

Abstract
Subdenaturing concentrations of guanidine hydrochloride (GdnHCl) stabilize proteins. For ferrocytochrome c the stabilization is detected at subglobal level with no measured change in global stability. These deductions are made by comparing observed rates of thermally driven ferrocytochrome cHCO reactions with global unfolding rates of ferrocytochrome c measured by stopped flow and NMR hydrogen exchange in the presence of a wide range of GdnHCl concentrations at pH 7, 22°C.
INTRODUCTION
The exact mechanism of denaturant-induced protein unfolding is not fully understood. Both stoichiometric binding theory for direct protein-denaturant interaction and denaturant-mediated alteration in protein-solvent interactions afford interpretations of unfolding transitions (Schellman, 1978, 1987). Recent reports also indicate that subdenaturing concentrations of GdnHCl can stabilize proteins by committing GdnH+ and Cl− ions to screen charge-charge interactions in the native state of the protein (Pace et al., 1990; Mayr and Schmid, 1993; Morjana et al., 1993; Monera et al., 1994; Makhatadze et al., 1998; Iberra-Molero et al., 1999; Bhuyan, 2002). A more recent demonstration that even urea, when used in subdenaturing concentrations, stabilizes proteins implies that protein-denaturant interactions also lower the conformational entropy of the protein (Bhuyan, 2002). These observations underscore the complexity of the mechanism by which denaturants achieve their protein-unfolding action.
In an earlier study GdnHCl, urea, and salt were used to establish that the GdnHCl stabilization originates from both entropic effect due to intramolecular protein cross-linking action of GdnH+ and electrostatic effect due to the interaction of Cl−, and also possibly of GdnH+, with charged groups of ferrocytochrome c (Bhuyan, 2002). It was, however, not clear whether the stabilization adds on to the global stability of the protein or is manifested at subglobal level. The major endeavor of this work is to demonstrate that energetic stabilization of cytochrome c by subdenaturing concentrations of GdnHCl is detected only at subglobal level, and there is apparently no increase in the global stability of the protein. A tentative structural interpretation of the observed effect is presented.
The strategy used for cytochrome c experiments is different from the usual method of monitoring thermal unfolding with subdenaturing increments of GdnHCl (Mayr and Schmid, 1993). Instead, the stabilizing effect is deduced from thermodynamic and kinetic parameters measured for ferrocytochrome c−CO reaction under conditions of varying protein stability (Bhuyan, 2002). Briefly, GdnHCl-unfolded ferrocytochrome c is allowed to bind CO (UCO). When the initial UCO solution is diluted to a final concentration of GdnHCl that supports refolding, the UCO→NCO process occurs, where NCO is native (or native-like) ferrocytochrome c in which the CO is trapped. Since the concentration of CO in the refolding medium is substantially low, and because the Fe2+−M80 interaction is preferred over Fe2+−CO, the trapped CO escapes as thermal motions facilitate the dissociation of the Fe2+−CO bond. The rate of this slow thermal process, NCO→N + CO, is studied with systematic increments of GdnHCl concentration. This study also employs the CO association reaction, N + CO→NCO. It is found that under mildly destabilizing conditions ferrocytochrome c can be driven to bind CO when the latter is used in saturating concentration (≈1 mM). Rate coefficients of CO association and dissociation reactions have been measured in a wide range of GdnHCl concentration by optical spectroscopy and real-time NMR methods. These data in conjunction with backbone hydrogen exchange rates for global unfolding of ferrocytochrome c show a progressive decrease in spatial displacement of thermal fluctuations at the subglobal level as the protein is taken from the native to subdenaturing milieu. As denaturing to unfolding conditions are approached, large-scale unfolding fluctuations set in that eventually connect with global unfolding motion. The GdnHCl stabilization is not registered at the global level of ferrocytochrome stability.
MATERIALS AND METHODS
Horse heart Cyt c (type VI) was from Sigma-Aldrich (St. Louis, MO), ultrapure GdnHCl from USB (Cleveland, OH), 99.9% D2O from Sigma-Aldrich, and other chemicals from Sigma-Aldrich or Merck (Mumbai, India). All experiments were done in 0.1 M sodium phosphate buffer, pH 7, 22°C, under strictly anaerobic conditions.
Preparation of native carbonmonoxycytochrome c and measurement of CO dissociation kinetics
Cytochrome c, initially dissolved in 6.35 M GdnHCl, was deaerated and reduced by adding sodium dithionite to a final concentration of 1.8 mM. Unfolded ferrocytochrome c (U) thus obtained was liganded with CO. Unfolded carbonmonoxycytochrome c (UCO) was then diluted 101-fold into a degassed and dithionite-reduced CO-free refolding buffer containing a desired concentration of GdnHCl. This procedure allows complete refolding of UCO to generate native carbonmonoxycytochrome c (NCO). The fast UCO→NCO process, measurable by stopped flow, precedes the slow NCO→N + CO dissociation. Kinetics of CO dissociation were generally monitored by both 549.5-nm heme absorbance in a Shimadzu (Kyoto, Japan) UV-Vis-NIR spectrophotometer (UV-3101 PC) and real-time NMR. Under denaturing conditions, where the CO dissociation reaction is relatively faster, kinetics were measured in a stopped-flow spectrometer in optical absorption (549.5 nm) mode.
Measurement of CO association kinetics
Small volumes of ferrocytochrome c solution were diluted 101-fold into CO-saturated buffer containing 1.8 mM dithionite and varying concentrations of GdnHCl. The final protein concentration was 10 μM. CO association kinetics were measured by both heme absorbance at 549.5 nm and real-time NMR. In NMR experiments the protein concentration was raised to ∼0.05 mM. In the presence of destabilizing concentrations of GdnHCl, rates were measured by stopped flow.
Stopped-flow measurement of folding-unfolding kinetics of ferrocytochrome c
Cytochrome c (0.35 mM), initially unfolded in 7 M GdnHCl, pH 7, was reduced under nitrogen by the addition of a concentrated solution of sodium dithionite to a final concentration of 3.2 mM. This is unfolded ferrocytochrome c, and was mixed with the refolding buffer in the stopped flow in the desired ratio to record refolding kinetics. For refolding of carbonmonoxycytochrome c, unfolded ferrocytochrome c was liganded with CO before loading into the mixing module. In each case, an equilibration time of ∼10 min was allowed before mixing.
For unfolding, native ferrocytochrome c was prepared simply by reducing the protein with sodium dithionite added to a final concentration of 3.2 mM. Whenever desired, the native protein solution contained low concentrations of GdnHCl. This was done to check if the unfolding rate under a given condition depends on the guanidine content in the initial native-state protein solution.
Stopped-flow experiments used a Biologic SFM400 module regulated at 22°C by the use of an external water bath. The spectrometer was configured for fluorescence detection (excitation: 280 nm; emission: 335 nm). A two-syringe mixing (1:7, protein/buffer) at a total flow rate of 8 ml s−1 was employed. Under these conditions the instrument dead time for a high-density mixer and a 0.8-mm flow cell was determined to be 2.9 (±0.1) ms. Typically, 10–20 shots were averaged.
Equilibrium hydrogen exchange (HX) and NMR
Cytochrome c dissolved in D2O was reduced by ascorbate and exposed to 50°C for ∼3 h to exchange out the labile fast hydrogens for deuterium. This preexchanged protein solution was transferred to a deuterated denaturing medium by passing through a Sephadex G-25 column equilibrated in a given concentration of GdnDCl, pD 6.8, 25 mM dithionite. The eluate was incubated under argon or nitrogen at 22°C for a desired time period, after which HX was quenched by passing the sample through another Sephadex G-25 column equilibrated with the quenching buffer (50 mM phosphate, 50 mM ascorbate, pH 5.3) in the absence of GdnDCl. Care was taken to minimize air exposure of the samples all through these steps. 1D and 2D correlated spectra were recorded at 20°C in a 600 MHz Varian Unity Plus spectrometer (Varian, Salt Lake City, UT).
HX measurement by stopped-flow NMR
Under strongly destabilizing conditions the kinetics of HX are too fast, and are immeasurable if the exchange reaction is initiated by the solvent exchange technique described above. In GdnDCl >4 M, HX was initiated by mixing the protein solution with the GdnDCl-containing buffer within the NMR magnet. The procedure has been described before (Bhuyan and Udgaonkar, 1998). Briefly, the required solutions, contained separately in two gas-tight syringes, are rapidly injected via two flow lines into the NMR tube positioned inside the magnet. A third flow line delivers a gentle stream of argon over the mixed solution to provide an anaerobic atmosphere. Arrays of 1D spectra were acquired at 600 MHz 1H frequency. All spectra were processed using FELIX software.
RESULTS
Thermal dissociation of CO from NCO
Within the limit of the stopped-flow resolution (2.9 ± 0.1 ms dead time), unfolded carbonmonoxycytochrome c refolds extremely fast. The kinetics exemplified in Fig. 1 a give an observed rate of 305 s−1 for the major refolding phase in the presence of 1.3 M GdnHCl. This process indeed is the UCO→NCO reaction. Since the concentration of CO in the refolding milieu is substantially reduced, and because the affinity of native ferrocytochrome c for CO is much lower relative to that for the intrinsic M80 ligand, the UCO→NCO refolding reaction is followed by the NCO→N + CO conversion yielding to the formation of the Fe2+−M80 bond. It is the rate of this nonequilibrium process the GdnHCl dependence of which provides a clue to the protein stabilizing effect of the denaturant.
FIGURE 1.

Representative kinetic traces. (a) Unfolded carbonmonoxycytochrome c (UCO) refolds fast (UCO→NCO; k = 305 s−1, 1.3 M GdnHCl, 22°C). Seven percent of the observed amplitude is due to a slow minor phase (k = 38 s−1). The normalized fluorescence signals (inset) indicate occurrence of processes faster than what is seen in the stopped flow. (b) The slow single-phase dissociation of CO, NCO→N + CO (τ = 43.3503 min, 0.4 M GdnHCl, 22°C).
Fig. 1 b typifies the kinetics of the NCO→N + CO dissociation recorded after diluting the UCO solution 101-fold into a CO-free refolding buffer to obtain 0.4 M GdnHCl finally. The slow increase in absorbance at the heme π→π* α-band (549.5 nm) in a single exponential is due to dissociation of CO. Because of significantly low concentration of CO in the refolding medium, the reverse reaction of CO binding back to N is negligible. Thus the rate constant obtained can be equated directly to the CO dissociation rate coefficient, kdiss. The slowness of the reaction has allowed accurate determination of kdiss (τ = 43.4 min, Fig. 1 b) by all of the methods—spectrophotometric, real-time NMR, and stopped-flow.
Association of CO with ferrocytochrome c
Ferrocytochrome c can be forced to bind CO under mildly destabilizing conditions when the latter is used in saturating concentration (≈1 mM), and the same methods used to monitor the dissociation process were employed to extract the association rate coefficients, kass. As an example, Fig. 2 shows the evolution of M80 side-chain resonances during the course of the N + CO→NCO reaction in the presence of 2.35 M GdnDCl. This is a Fe2+−M80→Fe2+−CO replacement process, the time constant for the single exponential decay of the CɛH3 resonance being 42.5 min (Fig. 2, inset). Decay of peak areas of certain other side-chain resonances is also observed, a detailed analysis of which will be presented elsewhere. In the course of both dissociation and association of CO reactions, no significant NMR line broadening or shift of resonances was observed, indicating that the N and NCO conformations are not different. Other evidence, including fluorescence and circular dichroism spectra (data not shown), also indicate that the NCO conformation is just the native conformation of ferrocytochrome c except for possible minor readjustments of some side chains in the former.
FIGURE 2.

Real-time NMR monitoring of M80 side-chain resonances during association of CO to ferrocytochrome c, N + CO→NCO (τ = 42.46 min, 2.35 M GdnHCl, 22°C). Use of very low concentration of the protein (∼50 μM) to achieve pseudo-first order condition with respect to CO (∼1 mM) rendered 1D NMR acquisition more useful (inset) than 2D acquisition.
GdnHCl dependence of kdiss and kass, and the motional mode affected by the denaturant
Fig. 3 a shows the GdnHCl-induced equilibrium unfolding transition of ferrocytochrome c, and Fig. 3 b presents the rate coefficients for dissociation of CO from NCO and association of CO to N in the 0–5.4 M range of GdnHCl concentration. Given Cm ≈ 5 M GdnHCl for the equilibrium unfolding, the rate-GdnHCl data provide an opportunity to analyze the thermodynamic properties of the protein in subdenaturing and denaturing limits. Data in Fig. 3 b provide two major results. 1), The congruence of kdiss and kass: within experimental error kdiss and kass values are superimposable under all conditions of protein stability, indicating that it is the same mode of motion that operates in both CO association and dissociation processes. The congruence also implies that the activation barriers for association and dissociation in a given concentration of GdnHCl have the same height. 2), The variation in magnitudes of kdiss and kass with denaturant: as the final concentration of GdnHCl in the reaction medium is raised starting from strongly native-like conditions, kdiss/ass initially decreases and then increases, displaying a broad minimum around 2.1 M GdnHCl, consistent with the earlier report (Bhuyan, 2002). In going from 0.05 to 2.1 M GdnHCl the value of kdiss/ass decreases 2.3 (±0.3)-fold, and increases thereafter. A linear increase up to ∼4.3 M GdnHCl is followed by apparently stronger dependence on the denaturant concentration. The decrease in the subdenaturing limit suggests that GdnHCl tends to block both dissociation of CO from NCO and association of CO to N. The increase in rate coefficients in GdnHCl concentrations higher than ∼2.1 M can be interpreted to arise from protein destabilization and structural unfolding that would facilitate dissociation and association processes.
FIGURE 3.

Equilibrium stability and rate-denaturant spaces. (a) GdnHCl-induced equilibrium unfolding of ferrocytochrome c at 23 (±1)°C. The continuous curve is fit to the data using procedures described in the Results section. Values of ΔG(H2O) and mg obtained are 18 (±0.5) kcal mol−1 and 3.6 kcal mol−1 M−1, respectively. (b) Dependence of the rates of CO association, kass (▿, manually measured OD549.5 nm; ♦, stopped-flow OD549.5 nm; ⋄, NMR-monitored M80 CɛH3 resonance), and dissociation, kdiss (▴, manually measured OD549.5 nm; ▵, stopped-flow OD549.5 nm; □, NMR-monitored M80 CɛH3 resonance). (c) GdnHCl dependence of kass and kdiss presented in the perspective of global folding-unfolding kinetics, U N; folding (kf, •) and unfolding (ku, ○) rates. Values of ku below the Cm was derived from hydrogen exchange data for L98 amide proton (▪) as described in the text. The meaning of other symbols is the same as in b.
Results set in Fig. 3 c distinguish the GdnHCl dependence of the motional mode that controls the protein-CO reaction from the global unfolding mode of ferrocytochrome c. The upper chevron shows the denaturant dependence of folding-unfolding rates of ferrocytochrome c. The kinetics are apparently two-state (N U), consistent with earlier reports (Bhuyan and Udgaonkar, 2001; Bhuyan and Kumar, 2002; Prabhu et al., 2004). The GdnHCl concentration corresponding to the minimum in the folding chevron is the Cm (Fig. 3, a and c). Values of ku in GdnHCl higher than the Cm are obtained directly from stopped-flow unfolding experiments. Values below Cm were extracted from HX data treated under EX2 condition; for a two-state N U kinetics, ku = (kexkf)/kch, where kex, kf, and kch are, respectively, observed HX rate, protein folding rate, and the free peptide rate. Plotted in Fig. 3 c is the GdnHCl dependence of HX-derived ku values for L98 that provides a marker for global unfolding of cytochrome c (Xu et al., 1998). Errors in values of kf read from the curved left limb of the chevron together with some uncertainties in the values of pD of the HX medium that arise from the required manipulations in the presence of dithionite, introduce some difficulties in accurate determination of ku values in the region below Cm. Keeping the protein fully reduced throughout the hydrogen exchange time is another experimental difficulty, which is the reason why HX experiments were not carried out under strongly native-like conditions. Nevertheless, the data presented provide adequately a marker that tracks linear functional dependence of the logarithm of ku on denaturant. As GdnHCl concentration increases, the kdiss/ass values merge with the ku values. This happens around 4.3 M GdnHCl, indicating that the motional mode of ferrocytochrome c that controls CO association and dissociation reactions preserves its identity as a local mode in the subdenaturing to mildly denaturing conditions, but is dominated by the global unfolding motions as higher concentrations of the denaturant strongly destabilize the protein. An important conclusion emerges: in its subdenaturing limit GdnHCl affects the local motional mode or the particular thermal fluctuations involved in the protein-CO reactions; the global unfolding mode is not affected.
Thermal unfolding of cytochrome c in the presence of subdenaturing concentrations of GdnHCl
To establish that the global unfolding is not affected by subdenaturing concentrations of the denaturant a series of thermal melts were recorded. For this set of experiments the oxidized form of the protein (ferricytochrome c) was chosen since ferrocytochrome c is so stable a protein that it is not amenable at all to thermal melting experiments. Fig. 4 presents a representative set of thermal transition data taken at pH 5.8. The dependence of the midpoint of the thermal transition (Tm) on subdenaturing concentrations of GdnHCl is shown in the inset. Clearly, the protein stability decreases as the denaturant concentration is increased.
FIGURE 4.

Thermal unfolding of ferricytochrome c at pH 5.8. Transitions were monitored by the increase in optical density at 399 nm. Curves from left to right are in the presence of 0, 0.04, 0.08, 0.12, 0.17, 0.25, and 0.4 M GdnHCl, respectively. Plotted in the inset is the dependence of the midpoint of the thermal transition on GdnHCl concentration.
Denaturant dependence of activation enthalpy and entropy of CO dissociation and association reactions
The observation that the logarithm of kdiss/ass linearly decreases with increments of subdenaturing concentration of GdnHCl up to ∼2.1 M and then increases gradually with destabilizing concentrations of the denaturant (Fig. 3, b and c) warrants a thermodynamic analysis of the reaction. The pertinent reactions monitored are NCO→N + CO and N + CO→NCO, and the analysis is based on thermodynamic formulation of the conventional transition state theory (Laidler, 1987), with the assumption that GdnHCl has no effect at the transition state level. If the decrease in rate coefficients in the subdenaturing zone is due to stabilization of the reactant states, the decrease in entropy of the ground state in the presence of the denaturant must be compensated by an increase in activation enthalpy of the respective reaction. This follows from
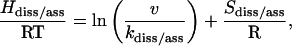 |
(1) |
where ν is the vibrational frequency, and Hdiss/ass and Sdiss/ass are changes in enthalpy and entropy, respectively, between the relevant reactant and transition states. By comparing Eq. 1 with the Arrhenius expression, kdiss/ass = Aexp(−Ea/RT), where A is the front factor and Ea is the activation energy, the following equalities are obtained:
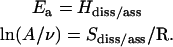 |
(2) |
Fig. 5 a shows the denaturant distribution of Hdiss/ass determined from temperature dependence of a series of association and dissociation reactions in the 0–4 M range of GdnHCl. Both Hdiss and Hass peak at ∼2.2 M GdnHCl, which is also the concentration at which the kdiss/ass-denaturant space shows the minimum (Fig. 3 b). This result strongly suggests that in subdenaturing concentrations of GdnHCl the free energy of the protein is lowered, leading to a linear decrease in the logarithm of kdiss/ass. When present in destabilizing concentrations the unfolding role of the denaturant outdoes its stabilizing role. kdiss/ass now increases because of an increase in conformational entropy of the protein that leads to a decrease in Hdiss/ass and Sdiss/ass.
FIGURE 5.

Denaturant distribution of (a) activation enthalpy (Hdiss/ass), and (b) conformational entropy loss by NCO relative to the entropy of the protein in the absence of the denaturant (ΔS, Eq. 3) for CO dissociation and association reactions (•, NCO→N + CO; ○, N + CO→NCO). The peaks of both distributions appear at the same GdnHCl concentration where the minimum of the kdiss/ass-GdnHCl space (Fig. 3 b) occurs.
Data in Fig. 5 b quantify the GdnHCl dependence ΔS, defined as the conformational entropy loss by NCO relative to the entropy of the protein in the absence of the denaturant. Ordinate values were calculated according to
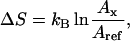 |
(3) |
where Aref and Ax are Arrhenius front factors for temperature dependence of the reactions—Aref in the absence of GdnHCl, and Ax in the presence of x concentration of GdnHCl. ΔS is expressed in Boltzmann units (kB). The entropy loss is apparently linear with denaturant concentration registering a peak value of ∼6.4 kB at 2.1 M GdnHCl. With further increments of GdnHCl, protein destabilization due to global unfolding leads to a gradual increase in entropy. ΔS ∼6.4 kB at 2.1 M GdnHCl is significant. It indicates a dramatic loss of motional freedom of the protein.
The analysis presented here considers only the relative properties of the reactant and the activated state. Such an analysis is afforded by the irreversible nature of the CO dissociation (NCO→N + CO) and association (N + CO→NCO) reactions under the experimental conditions employed. Otherwise (i.e, for reversible reactions), kinetics depend on the properties of not only reactants and activated states but also products.
DISCUSSION
Protein stiffening due to denaturant binding
Evidence for direct interactions between proteins and GdnH+ and urea comes from a number of studies, including x-ray crystallography (Hibbard and Tulinsky, 1978; Pike and Acharya, 1994; Dunbar et al., 1997) and isothermal calorimetry (Makhatadze and Privalov, 1992). When used in subdenaturing concentrations, GdnHCl and urea serve to cross-link different parts of the protein by establishing multiple variable-length hydrogen-bonding and van der Waals interactions with main-chain and side-chain groups of proteins (Pike and Acharya, 1994; Dunbar et al., 1997). The denaturant-mediated cross-linking is expected to reduce the motional freedom by increasing the barriers to motions. Such restrictions in the conformational freedom imposed by denaturants have been suggested through analyses of x-ray B-factors for lysozyme (Pike and Acharya, 1994), ribonuclease A, and dihydrofolate reductase (Dunbar et al., 1997). In the presence of subdenaturing concentrations of denaturants the reduction in fluctuations in positions of individual or clusters of protein atoms around their average has been called protein stiffening (Bhuyan, 2002).
Kinetic and thermodynamic consequences of protein stiffening
Intramolecular thermal collisions provide the energy for barrier crossing in the NCO→N + CO and N + CO→NCO reactions, and constrained dynamics must imply a reduction in the amplitude of thermal fluctuations. Thus, the rate coefficients, kdiss and kass, are expected to decrease as the protein is progressively stiffened. This is what is seen in the primary rate-denaturant data (Fig. 3 b). As denaturing conditions are approached (>2.1 M GdnHCl) the protein stiffening action of the denaturant is suppressed by its own structural unfolding effect, and the rate coefficients begin to increase. By corollary, the conformational entropy of the protein is lost as far as the structural unfolding effect does not dominate the protein stiffening action of the denaturant (Fig. 5 b).
Entropic stabilization and electrostatic screening effects of GdnHCl
The association and dissociation reactions of ferrocytochrome c and CO have been designed to underline the changes in conformational entropy of proteins in the presence of subdenaturing concentrations of denaturant. As noted earlier the charge screening effect of ionic GdnHCl could also add on to the stability of native proteins (see, for example, Mayr and Schmid, 1993; Monera et al., 1994). Indeed, both entropic and electrostatic effects contribute toward the stability of ferrocytochrome c. Purely entropic stabilization can be isolated by using nonionic urea as reported earlier (Bhuyan, 2002). Unfortunately, experiments over a wide range of ferrocytochrome stability as presented here could not be conducted by using urea.
GdnHCl-induced stabilization: local or global?
The observed changes in the thermodynamic character of the protein in the presence of subdenaturing concentrations of the denaturant warrants a check for stabilization at both global and subglobal levels. Extensive thermal unfolding experiments with ferrocytochrome c in the presence of a number of low concentrations of GdnHCl did not hint at global energetic stabilization (Fig. 4). This fact is substantiated by the observation that the logarithm of the global unfolding rate, ku, has linear dependence on GdnHCl, apparent even in the presence of mildly denaturing concentrations (Fig. 3 c). Furthermore, in stopped-flow experiments where the protein was unfolded to a given concentration of GdnHCl starting from the native state poised in different nondenaturing concentrations of the denaturant, the unfolding rate measured was the same (Fig. 3 c). In other words, the value of ku in a given final concentration of the denaturant is independent of the initial concentration of the denaturant. This evidence does not indicate global stabilization. The reason for this cannot be ascertained from the available data. It is possible that the number of GdnHCl molecules that interact with the protein in the subdenaturing limit are insufficient to cross-link a large part of the molecule. Using the numbers of Makhatadze and Privalov (1992) for native-state binding sites it is estimated that just ∼12 molcules of the denaturant are bound to cytochrome c when the protein is poised in 2.1 M GdnHCl. These may not produce extensive cross-linking to bring about global stabilization.
It follows that the observed stabilization is localized to a subglobal part of the protein, the dynamics of which affect the NCO→N + CO and N + CO→NCO reactions. It can only be speculated that the M80-resident segment of the polypeptide, which is linked to the heme iron through the Fe2+−M80 bond in N but is free in NCO, represents this subglobal part. The segment of the polypeptide between residues 70 and 85 forms a small Ω-loop (Leszczynski and Rose, 1986), and Englander and coworkers have identified this as a partially unfolded form (“red” PUF) (Xu et al., 1998; Hoang et al., 2003). Because the local mobility of the heme ring is suppressed by the intrinsic size and the rigidity of the ring system (Morgan and McCammon, 1983), and given that the neighboring residues of M80 have significantly higher thermal factors in the x-ray structure of cytochrome c (Berghuis and Brayer, 1992), the collective motion of the Ω-loop is expected to be the leading determinant of the CO association and dissociation processes. If so, the denaturant modulation of kdiss/ass (Fig. 3, b and c) may reveal the way the collective motion of the loop, or of a part of it, is constrained in response to GdnHCl content in the reaction medium. Experimental support for this conjectured structural interpretation remains to be seen.
Denaturant-induced entropic stabilization of proteins: a general effect
Proteins in which GdnH+ and Cl− ions do not participate in charge-screening type of electrostatic interactions either due to the absence of relevant charge pairs or possibly for steric reasons, will not be stabilized by the electrostatic mechanism. However, stiffening due to direct protein-denaturant interaction is expected to stabilize all proteins through lowering of the conformational entropy. Although the entropic effect operates invariably, the electrostatic effect would contribute under situations favorable for charge screening. As suggested earlier, the urea stabilization is purely entropic (Bhuyan, 2002). The results also indicate that the general stabilizing action of denaturants need not necessarily show up at the global level of proteins. In the subdenaturing limit the number of denaturant molecules available for interaction with the protein may not be enough to provide global stability. On the other hand, when the concentration of the denaturant is increased its structural unfolding action overrides its own stabilizing effect.
SUMMARY AND CONCLUSION
In the subdenaturing limit, noncovalent polyfunctional interactions between protein groups and denaturants serve to cross-link different parts of the protein. The resultant conformation is dynamically constrained and lower in entropy. The stability conferred may or may not add on to the global stability of the protein. In the case of ferrocytochrome c the stabilization is detected at the subglobal level. When the concentrations of denaturants are increased beyond subdenaturing limits their structure-unfolding action runs over the initial stabilizing effect.
Acknowledgments
This work was supported by the Department of Biotechnology (grant BRB/15/227/2001) and University with Potential for Excellence funding of the University Grants Commission. A.K.B. is the recipient of a Swarnajayanti Fellowship from the Department of Science and Technology, government of India.
This article is dedicated to the memory of Prof. Bhaskar G. Maiya.
References
- Berghuis, A. M., and G. D. Brayer. 1992. Oxidation state-dependent conformational changes in cytochrome c. J. Mol. Biol. 223:959–976. [DOI] [PubMed] [Google Scholar]
- Bhuyan, A. K. 2002. Protein stabilization by urea and guanidine hydrochloride. Biochemistry. 41:13386–13394. [DOI] [PubMed] [Google Scholar]
- Bhuyan, A. K., and R. Kumar. 2002. Kinetic barriers to the folding of horse cytochrome c in the reduced state. Biochemistry. 41:12821–12834. [DOI] [PubMed] [Google Scholar]
- Bhuyan, A. K., and J. B. Udgaonkar. 1998. Stopped-flow NMR measurement of hydrogen exchange rates in reduced horse cytochrome c under strongly destabilizing conditions. Proteins. 32:241–247. [PubMed] [Google Scholar]
- Bhuyan, A. K., and J. B. Udgaonkar. 2001. Folding of horse cytochrome c in the reduced state. J. Mol. Biol. 312:1135–1160. [DOI] [PubMed] [Google Scholar]
- Dunbar, J., H. P. Yennawar, S. Banerjee, J. Luo, and G. K. Farber. 1997. The effect of denaturants on protein structure. Protein Sci. 6:1272–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbard, L. S., and A. Tulinsky. 1978. Expression of functionality of α-chymotrypsin. Effects of guanidine hydrochloride and urea on the onset of denaturation. Biochemistry. 17:5460–5468. [DOI] [PubMed] [Google Scholar]
- Hoang, L., H. Maity, M. M. G. Krishna, Y. Lin, and S. W. Englander. 2003. Folding units govern the cytochrome c alkaline transition. J. Mol. Biol. 331:37–43. [DOI] [PubMed] [Google Scholar]
- Iberra-Molero, B., V. V. Loladze, G. I. Makhatadze, and J. M. Sanchez-Ruiz. 1999. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 38:8138–8149. [DOI] [PubMed] [Google Scholar]
- Laidler, K. J. 1987. Chemical Kinetics. 3rd ed. Harper & Row, New York.
- Leszczynski, J. F., and G. D. Rose. 1986. Loops in globular proteins: a novel category of secondary structure. Science. 234:849–855. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G. I., M. M. Lopez, J. M. Richardson, and S. T. Thomas. 1998. Anion binding to the ubiquitin molecule. Protein Sci. 7:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhatadze, G. I., and P. L. Privalov. 1992. Protein interactions with urea and guanidinium chloride. J. Mol. Biol. 226:491–505. [DOI] [PubMed] [Google Scholar]
- Mayr, L. M., and F. X. Schmid. 1993. Stabilization of a protein by guanidine chloride. Biochemistry. 32:7994–7998. [DOI] [PubMed] [Google Scholar]
- Monera, O. D., C. M. Kay, and R. S. Hodges. 1994. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 3:1984–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, J. D., and J. A. McCammon. 1983. Molecular dynamics of ferrocytochrome c: time dependence of the atomic displacements. Biopolymers. 22:1579–1593. [Google Scholar]
- Morjana, N. A., B. J. McKeone, and H. F. Gilbert. 1993. Guanidine hydrochloride stabilization of a partially unfolded intermediate during the reversible denaturation of protein disulfide isomerase. Proc. Natl. Acad. Sci. USA. 90:2107–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C. N., D. V. Laurents, and J. A. Thomson. 1990. pH dependence of the urea and guanidine hydrochloride denaturation of ribonuclease A and ribonuclease T1. Biochemistry. 29:2564–2572. [DOI] [PubMed] [Google Scholar]
- Pike, A. C. W., and R. Acharya. 1994. A structural basis for the interaction of urea with lysozyme. Protein Sci. 3:706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu, N. P., R. Kumar, and A. K. Bhuyan. 2004. Folding barrier in horse cytochrome c: support for a classical folding pathway. J. Mol. Biol. 337:195–208. [DOI] [PubMed] [Google Scholar]
- Schellman, J. A. 1978. Solvent denaturation. Biopolymers. 17:1305–1322. [DOI] [PubMed] [Google Scholar]
- Schellman, J. A. 1987. The thermodynamic stability of proteins. Annu. Rev. Biophys. Biophys. Chem. 16:115–137. [DOI] [PubMed] [Google Scholar]
- Xu, Y., L. C. Mayne, and S. W. Englander. 1998. Evidence for an unfolding and refolding pathway in cytochrome c. Nat. Struct. Biol. 5:774–778. [DOI] [PubMed] [Google Scholar]