

Abstract
Kinetics of intermolecular interaction between reduced cytochrome c (Cyt c) protein and solvent during the protein-refolding process is studied by monitoring the time dependence of apparent diffusion coefficient (D) using the pulsed-laser-induced transient grating technique. The refolding was triggered by photoinduced reduction of unfolded Fe(III) Cyt c in 3.5 M guanidine hydrochloride (GdnHCl) solution and the change in the diffusion coefficient was monitored in time domain. The relationship between D and the protein conformations under equilibrium condition were investigated at various GdnHCl concentrations using a photolabeling reagent. The time dependence of the observed transient grating signal was analyzed using these data and two models: a continuous change model of the intermolecular interaction and a two-state model. It was found that the TG signals in various time ranges can be consistently reproduced well by the two-state model. The dynamics of D is expressed well by a single exponential function with a rate constant of 22 ± 7 s−1 in a whole time range. The folding process of Cyt c is discussed based on these observations.
INTRODUCTION
Understanding the mechanism of protein folding has continuously been a challenge for theory and experiment (Pain, 2000). In particular, investigations of the kinetics of the folding processes have been central topics in the study. For that purpose, it has been important to develop methods to trigger the folding reaction and to detect the dynamics. To trigger the folding dynamics, various methods, such as the stopped flow, the rapid mixing, the laser-induced temperature jump, or light-induced electron injection methods, have been developed. The protein-folding kinetics sometimes depends on the triggering method, because the required environment could be different depending on the method.
Monitoring method of the kinetics is further important to understand the dynamics. Because proteins are large macromolecules, there are huge numbers of degrees of freedom. Hence, during the refolding process, drastic changes of protein conformations including environment around the protein occur. The intermolecular interaction between the protein and solvent should be significantly rearranged. For monitoring such complex refolding dynamics in time domain, any single detection method is not enough. The dynamics of macromolecules such as proteins should depend on monitored properties. In other words, we have to consider the dynamics from view points of various properties of the protein. For example, the emission intensity or absorbance change during the protein-folding reaction reflects rather local protein structural changes around the chromophores associated with the optical transition. This time dependence of this property provides us information on how the specific distance between two chromophores is changing. The circular dichroism (CD) signal intensity represents the formation of the secondary structure (e.g., α-helices or β-sheets). Hence, the kinetics of secondary structure construction can be monitored. Small angle x-ray scattering data represent the radius of gyration of the protein. Combining the dynamics of these properties, we may construct a total view of the protein folding. However, there has been missing information in the time-domain measurement: the intermolecular interaction, in particular, the protein-solvent interaction, e.g., hydrogen bonding interaction. During the protein folding, many atoms relocate the position and the dynamics should reflect the energetic changes. One of the sources of the change comes from the intermolecular interaction terms. However, there has been no experimental technique to monitor the change in the intermolecular interaction in time domain.
From extensive researches on the diffusion process over more than 100 years, it is recognized that the diffusion coefficient (D) reflects not only the molecular size or solution viscosity but also the shape of the molecule as well as the intermolecular interaction (Cussler, 1984; Tyrrell and Harris, 1984). We recently showed that D of a protein is an important quantity to monitor the intermolecular interaction (Takeshita et al., 2002; Choi and Terazima, 2002). We reported in preliminary findings the time profile of the transient grating (TG) signal during the protein-folding reaction of cytochrome c (Cyt c) and found that the TG signal should be interpreted in terms of the time-dependent apparent D (Nada and Terazima, 2003). However, detailed analysis has not been developed. In this article, combining D data under equilibrium conditions at various conformations, we analyze the time dependence of the signal in whole time range during the Cyt c folding process at 3.5 M guanidine hydrochloride (GdnHCl) to determine the dynamics of the intermolecular interaction. This is the first attempt to analyze the folding dynamics in terms of time-dependent D.
Cyt c is a small protein (104 amino acids) with a heme group covalently bound to residues Cys-14 and Cys-17. The folding process of this protein has been studied from many view points under various conditions including the trigger methods (Jones et al., 1993; Pascher et al., 1996; Telford et al., 1998, 1999; Hagen et al., 1997; Takahashi et al., 1997; Yeh et al., 1997; Englander et al., 1998; Shastry and Roder, 1998; Shastry et al., 1998; Segel et al., 1998, 1999; Godbole and Bowler, 1999; Chen et al., 1999; Okuno et al., 2000; Abbruzzettri et al., 2001; Bhuyan and Udgaonkar, 2001; Pascher, 2001; Akiyama et al., 2002). In this study, we started the refolding process of Cyt c by the laser-induced photoreduction method. Because the reduced form of Cyt c (Fe(II) Cyt c) is more stable than oxidized Cyt c (Fe(III) Cyt c) against GdnHCl, the pulsed laser-induced reduction of unfolded Fe(III) Cyt c can initiate the refolding process (Jones et al., 1993; Pascher et al., 1996; Telford et al., 1998). Hence, the refolding dynamics of reduced Cyt c can be detected by the time-resolved spectroscopy. The TG method is a spectroscopy such that the spatial modulation of the refractive index change induced by the photoexcitation with the interference pattern of two light waves can be sensitively detected in time domain. The previously reported TG signal for the Cyt c folding dynamics showed prominent features, which have never been observed before (Nada and Terazima, 2003). Based on several characteristic points summarized later, we concluded that the apparent D of Fe(II) Cyt c after the reduction is time dependent, which must be associated with the protein-folding dynamics. A simple analysis from the curvature of the signal at a specific time clearly indicated the time dependence of apparent D for the first time. In this article, to obtain the temporal development of D in more detail, the TG signal in a wide range is analyzed based on two models: a continuous D change model and a two-state model. In the continuous model, the intermolecular interaction, e.g., hydrogen bond network, of each protein is assumed to gradually change in the observation time window. In the two-state model, we consider that there are two conformations with different D and the fraction of each conformation is changed. For the detailed analysis of the TG signal without ambiguity and also for studying the relationship between the conformation and D, we measured D of Cyt c under equilibrium conditions at various GdnHCl concentrations. Combining these data, we found that the signal in a wide time range can be satisfactorily analyzed by the two-state model, whereas the continuous model cannot consistently simulate the data. Furthermore, it is found that the change in D in the whole time region until the complete folding can be expressed well by a single exponential manner with a rate constant of 24 ± 7 s−1 at 3.5 M of GdnHCl. We discuss the protein-folding scheme of Cyt c based on our results as well as previously reported data.
EXPERIMENTAL
The experimental setup and the principle of the measurement were similar to that reported previously (Terazima and Hirota, 1993; Terazima et al., 1993, 1995; Okamoto et al., 1995, 1997, 1998; Ukai et al., 2000a,b; Terazima, 2000; Nada and Terazima, 2003). Briefly, the third harmonic of an Nd-YAG laser (Quantum-Ray model GCR-170-10, Spectra-Physics, Mountain View, CA) with a 10-ns pulse was used as an excitation beam and a photodiode laser (840 nm) as a probe beam for the refolding experiment. For the D measurement of Cyt c at various GdnHCl concentrations under equilibrium conditions, an excimer laser (Lamda Physik, Göttingen, Germany) was used as the excitation light for N-hydroxysulfosuccinimidyl-4-azidobenzoate (sulfo-HSAB). The excitation beam was split into two by a beam splitter, and crossed inside a sample cell. The sample is photoexcited by the created interference pattern to induce the refractive index modulation in the sample. A part of the probe beam was diffracted by the modulation (TG signal). The signal was isolated from the excitation laser beam with a glass filter and a pinhole, detected by a photomultiplier tube, and recorded by a digital oscilloscope. The spacing of the fringe was measured by the decay rate constant of the thermal grating signal from a calorimetric standard sample, which releases all the photon energy of the excitation as the thermal energy within a time response of our system. All measurements were carried out at room temperature under the oxygen-purged condition by a nitrogen bubbling method.
Horse heart Cyt c and ultrapure GdnHCl were purchased from Nakalai Tesque (Kyoto, Japan). NADH and sulfo-HSAB were obtained from Sigma (St. Louis, MO) and Pierce Biotechnology (Rockford, IL), respectively. They were used without further purification. Cyt c (66 μM) with NADH (650 μM) sample was prepared in a 100-mM phosphate buffer (pH = 7). The sample solution was filtered with a 0.2-μm filter to remove dusts and transferred to a 1-cm optical path quartz cell. Because NADH is very sensitive to the light irradiation, the sample solution was changed to a fresh one after ∼150 shots of the excitation laser pulses. The photoinduced reduction of Fe(III) Cyt c was confirmed by the transient absorption signal monitored at 550 nm. The unfolded and folded conformations of Fe(III) and Fe(II) Cyt c, respectively, under this condition was checked by the CD spectrum.
PRINCIPLE AND THEORETICAL
The principle of the TG measurement has been reported previously (Terazima and Hirota, 1993; Terazima et al., 1993, 1995; Okamoto et al., 1995, 1997, 1998; Ukai et al., 2000a,b; Terazima, 2000). In the TG experiment, a photoinduced reaction is initiated by the spatially modulated light intensity that is produced by the interference of two excitation light waves. The sinusoidal modulations of the concentrations of the reactant and the product lead to the sinusoidal modulation in the refractive index (δn). This modulation can be monitored by the diffraction efficiency of a probe beam (TG signal).
The refractive index change mainly comes from the thermal energy releasing (thermal grating) and created (or depleted) chemical species by the photoreaction (species grating). The species grating signal intensity is given by the difference of the refractive index changes due to the reactant (δnr) and product (δnp):
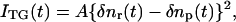 |
(1) |
where A is a constant. The sign of δnp is negative because the phase of the spatial concentration modulation is 180°-shifted from that of the reactant. The species grating signal intensity becomes weaker as the spatial modulations of the refractive index become uniform, which is accomplished by the translational diffusion. Temporal development and the spatial distribution of concentration of chemical species (C(x,t)) is calculated by solving a diffusion equation:
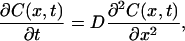 |
(2) |
where x is a direction along the grating wave vector and D is the diffusion coefficient of the chemical species. Solving this diffusion equation by using the Fourier transform method under an assumption of constant D and the initial condition of the concentration:
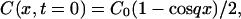 |
(3) |
where C0 is the initial concentration of the product and q is the grating wavenumber, we may find that the q-Fourier component of the concentration decays with a rate constant of Dq2. Hence, the time development of the TG signal can be expressed by a biexponential function (Terazima and Hirota, 1993; Terazima et al., 1993, 1995; Okamoto et al., 1995, 1997, 1998; Ukai et al., 2000a,b; Terazima, 2000).
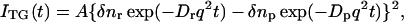 |
(4) |
where Dr and Dp are diffusion coefficients of the reactant and the product, respectively. Furthermore, δnr(>0) and δnp(>0) are, respectively, the initial refractive index changes due to the presence of the reactant and the product. Similarly, the thermal grating signal decays with a rate constant of Dthq2 (Dth is the thermal diffusivity of the solution).
When the apparent D is time dependent, the observed TG signal should be significantly different from that predicted by Eq. 4. For analyzing the observed TG signal that represents the time-dependent apparent D, the following two models were used.
Continuous model
In this model, D of Fe(II) Cyt c is considered to change continuously. We assume that the change is expressed by a single exponential function with a rate constant of k:
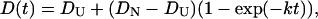 |
(5) |
where DU and DN are D of the unfolded and the native Cyt c, respectively. The time profile of the concentration of the product (Fe(II) Cyt c) is governed by the diffusion equation of
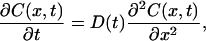 |
(6) |
where D is now time dependent. This equation can be solved by the Fourier transformation method under the same initial condition as Eq. 3. Solving this equation with D(t) of Eq. 5, one may find that the refractive index change due to the reactant and product at the q-Fourier component are expressed by
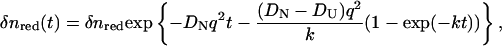 |
(7-a) |
where δnred is the refractive index change due to the presence of Fe(II) Cyt c. The time development of reactant (Fe(III) Cyt c) is given by Eq. 2 with constant D, so that
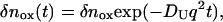 |
(7-b) |
where δnox is the refractive index change due to the presence of Fe(III) Cyt c. The temporal profile of the TG signal based on this model is calculated by Eqs. 1 and 7.
Two-state model
In this model, we consider that there are two types of conformations of Cyt c having different D (DU for unfolded protein and DN for the native protein). By the populational transition between these two states, D of every Cyt c changes suddenly between two states, and the observed D (D(obs)) may be expressed by the population-weighted average of D
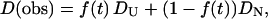 |
(8) |
where f(t) is a time-dependent fraction of unfolded species having a diffusion coefficient of DU. If we assume that the conformational transition between two states occurs with a rate constant k (f(t) = exp(−kt)), the time dependence of the concentrations of unfolded ([U]) and native ([N]) Cyt c are governed by diffusion equations of
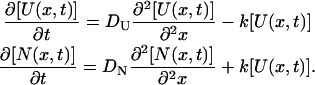 |
Solving these equations, we may find the time dependence of the refractive index as
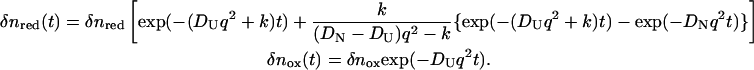 |
(9) |
The temporal profile of the TG signal based on this model is calculated from Eqs. 1 and 9.
RESULTS
TG signal of refolding
First, we describe the initiation process of the refolding of Cyt c. The GdnHCl-induced unfolding transitions of Fe(III) and Fe(II) Cyt c at pH = 7.0 are monitored by the CD intensity at 222 nm under equilibrium condition, and are depicted in Fig. 1 a. The transition midpoint concentrations of GdnHCl for the unfolding of the protein are 2.9 M and 5.6 M for the Fe(III) and Fe(II) Cyt c, respectively. The CD spectra suggest that all secondary structure of Fe(III) Cyt c is lost above [GdnHCl] > 3.5 M. These denaturation curves agree well with the previously reported ones (Pascher et al., 1996; Telford et al., 1998). These data show that the protein structure is more stable for the reduced form than for the oxidized form. Hence, by the photoreduction of Cyt c at a GdnHCl concentration, under which the oxidized form is unfolded but the reduced form is folded, the refolding process can be initiated.
FIGURE 1.

The circular dichroism intensity at 222 nm (a, Fe(III) Cyt c (▪) and Fe(II) Cyt c (▴)) and the diffusion coefficients (b, Fe(III) Cyt c (•)) against various concentrations of GdnHCl in phosphate buffer solution. The best-fitted curves in panel a by the two-state transition model are shown by the solid lines. In panel b, D was monitored by the sulfo-HSAB method described in text. The D values corrected for the increase of the solution viscosity by adding GdnHCl (corresponding to D at [GdnHCl] = 0 M) are shown (□), and the fitted curve for these data is shown by the dashed line. D of native Cyt c at various [GdnHCl] (solid line in b) is calculated from D in buffer solution without GdnHCl and the [GdnHCl] dependence of the solution viscosity.
The photoreduction is achieved by using the multiphoton excitation of NADH at 355 nm followed by the electron ejection to yield NAD radical (·NAD). The reaction finally produces the dimer (NAD)2 (Orii, 1993):
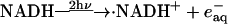 |
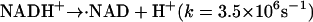 |
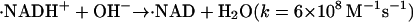 |
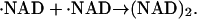 |
The solvated electron (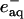 ) and ·NAD can efficiently reduce Cyt c. The kinetics of the reduction was traced by the transient absorption signals after the photoexcitation of NADH, and we found the reduction of Cyt c with a rate constant of 3 × 104 s−1 under these experimental conditions (Nada and Terazima, 2003).
) and ·NAD can efficiently reduce Cyt c. The kinetics of the reduction was traced by the transient absorption signals after the photoexcitation of NADH, and we found the reduction of Cyt c with a rate constant of 3 × 104 s−1 under these experimental conditions (Nada and Terazima, 2003).
The TG signals after photoexcitation of NADH in phosphate buffer solution as well as NADH + Cyt c in the same buffer solution were described previously (Nada and Terazima, 2003). Here we briefly summarize the essential features first. The TG signal after the photoexcitation of NADH without Cyt c rises with our instrumental response time (∼10 ns) and it decays with a lifetime of 200 ns and 1.8 μs under this condition in microseconds time range. The fast decay component (200 ns) represents the dynamics of the solvated electron released from NADH. The longer lifetime (1.8 μs) depends on q2 and it agrees well with the rate of Dthq2. This is the thermal grating component created by the thermal energy due to the nonradiative transition from the excited state. The inhomogeneity of temperature of the solution becomes uniform with Dthq2, and therefore, the temperature inhomogeneity cannot affect the molecular dynamics on longer timescale than (Dthq2)−1, which is typically 1 ∼ 10 μs. In the millisecond time range, a slower rise-decay signal was observed (Fig. 2 b in Nada and Terazima, 2003). The signal was reproduced well by a biexponential function (Eq. 4), and the rate constants of the rise and decay components are proportional to q2. This signal represents the diffusion process of the reactant (NADH) and the product ((NAD)2). D values of these species were determined (D(NADH) = 4.0 ± 0.4 × 10−10 m2/s and D((NAD)2) = 2.5 ± 0.3 × 10−10 m2/s).
FIGURE 2.

(a) Time profiles of the TG signal (dotted line) after the photoexcitation of NADH with Fe(III) Cyt c (66 μM) and GdnHCl (3.5 M) in phosphate (100 mM) buffer at various grating wavenumbers; q2 = (1) 1.4 × 1012 m−2, (2) 6.4 × 1011 m−2, (3) 2.5 × 1011 m−2, and (4) 5.7 × 1010 m−2 probed at 840 nm. The solid line is the best-fitted curve based on the two-state model with δnox = δnred. The rate constants k from the fitting of 1–4 are 24 s−1, 19 s−1, 20 s−1, and 23 s−1, respectively. All the signal intensities are normalized. (b) Time profiles of the TG signal (dotted line) at q2 = 7.1 × 1012 m−2 probed at 543 nm. The best-fitted signal by a single exponential function is shown by the solid line. The sharp spike-like signal at t ∼ 0 is the thermal grating signal. (c) Time profile of the TG signal at q2 = 1.32 × 1010 m−2 probed at 840 nm (dotted line), and the fitted curve by the biexponential function (Eq. 4) after 200 ms (solid line).
When Cyt c is added to NADH + GdnHCl (3.5 M)/buffer solution, a much stronger species grating signal was observed on a much longer timescale (Fig. 2). The origin of the signal was assigned based on the following facts (Nada and Terazima, 2003).
Because this signal was not observed without NADH, we can neglect the effect of direct photoexcitation of Cyt c.
Because the time range of this signal is much longer than that expected from NADH or (NAD)2, the chemical species contributed to the signal is not NADH but it should be a much larger one, i.e., Cyt c.
The rise-decay curve clearly indicates that two species having different D contribute to the signal, and they have positive and negative signs of the δn components. Hence, we concluded that the rise and decay components correspond to the presence of the product (Fe(II) Cyt c) and the reactant (Fe(III) Cyt c), respectively.
This signal does not appear at a low concentration of GdnHCl and the intensity gradually increases in a range of 2.5 M < [GdnHCl] < 3.5 M. This range coincides with a range where Fe(III) Cyt c is unfolded and Fe(II) Cyt c is folded. Hence, this signal appears due to the folding reaction of Cyt c.
Contrary to the TG signal of NADH/buffer solution, the signal of the solution containing Cyt c cannot be analyzed by two diffusing species with constant D (Eq. 4), because of the following two characteristic features (Nada and Terazima, 2003). First, the time profile cannot be expressed by the biexponential function of Eq. 4 (Fig. 2 c in Nada and Terazima, 2003). Second, the time profile and the signal intensity depend on the observation time range significantly. The TG signal in a fast timescale (with a large q2) is weak, but it increases with increasing the observation time (Fig. 3 a in Nada and Terazima, 2003). We have concluded that these features should be interpreted in terms of the time-dependent apparent D. Because D of the reactant (Fe(III) Cyt c) is constant, the time dependence of D should come from the time dependence of D of Fe(II) Cyt c. We extended similar measurements at various q2 for the detailed analysis described later.
FIGURE 3.

Reaction scheme of N-hydroxysulfosuccinimidyl-4-azidobenzoate (sulfo-HSAB) and Cyt c. The size of Cyt c is much larger than that of sulfo-HSAB. The photoproduct with nitrene may react (a) intermolecularly to yield the dimer of Cyt c, or (b) intramolecularly.
Previously, the profile was analyzed only from the curvatures at the top of the rise-decay curve to obtain averaged D at one specific time. However, the effect of the time-dependent D should be observed in any time region of the signal. Here, we analyze the time profiles in the whole time range.
This time-dependent apparent D may be interpreted based on two possible models. First, the conformational change and/or hydrogen-bonding network rearrangement could occur gradually and continuously. In this case, D of each protein gradually changes in time (continuous model). Second, D could change by the two-state manner (two-state model); that is, the conformation of each protein changes suddenly from unfolded conformations having DU to compact ones having DN, but the relative population of these conformation changes with a rate constant of k. The theoretical expressions of the time dependences of the TG signals based on these two models are derived and given in the “Principle and theoretical” section.
By the fitting of the observed TG signal using these equations, we expected to find the appropriate model to describe this folding reaction and the rate constant of the D change. However, contrary to this expectation, we found that relatively good fittings were possible using both models, because the fitting functions (Eqs. 7 and 9) contain many unknown quantities (δnred, δnox, DN, DU, and k), which should be used as the adjustable parameters. Therefore, we tried to reduce the number of the fitting parameters by measuring DN and DU independently as described in the next section.
Diffusion of Cyt c
To elucidate the relationship between D and the protein conformations, we measured D of Cyt c under equilibrium condition at various concentrations of GdnHCl. Furthermore, as described in the previous section, it is desirable to know D of the unfolded and native Cyt c under the same experimental conditions as that in this study for detailed analysis. For the measurement of D by the TG method, however, the target molecule should be photoreactive to induce the refractive index modulation. Although Cyt c itself is not photoreactive, we succeeded in measuring the TG signal by utilizing a photolabeling compound as follows (Baden et al., 2004).
We used sulfo-HSAB to photolabel Cyt c. Sulfo-HSAB has been developed and used for a photoactive cross-linking reagent. When this reagent was mixed with the Cyt c solution, the N-hydroxysuccinimide (NHS) end reacts with Cyt c (Fig. 3). This reaction should take place in darkroom conditions to protect the aryl azide from photolysis. The photoactive end (azide) releases a nitrogen molecule upon photoexcitation. This photochemical reaction induces the refractive index modulation to produce the TG signal. From the time profile of the signal, D can be determined. (By this method, D of Cyt c-NHS not Cyt c is measured. However, because the molecular size of Cyt c (molecular weight = 1.2 × 104 g/mol) is significantly larger than that of NHS (146 g/mol), the difference between D of Cyt c-NHS and Cyt c can be neglected).
First, we show the TG signal after photoexcitation of Cyt c (2 mM) with Sulfo-HASB (200 μM) and [GdnHCl] = 1 M in buffer solution (Fig. 4 a). Under this condition, Cyt c possesses the native structure. The TG signal rises quickly (within 50 ns) and this thermal grating signal decays with the rate constant of Dthq2. After the thermal grating signal, a species grating signal appears. The species grating signal rises slowly and then turns to decay. The species grating signal after 2 ms was fitted by a biexponential function (Eq. 4) with a rise rate constant of krise and decay rate constant of kdecay (Fig. 4 a). (We used the data after 2 ms for the fitting, because the diffusion of free sulfo-HSAB (not bound to Cyt c) contributes to the early part of the species grating signal (t < 2 ms), which was confirmed by the TG signal of sulfo-HSAB solution without Cyt c (data are not shown)). From the q2 dependence, we found that these rate constants represent the molecular diffusion process. D values corresponding to the rise and decay components are calculated from the relationships of krise = Driseq2 and kdecay = Ddecayq2 to be Drise = 1.18 × 10−10 m2/s and Ddecay = 0.9 × 10−10 m2/s at [GdnHCl] = 1.0 M. Because both D values are significantly smaller than D of sulfo-HSAB itself (3 × 10−10 m2/s), they should represent D of proteins. D of the native Cyt c has been reported to be 1.2 ∼ 1.3 × 10−10 m2/s (Fuh et al., 1993). Therefore, Drise in this experiment should be attributed to D of Cyt c.
FIGURE 4.

Time profiles of the TG signal (dotted lines) after the photoexcitation of sulfo-HSAB with Fe(III) Cyt c (1 mM) in (a) [GdnHCl] = 1 M, and (b) [GdnHCl] = 3.5 M phosphate buffer solution. The best-fitted curves by a biexponential function are shown by the solid line. The sharp spike-like signal at t ∼ 0 is the thermal grating signal.
What is the diffusing species of the decay component? For the assignment, one should note that Drise is ∼1.3 times larger than Ddecay. According to the Stoke-Einstein relationship, D is given by
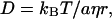 |
(10) |
where kB, T, η, a, and r are Boltzmann constant, temperature, viscosity, a constant representing the boundary condition between the diffusing molecule and the solvent, and radius of the molecule, respectively. If we assume that the diffusing species is spherical, the 1.3 times difference in D corresponds to ∼2 times difference in the molecular volume. This factor 2 suggests that the diffusing species corresponding to the decay component could be the dimer form of Cyt c. This assignment may be reasonable, because sulfo-HSAB is a nonspecific linker of proteins (Fig. 3 a). Therefore, we attribute Drise to D of Cyt c and Ddecay to that of Cyt c dimer produced by the interprotein linkage by the nitrene group of the photodecomposed sulfo-HSAB.
A similar signal on a much longer timescale is observed for Cyt c with [GdnHCl] = 3.5 M solution and similar analysis was performed (Fig. 4 b). The slower dynamics than that with [GdnHCl] = 1.0 M reflects the slower diffusion rate of the unfolded Cyt c, and D under this condition is determined to be 0.65 × 10−10 m2/s.
For further testing this assignment (Drise corresponds to D of Cyt c), we tried to measure D of Cyt c by a different method. When we measure the TG signal of Cyt c + NADH in the buffer solution under a large q condition, the concentration modulation is smeared out quickly by the diffusion due to the short distance of the fringes. If the decay rate is faster than the folding rate or at least faster than the changing rate of D, the decay of the grating signal should represent the diffusion process of only the unfolded Cyt c. In other words, Fe(III) Cyt c is photoreduced to Fe(II) Cyt c by the photoexcitation of NADH, but the product, Fe(II) Cyt c, diffuses with the same rate as the unfolded protein before D changes. Hence, we expect a single exponential decay (Dr ∼ Dp) in Eq. 4 and D of unfolded Cyt c should be determined uniquely from the rate and the q2 value.
We have tried this method under the same experimental conditions for Cyt c folding experiment. However, we found that the signal is very weak under this large q2 condition, indicating that δnr ∼ δnp and Dr ∼ Dp in Eq. 4. To overcome this weak signal, we used the probe wavelength of 543 nm for monitoring the absorption change (amplitude grating term), which provides a strong signal due to the resonance enhancement. As expected, the TG signal at q2 = 7.1 × 1012 m2 is expressed well by a single exponential function (Fig. 2 b). From the relation of k = Dq2, D of the native Cyt c at [GdnHCl] = 1 M and the unfolded Cyt c at [GdnHCl] = 3.5 M are obtained to be 1.2 × 10−10 m2/s and 0.66 × 10−10 m2/s, respectively. These values agree well with that measured by the sulfo-HSAB method, supporting the validity of this method.
D of Cyt c at various GdnHCl concentrations are measured by the sulfo-HSAB method and it was found that D decreases with increasing [GdnHCl] (Fig. 1 b). For comparing the [GdnHCl] dependence with the unfolding curve monitored by the CD intensity (Fig. 1 a), we should note the viscosity increases with increasing [GdnHCl]. Some part of the gradual change of D in Fig. 1 b is due to the [GdnHCl] dependence of the solution viscosity. For correcting the viscosity effect, D at [GdnHCl] = 0 M is calculated by using the Stoke-Einstein relationship and the reported viscosity data (Kawahara and Tanford, 1966). We found that the [GdnHCl] dependence of corrected D up to [GdnHCl] < 5 M is described well by the two-state model with a midpoint of [GdnHCl] = 2.9 M. The changes of D as a function of [GdnHCl] are similar to those from the CD signal intensity. (D increases in a region of [GdnHCl] > 4.5 M. This unusual behavior after the denaturation transition may be due to the contribution of different intermediate species that has been suggested from the small angle x-ray scattering data (Segel et al., 1998). We will investigate this behavior in the future in detail. In this article, we will limit our discussion in a range of 0 M < [GdnHCl] < 4 M.
D of the native Cyt c is measured by the same method at [GdnHCl] = 0 M to be 1.25 × 10−10 m2/s. DN at [GdnHCl] = 3.5 M was calculated from this value and the viscosity at [GdnHCl] = 3.5 M based on the Stoke-Einstein relationship.
Analysis of TG signal
The TG signals for the Cyt c refolding reaction are fitted based on the two different models described in the “Principle and theoretical” section using the diffusion coefficients of unfolded (DU) and native (DN) proteins determined in the previous section. Using the continuous model (Eq. 7), we found that the observed signals cannot be fitted well. A typical example of the fitted curve using this model is shown in Fig. 5. On the other hand, the observed TG signals can be reproduced well based on the two-state model (Figs. 2 and 5). The remarkable agreements between the fitted and the observed signals indicate that the D change during the folding process should be expressed by the two-state model.
FIGURE 5.

An example of the best-fitted curve (dashed line) of the observed TG signal (dotted line) at q2 = 2.3 × 1011 m−2 based on the continuous model. For comparison, the best-fitted curve based on the two-state model is shown by the solid line.
This agreement is further interesting, if we notice that the best fitting can be achieved with δnox ∼ δnred. The similar amplitudes of the species grating between Fe(III) Cyt c and Fe(II) Cyt c can be seen from the very weak signal intensity on an early timescale (e.g., t < 20 ms in Fig. 2 c). What does this similarity mean? The refractive index change of the species grating consists of the refractive index difference between the reactant and products due to the change of the absorption spectrum (δnpop, population grating), and the density change caused by the reaction volume (δnv, volume grating), i.e., the weak grating signal intensity indicates δnspe = δnpop + δnvol ∼ 0. For further discussion, we estimate the population grating contribution from the signal intensity under a condition such that the volume change is not expected. The TG signal without volume change may be obtained under a condition that the refolding does not occur; that is, either at the low concentration of GdnHCl or in a fast time region. As described in the “TG signal of refolding” section, the species grating signal in [GdnHCl] < 2.0 M is very weak. We also found that the species grating intensity at a large q2 (t < 2 ms) is very weak as described in the “Diffusion of Cyt c” section. These weak signals without the folding reaction indicate that the population grating contribution by the photoreduction of Cyt c from Fe(III) to Fe(II) is negligibly small. Considering these two facts (δnspe ∼ 0 and δnpop ∼ 0), we conclude that the volume grating contribution is also negligible (δnvol ∼ 0) in this time range. In other words, the volume change by the folding reaction is negligible in t < 20 ms (Fig. 2 c). This point will be discussed later.
In Fig. 2 a, the observed TG signals and the fitted curves at various q2 with the fitting parameters of only δnred( = δnox) and k are depicted. The determined rate constant, k, from the fitting of Fig. 2 a (1–4) are 24 ± 3 s−1, 19 ± 3 s−1, 20 ± 3 s−1, and 23 ± 3 s−1, respectively. They are close to each other on these different timescales from 2 ms to 2 s. If the folding pathway is described by more than two states in this time range, we expect that the observed TG signal cannot be fitted by the two-state model or the obtained rate constant should be significantly altered depending on the fitting time range, i.e., at different q2. The small deviation of k in a time range of from 2 ms to 2 s indicates that the folding process is really two state. The rate constant determined by the fitting under various q2 is 22 ± 7 s−1.
We next examined if there is any change in D outside of our observation time range (t < 2 ms or t > 2 s). First, if D has already changed before 2 ms, the TG signal having two different D should appear in the signal at a large q2. However, the single exponential behavior of the signal at q2 = 7 × 1012 m−2 (Fig. 2 b) in a range of t < 2 ms clearly indicates a single D before this time. Furthermore, the determined D agrees well with that of the unfolded (initial) protein. If D within 2 ms after the folding trigger is different from DU even by 10%, the signal should be significantly deviates from the single exponential behavior. Although we cannot estimate the upper limit of the D change within 2 ms accurately, the qualitative different feature between the single- and biexponential functions and the good agreement of the single-exponential fitting for the observed signal (Fig. 2 b) suggest that the D change within 2 ms should be smaller than 10% of DU. Therefore, we conclude that there is almost no change in D before 2 ms.
Second, if the dynamics of D is completed with the rate constant of 22 s−1, the signal sufficiently after this process, for instance, after ∼200 ms should be analyzed by two time-independent D values of native and unfolded Cyt c. We examined this possibility by fitting the TG signal at q2 = 1.3 × 1010 m−2 with a biexponential function (Eq. 4) after 200 ms (Fig. 2 c). The D values from the fitting are 1.05 × 10−10 m2/s and 0.64 × 10−10 m2/s, which agrees with DN and DU at [GdnHCl] = 3.5 M determined independently in the “Diffusion of Cyt c” section. Hence, we conclude that the change in D finishes completely until this time and there is no dynamics of D after the 22 s−1 process.
DISCUSSION
There are several factors that govern the diffusion process of molecules in solution, such as the molecular size, shape, and the intermolecular interaction between the solvent and the diffusing molecules. To investigate the origin of the diffusion, we first examine the effect of the molecular shape on D. The Stokes-Einstein equation was derived for the diffusion process of spherical molecules. The native Cyt c may be treated as a spherical shape (Bushnell et al., 1990). When the shape is deformed from the spherical symmetry, the friction should be different from Eq. 10. For example, a frictional ratio (f/f0) for a prolate molecule was theoretically derived as (Van Holde et al., 1998):
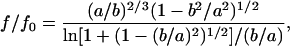 |
(11) |
where f0 is the friction of a spherical molecule, a is the semimajor axis and b is the minor axis. If the molecule possesses a rodlike shape, f/f0 is given by
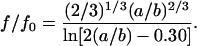 |
(12) |
If the denatured Cyt c structure possesses a/b = 2, f/f0 of this protein calculated from Eqs. 11 and 12 are 1.08 and 1.06, respectively. This ratio is much smaller than the ratio of DN/DU (DN/DU = 1.9). Furthermore, although we do not know the exact shape of the denatured Cyt c, it may be reasonable to consider that the conformation of the random coil or molten globule state does not deviate from the spherical symmetry so much larger than a/b > 2. Therefore, we think that the change of the shape of the protein is not the main origin of the smaller D under the denatured condition.
On the other hand, the interaction between a protein and water molecules shall affect the friction. In the high concentration of GdnHCl, the protein is completely denaturated and consequently the solvent-exposed surface area of the protein increases, which should cause the increase in the binding site of GdnHCl and water upon a protein. Hence the solvent molecules will interact with a more corrugated protein surface and consequently the protein molecule feels more frictional drag in the solvents and its dynamic flexibility is more restrained. The denaturation curve monitored by D (Fig. 1 b) is considered to represent this change. The similarity of the denaturation curves monitored by the CD intensity (Fig. 1 a) and D may reflect that the intermolecular interaction enhanced by the destruction of the secondary structure.
A small angle x-ray scattering (SAXS) measurement of the Fe(II) Cyt c radius of gyration showed a cooperative unfolding transition at [GdnHCl] = 2.6 M (Segel et al., 1998). Above 3.5 M, the SAXS data suggest that Fe(II) Cyt c is a random coil. The radius increases from 1.38 nm to 3.0 nm as the protein unfolds. A rapid mixing experiment of pH jump on Fe(III) Cyt c solution using the SAXS detection indicates that the radius of gyration of Cyt c decreases 2.05 nm → 1.77 nm → 1.39 nm with a rate constant of 2400 s−1 and 68 s−1 (Akiyama et al., 2002). The initial collapse with 2400 s−1 is sufficiently faster than the rate constant in this D-change dynamics (22 s−1). This discrepancy in the rates might be explained by the different experimental conditions (pH jump versus [GdnHCl] = 3.5 M) and the different target proteins (Fe(III) versus Fe(II) Cyt c). It is probable that the initial fast collapse does not occur for the Fe(II) Cyt c at this GdnHCl concentration. However, the previous time-resolved CD experiment showed three phases with lifetimes of 5 μs, 6 ms, and 110 ms (Chen et al., 1999). The fastest 5-μs process is a rapid formation of ∼20% secondary structure, and could be interpreted in terms of the initial collapse. Furthermore, based on accumulated kinetic data of the protein-folding researches (Pain, 2000), the “initial burst phase” has been observed for many systems, and it is plausible that the initial collapse exists faster than 22 s−1 s under our experimental conditions, too. Assuming that there exists the initial collapse, we here consider two aspects on the protein dynamics.
First, why does not this initial collapse lead to the D change? The collapse without the accompanying D change is consistent with the previous conclusion that a small change in the protein shape does not result in the D change. It also indicates that the initial collapse does not change the intermolecular interaction, such as the hydrogen-bonding network, i.e., the protein still forms the hydrogen bonding with the solvent after this collapse. Indeed, the radius of gyration after the initial collapse detected by the rapid mixing is 1.78 nm, which is still much larger than that of the native form (1.39 nm) (Akiyama et al., 2002). Therefore, it is reasonable to consider that the intermolecular hydrogen network does not change for this swelled protein, and that D should be similar to that of the initial much-swelled (larger radius of gyration) protein before the collapse. Furthermore, it should be noted that D reflects not only the size of the molecule but also the intermolecular interaction. Frequently, the D value is converted to the hydrodynamic radius based on the Stoke-Einstein relationship and discussed in terms of the molecular “size”. However, contrary to the radius of gyration, which is defined solely from the geometric structure, many factors contribute to the hydrodynamic radius. For example, even the molecular size is the same, D (or the hydrodynamic radius) could be different depending on the intermolecular interaction between the solute and the solvent (Terazima and Hirota, 1993; Terazima et al., 1993, 1995; Okamoto et al., 1995, 1997, 1998; Ukai et al., 2000a,b).
Second, because the TG signal does not appear after the initial collapse, we should consider that this collapse does not induce the partial molar change. This aspect can be explained consistently as follows. There are several contributions in the partial molar volume of protein (Chalikian and Breslauer, 1996; Terazima, 2002). Major contributions may be classified as: 1), constitutive volume, which is the sum of the van der Waals volumes of all of the constitutive atoms; 2), void volume, which is the volume of the structural voids within the solvent-inaccessible core that results from imperfect atomic packing; 3), solvent exclusion volume, which is a volume that cannot be entered by solvent molecules; and 4), interaction volume, which is the effects of solute-solvent interactions.
If two spherical molecules contact each other, the partial molar volume should decrease due to the loss of solvent exclusion volume around the molecule. Similarly, we expect that the making contacts of many atoms of the protein result in the volume decrease. However, the fact that we do not observe such volume change within t < 20 ms instead of the collapse in the fast time range (∼400 μs) suggests that solvent molecules exist between the residues of the protein even after the initial collapse; in other words, the residues do not contact each other directly during this initial collapse. This picture is consistent with the above finding on D change; i.e., the protein still forms the hydrogen bonding with the solvent after this collapse. We may consider that the solvent molecules between the residues are finally squeezed out by the step of the D-change dynamics.
Similarly, although the time-resolved CD experiment showed 6-ms dynamics, the D change does not occur with this rate (Chen et al., 1999). This seemingly inconsistency may be also explained by the similar argument as above.
Here, it is important to stress that the TG signal can be fitted by the two-state model with a single exponential function of f(t) in a whole time range. The two-state behavior is the same as that reported previously for explaining experimental results (Pascher et al., 1996; Telford et al., 1999). However, the nature of the two states from this experiment should be different from those in previous reports, because the two states in this work are distinguished in terms of the diffusion coefficient. Therefore, the rate from this experiment should not be the same as the rate obtained by other experiments that do not probe the diffusion; i.e., the structural change that does not change D should not appear as the TG signal as discussed above in detail. Only the population change between chemical species having different intermolecular interaction, such as the hydrogen-bonding interaction or hydrophobic interaction, manifests itself. We next compare the folding kinetics with other ones reported previously.
The folding kinetics is sensitive to the refolding conditions. It is desirable to compare the data measured under similar conditions. However, not so many experiments have been reported on the folding dynamics of Fe(II) Cyt c so far. Below, we compare the dynamics in previous results investigated under the similar conditions. Previous time-resolved CD experiments have shown a change in the amplitude of the α-helix CD signal by three phases with lifetimes of 5 μs, 6 ms, and 110 ms (and 180-ms phase with decreasing the amplitude of the CD signal) (Chen et al., 1999). As described before, the 5-μs process is a rapid formation of ∼20% secondary structure after the photoreduction. The 6-ms phase has a small amplitude in the secondary structure refolding. A major change was observed in the 110-ms time range. About 65% of native reduced Cyt c secondary structure is detected with this phase, and this observation is consistent the 100-ms rate of 70% recovery of Fe(III) Cyt c detected by a stopped-flow CD experiment (Elöve et al., 1992). Refolding rate under similar experimental conditions measured by the tryptophan fluorescence quenching method indicates 680-ms dynamics (Pascher, 2001). Because Trp fluorescence is quenched by the heme, this dynamics should represent the movement of approaching Trp to the heme. In native state, the heme iron has axial ligands His-18 and Met-80. Under typical denaturation conditions, such as high [GdnHCl] at neutral pH, Met-80 dissociates from the heme. Telford et al. (1999) measured the ligand substitution reaction in Fe(II) Cyt c at [GdnHCl] = 3.2 M. Above pH = 5.5, Fe(II) Cyt c folding is monophase with a lifetime of 100 ms at pH = 7 (Telford et al., 1999). This rate was attributed to the conversion of five coordinate high-spin His-18-Fe(II) to six coordinate low-spin Met-80-Fe(II)-His-18 Cyt c. At neutral pH, it was agreed that the unfolded peptide collapses around the heme before the replacement of Met-80 from His. The folding pathway based on these reports and this finding may be summarized as Fig. 6. The lifetimes of D change, 41 ms, is slightly faster or rather close to the major change of the α-helix formation detected by the time-resolved CD experiment (110 ms) and the ligand substitution reaction (100 ms). The slightly faster rate of D change (41 ms) may indicate that the creation of the intramolecular hydrogen bonding is accomplished before the major formation of the α-helix (100 ms). This α-helix formation then leads the ligand exchange. It is important to stress that the dynamics of the intermolecular interaction occurs by one phase with the rate of 22 s−1 in the entire time region.
FIGURE 6.

Schematic illustrations of refolding dynamics of Fe(II) Cyt c monitored by various properties. Rough timescales for these dynamics are shown by arrows together with the detection method. The rates of protein collapse measured by the rapid mixing experiment using the pH jump for Fe(III) Cyt c is also shown in parentheses just for comparison. Structures in bottom of the figure indicate: (I) unfolded Fe(II) Cyt c, the dominant part of the hydrogen bonding is the intermolecular one between the protein and solvent molecules; (II) a small amount of α-helix is formed; (III) hydrogen bonding is rearranged from the intermolecular one between the protein and solvent to the intramolecular one; (IV) formation of the α-helix in the major part and the ligand exchange; (V) native Fe(II) Cyt c. The thick lines indicate the protein backbone and the thin lines represent the hydrogen bonding. See text for detail.
CONCLUSION
Kinetics of intermolecular interaction between cytochrome c (Cyt c) protein and solvent is studied during the protein-refolding process from a view point of diffusion coefficient (D) using the pulsed-laser-induced transient grating technique. D of Cyt c after triggered by photoinduced reduction of unfolded Fe(III) Cyt c in 3.5 M GdnHCl solution is monitored in time domain. D of Cyt c at various GdnHCl concentrations is measured by the TG method using a photolabeling reagent. The denaturation curve monitored by D is similar to that monitored by the CD intensity. Using these data, it was found that the time dependence of the observed TG signal in a whole time range can be consistently reproduced well by the two-state model. The dynamics of D is expressed well by a single exponential function with a rate constant of 22 ± 7 s−1. The folding process of Fe(III) Cyt c in 3.5 M GdnHCl solution triggered by the laser-induced electron injection method is discussed based on these observations and on the data reported previously. After the electron injection, a minor α-helix is formed as detected by the time-resolved CD measurement, which could reflect possible initial protein collapse. Until 25 ms, the hydrogen-bonding network retains between the protein and the solvent. With the rate constant of 22 s−1, the hydrogen bonding changes from the intermolecular to intramolecular one. Slightly after this rearrangement or with a similar rate, the α-helix is formed in the major part and the ligand exchange occurs. This TG technique will be a unique and powerful tool to elucidate the time dependence of the intermolecular interaction in many systems in the future.
Acknowledgments
This work is supported by the Grant-in-Aid (Nos. 13853002 and 15076204) from the Ministry of Education, Science, Sports and Culture in Japan.
References
- Abbruzzettri, S., C. Viappiani, J. R. Small, L. J. Libertini, and E. W. Small. 2001. Kinetics of histidine deligation from the heme in GuHCl-unfolded Fe(III) cytochrome c studied by a laser induced pH-jump technique. J. Am. Chem. Soc. 123:6649–6653. [DOI] [PubMed] [Google Scholar]
- Akiyama, S., S. Takahashi, T. Kimura, K. Ishimori, I. Morishima, Y. Nishikawa, and T. Fujisawa. 2002. Conformational landscape of cytochrome c folding studied by microsecond-resolved small angle x-ray scattering. Proc. Natl. Acad. Sci. U.S.A. 99:1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden, N., and M. Terazima. 2004. A novel method for measurement of diffusion coefficients of proteins and DNA in solution. Chem. Phys. Lett. 393:539–545. [Google Scholar]
- Bhuyan, A. K., and J. B. Udgaonkar. 2001. Folding of horse cytochrome c in the reduced state. J. Mol. Biol. 312:1135–1160. [DOI] [PubMed] [Google Scholar]
- Bushnell, G. W., G. V. Louie, and G. D. Brayer. 1990. High-resolution three-dimensional structure of horse heart cytochrome c. J. Mol. Biol. 214:585–595. [DOI] [PubMed] [Google Scholar]
- Chalikian, T. V., and K. J. Breslauer. 1996. On volume changes accompanying conformational transitions of biopolymers. Biopolymers. 39:619–626. [DOI] [PubMed] [Google Scholar]
- Chen, E., P. Wittung-Stefshede, and D. S. Kliger. 1999. Far-UV time-resolved circular dichroism detection of electron-transfer-triggered cytochrome c folding. J. Am. Chem. Soc. 121:3811–3817. [Google Scholar]
- Choi, J., and M. Terazima. 2002. Denaturation of a protein monitored by diffusion coefficients:myoglobin. J. Phys. Chem. B. 106:6587–6593. [Google Scholar]
- Cussler, E. L. 1984. Diffusion. Cambridge University Press., Cambridge, UK.
- Elöve, G. A., A. F. Chaffotte, H. Roder, and M. E. Goldberg. 1992. Biochemistry. 31:6876–6883. [DOI] [PubMed] [Google Scholar]
- Englander, S. W., T. R. Sosnick, L. C. Mayne, M. Shtilerman, P. X. Qi, and Y. Bai. 1998. Fast and slow folding in cytochrome c. Acc. Chem. Res. 31:737–744. [Google Scholar]
- Fuh, C. B., S. Levin, and J. C. Giddings. 1993. Rapid diffusion coefficient measurements using analytical SPLITT fractionation: application to proteins. Anal. Biochem. 208:80–87. [DOI] [PubMed] [Google Scholar]
- Godbole, S., and B. E. Bowler. 1999. Effect of pH on formation of a nativelike intermediate on the unfolding pathway of a Lys73->His variant of yeast iso-1-cytochrome c. Biochemistry. 38:487–495. [DOI] [PubMed] [Google Scholar]
- Hagen, S. J., J. Hofrichter, and W. A. Eaton. 1997. Rate of intrachain diffusion of unfolded cytochrome c. J. Phys. Chem. B101:2352–2365. [Google Scholar]
- Jones, C. M., E. R. Henry, Y. Hu, C. Chan, S. D. Luck, A. Bhuyan, H. Roder, J. Hofrichter, and W. A. Eaton. 1993. Fast events in protein folding initiated by nanosecond laser photolysis. Proc. Natl. Acad. Sci. USA. 90:11860–11864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara, K., and C. Tanford. 1966. Viscosity and density of aqueous solutions of urea and guanidine hydrochloride. J. Biol. Chem. 241:3228–3232. [PubMed] [Google Scholar]
- Nada, T., and M. Terazima. 2003. A novel method for study of protein folding kinetics by monitoring diffusion coefficient in time domain. Biophys. J. 85:1876–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, K., N. Hirota, and M. Terazima. 1997. Diffusion process of the benzyl radical created by photodissociation probed by the transient grating method. J. Phys. Chem. A. 101:5269–5277. [Google Scholar]
- Okamoto, K., N. Hirota, and M. Terazima. 1998. Diffusion of photochemical intermediate radicals in water/ethanol mixed solvents. J. Phys. Chem. A. 102:3447–3454. [Google Scholar]
- Okamoto, K., M. Terazima, and N. Hirota. 1995. Temperature dependence of diffusion processes of radical intermediates probed by the transient grating method. J. Chem. Phys. 103:10445–10452. [Google Scholar]
- Okuno, T., S. Hirota, and O. Yamauchi. 2000. Folding character of cytochrome c studied by o-nitrobenzyl modification of methionine 65 and subsequent ultraviolet light irradiation. Biochemistry. 39:7538–7545. [DOI] [PubMed] [Google Scholar]
- Orii, Y. 1993. Immediate reduction of cytochrome c by photoexcited NADH: reaction mechanism as revealed by flow-flash and rapid-scan studies. Biochemistry. 32:11910–11914. [DOI] [PubMed] [Google Scholar]
- Pain, R. H. 2000. Mechanism of Protein Folding, 2nd Ed. Oxford University Press, Oxford, UK.
- Pascher, T. 2001. Temperature and driving force dependence of the folding rate of reduced horse herat cytochrome c. Biochemistry. 40:5812–5820. [DOI] [PubMed] [Google Scholar]
- Pascher, T., J. P. Chesick, J. R. Winkler, and H. B. Gray. 1996. Protein folding triggered by electron transfer. Science. 271:1558–1560. [DOI] [PubMed] [Google Scholar]
- Segel, D. J., D. Eliezer, V. Uversky, A. L. Fink, K. O. Hodgson, and S. Doniach. 1999. Transient dimer in the refolding kinetics of cytochrome c characterized by small angle X-ray scattering. Biochemistry. 38:15352–15359. [DOI] [PubMed] [Google Scholar]
- Segel, D. J., A. L. Fink, K. O. Hodgson, and S. Doniach. 1998. Protein denaturation: a small-angle X-ray scattering study of the ensemble of unfolded states of cytochrome c. Biochemistry. 37:12443–12451. [DOI] [PubMed] [Google Scholar]
- Shastry, M. C. R., and H. Roder. 1998. Evidence for barrier-limited protein folding kinetics on the microsecond time scale. Nat. Struct. Biol. 5:385–392. [DOI] [PubMed] [Google Scholar]
- Shastry, M. C. R., J. M. Sauder, and H. Roder. 1998. Kinetic and structural analysis of submillisecond folding events in cytochrome c. Acc. Chem. Res. 31:717–725. [Google Scholar]
- Takahashi, S., S.-R. Yeh, T. K. Das, C.-K. Chan, D. S. Gettfried, and D. L. Rousseau. 1997. Folding of cytochrome c initiated by submillisecond mixing. Nat. Struct. Biol. 4:44–50. [DOI] [PubMed] [Google Scholar]
- Takeshita, K., Y. Imamoto, M. Kataoka, F. Tokunaga, and M. Terazima. 2002. Themodynamical and transport properties of intermediate states of photo-cyclic reaction of photoactive yellow protein. Biochemistry. 41:3037–3048. [DOI] [PubMed] [Google Scholar]
- Telford, J. R., F. A. Tezcan, H. B. Gray, and J. R. Winkler. 1999. Role of ligand substitution in ferrocytochrome c folding. Biochemistry. 38:1944–1949. [DOI] [PubMed] [Google Scholar]
- Telford, J. R., P. Wittung-Stafshede, H. B. Gray, and J. R. Winkler. 1998. Protein folding triggered by electron transfer. Acc. Chem. Res. 31:755–763. [Google Scholar]
- Terazima, M. 2000. Translational diffusion of organic radicals in solution. Acc. Chem. Res. 33:687–694. [DOI] [PubMed] [Google Scholar]
- Terazima, M. 2002. Molecular volume and enthalpy changes associated with irreversible photo-reactions. J. Photochem. Photobiol. C. 24:1–28. [Google Scholar]
- Terazima, M., and N. Hirota. 1993. Translational diffusion of a transient radical studied by the transient grating method: pyrazinyl radical in 2-propanol. J. Chem. Phys. 98:6257–6262. [Google Scholar]
- Terazima, M., K. Okamoto, and N. Hirota. 1993. Transient radical diffusion in photoinduced hydrogen abstraction reactions of benzophenone probed by the transient grating method. J. Phys. Chem. 97:13387–13393. [Google Scholar]
- Terazima, M., K. Okamoto, and N. Hirota. 1995. Translational diffusion of transient radicals created by the photo-induced hydrogen abstraction reaction in solution: anomalous size dependence in the radical diffusion. J. Chem. Phys. 102:2506–2515. [Google Scholar]
- Tyrrell, H. J. V., and K. R. Harris. 1984. Diffusion in Liquids. Butterworth, London, UK.
- Ukai, A., N. Hirota, and M. Terazima. 2000a. Radical diffusion measured by the transient grating in a short time scale. Chem. Phys. Lett. 319:427–433. [Google Scholar]
- Ukai, A., N. Hirota, and M. Terazima. 2000b. Diffusion of organic molecules in the excited triplet states detected by the transient grating with a high wavenumber. J. Phys. Chem. A. 104:6681–6688. [Google Scholar]
- Van Holde, K. E., W. C. Johnson, and P. Shing Ho.1998. Physical Biochemistry. Prentice- Hall, Saddle River, NJ.
- Yeh, S.-R., S. Takahashi, B. Fan, and D. L. Rousseau. 1997. Ligand exchange during cytochrome c folding. Nat. Struct. Biol. 4:51–56. [DOI] [PubMed] [Google Scholar]