

Abstract
Time-resolved flavin fluorescence anisotropy studies on glutathione reductase (GR) have revealed a remarkable new phenomenon: wild-type GR displays a rapid process of fluorescence depolarization, that is absent in mutant enzymes lacking a nearby tyrosine residue that blocks the NADPH-binding cleft. Fluorescence lifetime data, however, have shown a more rigid active-site structure for wild-type GR than for the tyrosine mutants. These results suggest that the rapid depolarization in wild-type GR originates from an interaction with the flavin-shielding tyrosine, and not from restricted reorientational motion of the flavin. A novel mechanism of fluorescence depolarization is proposed that involves a transient charge-transfer complex between the tyrosine and the light-excited flavin, with a concomitant change in the direction of the emission dipole moment of the flavin. This interaction is likely to result from side-chain relaxation of the tyrosine in the minor fraction of enzyme molecules in which this residue is in an unsuitable position for immediate fluorescence quenching at the moment of excitation. Support for this mechanism is provided by binding studies with NADP+ and 2′P-5′ADP-ribose that can intercalate between the flavin and tyrosine and/or block the latter. Fluorescence depolarization analyses as a function of temperature and viscosity confirm the dynamic nature of the process. A comparison with fluorescence depolarization effects in a related flavoenzyme indicates that this mechanism of flavin fluorescence depolarization is more generally applicable.
INTRODUCTION
Polarized time-resolved fluorescence is a well-established method to investigate the dynamics of molecular systems (Lakowicz, 1999). Whereas fluorescence lifetime experiments yield information on the microenvironment of the fluorophore, fluorescence depolarization studies are widely applied for investigating rotational diffusion and segmental motions. For proteins, processes such as aggregation, dissociation, denaturation, and folding may be easily detected through fluorescence depolarization. In addition, the technique can be used to monitor the viscosity of the environment and changes therein, such as in membranes or cell systems. Fluorescence labeling is generally used to attach suitable fluorophores to lipids, DNA, and proteins. For the latter, one could in principle use the intrinsic fluorescence of tryptophan residues. In practice, using tryptophan fluorescence is complicated by interconversion between two electronic levels in the first absorption band that are nearly degenerate (Creed, 1984), resulting in fast fluorescence depolarization (Ruggiero et al., 1990). In addition, tryptophan fluorescence is often difficult to interpret owing to the presence of more than one Trp residue. However, external fluorescence labeling is not suitable for all systems. Besides inconvenient mobility of the label itself, which may be avoided by bimodal labeling techniques, steric aspects may not always allow the introduction of a fluorescence label. The delicacy of the molecular structure and of the dynamics may also play a role. Intrinsic protein fluorescence is therefore preferable for studying processes such as the active-site dynamics of enzymes. Preeminently suited for this are enzymes that contain a single naturally fluorescent reporter group in the active-site region, such as flavoenzymes.
The characteristic green fluorescence of the flavin cofactor has provided detailed information on the active-site dynamics of a variety of enzymes that contain flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN) as a redox-active prosthetic group (for an overview, see van den Berg and Visser, 2001). Flavin fluorescence depolarization studies have shed light on possible dissociation of the cofactor, local mobility of the isoalloxazine ring, and—if fluorescence lifetime and molecular mass allowed it—overall protein tumbling. In addition, structural information on multimeric flavoproteins has been obtained from time-resolved anisotropy studies through (homo)energy transfer between the flavin cofactors (Bastiaens et al., 1992a,b; Visser et al., 1998). For the dimeric Azotobacter vinelandii lipoamide dehydrogenase, a small rapidly depolarizing process was originally interpreted as local mobility of the fluorophore (de Kok and Visser, 1987). Later studies with more advanced techniques and comparison of the results with the interflavin distance and orientation of the flavins, as became available from structural data of the enzyme (Mattevi et al., 1991), showed that not mobility but homoenergy transfer between the flavin cofactors from the two different subunits is the source of depolarization (Bastiaens et al., 1992a,b). Bastiaens et al. (1992a) compared the active-site dynamics of A. vinelandii lipoamide dehydrogenase with those of glutathione reductase (GR) from human erythrocytes, an enzyme with a largely similar active-site structure. Fluorescence depolarization studies on erythrocyte GR revealed in addition to a small amount of homoenergy transfer as in lipoamide dehydrogenase, a more rapidly depolarizing process with large amplitude that was assigned to restricted reorientational mobility of the isoalloxazine ring (Bastiaens et al., 1992b).
In the active-site region, the most striking difference between glutathione reductase and lipoamide dehydrogenase is that GR contains a tyrosine residue adjacent to the flavin (Tyr-197 in human erythrocyte GR, Tyr-177 in Escherichia coli GR; Thieme et al., 1981; Karplus and Schulz, 1987; Mittl and Schulz, 1994). Whereas the active site of lipoamide dehydrogenase is easily accessible for the nicotinamide cofactor, the binding cleft for NADPH in GR is blocked by this tyrosine residue (Karplus and Schulz, 1987). The catalytic mechanism of GR therefore includes a movement of this tyrosine away from the flavin (Pai and Schulz, 1983). Crystallographic analysis of complexes of GR with NADPH, and several analogs and fragments thereof, indeed showed a full or partial flip of the tyrosine side chain toward an “out” position that allows flavin reduction (Pai et al., 1988; Karplus and Schulz, 1987, 1989; Mittl et al., 1994; Fig. 1).
FIGURE 1.

Representation of part of the active-site structure of E. coli glutathione reductase. Shown are the relative positions of the isoalloxazine ring of FAD and Tyr-177 in the free enzyme (top panel; Mittl and Schulz, 1994) and in the enzyme complexed with NADP+ (bottom panel; Mittl et al., 1994). Note that in the latter case Tyr-177 has moved away from the isoalloxazine.
More recently, a detailed time-resolved fluorescence investigation was performed on the role of the flavin-shielding tyrosine residue in the active-site dynamics of E. coli GR (van den Berg et al., 1998). The tertiary structure of E. coli GR is nearly identical to the enzyme from human erythrocytes (Mittl and Schulz, 1994), although only 52% sequence homology exists (Greer and Perham, 1986), and the enzyme lacks the 16 N-terminal residues of erythrocyte GR. To reveal the role of the flavin-shielding Tyr-177 (equivalent to Tyr-197 in erythrocyte GR), wild-type E. coli GR was compared with mutants in which this residue had been replaced by site-directed mutagenesis. Fluorescence lifetime data revealed that in ∼90% of the free enzyme molecules, Tyr-177 directly interacts with the light-excited flavin resulting in flavin fluorescence quenching with a lifetime constant of only 7 ps (van den Berg et al., 1998). Based on the temperature invariance of this process and absorption spectra, and supported by redox calculations and observations of tyrosine radicals in other flavoenzymes, the ultrafast fluorescence quenching was explained by photoinduced electron transfer from the tyrosine to the adjacent flavin. The temperature and viscosity dependencies of the fluorescence lifetime data clearly showed that the mutants GR Y177F and GR Y177G have a more flexible protein structure than wild-type GR.
Preliminary experiments have shown a different time-dependent fluorescence anisotropy decay of wild-type E. coli GR as compared to the tyrosine mutants GR Y177F and GR Y177G (van den Berg et al., 1998). In this article, a more detailed time-resolved fluorescence anisotropy investigation of wild-type E. coli GR and the tyrosine mutants GR Y177F and GR Y177G is presented. To unravel the nature of the processes inducing fluorescence depolarization, wild-type E. coli GR is studied as a function of temperature and viscosity, and in complex with substrates and analogs thereof. Based on the results, a novel mechanism of fluorescence depolarization is proposed that involves a shift in the direction of the emission dipole moment of the flavin resulting from a transient charge-transfer interaction with the adjacent tyrosine.
MATERIALS AND METHODS
Enzyme material and sample preparation
Wild-type and mutant E. coli glutathione reductases were purified from the gor-deleted E. coli strains NS3 and SG5, respectively, transformed with the appropriate expression plasmid (Scrutton et al., 1987). The purification was based on the method described by Berry et al. (1989) and is in detail described elsewhere (van den Berg et al., 1998). Wild-type and mutant glutathione reductase from human erythrocytes were a kind gift from Professor R. H. Schirmer, Heidelberg University, Germany. Pure enzymes were stored in 80% ammonium sulfate at 277 K. Before use, possible traces of free FAD were removed by chromatography on a Biogel PGD-6 column (Biorad, Hercules, CA) equilibrated with the appropriate measuring buffer. Measurements were carried out in 50 mM potassium phosphate buffer pH 7.6, at 293 K, except for the titrations with 2′P-5′ADP-ribose for which 50 mM MOPS buffer pH 7.6, was used. The enzyme concentration was kept between 9 and 11 μM with respect to FAD (maximum optical density of 0.10 at the excitation wavelength, with ɛ463 GR = 11.3 mM−1 cm−1 (Williams, 1976). Buffers were made from nanopure-grade water (Millipore, Billerica, MA) and were filtered through a 0.22-μm filter (Millipore). 2′P-5′ADP-ribose and NADP+ were purchased from Sigma (St. Louis, MO). All chemicals used were of the highest purity available. Samples containing glycerol were prepared by gently mixing the eluted protein with fluorescent-grade glycerol (Merck, Whitehouse Station, NJ). Before and after a series of fluorescence experiments, flavin absorption spectra (Aminco DW2000 spectrophotometer, SLM Instruments, Urbana, IL) were measured at 298 K to check the quality of the samples.
Time-resolved fluorescence and fluorescence anisotropy measurements
Time-resolved polarized fluorescence experiments were carried out using the time-correlated single-photon counting technique (TCSPC) (O'Connor and Phillips, 1984). The TCSPC setup and the measurement procedures used were described in detail elsewhere (van den Berg et al., 1998), and will only be shortly outlined below. A mode-locked CW Nd:YLF laser was used for the synchronously pumping of a cavity-dumped dye laser. Stilbene 420 and Coumarin 460 (Exciton, Dayton, OH) were used as dyes for excitation at 450 nm and 460 nm, respectively. The samples were excited with vertically polarized light with an excitation frequency of 594 kHz (duration 4 ps, full-width at half maximum (FWHM)) and both parallel and perpendicularly polarized fluorescence was detected. At 450-nm excitation, fluorescence was detected with a 557.9-nm interference filter (Schott, Mainz, Germany) (FWHM of 11.8 nm) in combination with a KV 520 cutoff filter (Schott). At 460-nm excitation, fluorescence was detected with a 526.0-nm interference filter (Schott, FWHM of 12.6 nm) in combination with a KV 520 cutoff filter (Schott), to avoid interference from Raman scattering of water (located at 551 nm at 460-nm excitation). The use of filters for fluorescence selection allows an optical scheme with high collection efficiency. A disadvantage is that reflections of the filters from and toward the highly reflective (and also flat) cathode of the photomultiplier cannot always be avoided, depending on the possible angle of the filter with respect to the optical axis. These reflections may show up in the fluorescence and anisotropy decay as shoulders in the graph of these decays (see, for instance, Fig. 2). In Figs. 3 and 4 no shoulders were found because the filter angle was different in that particular measurement. Reflections are similarly present in the decay curves of the reference compound. Therefore with the application of a deconvolution procedure these reflections do not affect the recovered decay times. The data were collected in a multichannel analyzer with a time window of 1024 channels at typically 15 ps per channel. For better time resolution in the picosecond domain, additional data of wild-type GR were collected at 7.0 ps per channel. The dynamic instrumental response function of the setup is ∼40–50 ps FWHM, and was obtained at the emission wavelength using erythrosine B in water (τ = 80 ps at 293 K) as a reference compound (Bastiaens et al., 1992b).
FIGURE 2.

(a) Experimental fluorescence anisotropy decay (noisy gray line) of wild-type GR at 293 K (50 mM potassium phosphate buffer pH 7.6, data acquired at 7.0 ps/channel), and corresponding theoretical data as obtained from the nonassociative fitting with five lifetime components and two rotational correlation times (solid gray line), and from the associative fitting with five lifetime components and a model for anisotropy decay as described in Table 1 (solid black line). The total fluorescence decay normalized to 0.5 (dotted line) has been presented for clarity. Note the difference in the quality of both fits in the first 600 ps of the anisotropy decay as illustrated by the weighted residuals presented for nonassociative (b) and associative fitting (c). The excitation wavelength was 450 nm and the emission was detected at 557.9 nm.
FIGURE 3.

Fluorescence depolarization of wild-type E. coli GR at 293 K (in 50 mM potassium phosphate buffer, pH 7.6) as a function of the NADP+ concentration. The excitation wavelength was 460 nm and the emission was detected at 526.0 nm.
FIGURE 4.

Fluorescence depolarization of wild-type E. coli GR at 293 K (in 50 mM MOPS, pH 7.6) as a function of the concentration 2′P-5′ADP ribose (ADPR). The excitation wavelength was 460 nm and the emission was detected at 526.0 nm.
Data analysis
Data analysis was performed using a model of discrete exponential terms. Global, nonassociative, and associative fitting of the experimental data was performed using the TRFA data-processing package of the Scientific Software Technologies Center (Belarusian State University, Minsk, Belarus) (Digris et al., 1999).
The total fluorescence intensity decay I(t) and anisotropy decay r(t) are obtained from the measured parallel I‖(t) and perpendicular I⊥(t) fluorescence intensity components through the relations:
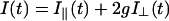 |
(1) |
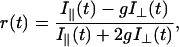 |
(2) |
in which the g-factor describes the sensitivity of the detection system for the perpendicular component with respect to the parallel one. For the setup used, the g-factor equals unity (van Hoek et al., 1987). The fluorescence lifetime profile consisting of a sum of discrete exponentials with lifetime τi and amplitude αi can be retrieved from the total fluorescence I(t) through:
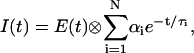 |
(3) |
where E(t) is the instrumental response function. Fluorescence lifetime analysis of the GR enzymes was employed using a five-component model as confirmed by the fluorescence lifetime distributions obtained with the maximum entropy method (MEM) described by van den Berg et al. (1998).
In fluorescence anisotropy analysis, after deconvolution the time-dependent fluorescence anisotropy r(t) is calculated from the parallel I‖(t) and perpendicular I⊥(t) components through the relations (Lakowicz, 1999):
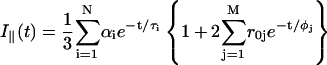 |
(4) |
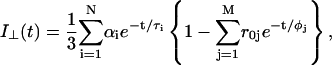 |
(5) |
in which i = j for correlated systems in which particular lifetime components τi are associated with particular correlation times φi (correlated or associative model), and i ≠ j for systems in which all lifetimes equally contribute to the anisotropy (uncorrelated or nonassociative model). In the case of uncorrelated fluorescence lifetimes and rotational correlation times the time dependence of the anisotropy after δ-pulse excitation can thus be described as:
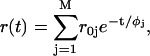 |
(6) |
where the sum of r0j is the fundamental initial anisotropy r0.
In a first approach, the nonassociative model was assumed in analyzing the anisotropy decay of the GR enzymes. Global analysis (Beechem, 1992) in which data sets were fitted simultaneously with a sum of discrete exponentials, were performed through linking the fluorescence lifetimes constants and/or the rotational correlation times for multiple data sets.
Further analysis of the anisotropy decay of wild-type GR was performed using the associative fitting model. Hereto, each fluorescence lifetime was considered to be related to an independent quenching process with a potentially process-specific anisotropy behavior (five models because of the presence of five lifetimes). First estimates of the rotational correlation parameters were obtained using a model of a single rotational correlation time per fluorescence lifetime, with fixed values for the lifetime parameters as obtained from the total decay analysis of the respective data set. To optimize the curve fitting at the very beginning of the anisotropy decay (including the leading edge in the first 150 ps), and in the region above ∼3 ns, two rotational correlation times were allowed for the fluorescence lifetime components of 7 ps and 2.6 ns. Theoretical rotational correlation times calculated for overall protein tumbling and homoenergy transfer were used to find proper starting conditions. In a final fitting procedure, freedom was allowed for all parameters from both fluorescence intensity and anisotropy decays.
Theoretical models for fluorescence depolarization
The rotational correlation time data were evaluated according to the well-established relations for overall protein tumbling and homoenergy transfer (Lakowicz, 1999). For a spherical protein, the correlation time for rotational diffusion φr is given by the Stokes-Einstein relation:
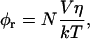 |
(7) |
where V is the molecular volume (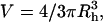 Rh is the Stokes radius), η the viscosity, N Avogadro's number, k the Boltzmann's constant, and T the absolute temperature. For a globular protein at 293 K, this relation is approximated by Visser and Lee (1980):
Rh is the Stokes radius), η the viscosity, N Avogadro's number, k the Boltzmann's constant, and T the absolute temperature. For a globular protein at 293 K, this relation is approximated by Visser and Lee (1980):
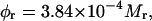 |
(8) |
in which Mr is the relative molecular mass of the protein in Da, and φr is in ns.
The correlation time for energy transfer φT between a donor and acceptor in a homodimer is defined as:
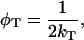 |
(9) |
in which the rate constant for energy transfer kT is given by the Förster equation (Förster, 1948):
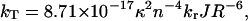 |
(10) |
where κ2 is the orientation factor for the relevant transition dipole moments, n the refractive index, kr the radiative fluorescence rate constant (ns−1) of the flavin, J the integrated spectral overlap of the flavin absorbance and fluorescence spectra (M−1 cm3), and R is the distance (nm) between the donor and acceptor. The value for the radiative fluorescence rate of the flavin is kr = 0.056 ns−1 (Visser and Müller, 1979), the refractive index was estimated to be n = 1.4 (Steinberg, 1971; Bastiaens et al., 1992a). The orientation factor κ2 was calculated from the orientation between the emission dipole moment of the donor flavin (Bastiaens et al., 1992a) and the absorption dipole moment of the acceptor flavin (Johansson et al., 1979) appearing in the crystal structure of GR, according to the method described by Dale et al. (1979).
RESULTS
Fluorescence anisotropy decay of glutathione reductase
Time-resolved flavin fluorescence anisotropy analysis of the E. coli GR enzymes showed that the polarization behavior of wild-type GR differs strongly from the tyrosine mutants GR Y177F and GR Y177G. For wild-type GR, a large depolarizing process was observed that is absent in the mutant enzymes where only little fluorescence depolarization occurs (van den Berg et al., 1998). In a first MEM approach, the fluorescence anisotropy decays were analyzed treating fluorescence lifetimes and rotational correlation times as uncorrelated parameters (van den Berg et al., 1998). Overall protein tumbling was not observed due to highly efficient flavin fluorescence quenching in the E. coli GR enzymes. Lifetime analysis of the highly heterogeneous flavin fluorescence intensity decays of these enzymes yields lifetime distributions with five components (van den Berg et al., 1998): for the mutant enzymes, lifetime components in the range between 0.1 and 2.5 ns yield an average fluorescence lifetime of 0.15 ns, whereas predominant ultrarapid quenching with a time constant of 7 ps resulted in an average fluorescence lifetime of only 27 ps for wild-type E. coli GR. The intensity of the fluorescence signal is therefore insufficient to clearly resolve a rotational correlation time of 38 ns, as can be expected from the modified Stokes-Einstein relation (Eq. 8) for a protein of 100 kDa at 293 K. For GR Y177F and GR Y177G, a correlation time in the range between 2 and 7 ns was resolved, but this process only contributed with very small amplitude (van den Berg et al., 1998). In 80% v/v glycerol between 293 K and 223 K, this depolarizing effect was invariably described by a correlation time of 6–7 ns. This time constant agrees with the small depolarizing process reported for human erythrocyte GR, and can be assigned to intramolecular energy transfer between the two flavins (Bastiaens et al., 1992b; Eqs. 9 and 10).
In wild-type E. coli GR, the large fluorescence depolarization can be described by a rotational correlation time of ∼2 ns at 293 K using the nonassociative model (van den Berg et al., 1998). In the uncorrelated fitting approach, however, the first half nanosecond of the anisotropy decay of wild-type GR could not be described adequately (Fig. 2). For this reason, associative analysis of the fluorescence lifetimes and rotational correlation times was performed to yield better insight in the fluorescence depolarization behavior of wild-type GR. Associative analysis is, however, severely complicated by the large number of potential parameters resulting from the five lifetime components of GR that may in principle be correlated with one or more different depolarization processes. Moreover, as a consequence of the extremely short average fluorescence lifetime of GR, the anisotropy signal in the nanosecond region is carried by a very low number of photons yielding a rather poor signal/noise ratio. Results of the associative analysis are therefore limited to a more qualitative description of clear tendencies and potential correlations.
Associative analysis of the anisotropy decay of wild-type GR drastically improved the quality of the fits within the first 600 ps of the anisotropy decay (Fig. 2). Unambiguous associations between lifetime and correlation time constants were obtained for the fluorescence lifetime components of 0.1 ns and 0.3 ns (Table 1). The 0.1-ns component does not show any significant depolarization and may be connected with overall protein tumbling. The 0.3-ns lifetime fully corresponds with a rapidly depolarizing process with a time constant of ∼2 ns at 293 K. Particularly this fluorescence lifetime seems responsible for the rapid depolarization that can easily be inspected in the experimental data by eye. For the longer lifetime components, it was more difficult to obtain clear correlations. Although the 1.0-ns and 2.6-ns fluorescence lifetime components only contribute to the total fluorescence decay for <1% each, they are the main carriers of the anisotropy signal in the nanosecond time region. Given the poor signal/noise ratio in this region, discrimination between a single or double depolarizing event, and an estimate of the time intervals for the corresponding correlation times could be made reasonably well. For the 1.0-ns lifetime component such analysis resulted in a single correlation time with a time constant between ∼2 and 3.5 ns. For the 2.6-ns component, two correlation times (φ1 ≈ 3–7 ns and φ2 > 30 ns) were fitted. For an adequate description of the “pseudoplateau” between 150 and 300 ps, and in particular, the slow ingrowth of anisotropy near 150 ps, also two rotational correlation times had to be allowed; one corresponding to the rotational diffusion of the protein, and a faster process with an ill-defined time constant in the region between 0.1 and 5 ns. The effect of changes in relative amplitude of the correlation times corresponding to the 7-ps lifetime was surprisingly large: not only the anisotropy features of the leading edge were determined by these amplitudes, but also proper fitting up to the first hundreds of picoseconds of the anisotropy decay. The presence of a rapidly depolarizing process connected with the ultrashort lifetime can thus be firmly established, although the time constant cannot be determined accurately enough to discriminate between a protein relaxation-related process or other sources of rapid depolarization. Although we cannot definitely exclude the presence of (sub)nanomolar traces of free FAD (φFAD = 0.2 ns and τ1−FAD (∼80%) = 7 ps at 293 K; van den Berg et al., 2002), we have no indications for it from experiments, nor from the associative analyses of the other fluorescence lifetime parameters; besides the predominant ultrashort lifetime of the conformation in which the flavin and adenine moieties are stacked, free FAD has a second lifetime component (τ = 2.7 ns) with considerable amplitude (∼15%) related to the “open” conformation (van den Berg et al., 2002). The minor 2.6-ns lifetime component obtained for E. coli GR, however, did not reveal fast depolarization of the order of 0.2 ns.
TABLE 1.
Potential relations between the fluorescence lifetime parameters and fluorescence depolarization processes in wild-type E. coli GR as retrieved from associative fitting
| Lifetime parameters (τi in ns, αi in %) | Correlation times (order of magnitude in ns) | Relative contributions (normalized per τi) |
|---|---|---|
| 0.007 (∼90%) | >30 | large (∼75%) |
| ∼0.1–5 | small (∼25%) | |
| 0.09 (∼8%) | >30 | – |
| 0.3 (∼2%) | 1.8–2.2 | – |
| 1.0 (<1%) | ∼2–3.5 | – |
| 2.6 (<1%) | ∼3–7 | large (∼85%) |
| >30 | small (∼15%) |
At 293 K, pH 7.6, in 50 mM potassium phosphate buffer.
The correlation time of 2 ns in E. coli GR is close to the time constant reported for the dominating rapid anisotropy decay in human erythrocyte GR (φ ≈ 1.5 ns; Bastiaens et al., 1992b) that was explained by assuming a restricted reorientational motion of the flavin. The fact that no such rapid depolarization is observed in the tyrosine mutants of E. coli GR, renders the interpretation of internal mobility questionable, the more as fluorescence lifetime data rather indicated a more mobile flexible structure for the mutant enzymes than for wild-type GR (van den Berg et al., 1998). To rule out the possibility of species-specific effects, human erythrocyte GR was reevaluated and compared with its mutant enzyme GR Y197S, in which the equivalent flavin-shielding tyrosine was replaced. Time-resolved fluorescence anisotropy experiments on these enzymes revealed the same phenomenon: the large rapid depolarization found in wild-type erythrocyte GR is absent in the mutant Y197S lacking the juxtaposed tyrosine, indicating a role for this particular residue in the mechanism of fluorescence depolarization (data not shown).
The role of Tyr-177 in flavin fluorescence depolarization
To unravel the nature of rapid fluorescence depolarization in GR and to validate the involvement of the flavin shielding tyrosine, wild-type E. coli GR was further tested by titration with substrate analogs that bind via the NADPH-binding cleft, but do not reduce the flavin. Crystal structures of the E. coli GR/NADP+ complex had shown that in this complex, the oxidized nicotinamide cofactor intercalates between the tyrosine and the flavin, so that the tyrosine side chain is shifted toward an “out” position. (Fig. 1; Mittl et al., 1994). Titrations of wild-type GR with NADP+ showed that upon complex formation, the amplitude of the rapidly depolarizing process diminishes, until full saturation occurs (Fig. 3). A similar effect was obtained with the substrate analog 2′P-5′ADP-ribose (Fig. 4). Analysis of crystals of human erythrocyte GR soaked with this analog showed the tyrosine in the “out” position for ∼50% of the enzyme/substrate complexes, whereas in the remaining ones the substrate analog blocked the NADPH entrance with the tyrosine side chain in the “in” position adjacent to the flavin (Pai et al., 1988). At full saturation with either NADP+ or 2′P-5′ADP-ribose, the anisotropy decay of wild-type GR was identical to that of the tyrosine mutants in the unliganded state. These results are consistent with the proposed interaction between the flavin and Tyr-177 causing rapid fluorescence depolarization. A prominent effect on the fluorescence anisotropy was observed for concentrations of 1–10 μM 2′P-5′-ADP-ribose, and 0.1–3 mM NADP+, in accordance with the lower binding affinity of the enzyme for the latter (Pai et al., 1988), in particular in the presence of free phosphate. Although beyond the scope of this investigation, accurate binding constants for the above-mentioned ligands could in principle be determined from the (steady-state) anisotropy. In titrations, a significant effect on the fluorescence lifetime patterns was observed (Table 2). NADP+ binding induced a decrease in amplitude of the ultrafast component and some shifts in the time constants of the components. Experiments with 2′P-5′ADP-ribose were performed in MOPS buffer, which itself caused a slight shift of the 0.1 and 0.3 lifetime constants (Table 2). Binding of 2′P-5′ADP-ribose reduced the amplitude of the ultrafast component with 20 ∼ 25%, and changed the lifetime components toward time constants as found for the enzyme/NADP+ complex. This decrease in amplitude is consistent with crystallographic data on 2′P-5′ADP-ribose (see Discussion). In the complex with NADP+, the remaining ultrafast lifetime component may originate from an efficient quenching interaction with the bound nicotinamide cofactor.
TABLE 2.
Fluorescence lifetime constants (τi) and fractional contributions (αi) of wild-type E. coli GR
| Potassium phosphate
|
MOPS
|
||||||
|---|---|---|---|---|---|---|---|
| wt GR
|
wt GR + NADP+
|
wt GR
|
wt GR + ADPR
|
||||
| τi (ns) | αi | τi (ns) | αi | τi (ns) | αi | τi (ns) | αi |
| 0.007 ± 0.001 | 0.90 | 0.007 ± 0.002 | 0.76 | 0.007 ± 0.001 | 0.82 | 0.005 ± 0.001 | 0.67 |
| 0.090 ± 0.008 | 0.076 | 0.12 ± 0.01 | 0.18 | 0.061 ± 0.009 | 0.13 | 0.11 ± 0.03 | 0.15 |
| 0.29 ± 0.04 | 0.020 | 0.37 ± 0.04 | 0.04 | 0.22 ± 0.02 | 0.04 | 0.34 ± 0.02 | 0.10 |
| 1.0 ± 0.2 | 0.003 | 0.76 ± 0.05 | 0.01 | 0.86 ± 0.2 | 0.004 | 0.73 ± 0.1 | 0.053 |
| 2.6 ± 0.3 | 0.002 | 2.1 ± 0.2 | 0.01 | 2.4 ± 0.1 | 0.002 | 2.0 ± 0.4 | 0.013 |
At 293 K, pH 7.6, in 50 mM potassium phosphate buffer, unliganded and saturated with NADP+; and 50 mM MOPS buffer, unliganded and saturated with 2′P-5′ADP-ribose (ADPR).
The mechanism of interaction between the flavin and Tyr-177 leading to rapid fluorescence depolarization was further investigated as a function of both temperature and solvent viscosity, using sucrose as a cosolvent. The fast correlation time of E. coli GR appeared to be strongly dependent on viscosity and temperature (Fig. 5). Increasing temperature from 277 K to 303 K caused a decrease in the fast correlation time by a factor of 2 (irrespective of the sucrose concentration used), whereas increasing the sucrose concentration from 0% to 60% resulted in a 2.5–3 times longer correlation time. In aqueous solution, the change in rotational correlation time roughly corresponded with the viscosity effect of changing temperature. The bulk viscosity increase induced with sucrose was larger than the change in correlation time corresponding to the rapid depolarization process. In high percentages of glycerol, no rapid fluorescence depolarization was observed. These results clearly show the dynamic nature of the interaction leading to rapid fluorescence depolarization.
FIGURE 5.

Temperature and viscosity dependencies of the fast correlation time of wild-type E. coli GR. Measurements were performed in the range between 277 and 303 K, in 50 mM potassium phosphate buffer, pH 7.6, with a percentage (w/w) sucrose varying from 0 to 60%.
DISCUSSION
Time-resolved flavin fluorescence anisotropy studies on glutathione reductase revealed a remarkable new phenomenon: wild-type GR exhibits rapid fluorescence depolarization that is absent in mutant enzymes lacking the flavin-shielding tyrosine. In accordance with the common interpretation of rapid fluorescence depolarization that cannot be explained by rotational diffusion or energy transfer, restricted segmental mobility was proposed for the isoalloxazine ring of the flavin cofactor in human erythrocyte GR (Bastiaens et al., 1992b). Based on fluorescence lifetime experiments of the E. coli GR enzymes (van den Berg et al., 1998), however, there is no reason to assume the flavin to be more mobile in wild-type GR than in the mutants lacking the tyrosine. In fact, temperature and viscosity dependencies of the fluorescence lifetime data indicated a more flexible protein structure for the GR Tyr-177 mutant enzymes. The rigidity of the microenvironment of the isoalloxazine ring of the flavin that was observed in fluorescence lifetime data of wild-type GR is in agreement with crystallographic studies revealing very low B-factors for the entire active center of GR (〈B〉 ≈ 10 Å2 in erythrocyte GR; Karplus and Schulz, 1987; Mittl and Schulz, 1994). For E. coli GR as well as human erythrocyte GR, the isoalloxazine ring of FAD is very rigidly bound (〈B〉 = 9.1/7.9 Å2 and 〈B〉 = 8.7 Å2, respectively), and cofactor mobility gradually increases toward the adenine end (〈B〉 = 22.9/17.1 Å2 and 〈B〉 = 16.2 Å2, respectively). On the basis of these preliminary data, the difference in fluorescence depolarization behavior between wild-type GR and the tyrosine mutants has led us earlier to propose a novel mechanism for fast anisotropy decay: flavin fluorescence depolarization may arise from the formation of a charge-transfer (CT) complex between the light-excited flavin and the adjacent tyrosine, under condition that the direction of the emission dipole moment in the CT complex is shifted (van den Berg et al., 1998).
Firm support for this depolarization mechanism has been obtained from these studies on binary complexes between wild-type E. coli GR and substrate analogs. The absence of fast depolarization upon blocking the position of the flavin-shielding tyrosine in either the “out” or the “in” conformation provides further evidence for the involvement of an interaction with this particular tyrosine residue. In crystallographic studies on the E. coli GR/NADP+ complex, the displacement of Tyr-177 to the “out” position (Fig. 1) was directly related to the degree of occupation of the nicotinamide binding site (Mittl et al., 1994). Crystals of human erythrocyte GR soaked with the analog 2′P-5′ADP-ribose lacking the nicotinamide moiety, showed Tyr-197 in the “out” conformation in ∼30% of the molecules (Pai et al., 1988). As the pyrophosphate moiety of this analog bound with an occupancy of ∼60%, it was concluded that the “ribose-in/Tyr-197-out” conformation and the “ribose-out/Tyr-197-in” conformation of the complex with 2′P-5′ADP-ribose are close in energy under crystallization conditions. Based on the crystallographic analysis of a series of fragments and analogs Pai et al. (1988) concluded that for movement of the tyrosine a ligand extending from adenine to at least the N-ribose was required. As the binding affinity for the ligands decreased from the adenine end toward the nicotinamide end, they suggested that productive binding of NADPH starts with binding of the adenine moiety to the preformed 2′-phosphate binding site, followed by an “induced fit” movement of Tyr-197 by the nicotinamide moiety. Important for the displacement of the flavin shielding was the small rotation of the α-helical residues 196–201 observed: because of steric effects, the ribose cannot bind properly without a movement of the side chains of Tyr-197 and in particular Ile-198. Although for E. coli GR, no crystallographic data of complexes with fragments of NADPH are available, similar effects and mechanisms are expected on the basis of the very high similarity of the active site structures of the E. coli and human erythrocyte enzyme (Mittl and Schulz, 1994). The fact that human erythrocyte GR and its mutant Y197S behaved identical to the corresponding E. coli enzymes in both fluorescence intensity decay and fluorescence depolarization (albeit with somewhat different time constants and amplitudes) is in accordance with this.
The dynamic origin of the fluorescence depolarization process in GR is clearly confirmed by its strong temperature and viscosity dependencies. Whereas increasing temperature accelerated fluorescence depolarization, increasing solvent viscosity slowed the process down. The rapid fluorescence depolarization in GR may thus reflect a relaxational process during the lifetime of the excited state of the flavin involving the flavin-shielding tyrosine. Light excitation itself is known to induce rapid (vibrational) relaxation of the protein environment near the fluorophore. This effect, similar to solvent relaxation of chromophores free in solution, generally takes place on timescales of femtoseconds to several picoseconds and affects the direct environment of the chromophore. The nanosecond fluorescence depolarization reported in this study is therefore likely to originate from a process that is either not, or indirectly influenced (e.g., through dipolar effects or long-range interactions) by light excitation. As the fluorescence depolarization signal in E. coli GR is only carried by photons from the minor fraction of GR molecules in which flavin fluorescence is not quenched almost instantaneously (α ≈ 90% for τ = 7 ps), an explanation for the nanosecond time constant may be relaxation from Tyr-177 from a more “out” position at the moment of excitation, to the “in” position in which the flavin and tyrosine can interact. It may be that in a fraction of the enzyme molecules the Tyr-177 remains in an “out” position throughout the lifetime of the excited state. Although the results of the associative fitting are certainly not conclusive, the (partial) correlation of the longest lifetime with overall protein tumbling and homoenergy transfer may be indicative of this.
All together, the fluorescence lifetime and fluorescence depolarization experiments on wild-type GR and the tyrosine mutants, supported by the substrate-binding, temperature, and viscosity experiments as well as crystallographic data, provide evidence for a novel mechanism of flavin fluorescence depolarization; during the lifetime of the excited state the flavin interacts with the nearby tyrosine and a CT complex is generated in which the emission dipole moment of the flavin is shifted. The basis for a charge-transfer mechanism for ultrafast fluorescence quenching in E. coli GR was discussed thoroughly (van den Berg et al., 1998, and references therein). Firm evidence for the formation of CT complexes between the light-excited isoalloxazine ring and juxtaposed tyrosine and tryptophan residues through photoinduced electron transfer was more recently also provided in time-resolved fluorescence and absorption studies with femtosecond resolution on the flavoproteins glucose oxidase and riboflavin-binding protein (Zhong and Zewail, 2001).
To observe fluorescence depolarization in a rigid CT complex, two prerequisites should be met. First, the direction of the emission dipole moment of the CT complex should shift with respect to that of the fluorophore itself. The relation between the direction of the emission dipole moment and the initial anisotropy r0, which is equivalent to the time-dependent anisotropy r(t) in a rigid system without further interactions, is given by (Lakowicz, 1999):
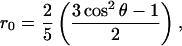 |
(11) |
where θ is the angle between the absorption and the emission dipole moments, and cos2 θ describes the average value for the angular displacement. Under the assumption that the end value of the anisotropy decay only reflects the CT complex, a very crude estimation for the angular displacement in wild-type GR (293 K) is ∼36–48° (Fig. 6). This estimation almost certainly oversimplifies the system: it neglects the possibility of molecules with a long lifetime of the excited state that do not form a CT complex, but show (little) fluorescence depolarization through energy transfer or overall rotation. However, as the tyrosine mutants of E. coli GR point out, the depolarization through homoenergy transfer is only very small (van den Berg et al., 1998), and the decrease in anisotropy as a result of overall protein tumbling is low due to the much longer time constant (∼38 ns). A similar shift in the direction of the emission dipole moment is likely to occur in the rapidly formed CT complex that leads to the 7 ps fluorescence lifetime constant. Because of the very short fluorescence lifetime, this major population hardly contributes to the fluorescence depolarization signal, but in principle, it may show up in a decreased value for r0, or in the leading edge of the depolarization curve. In the time-resolved fluorescence data, the leading edge of the fluorescence decay shows an increase in anisotropy with increasing fluorescence intensity, but significant differences between wild-type E. coli GR and the tyrosine mutants could not be resolved. A setup with a better time resolution may yield more insight into such effects.
FIGURE 6.

Schematic representation of the interaction causing depolarization by a change in direction of the emission transition moment from the initial “in-plane” (of the isoalloxazine ring) direction (μem) to an “out-of-plane” direction corresponding to the emission transition moment of the charge-transfer excited state (μct). The angle between the absorption and emission dipole moments in the CT complex is roughly estimated between ∼36 and 48°. The example is taken from the structure of E. coli GR.
A second prerequisite for observing fluorescence depolarization from a CT complex is that the complex is (partially) emissive. It was discussed previously that the formation of a CT complex may lead to very efficient nonradiative deexcitation pathways (van den Berg et al., 1998). This leaves unimpaired that for the nanosecond depolarization effect, at least a fraction of molecules contributing to the phenomenon, must have emitted light. A recent absorption and fluorescence study with femtosecond resolution on the flavoproteins glucose oxidase and riboflavin-binding protein leaves such possibility open; for the enzyme glucose oxidase, in which the flavin is surrounded by two tyrosines and two tryptophans (Hecht et al., 1993), transient absorption spectra revealed a rise time for the CT complex comparable to the dominant fluorescence lifetime (τ = 1.8 ps, α = 75%; Zhong and Zewail, 2001). The absorption decay of the CT state showed two components; one of ∼30 ps and one of nanoseconds or longer. Zhong and Zewail (2001) attributed the decay times of the FAD˙−/Tyr˙+/Trp˙+ complexes to possible charge recombination of the FAD˙−/Trp˙+ complex in ∼30 ps, and that of the FAD˙−/Tyr˙+ complex to a time constant of more than a nanosecond. As basis for the proposed slower charge recombination in the flavin/tyrosine CT complex, it was suggested that after photoinduced electron transfer, proton transfer from the oxidized tyrosine radical to the FAD anion may occur, leading to much longer times necessary for recombination of both the electron and the proton. Although in our opinion, the experiments do certainly not rule out back-electron transfer from the flavin to the tyrosine in ∼30 ps, the existence of long-lived CT interactions and the observation that CT complexes can also absorb light in the blue spectral region, are certainly interesting. In fluorescence, glucose oxidase showed besides the dominant fluorescence lifetime time of 1.8 ps, a second lifetime component of 10 ps, and minor components (1–3%) in the nanosecond region (Zhong and Zewail, 2001). The latter were attributed to the presence of free flavin in the samples. As the fluorescence up-conversion experiments did not yield anisotropy information, comparison with the fluorescence depolarization effects reported in this study is not possible.
Similar fluorescence depolarization phenomena as reported here have also been obtained for NADH peroxidase (Visser et al., 1998). The active-site structure of this related tetrameric peroxide reductase shows strong similarities with that of GR; on the re-side of the isoalloxazine ring, a nearby tyrosine residue (Tyr-159) blocks the NADH-binding site (Stehle et al., 1991). As with GR, crystallographic analysis revealed that binding of the nicotinamide cofactor is coupled with a movement of this tyrosine away from the flavin (Stehle et al., 1993). Time-resolved fluorescence anisotropy experiments on wild-type NADH peroxidase revealed a rapidly depolarizing process with a time constant similar to that of GR (Visser et al., 1998). In the mutant enzyme NADH peroxidase Y159A, this rapid depolarization no longer appeared and the remaining small amount of depolarization was only determined by homoenergy transfer between the four flavins in the tetramer. Besides temperature dependence, the rapid depolarization in wild-type enzyme also showed a clear wavelength dependence. On the red side of the emission spectrum (567 nm), depolarization occurred faster than on main-band detection (526 nm), which confirms the relaxational character of the process; the formation of a transient complex between the light-excited flavin and the tyrosine results in a more stabilized charge-transfer excited state that is hence red-shifted compared to the first-excited singlet state. For wt GR, no significant differences were observed between data obtained at 526 nm and 558 nm. It may well be that a wavelength dependency connected with the ultrashort lifetime component is only visible at the blue side of the spectrum. Such behavior has been observed in decay-associated spectra for the mutant enzyme thioredoxin reductase C138S, in which the spectra of ultrashort lifetime components of 1.2 ps and 7.3 ps were ∼8 nm blue-shifted compared to the spectra of two longer components (van den Berg et al., 2001). A thorough investigation on GR using a streak camera setup can elucidate such wavelength dependent behavior of the enzyme. In analogy with GR, the rapid depolarization in NADH peroxidase was attributed to a transient interaction between the tyrosine and the light-excited flavin. The similarity of the phenomena in both flavoproteins indicates that the formation of a charge-transfer complex, in which the emission dipole moment moves out of the plane of the isoalloxazine ring may well be a general mechanism of flavin fluorescence depolarization in systems that allow relaxation of an aromatic amino acid positioned at Van der Waals distance of the isoalloxazine ring.
Acknowledgments
We are very much obliged to Professor Dr. R. H. Schirmer and Dr. K. Becker (Heidelberg University, Germany), for the kind gift of the GR enzymes from human erythrocytes, and for stimulating discussions and fruitful suggestions. We thank Professor Dr. R. N. Perham for the pleasant cooperation on the E. coli GR enzymes and the hospitality in his laboratory. We thank Dr. W. J. H. van Berkel and Professor Dr. C. Laane for discussions and support.
This work was financially supported by the Netherlands Foundation for Chemical Research with financial aid from the Netherlands Organization for Scientific Research (NWO) and the E. C. Human Capital and Mobility Programme CHRX-CT93-0166 (“Flavoproteins, Structure and Activity”).
References
- Bastiaens, P. I. H., A. van Hoek, J. A. E. Benen, J. C. Brochon, and A. J. W. G. Visser. 1992a. Conformational dynamics and intersubunit energy transfer in wild-type and mutant lipoamide dehydrogenase from Azotobacter vinelandii. Biophys. J. 63:839–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaens, P. I. H., A. van Hoek, W. F. Wolkers, J. C. Brochon, and A. J. W. G. Visser. 1992b. Comparison of the dynamical structures of lipoamide dehydrogenase and glutathione reductase by time-resolved polarized flavin fluorescence. Biochemistry. 31:7050–7060. [DOI] [PubMed] [Google Scholar]
- Beechem, J. M. 1992. Global analysis of biochemical and biophysical data. Methods Enzymol. 210:37–54. [DOI] [PubMed] [Google Scholar]
- Berry, A., N. S. Scrutton, and R. N. Perham. 1989. Switching kinetic mechanism and putative proton donor by directed mutagenesis of glutathione reductase. Biochemistry. 28:1264–1269. [DOI] [PubMed] [Google Scholar]
- Creed, D. 1984. The photophysics and photochemistry of near-uv absorbing amino acids-1. tryptophan and its simple derivatives. Photochem. Photobiol. 39:537–562. [Google Scholar]
- Dale, R. E., J. Eisinger, and W. E. Blumberg. 1979. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 26:161–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kok, A., and A. J. W. G. Visser. 1987. Flavin binding site differences between lipoamide dehydrogenase and glutathione dehydrogenase as revealed by static and time-resolved flavin fluorescence. FEBS Lett. 218:135–138. [DOI] [PubMed] [Google Scholar]
- Digris, A. V., V. V. Skakoun, E. G. Novikov, A. van Hoek, A. Claiborne, and A. J. W. G. Visser. 1999. Thermal stability of a flavoprotein assessed from associative analysis of polarized time-resolved fluorescence spectroscopy. Eur. Biophys. J. 28:526–531. [DOI] [PubMed] [Google Scholar]
- Förster, T. 1948. Zwischenmolekulare Energiewanderumg und Fluoreszenz. Ann. Physik. 2:55–75. [Google Scholar]
- Greer, S., and R. N. Perham. 1986. Glutathione reductase from Escherichia coli: cloning and sequence analysis of the gene and relationship to other flavoprotein disulfide oxidoreductases. Biochemistry. 25:2736–2742. [DOI] [PubMed] [Google Scholar]
- Hecht, H. J., H. M. Kalisz, J. Hendle, R. D. Schmid, and D. Schomburg. 1993. Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 Å resolution. J. Mol. Biol. 229:153–172. [DOI] [PubMed] [Google Scholar]
- Johansson, L. B.-Å., Å. Davidsson, G. Lindblom, and K. Razi Naqvi. 1979. Electronic transitions in the isoalloxazine ring and orientation of flavins in model membranes studied by polarized light spectroscopy. Biochemistry. 18:4249–4253. [DOI] [PubMed] [Google Scholar]
- Karplus, P. A., and G. E. Schulz. 1987. Refined structure of glutathione reductase at 1.54 Å resolution. J. Mol. Biol. 195:701–729. [DOI] [PubMed] [Google Scholar]
- Karplus, P. A., and G. E. Schulz. 1989. Substrate binding and catalysis by glutathione reductase as derived from refined enzyme-substrate crystal structures at 2 Å resolution. J. Mol. Biol. 210:163–180. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd Ed. Kluwer Academic, Plenum Publishers, New York.
- Mattevi, A., A. J. Schierbeek, and W. G. J. Hol. 1991. Refined crystal structure of lipoamide dehydrogenase from Azotobacter vinelandii at 2.2 Å resolution. A comparison with the structure of glutathione reductase. J. Mol. Biol. 220:975–994. [DOI] [PubMed] [Google Scholar]
- Mittl, P. R. E., A. Berry, N. S. Scrutton, R. N. Perham, and G. E. Schulz. 1994. Anatomy of an engineered binding site. Protein Sci. 3:1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittl, P. R. E., and G. E. Schulz. 1994. Structure of glutathione reductase from Escherichia coli at 1.86 Å resolution: comparison with the enzyme from human erythrocytes. Protein Sci. 3:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, D. V., and D. Phillips. 1984. Time-Correlated Single Photon Counting. Academic Press, London, UK.
- Pai, E. F., P. A. Karplus, and G. E. Schulz. 1988. Crystallographic analysis of the binding of NADPH, NADPH fragments, and NADPH analogues to glutathione reductase. Biochemistry. 27:4465–4474. [DOI] [PubMed] [Google Scholar]
- Pai, E. F., and G. E. Schulz. 1983. The catalytic mechanism of glutathione reductase as derived from X-ray diffraction analyses of reaction intermediates. J. Biol. Chem. 258:1752–1757. [PubMed] [Google Scholar]
- Ruggiero, A. J., D. C. Todd, and G. R. Fleming. 1990. Subpicosecond anisotropy studies of tryptophan in water. J. Am. Chem. Soc. 112:1003–1014. [Google Scholar]
- Scrutton, N. S., A. Berry, and R. N. Perham. 1987. Purification and characterization of glutathione reductase encoded by a cloned and over-expressed gene in Escherichia coli. Biochem. J. 245:875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle, T., S. A. Ahmed, A. Claiborne, and G. E. Schulz. 1991. Structure of NADH peroxidase from Streptococcus faecalis 10C1 refined at 2.16 A resolution. J. Mol. Biol. 221:1325–1344. [PubMed] [Google Scholar]
- Stehle, T., A. Claiborne, and G. E. Schulz. 1993. NADH binding site and catalysis of NADH peroxidase. Eur. J. Biochem. 211:221–226. [DOI] [PubMed] [Google Scholar]
- Steinberg, I. Z. 1971. Long-range nonradiative transfer of electronic excitation energy in proteins and polypeptides. Annu. Rev. Biochem. 40:83–114. [DOI] [PubMed] [Google Scholar]
- Thieme, R., E. F. Pai, R. H. Schirmer, and G. E. Schulz. 1981. Three-dimensional structure of glutathione reductase at 2 Å resolution. J. Mol. Biol. 152:763–782. [DOI] [PubMed] [Google Scholar]
- van den Berg, P. A. W., K. A. Feenstra, A. E. Mark, H. J. C. Berendsen, and A. J. W. G. Visser. 2002. Dynamic conformations of flavin adenine dinucleotide: simulated molecular dynamics of the flavin cofactor related to the time-resolved fluorescence characteristics. J. Phys. Chem. B. 106:8858–8869. [Google Scholar]
- van den Berg, P. A. W., S. B. Mulrooney, B. Gobets, I. H. M. van Stokkum, A. van Hoek, C. H. Williams, Jr., and A. J. W. G. Visser. 2001. Exploring the conformational equilibrium of E. coli thioredoxin reductase: characterization of two catalytically important states by ultrafast flavin fluorescence spectroscopy. Protein Sci. 10:2037–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg, P. A. W., A. van Hoek, C. D. Walentas, R. N. Perham, and A. J. W. G. Visser. 1998. Flavin fluorescence dynamics and photoinduced electron transfer in Escherichia coli glutathione reductase. Biophys. J. 74:2046–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg, P. A. W., and A. J. W. G. Visser. 2001. Tracking molecular dynamics of flavoproteins with time-resolved fluorescence spectroscopy. In New Trends in Fluorescence Spectroscopy. Applications to Chemical and Life Sciences, B. Valeur and J. C. Brochon, editors. Springer, Berlin, Germany. 457–485.
- van Hoek, A., K. Vos, and A. J. W. G. Visser. 1987. Ultrasensitive time-resolved polarized fluorescence spectroscopy as a tool in biology and medicine. IEEE J. Quantum Electron. QE-23:1812–1820. [Google Scholar]
- Visser, A. J. W. G., P. A. W. van den Berg, N. V. Visser, A. van Hoek, H. A. van den Burg, D. Parsonage, and A. Claiborne. 1998. Time-resolved fluorescence of flavin adenine dinucleotide in wild-type and mutant NADH peroxidase. Elucidation of quenching sites and discovery of a new fluorescence depolarization mechanism. J. Phys. Chem. B. 102:10431–10439. [Google Scholar]
- Visser, A. J. W. G., and J. Lee. 1980. Lumazine protein from the bioluminescent bacterium Photobacterium phosphoreum. A fluorescence study of the protein-ligand equilibrium. Biochemistry. 19:4366–4372. [DOI] [PubMed] [Google Scholar]
- Visser, A. J. W. G., and F. Müller. 1979. Absorption and fluorescence studies on neutral and cationic isoalloxazines. Helv. Chim. Acta. 62:593–608. [Google Scholar]
- Williams, C. H., Jr. 1976. Flavin-containing dehydrogenases. In The Enzymes, 3rd Ed, Vol. 13. P. D. Boyer, editor. Academic Press, New York. 89–173.
- Zhong, D., and A. H. Zewail. 2001. Femtosecond dynamics of flavoproteins: charge separation and recombination in riboflavin (vitamin B2)-binding protein and in glucose oxidase enzyme. Proc. Natl. Acad. Sci. USA. 98:11867–11872. [DOI] [PMC free article] [PubMed] [Google Scholar]