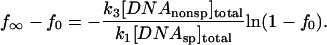 |
(5) |
For [DNAnonsp]total ≫ [DNAsp]total, f0 is small and can be approximated by,
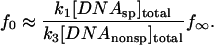 |
(6) |
Equation 4 for the rate of appearance of DNA fragment with bound protein can be rewritten as,
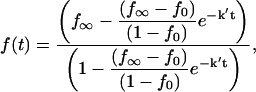 |
(7) |
where
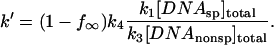 |
(8) |
These expressions are somewhat more complicated than most dissociation rate equations because the second-order nature of the reaction of protein with specific sequence fragment is included. The apparent rate constant, k′, depends on the ratio of DNA concentrations because after dissociation of nonspecifically bound protein the probability of associating with the specific sequence DNA fragment rather than with another oligonucleotide is approximately proportional to k1[DNAsp]total/k3[DNAnonsp]total. The ratio of association constants k1/k3 can be well estimated from the initial partitioning of EcoRI between the 360-bp specific site DNA fragment and specific recognition sequence oligonucleotide, i.e., under conditions such that the association time is much faster and the dissociation rate much slower than the incubation time. We find that k1/k3 = 3.7 ± 0.6 for the pressure range 1 and 5 Osm as calculated from Eq. 1.
RESULTS
Equilibrium competition
The general strategy is the same as described earlier (Sidorova and Rau, 1996, 1999). EcoRI binding free energies of different DNA sequences relative to the specific sequence were measured using a competition assay. The ratio of binding constants for the specific fragment and competing oligonucleotide were extracted from the decrease in binding to the specific sequence DNA fragment due to the presence of the competitor. Changes in specific EcoRI binding were determined using the gel mobility shift assay. Fig. 1 a shows a typical gel image illustrating the competition assay. The fluorescently stained bands show the variation in free DNA fragment and protein-DNA complex as betaine glycine osmotic pressure is increased at constant competitor oligonucleotide concentration. EcoRI binding to the specific sequence fragment is enhanced with higher pressures. Fig. 1 b shows the osmotic stress dependence of the relative binding free energies for the TAATTC “star” sequence that differ from the canonical site by a single incorrect basepair and the CTTAAG inverted sequence competitor oligonucleotides calculated with Eq. 2. In the absence of added osmolyte, the association binding constant to the inverted sequence oligonucleotide is ∼1.2 × 104 times smaller than the binding constant to the specific site. The TAATTC “star” sequence oligonucleotide binds EcoRI ∼2.3-fold more strongly than the inverted sequence oligonucleotide. The dependence of ln(Ksp/Knonsp) on osmotic pressure for the inverted sequence oligonucleotide-specific site competition is linear throughout the entire 2-Osm pressure range for both betaine glycine and α-methyl glucoside including the no-added solute, Π = 0, limit. This linearity along with our previous observation of the linearity of ln(kd) vs. Π for specific complex dissociation (Sidorova and Rau, 2001) again including the Π = 0 limit indicates that either solutes do not bind to the specific or nonspecific complexes or that solute binding is inconsequential for DNA-EcoRI binding. The slope translates into ΔNw = 108 ± 5 water molecules in a good agreement with previous results (Sidorova and Rau, 1996, 2001). Relative binding energies for competition between the TAATTC “star” sequence oligonucleotide and the specific site fragment are linear only through ∼1–1.5 Osm. The slope in this region corresponds to a difference of 105 ± 7 waters. The twofold stronger binding to this “star” sequence oligonucleotide compared to the inverted sequence oligonucleotide means that at least half the protein bound to the “star” sequence oligonucleotide at low stresses is directly associated with the TAATTC sequence. Thus, half of the osmotic dependence of ln(Ksp/Knonsp) is the contribution from the “star” sequence complex itself. There is, therefore, no significant difference between water sequestered by a nonspecific complex and the TAATTC “star” sequence complex at low stresses.
FIGURE 1.

(a) A polyacrylamide gel image illustrating that osmotic pressure favors specific sequence EcoRI binding over nonspecific binding in competition experiments. The gel mobility shift assay is used to monitor EcoRI binding to a 360-bp fragment containing its recognition sequence. The concentrations of protein, specific site fragment, and inverted sequence oligonucleotide competitor are constant as the concentration of the osmolyte betaine glycine is increased. The fraction of specific sequence complex is determined from fluorescent intensities of the SYBR Green I stained DNA bands. The short 30-bp competitor oligonucleotide is not seen on the gel. The particular conditions were: 120 mM NaCl, 20 mM imidazole pH 7.6, 20°C. Competitor oligonucleotide concentration is 14 μM; EcoRI and specific fragment concentrations are 0.75 and 1.5 nM, respectively. (b) The dependence of ln(Ksp/Knonsp) on betaine glycine or methyl glucoside osmolal concentration is shown for two noncognate oligonucleotides. The ratio of association binding constants for specific and nonspecific sequences, Ksp/Knonsp, was calculated from experiments as shown in Fig. 1 a using Eq. 2. The nonspecific competitors are the TAATTC “star” sequence oligonucleotide (•, betaine; ○, methyl glucoside) and the nonspecific CTTAAG inverted sequence oligonucleotide (▪, betaine; □, methyl glucoside). Except for the central six basepairs, the two 30-bp oligonucleotides are otherwise identical; full sequences are given in Methods and Materials. The error bars (shown for betaine) are calculated from three to four experiments at each osmotic pressure. Experimental conditions are given in Methods and Materials. The slopes at low pressures correspond to 105–110 more waters sequestered by the nonspecific sequence complexes compared with the specific site complex. The linear dependence on osmotic pressure for competition with the inverted sequence oligonucleotide indicates there is no change in the number of water sequestered by this nonspecific complex throughout the entire pressure range. The TAATTC sequence complex, however, apparently begins to lose water at osmotic pressures higher than 1–1.5 Osm.
At higher stresses the slope of the plot for the TAATTC “star” sequence oligonucleotide complex decreases. This nonlinearity indicates either a differential loss of water from the “star” sequence complex or osmolyte binding. The solute binding explanation demands that both betaine glycine and α-methyl glucoside bind not only with very similar energies and extents but also specifically to the “star” sequence complex, because both solutes continue to act osmotically on the competition between the inverted and specific sequence complexes. We believe this unlikely and consider probable that the nonlinearity reflects a loss of water from the “star” sequence complex. The average slope in the 1–2 osmolal range corresponds to ∼85 waters. Competition with the CAATTC “star” site oligonucleotide was 1.6-fold stronger than with the inverted sequence oligonucleotide and showed similar nonlinear behavior at higher osmotic stresses (data not shown).
Nonspecific complex dissociation kinetics
Measurement of competitive equilibrium binding at even higher pressures is difficult due to the very slow dissociation rates of the specific sequence complex and the impractically high oligonucleotide DNA concentrations needed for competition. Dissociation rates of EcoRI complexes, however, can be measured at very high osmotic pressures and give nearly the same osmotic stress information as equilibrium measurements. The dissociation rate of noncognate complexes can be measured using the gel mobility shift assay from the kinetics of the specific sequence complex formation after EcoRI is added to a mixture of the specific sequence fragment and noncognate oligonucleotide. Fig. 2 a shows a gel image illustrating the time course of specific complex appearance in the presence of 250-fold excess of the TAATTC oligonucleotide at 3.6 Osm betaine glycine. Fig. 2 b shows the dependence of the specific complex fraction on time and the fit to Eq. 7, allowing the initial and final fractions of EcoRI bound DNA fragment, f0 and f∞, respectively, and the apparent rate constant for specific site binding, k′, to vary. Setting f0 = 0 affected the fit negligibly. Enough protein was added such that f∞ is ∼0.5. The nonspecific dissociation rate constant, k4, calculated from Eq. 8 is 2.4 min−1. Under the same conditions, no dissociation from the specific recognition sequence complex can be observed within the time course of the experiment. If the rate is estimated by extrapolating our previous data (Sidorova and Rau, 2001), k2 ∼ 8 × 10−6 min−1.
FIGURE 2.

(a) A polyacrylamide gel illustrating the kinetics of specific EcoRI binding to the 360-bp DNA fragment in the presence of the TAATTC “star” sequence oligonucleotide. In the absence of oligonucleotide, specific sequence binding is complete within 5 min. The dissociation rate of EcoRI from the TAATTC oligonucleotide complex is determined from the time dependence. The gel mobility shift assay is used to monitor the gain of specific EcoRI-DNA complex. EcoRI (1.5 nM) was incubated for different times with a mixture of specific sequence DNA fragment (3 nM) and the TAATTC sequence oligonucleotide. The particular conditions used in this experiment were 90 mM NaCl, 3.95 Osm betaine glycine, 20 mM imidazole (pH 7.0), and 20°C. The TAATTC sequence oligonucleotide was present in 250-fold molar excess over the specific sequence fragment. (b) The fraction of the specific EcoRI-DNA fragment complex is shown plotted against time for the gel shown in panel a. The solid line is the best fit of Eq. 7 to the data. The dissociation rate constant k4 for this complex was calculated as 2.4 min−1.
Equation 4 predicts a dependence of the rate of appearance of specific complex on the ratio of specific sequence fragment and oligonucleotide concentrations because this determines the probability for a free protein to encounter the specific sequence fragment or noncognate oligonucleotide. We have explicitly confirmed this DNA concentration dependence. Fig. 3 a shows a gel image illustrating the decrease in specific fragment complex formed after 1 h incubation at 3.6 Osm betaine glycine as the concentration of the TAATTC oligonucleotide increases. Fig. 3 b shows the dependence of the specific complex fraction on oligonucleotide concentration. In Fig. 3 c the data is plotted using Eq. 4. The observed linearity over a wide range of concentrations for this “star” sequence oligonucleotide is consistent with the kinetic scheme. This DNA concentration dependence was confirmed for all three noncognate oligonucleotides at betaine glycine concentrations varying from 1.5 to 5 Osm and for [DNAnonsp]total/[DNAsp]total ratios ranging from 50 to 1000.
FIGURE 3.

(a) The fraction of specific sequence complex formed after one hour incubation decreases as the concentration of noncognate oligonucleotide increases. EcoRI (1.5 nM) was incubated with specific sequence DNA fragment (3 nM) and varying concentrations of noncognate oligonucleotides in 75 mM NaCl, 20 mM imidazole (pH 7.0), 3.6 Osm betaine, and at 20°C. The gel mobility shift assay was used to monitor specific complex formation. (b) The dependence of the specific EcoRI-DNA complex fraction on TAATTC sequence oligonucleotide concentration for the gel shown in panel a. (c) The linear dependence of 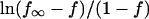 on
on 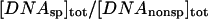 confirms the concentration dependence predicted by Eq. 4. The dissociation rate constant, k4, of the TAATTC “star” sequence oligonucleotide complex can be calculated from the slope. For this particular experiment, k4 is 0.86 min−1.
confirms the concentration dependence predicted by Eq. 4. The dissociation rate constant, k4, of the TAATTC “star” sequence oligonucleotide complex can be calculated from the slope. For this particular experiment, k4 is 0.86 min−1.
Osmotic stress dependence of the nonspecific dissociation rate
A clear difference is seen in the effect of osmotic pressure on the dissociation rates of EcoRI from the three oligonucleotides that differ only in the central six basepairs. Fig. 4 shows kinetic curves for the appearance of the EcoRI-specific 360-bp fragment complex in the presence of the three noncognate oligonucleotides at two osmotic pressures. Fig. 4 a shows the kinetics at 2.0 Osm betaine glycine with a 1000-fold molar excess of each oligonucleotide over specific site fragment. The dissociation rates for the two “star” sequence oligonucleotide complexes are ∼50% slower than for the inverted sequence oligonucleotide complex. Under the same conditions, dissociation from the specific recognition site is too slow to be observed. We estimate from our previous work an expected reaction half-life time of ∼60 h. Fig. 4 b shows that at 4.3 Osm betaine glycine and with a 100-fold molar excess of oligonucleotide over specific sequence fragment, the dissociation rate of the TAATTC oligonucleotide complex is now ∼10-fold slower than for the inverted sequence oligonucleotide complex. The dissociation rate of the CAATTC star sequence oligonucleotide complex is intermediate.
FIGURE 4.

Noncognate complex dissociation rates are strongly dependent on osmotic stress and the oligonucleotide sequence. Dissociation of EcoRI from the noncognate oligonucleotides is monitored by the increase in protein binding to the specific 360-bp DNA fragment with the incubation time. (a) In 2 Osm betaine glycine, the kinetic curves are similar for the three oligonucleotide complexes. All three noncognate oligonucleotides are present at 1000-fold molar excess over the specific DNA fragment. The salt and pH conditions used in this experiment were 90 mM NaCl, 20 mM imidazole (pH 7.0), 20°C. (b) At 4.3 Osm betaine glycine, the dissociation rates are clearly different for the three oligonucleotides. Both of the “star” sequence complexes dissociate substantially more slowly than the nonspecific inverted sequence complex. Of the “star” sequence complexes, the dissociation rate of the TAATTC oligonucleotide complex is significantly slower than that of the CAATTC oligonucleotide complex. All three noncognate oligonucleotides are present in 100-fold molar excess over specific DNA fragment.
The osmotic sensitivities of the EcoRI dissociation rate constant for both “star” sequence complexes and for the inverted sequence complex are shown in Fig. 5. Each rate constant shown was determined from full kinetic curve as shown in Figs. 2 and 4 and is the average of two to four measurements. Error bars are shown for the inverted sequence and TAATTC “star” sequence oligonucleotides. The EcoRI dissociation rate of the nonspecific oligonucleotide with the central inverted six-basepair sequence depends linearly on osmotic pressure over the entire range of stresses with a slope corresponding to 15 waters. The extrapolated dissociation rate of the inverted sequence oligonucleotide complex at 0 Osm is 40 min−1.
FIGURE 5.

The dependence of ln(k4) for EcoRI dissociation on betaine glycine osmolal pressure is shown for three noncognate complexes, with the TAATTC and CAATTC “star” sequence oligonucleotides and the nonspecific inverted CTTAAG sequence oligonucleotide. Error bars are given for the TAATTC and inverted sequence oligonucleotide complexes. The solid lines show the best linear fit to the nonspecific inverted sequence complex data, quadratic fit to the CAATTC complex data, and cubic fit to the TAATTC complex data. The nonspecific inverted sequence complex shows a linear osmotic dependence over the entire range of pressures translating in a constant uptake of ∼15 waters typical for a normally hydrated nonspecific complex. The nonlinearity for the “star” sequence complexes indicates an apparent loss of water from these complexes or stabilization of alternate conformations that sequester fewer waters. At 5 Osm the osmotic sensitivity of the k4 translates into a loss of ∼90 waters from the amount initially present in the TAATTC complex, leaving only ∼20 waters sequestered in the complex. The dashed line shows the best fit to the TAATTC complex data using Eq. 9 for a two-state model. The particular salt and pH conditions used in these experiments were 90 mM NaCl, 20 mM imidazole (pH 7.0), 20°C. The ratio of specific sequence DNA fragment and noncognate oligonucleotide was adjusted to give a convenient timescale for each experiment. Data points for k4 and associated error bars were extracted from two to four full kinetic curves measured at a fixed betaine glycine concentration.
At low osmotic pressures, the dissociation rates for the two “star” sequence oligonucleotides are somewhat smaller than for the nonspecific, inverted sequence oligonucleotide complex, but the osmotic pressure dependence is similar. At 2 Osm the dissociation rate of the TAATTC oligonucleotide complex is ∼1.6-fold smaller than the dissociation rate of the nonspecific oligonucleotide complex compared with the ratio of 2.3 for binding constants seen in Fig. 1 b. A clear deviation from linearity is seen for the TAATTC oligonucleotide complex at high osmotic pressures. Although not as dramatic, the CAATTC oligonucleotide also shows substantial nonlinearity at high osmotic pressures. Once again this nonlinearity could either reflect a change in the number of water molecules associated with the complex or betaine glycine binding. Because this solute continues to act osmotically on the dissociation rate of the inverted sequence complex through the entire osmotic pressure range and because we have previously observed that it also continues to act osmotically on the specific sequence complex through the highest pressure measured (∼3 Osm) (Sidorova and Rau, 2001), we consider it likely that betaine glycine continues to act osmotically on the “star” sequence complexes.
The solid lines in Fig. 5 show cubic and quadratic fits to the TAATTC and CAATTC “star” sequence data, respectively. The slopes calculated from these fits at 1.5 Osm give 30 and 20 waters coupled with the dissociation of protein from the TAATTC and CAATTC oligonucleotides, respectively. At 5 Osm, the slopes calculated from the polynomial fits correspond to an uptake of some 105 waters coupled with the dissociation from the TAATTC oligonucleotide complex and 45 waters for the CAATTC oligonucleotide complex. Of these waters 15 can be attributed to water coupled to nonspecific complex dissociation. At 5 osmolal, therefore, the TAATTC “star” sequence complex has lost an apparent 90 water molecules compared with the inverted sequence complex. This is comparable to the difference of 105–110 water molecules between specific and nonspecific EcoRI binding seen from competition equilibrium experiments at low osmotic pressures (Fig. 1 b). The CAATTC “star” sequence complex has lost ∼30 water molecules compared with the inverted sequence complex at 5 Osm.
The data for the TAATTC oligonucleotide complex can also be fit to a two-state model. We consider in particular a scheme in which a hydrated “star” sequence complex is in equilibrium with a complex that is dehydrated,
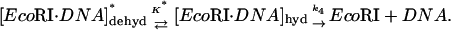 |
If the difference in associated waters between the two complexes is 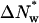 waters, then, at an osmotic pressure Π Osm,
waters, then, at an osmotic pressure Π Osm,
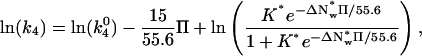 |
(9) |
where 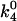 is k4 at 0 Osm. The 15 waters linked to the dissociation of protein from the hydrated complex have been explicitly incorporated. The dashed line in Fig. 5 shows the best fit of this equation to the TAATTC “star” sequence complex data. The best-fitting parameters are K* = 210 ± 80 and
is k4 at 0 Osm. The 15 waters linked to the dissociation of protein from the hydrated complex have been explicitly incorporated. The dashed line in Fig. 5 shows the best fit of this equation to the TAATTC “star” sequence complex data. The best-fitting parameters are K* = 210 ± 80 and 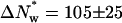 waters. From the equilibrium constant, the alternate, dehydrated complex is 3.2 kcal/mol less stable than the hydrated state within this model. Not enough water is lost from the CAATTC oligonucleotide complex to give a fit to a two-state model with <50% standard errors and reasonable dependencies.
waters. From the equilibrium constant, the alternate, dehydrated complex is 3.2 kcal/mol less stable than the hydrated state within this model. Not enough water is lost from the CAATTC oligonucleotide complex to give a fit to a two-state model with <50% standard errors and reasonable dependencies.
DISCUSSION
The binding of the restriction nuclease EcoRI to varying DNA sequences is exceptionally stringent. A change of even a single basepair from the recognition sequence, GAATTC, is sufficient to lower the binding free energy to values essentially characteristic of nonspecific complexes (Lesser et al., 1990). This is unlike many other sequence specific DNA-binding proteins, as the lac (Frank et al., 1997) and λ-Cro repressors (Takeda et al., 1989, 1992), that show a more gradual decrease in binding energy as the consensus recognition sequence is altered. As seen in Fig. 1, the binding competition at 20°C between a DNA fragment containing the specific recognition sequence and a 30-bp nonspecific inverted sequence CTTAAG oligonucleotide gives an association binding constant ratio of Ksp/Knonsp = 1.2 × 104. The same oligonucleotide but with a central TAATTC “star” sequence binds EcoRI only ∼2.3-fold stronger with no osmolyte. Another oligonucleotide with a central CAATTC “star” sequence binds ∼1.6-fold more tightly than the inverted sequence oligonucleotide. These 30-bp oligonucleotides, of course, contain several nonspecific binding sites. The EcoRI binding constants to these oligonucleotides relative to the specific sequence (Ksp/Knonsp) reflect binding competition for the whole oligonucleotide, not just the central six basepairs that we use to identify the oligonucleotide. Differences in binding constants and osmotic stress sensitivities among the oligonucleotides, however, do reflect difference in the binding properties of these central sequences. The approximate twofold increase in binding constant for the “star” sequence oligonucleotide complexes indicates that about half of the protein at low stresses is bound to the “star” sequence itself.
Accompanying the precipitous loss of binding energy with even a single basepair change is a large increase in the number of waters associated with the complex. The EcoRI complexes with all three oligonucleotides initially include 105–110 more waters than the specific complex as measurements at low osmotic pressures show (Fig. 1 b). The absence of any intermediate hydration state for the “star” sequence complexes would suggest there are two stable and distinct binding modes of the protein to DNA, perhaps analogous to the structures of the specific and nonspecific DNA complexes of BamHI (Newman et al., 1995; Viadiu and Aggarwal, 2000).
The equilibrium measurements of the binding competition between specific site and noncognate sequences in Fig. 1, however, reveal a difference between “star” sequence and nonspecific complexes at higher osmotic pressures. In contrast to the inverted sequence oligonucleotide complex that shows linear behavior through 2 Osm implying a constant number of sequestered waters, the TAATTC “star” sequence complex shows a decreasing slope at higher stresses suggesting an apparent loss of water by the complex or that an alternate, dehydrated conformation is stabilized. This apparent loss of water could be connected to other differences between EcoRI complexes with nonspecific and “star” sequences. It has long been known that as with many other type II restriction endonucleases EcoRI is capable of cleaving “star” sequences with low activity (Polisky et al., 1975; Goodman et al., 1977; Tikchonenko et al., 1978; Hsu and Berg, 1978; Malyguine et al., 1980; Gardner et al., 1982; Rosenberg and Greene, 1982; Lesser et al., 1990). This “star” activity can be promoted by modifying reaction conditions, in particular, by the addition of neutral solutes to the reaction (Goodman et al., 1977; Robinson and Sligar, 1993). Robinson and Sligar (1993) further showed that the increased cleavage of “star” sequences by EcoRI associated with neutral osmolytes directly correlates with water activity. One possible explanation for this effect is that osmotic stress modulates the equilibrium between the inactive binding mode of the enzyme and an energetically unfavorable, but enzymatically active mode of EcoRI binding to “star” sequences.
The apparent numbers of water molecules lost by the “star” sequence complexes, however, are quite modest at 2 Osm, ∼20 for the TAATTC sequence complex. The equilibrium experiments are quite difficult at higher osmotic stresses because dissociation rates of the protein from specific sequence become very slow and the concentrations of oligonucleotide DNA needed for competition become problematically large. The osmotic stress dependence of noncognate complex dissociation rates, however, can in principle give the same information about differences in associated water as equilibrium measurements, but can be measured to very high osmotic stresses. The osmotic sensitivity of the dissociation rate is determined by the difference in the number of waters between the initial binding state and the final dissociation transition state. The rate-limiting step for many protein-DNA complexes including EcoRI is the release of protein from a nonspecific complex. As we previously reported (Sidorova and Rau, 2001) the number of waters coupled with specific sequence dissociation is comprised of the 110 waters coupled to the difference between specific and nonspecific complexes. Another ∼10 waters for betaine glycine are linked to the dissociation of nonspecifically bound EcoRI, that is, there is a difference of ∼10 waters between the nonspecific complex and the dissociation transition state.
Dissociation rates of nonspecific complexes are not commonly measured. Typically, these rates are too fast for convenient measurement and can be monitored directly only with difficulty. The method we use might be of general interest and application. Dissociation rates of noncognate complexes can be extracted from the kinetics of appearance of specific site complex after EcoRI is added to a mixture of specific sequence fragment DNA and noncognate oligonucleotide under conditions of fast association rates. Three criteria indicate that we indeed are measuring nonspecific complex dissociation rates. The predicted dependence of the observed dissociation rate on noncognate sequence oligonucleotide concentration was confirmed (Fig. 3). The observed osmotic sensitivity of the inverted sequence oligonucleotide dissociation rate corresponding to ∼15 waters (Fig. 5) is consistent with 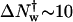 waters for betaine glycine extracted previously (Sidorova and Rau, 2001) from specific site off-rates for the release of protein from a nonspecific complex. Lastly and most importantly, the dissociation rate constant for an EcoRI specific site complex is determined by the nonspecific dissociation rate, k4, and the ratio of specific and nonspecific dissociation equilibrium constants (Sidorova and Rau, 2001),
waters for betaine glycine extracted previously (Sidorova and Rau, 2001) from specific site off-rates for the release of protein from a nonspecific complex. Lastly and most importantly, the dissociation rate constant for an EcoRI specific site complex is determined by the nonspecific dissociation rate, k4, and the ratio of specific and nonspecific dissociation equilibrium constants (Sidorova and Rau, 2001),
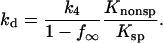 |
(10) |
The ratio of equilibrium constants can be calculated, therefore, from the dissociation rate constant for the inverted sequence oligonucleotide complex extrapolated to 0 Osm, k4 ∼ 4.1 × 101 min−1, and for the specific site dissociation rate at the same salt, pH, temperature, and osmotic pressure measured previously and corrected for the difference in length, kd(1 − f∞) ∼ 2.9 × 10−3 min−1. The ratio of equilibrium constants calculated from dissociation rates, 1.4 × 104, is in excellent agreement with the direct equilibrium measurement of 1.2 × 104 shown in Fig. 1.
We have used here a single osmolyte, betaine glycine, at high osmotic pressures to vary water activity. Betaine glycine is an excellent solute for these high osmotic pressure studies because of its high solubility and strong exclusion from proteins (Courtenay et al., 2000; Timasheff, 1993) coupled with its low viscosity. In our previous work (Sidorova and Rau, 1996) we investigated many solutes differing by size and nature and found that the action of all those examined on EcoRI-DNA binding is osmotic at least up to 1.6 Osm. Robinson and Sligar (1993, 1998) examined still other solutes and reported an osmotic effect on EcoRI binding and cleavage through at least 2 Osm. The nonlinear dependences of ln(Ksp/Knonsp) (Fig. 1 b) and of ln(k4) (Fig. 5) on osmotic pressure seen for “star” sequence complexes signifies that these complexes are either binding solute or losing water. We argue for the continued osmotic action of betaine glycine for several reasons. First, betaine glycine continues to act osmotically (linearly) on the nonspecific sequence complex competition with the specific complex at the same pressures that the “star” sequence complex competition begins showing nonlinearity (Fig. 1 b). A second solute, α-methyl glucoside shows the same nonlinearity at higher pressures for the “star” sequence complex in the competition assay as betaine glycine (Fig. 1 b). It is unlikely these two solutes would bind identically. We have previously seen that both α-methyl glucoside and betaine glycine continue to act osmotically on the dissociation of the specific sequence complex at least through 3 Osm (Sidorova and Rau, 2001). Betaine glycine continues to act osmotically on the dissociation of the nonspecific, inverted sequence oligonucleotide complex through the entire 6 Osm pressure range. It is improbable that betaine glycine binds differently to the “star” sequence oligonucleotide complexes than to specific or inverted sequence oligonucleotide complexes. The data indicate that betaine glycine very likely continues to act osmotically on the “star” sequence complex through high pressures and that the nonlinearity of the plots in Figs. 1 b and 5 indicates the “star” sequence complexes are losing water.
At low osmotic pressures (<2 Osm), the dissociation rate of the TAATTC “star” sequence complex is ∼1.6-fold slower than the nonspecific complex, comparable to the twofold change seen in the ratio of specific-noncognate equilibrium constants. Approximately 30 water molecules are seen coupled with the dissociation of this “star” sequence complex at low osmotic pressure, slightly larger than ∼15 waters seen for the inverted sequence oligonucleotide complex. The apparent loss of 15 more waters by the TAATTC complex is comparable to the decrease of ∼20 waters (from 105 to 85 waters) estimated for this complex from the osmotic dependence of the equilibrium measurements for the same pressure range (Fig. 1 b). Because the dissociation transition state remains unchanged as is indicated by the continued linearity of the inverted sequence dissociation rate data, the initial TAATTC complex is losing water with increasing osmotic pressure. A cubic fit to the data indicates that at 5-Osm pressure the TAATTC complex has lost some 90 waters compared with the inverted sequence oligonucleotide complex. The osmotic work, W, associated with the loss of ΔNw waters over the osmotic pressure range 0–Π Osm is given by:
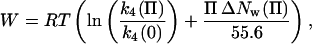 |
(11) |
where the thermal factor RT = 0.6 kcal/mol. The work needed to remove these 90 waters at 5 Osm from the TAATTC oligonucleotide complex is ∼3.5 kcal/mol. The osmotic energy gained through the release of sequestered water to the bulk solution is balanced by the resulting unfavorable interactions between DNA and protein surfaces.
The TAATTC oligonucleotide complex data can be well fit to a simple two-state model assuming equilibrium between the two states with different numbers of associated waters. The best fit gives a difference of ∼105 waters between the two states. Although we do not know where this water is located, it seems likely it is the same excess water in the nonspecific complex that is lost upon specific binding. The dehydrated state would then resemble the specific recognition sequence complex. The best fit also gives a free energy for the alternate dehydrated complex in the absence of solute that is ∼3.2 kcal/mol less favorable than the hydrated state. A more complicated two-state model that accounts for the fraction of protein actually bound to the “star” sequence itself changes the fitting parameters only slightly, ΔG = −2.8 kcal/mol and ΔNw = 90.
It is substantially more difficult to remove water from the CAATTC “star” sequence oligonucleotide complex. From a quadratic fit to the data, only ∼25 waters are apparently lost at 5 Osm from the hydrated complex initially present at low pressures. The osmotic work necessary to remove these 25 waters, ∼0.9 kcal/mol, compared with ∼0.45 kcal/mol required to remove the first 25 waters from the TAATTC “star” sequence complex. Although not enough water is lost from the CAATTC oligonucleotide complex to give a meaningful two-state model fit, an energy difference between the two states of ∼6.2 kcal/mol can be estimated if 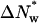 is fixed at 105 waters as for the TAATTC oligonucleotide complex.
is fixed at 105 waters as for the TAATTC oligonucleotide complex.
Of the “star” sequences, the TAATTC is the most readily cleaved by EcoRI and CAATTC the most cleavage resistant of the first basepair substitutions. Lesser et al. (1990) estimated energy differences for EcoRI binding to the cleavage transition state between the recognition sequence and various “star” sequences at 20°C. In particular, a free energy of 6.6 kcal/mol was reported for a TAATTC “star” sequence and 9.5 kcal/mol for a CAATTC sequence. We can compare these values with the energy differences we estimate for the dehydrated complexes of the TAATTC and CAATTC “star” sequences relative to specific sequence complex. This comparison is strictly valid only if the activation energy differences between the initial complexes with specific and “star” sequences and the cleavage transition state are small. The oligonucleotides used here and by Lesser et al. (1990) have different flanking sequences that can strongly affect activity (Jen-Jacobson, 1997) and also have different lengths and consequently different numbers of possible nonspecific sites that will affect relative binding energies. Nonetheless, from the competition experiments, with no osmolyte both TAATTC and CAATTC oligonucleotide complexes are ∼5 kcal/mol less stable than the specific recognition sequence complex. The dehydrated, TAATTC complex is an additional ∼3 kcal/mol less stable than the hydrated binding mode complex, or, therefore, ∼8 kcal/mol less stable than the specific recognition sequence complex. Similarly, we estimate that the dehydrated CAATTC complex is ∼11 kcal/mol less stable than the specific sequence complex. The agreement between our results and Lesser at al. (1990) is reasonable enough to suggest that the dehydrated “star” sequence complex is an enzymatically active form. In particular, both methods give ∼3 kcal/mol for the difference between TAATTC and CAATTA complexes. This difference should be much less sensitive to cleavage activation energies and the effect of flanking sequences and DNA length.
CONCLUSIONS
On the basis of the osmotic stress measurements, the nonspecific complex for EcoRI binding seems to be a well-defined structure. All noncognate sequence complexes we have investigated, even sequences that differ by only a single first basepair from the recognition sequence, sequester 105–110 waters more than the specific sequence complex. In analogy with the nonspecific complex of BamHI (Viadiu and Aggarwal, 2000), the protein likely contacts the sugar-phosphate backbone of DNA rather than bases in the major groove. The small differences in noncognate binding constants among sequences, e.g., the approximate twofold increase in binding for “star” sequences seen here to compare with inverted sequence could simply result from an indirect readout of sequence dependent helical parameters. A difference between “star” sequence and nonspecific complexes, however, is revealed at higher osmotic pressures. “Star” sequence complexes appear to lose water as the higher osmotic pressures or an alternate conformation with fewer sequestered waters is stabilized. If analyzed as two states, the alternate complex is almost as dehydrated as the specific sequence complex. Osmotic stress can be used to modulate the energy difference between a nonspecific complex and this specific-like binding mode. The work needed to remove virtually all the water from the “star” sequence complex, indicates that this dehydrated first basepair “star” sequence complex is destabilized by an estimated 8–11 kcal/mol from the specific sequence complex, depending on the “star” sequence. This large energy difference is consistent with a rigid and interconnected protein recognition surface that is unable to adapt to recognize mismatches. In this way the x-ray structure of the specific EcoRI complex (Kim et al., 1990; McClarin et al., 1986; Rosenberg, 1991) is different from lac (Lewis et al., 1996) or λ-Cro repressor-DNA (Albright and Matthews, 1998) structures that do not show the extensive DNA-protein and protein-protein bonding interconnections seen for EcoRI binding. The loss of water from the “star” sequence complexes provides a structural basis for the enzymatic “star” activity of EcoRI and a natural link between “star” activity and osmotic pressure as was anticipated by Pingoud and Jeltsch (1997). We cannot unambiguously discriminate between a gradual loss of water from the star sequence complexes and the two-state model. The calculated energies to remove water will depend somewhat on the actual mechanism, but only slightly. More importantly, the key component that differentiates EcoRI binding to various sequences is found in the water sequestered by the complexes.
Acknowledgments
We are deeply grateful to V. A. Parsegian for the valuable discussion and support, to L. Jen-Jacobson for the kind gift of highly purified EcoRI, and to Shakir Muradymov for his enthusiastic help with the experiments.
References
- Albright, R. A., and B. W. Matthews. 1998. Crystal structure of lambda-Cro bound to a consensus operator at 3.0 A resolution. J. Mol. Biol. 280:137–151. [DOI] [PubMed] [Google Scholar]
- Courtenay, E. S., M. W. Capp, C. F. Anderson, and M. T. Record, Jr. 2000. Vapor pressure osmometry studies of osmolyte-protein interactions: implications for the action of osmoprotectants in vivo and for the interpretation of osmotic stress experiments in vitro. Biochemistry. 39:4455–4471. [DOI] [PubMed] [Google Scholar]
- Davis-Searles, P. R., A. S. Morar, A. J. Saunders, D. A. Erie, and G. J. Pielak. 1998. Sugar-induced molten-globule model. Biochemistry. 37:17048–17053. [DOI] [PubMed] [Google Scholar]
- Ehbrecht, H. J., A. Pingoud, C. Urbanke, G. Maass, and C. Gualerzi. 1985. Linear diffusion of restriction endonucleases on DNA. J. Biol. Chem. 260:6160–6166. [PubMed] [Google Scholar]
- Frank, D. E., R. M. Saecker, J. P. Bond, M. W. Capp, O. V. Tsodikov, S. E. Melcher, M. M. Levandovski, and M. T. Record, Jr. 1997. Thermodynamic of the interactions of lac repressor with variants of the symmetric lac operator: effects of converting a consensus site to a non-specific site. J. Mol. Biol. 267:1186–1206. [DOI] [PubMed] [Google Scholar]
- Fried, M. G., and D. M. Crothers. 1981. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 9:6505–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried, M. G., D. F. Stickle, K. V. Smirnakis, C. Adams, D. MacDonald, and P. Lu. 2002. Role of hydration in the binding of lac repressor to DNA. J. Biol. Chem. 277:50676–50682. [DOI] [PubMed] [Google Scholar]
- Gardner, R. C., A. J. Howarth, J. Messing, and R. J. Shepherd. 1982. Cloning and sequencing of restriction fragments generated by EcoRI*. DNA. 1:109–115. [DOI] [PubMed] [Google Scholar]
- Garner, M. M., and D. C. Rau. 1995. Water release associated with specific binding of gal repressor. EMBO J. 14:1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner, M. M., and A. Revzin. 1981. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucl. Acids Res. 9:3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, H. M., P. J. Green, D. E. Garfin, and H. W. Boyer. 1977. DNA site recognition by the EcoRI restriction endonuclease and modification methylase. In Nucleic Acid: Protein Recognition. H. J. Vogel, editor. Academic Press, New York. 329–359.
- Hard, T., and T. Lundback. 1996. Thermodynamics of sequence-specific protein-DNA interactions. Biophys. Chem. 62:121–139. [DOI] [PubMed] [Google Scholar]
- Hsu, M., and P. Berg. 1978. Altering the specificity of restriction endonuclease: effect of replacing Mg2+ with Mn2+. Biochemistry. 17:131–138. [DOI] [PubMed] [Google Scholar]
- Jack, W. E., B. J. Terry, and P. Modrich. 1982. Involvement of outside DNA sequences in the major kinetic path by which EcoRI endonuclease locates and leaves its recognition sequence. Proc. Natl. Acad. Sci. USA. 79:4010–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch, A., J. Alves, H. Wolfes, G. Maas, and A. Pingoud. 1994. Pausing of the restriction endonuclease EcoRI during linear diffusion on DNA. Biochemistry. 33:10215–10219. [DOI] [PubMed] [Google Scholar]
- Jen-Jacobson, L. 1997. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 44:153–180. [DOI] [PubMed] [Google Scholar]
- Khrapunov, S., and M. Brenowitz. 2004. Comparison of the effect of water release on the interaction of the Saccharomyces cerevisiae TATA binding protein (TBP) with TATA box sequences composed of adenosine or inosine. Biophys. J. 86:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., J. C. Grable, R. Love, P. J. Greene, and J. M. Rosenberg. 1990. Refinement of EcoRI endonuclease crystal structure: a revised protein chain tracing. Science. 249:1307–1309. [DOI] [PubMed] [Google Scholar]
- Lesser, D. R., M. R. Kurpiewski, and L. Jen-Jacobson. 1990. The energetic basis of specificity in the EcoRI endonuclease-DNA interaction. Science. 250:776–786. [DOI] [PubMed] [Google Scholar]
- Lewis, M., G. Chang, N. C. Hortin, M. A. Kercher, H. C. Pace, M. A. Schumacher, R. G. Brennan, and P. Lu. 1996. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 271:1247–1254. [DOI] [PubMed] [Google Scholar]
- Li, L., and K. S. Matthews. 1997. Differences in water release with DNA binding by ultrabithorax and deformed homeodomains. Biochemistry. 36:7003–7011. [DOI] [PubMed] [Google Scholar]
- Lundback, T., H. Hansson, S. Knapp, R. Ladenstein, and T. Hard. 1998. Thermodynamic characterization of non-sequence-specific DNA-binding by the Sso7d protein from Sulfolobus solfataricus. J. Mol. Biol. 276:775–786. [DOI] [PubMed] [Google Scholar]
- Lynch, T. W., and S. G. Sligar. 2000. Macromolecular hydration changes associated with BamHI binding and catalysis. J. Biol. Chem. 275:30561–30565. [DOI] [PubMed] [Google Scholar]
- Malyguine, E., P. Vannier, and P. Yot. 1980. Alteration of the specificity of restriction endonucleases in the presence of organic solvents. Gene. 8:163–177. [DOI] [PubMed] [Google Scholar]
- McClarin, J. A., C. A. Frederick, B.-C. Wang, H. W. Boyer, J. Grable, and J. M. Rosenberg. 1986. Structure of the DNA-EcoRI endonuclease recognition complex at 3 A resolution. Science. 234:1526–1541. [DOI] [PubMed] [Google Scholar]
- Newman, M., T. Strzelecka, L. F. Dorner, I. Schildkraut, and A. K. Aggarwal. 1995. Structure of BamHI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science. 269:656–663. [DOI] [PubMed] [Google Scholar]
- Parsegian, V. A., R. P. Rand, and D. C. Rau. 1995. Macromolecules and water: probing with osmotic stress. Methods Enzymol. 259:43–94. [DOI] [PubMed] [Google Scholar]
- Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: a comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud, A., and A. Jeltsch. 1997. Recognition and cleavage of DNA by type-II restriction endonucleases. Eur. J. Biochem. 246:1–22. [DOI] [PubMed] [Google Scholar]
- Polisky, B., P. Greene, D. E. Garfin, B. J. McCarthy, H. M. Goodman, and H. W. Boyer. 1975. Specificity of substrate recognition by the EcoRI restriction endonuclease. Proc. Natl. Acad. Sci. USA. 72:3310–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, J., M. Bailey, D. J. Winzor, B. E. Davidson, and W. H. Sawyer. 1997. Effects of molecular crowding on the interaction between DNA and the Escherichia coli regulatory protein TyrR. Biophys. J. 73:3257–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisler, R. S., H.-H. Chen, M. F. Colombo, Y. Choe, B. J. Short, Jr., and D. C. Rau. 1995. The B-form to Z-form transition of polu(dG-m5dC) is sensitive to neutral solutes through an osmotic stress. Biochemistry. 34:14400–14407. [DOI] [PubMed] [Google Scholar]
- Robinson, C. R., and S. G. Sligar. 1993. Molecular recognition mediated by bound water. a mechanism for star activity of the restriction endonuclease EcoRI. J. Mol. Biol. 234:302–306. [DOI] [PubMed] [Google Scholar]
- Robinson, C. R., and S. G. Sligar. 1996. Participation of water in Hin recombinase-DNA recognition. Protein Sci. 5:2119–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, C. R., and S. G. Sligar. 1998. Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease. Proc. Natl. Acad. Sci. USA. 95:2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, J. M. 1991. Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol. 1:104–113. [DOI] [PubMed] [Google Scholar]
- Rosenberg, J. M., and P. Greene. 1982. EcoRI* specificity and hydrogen bonding. DNA. 1:117–124. [DOI] [PubMed] [Google Scholar]
- Schwabe, J. W. R. 1997. The role of water in protein-DNA interactions. Curr. Opin. Struct. Biol. 7:124–134. [DOI] [PubMed] [Google Scholar]
- Sidorova, N. Y., and D. C. Rau. 1996. Differences in water release for the binding of EcoRI to specific and nonspecific DNA sequences. Proc. Natl. Acad. Sci. USA. 93:12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova, N. Y., and D. C. Rau. 1999. Removing water from an EcoRI-noncognate DNA complex with osmotic stress. J. Biomol. Struct. and Dynam. 17:19–31. [DOI] [PubMed] [Google Scholar]
- Sidorova, N. Y., and D. C. Rau. 2000. The dissociation rate of the EcoRI-DNA specific complex is linked to water activity. Biopolymers. 53:363–368. [DOI] [PubMed] [Google Scholar]
- Sidorova, N. Y., and D. C. Rau. 2001. Linkage of EcoRI dissociation from its specific DNA recognition site to water activity, salt concentration, and pH: separating their roles in specific and non-specific binding. J. Mol. Biol. 310:801–816. [DOI] [PubMed] [Google Scholar]
- Swaminathan, C. P., U. T. Sankpal, D. N. Rao, and A. Surolia. 2002. Water-assisted dual mode cofactor recognition by HhaI DNA methyltranferase. J. Biol. Chem. 277:4042–4049. [DOI] [PubMed] [Google Scholar]
- Szwajkajzer, D., and J. Carey. 1997. Molecular and biological constraints on ligand-binding affinity and specificity. Biopolymers. 44:181–198. [DOI] [PubMed] [Google Scholar]
- Takeda, Y., P. D. Ross, and C. P. Mudd. 1992. Thermodynamics of Cro protein-DNA interactions. Proc. Natl. Acad. Sci. USA. 89:8180–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda, Y., A. Sarai, and V. M. Rivera. 1989. Analysis of the sequence-specific interactions between Cro repressor and operator DNA by systematic base substitution experiments. Proc. Natl. Acad. Sci. USA. 86:439–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry, B. J., W. E. Jack, and P. Modrich. 1985. Facilitated diffusion during catalysis by EcoRI endonuclease. Nonspecific interactions in EcoRI catalysis. J. Biol. Chem. 260:13130–13137. [PubMed] [Google Scholar]
- Tikchonenko, T. I., E. V. Karamov, B. A. Zavizion, and B. S. Naroditsky. 1978. EcoRI* activity: enzyme modification of activation of accompanying endonuclease. Gene. 4:195–212. [DOI] [PubMed] [Google Scholar]
- Timasheff, S. N. 1993. The control of protein stability and association by weak interactions with water: how do solvents affect these processes? Annu. Rev. Biophys. Biomol. Struct. 22:27–65. [DOI] [PubMed] [Google Scholar]
- Viadiu, H., and A. K. Aggarwal. 2000. Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol. Cell. 5:889–895. [DOI] [PubMed] [Google Scholar]
- Vossen, K. M., R. Wolz, M. A. Daugherty, and M. G. Fried. 1997. Role of macromolecular hydration in the binding of the Escherichia coli cyclic AMP receptor to DNA. Biochemistry. 36:11640–11647. [DOI] [PubMed] [Google Scholar]
- Wenner, J. R., and V. Bloomfield. 1999. Osmotic pressure effects on EcoRV cleavage and binding. J. Biomol. Struct. Dyn. 17:461–471. [DOI] [PubMed] [Google Scholar]
- Wright, D. J., W. E. Jack, and P. Modrich. 1999. The kinetic mechanism of EcoRI endonuclease. J. Biol. Chem. 274:31896–31902. [DOI] [PubMed] [Google Scholar]
- Wu, J., K. M. Parkhurst, R. M. Powell, M. Brenowitz, and L. J. Parkhurst. 2001. DNA bends in TATA-binding protein-TATA complexes in solution are DNA sequence-dependent. J. Biol. Chem. 276:14614–14622. [DOI] [PubMed] [Google Scholar]