

Abstract
Disagreement has remained about the spin state origin of the g = 4.1 EPR signal observed at X-band (9 GHz) from the S2 oxidation state of the Mn cluster of Photosystem II. In this study, the S2 state of PSII-enriched membrane fragments was examined at Q-band (34 GHz), with special interest in low-field signals. Light-induced signals at g = 3.1 and g = 4.6 were observed. The intensity of the signal at g = 3.1 was enhanced by the presence of F− and suppressed by the presence of 5% ethanol, indicating that it was from the same spin system as the X-band signal at g = 4.1. The Q-band signal at g = 4.6 was also enhanced by F−, but not suppressed by 5% ethanol, making its identity less clear. Although it can be accounted for by the same spin system, other sources for the signal are considered. The observation of the signal at g = 3.1 agrees well with a previous study at 15.5 GHz, in which the X-band g = 4.1 signal was proposed to arise from the middle Kramers doublet of a near rhombic S = 5/2 system. Zero-field splitting values of D = 0.455 cm−1 and E/D = 0.25 are used to simulate the spectra.
INTRODUCTION
Interest in the function of Photosystem II (PSII) from higher plants and cyanobacteria is high because of its importance in the life cycle of the planet as the main producer of atmospheric oxygen. The production of O2 is the result of electron donation from water to the electron transfer chain of photosynthetic centers within the thylakoid membrane and therefore involves a four-electron oxidation. The center at which this takes place, called the oxygen evolving complex (OEC) of PSII, includes a manganese cluster that cycles through a series of oxidation states, designated S0–S4, with oxygen evolution taking place from the highest oxidation state of S4. This process therefore represents a unique problem in catalysis as well as an important biological function.
The two electron paramagnetic resonance (EPR) signals from the S2 state of the OEC have been well characterized at X-band frequencies (9–10 GHz) since their discovery in the 1980s (Britt, 1996; Debus, 1992). The multiline signal, a signal at g = 2 with >19 main hyperfine lines, arises from an S = 1/2 ground state of the manganese cluster of the OEC. The other signal is known as the “g = 4.1 signal,” a title which describes its position as observed at X-band frequencies. At this band the signal has an isotropic appearance with width of 340−360 G and no resolved hyperfine structure in active samples. The signal was originally suggested to originate from a rhombic S = 5/2 system from iron because of its appearance (Casey and Sauer, 1984), but later work showed that it was associated with the S2 state of the OEC (de Paula et al., 1985; Zimmermann and Rutherford, 1986). Early studies of the signal also indicated that it was suppressed at higher temperatures by the presence of cryoprotectants, such as glycerol or ethylene glycol, and small alcohols (Zimmermann and Rutherford, 1986), and enhanced by the absence or substitution of Cl− (Casey and Sauer, 1984; Lindberg and Andréasson, 1996; Ono et al., 1986, 1987). Low-temperature (130–140 K) illumination of the S1 state in the presence of alcohols or the above cryoprotectants results in a g = 4.1 signal that disappears and shifts intensity to the multiline signal upon warming to 200 K (Casey and Sauer, 1984; de Paula et al., 1987). Higher temperature illumination (200 K) of the S1 state when using sucrose as the cryoprotectant (and in the absence of alcohols) results in a g = 4.1 signal that is accompanied by a multiline signal of similar size (de Paula et al., 1987; Zimmermann and Rutherford, 1986). The apparent interconversion between the two signals as a result of the presence of alcohol/cryoprotectant or anion has led to the general consensus that the two S2 state signals represent different coupled states of the manganese cluster. An early suggestion to explain this was that the g = 4.1 signal was due to an S = 3/2 Mn(IV) monomer in equilibrium with a Mn dimer which gave rise to the multiline signal (Hansson et al., 1987). Later it was found that NH3-treated PSII membrane fragments oriented on mylar sheets showed a g = 4.1 signal with hyperfine structure (Kim et al., 1990), indicating that it was due to a Mn multimer. More recent studies have shown that near-infrared (NIR) illumination of samples containing the S2 state multiline signal induces a g = 4.1 signal with properties similar to those observed after low-temperature illumination of the S1 state (Boussac et al., 1996, 1998b). Boussac and Rutherford (2000) also found that the g = 4.1 signals from PSII in sucrose solution after illumination at 200 K and from PSII after NIR illumination at 160 K showed similar properties, including Curie law temperature dependence, microwave power saturation at 4.2 K, and orientation dependence, leading to the conclusion that the signals have a common origin. It has also been found that infrared (IR) illumination at 65 K of PSII samples showing the S2 state multiline signal produced low-field signals at g = 10 and g = 6 attributed to an S = 5/2 state (Boussac et al., 1998a,b). This state is evidently representative of a transitional state between the S2 multiline signal and the IR-induced g = 4.1 signal. Upon warming, the g = 6 and 10 signals disappeared with the concurrent appearance of the g = 4.1 signal. Another low-field signal from the Mn cluster has been observed at about g = 4.7 after NIR illumination of samples in the S3 state at 50 K (Ioannidis and Petrouleas, 2000, 2002; Sanakis et al., 2001). A similar signal was observed in samples prepared in the S3 state after several days of incubation at 77 K (Nugent et al., 1997; Sanakis et al., 2001). Both were attributed to an S = 7/2 state arising from a modified S2 state, designated 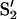 .
.
Despite these and many more recent studies, there is still an incomplete understanding of the relationship between the multiline and the g = 4.1 S2 state signals and even some disagreement regarding the spin state origin of the g = 4.1 signal. A study carried out by one of us on the g = 4 S2 state signal using 15.5 GHz EPR spectroscopy revealed g-anisotropy, which led to the conclusion that the signal arose from the middle Kramers doublet of an S = 5/2 system, with the zero-field splitting (ZFS) parameters of D = 0.43 cm−1 and λ = E/D = 0.25, corresponding to a near-rhombic system (Haddy et al., 1992). The S = 5/2 origin has been supported by other studies using pulsed EPR spectroscopy (Astashkin et al., 1994) and SQUID magnetization study of the IR-induced form of the g = 4 signal (Horner et al., 1998). The analysis of oriented NH3-treated PSII samples (Kim et al., 1990, 1992) have also been consistent with an S = 5/2 source for the signal. In contrast with these studies, the work of Pace and coworkers supports an S = 3/2 spin state origin for the signal at g = 4.1, based primarily on a study using Q-band (35 GHz) EPR spectroscopy in which a signal in the g = 4 region was observed (Smith et al., 1993; Smith and Pace, 1996). It is important to resolve this fundamental issue so that an understanding of these signals in terms of their function in the OEC cycle can be developed.
The approach of studying the signal at several frequencies should in principle lead to a clarification of the spin state. The results of the study at 15.5 GHz predicted a large shift in the g-factor with increasing frequency, such that it would appear around g = 3 at Q-band (34–35 GHz). In the study presented here, PSII-enriched membrane fragments from spinach have been examined using Q-band EPR spectroscopy. Light-induced resonances have been observed in the range g = 3–5 and are found to be consistent with the prediction from the 15.5 GHz study for the X-band g = 4.1 signal. The Q-band results provide information for further refinement of its zero field splitting parameters.
MATERIALS AND METHODS
Preparation of PSII-enriched membrane fragments
PSII-enriched thylakoid membrane fragments were prepared from fresh market spinach by extraction with Triton X-100 as described by Berthold et al. (1981) and modified in Ford and Evans (1983) and Franzén et al. (1985). All steps were carried out on ice or at 4°C, including subsequent treatments described below for EPR sample preparation. PSII preparations were suspended to a final concentration of 8–12 mg chlorophyll mL−1 (mgChl mL−1) in a buffer containing 20 mM Mes-NaOH, pH 6.3, 0.4 M sucrose, 15 mM NaCl, and 5 mM MgCl2 and stored in liquid N2. In some cases, MgCl2 was omitted from the last buffer, but in all cases, Mg2+ was removed from the sample by subsequent procedures.
O2 evolution rates were measured at 25°C with a Clark-type O2 electrode (model 5331, Yellow Springs Instruments, Yellow Springs, OH), using as electron acceptor 1 mM phenyl-p-benzoquinone (Aldrich, Milwaukee, WI) that had been purified by crystallization from ethanol. O2 concentration was calibrated using distilled water equilibrated with air, accounting for pressure and temperature variations. Samples were illuminated with a Dolan-Jenner (Lawrence, MA) model 180 Fiber-lite high-intensity illuminator (dual gooseneck fiber optics, EJV 21 V 150 W lamp) combined with a 300 W or higher projector lamp to ensure sample saturation. O2 evolution rates of control samples (i.e., with adequate chloride) were usually 500–600 μmol O2 mgChl−1 h−1. Rates given represent the averages of three or more separate measurements.
Subsequent treatments of PSII samples
Chloride depletion of PSII-enriched membrane fragments was carried out by dialysis using a procedure similar to that described in Lindberg and Andréasson (1996) and Lindberg et al. (1993), using a buffer containing 20 mM Mes-NaOH (Fisher Scientific, Fair Lawn, NJ), pH 6.3, and 0.40 M sucrose (Fisher Scientific, or J. T. Baker, Phillipsburg, NJ). We estimate that the concentration of chloride was 30–50 μM based on measurements using a Cl−-sensitive electrode. PSII-enriched membrane fragments were thawed from storage and suspended in the buffer to a concentration of 0.5–0.8 mgChl mL−1. The samples were then centrifuged at 20,000 × g for 8 min to pellet. The pellets were resuspended in the same buffer and centrifuged again to remove chloride from the buffer. The samples were then resuspended to a concentration of ∼2.3 mgChl mL−1 and placed in dialysis tubing (Spectra/Por 2.1, 15 kDa cutoff; Sigma, St. Louis, MO). The PSII samples were dialyzed against the chloride-depletion buffer at 4°C for 21–22 h in the dark. These PSII membrane fragments will be referred to as Cl−-depleted. For addition of anions, the samples were split up and diluted to 0.3–0.4 mgChl mL−1 in the chloride-depletion buffer. To each part was added either NaCl, NaN3, or NaF from a 1.0-M stock solution to a final concentration of 20 mM or water of the same volume (for no anion). After incubating on ice in the dark for ∼30 min, the membrane fragments were centrifuged to pellet and resuspended in the same medium (containing either 20 mM Cl−, 20 mM 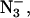 20 mM F−, or no anion) to 5–6 mgChl mL−1 or more. EPR samples for both X-band and Q-band were prepared from the same anion-treated PSII sample.
20 mM F−, or no anion) to 5–6 mgChl mL−1 or more. EPR samples for both X-band and Q-band were prepared from the same anion-treated PSII sample.
For some samples, chloride was replaced by other anions without prior Cl− depletion. In this case PSII-enriched membrane fragments were thawed from storage and suspended in buffer containing 50 mM Mes-NaOH (Fisher Scientific), pH 6.3, 0.40 M sucrose (Fisher Scientific) and 20 mM NaCl, NaF, or NaN3 to a concentration of ∼0.5 mgChl mL−1. The samples were centrifuged at 20,000 × g for 10 min to pellet. Each sample was resuspended in the same buffer and centrifuged again as above. Pellets were then resuspended to ∼0.5 mgChl mL−1 in buffer containing the appropriate anion and kept covered on ice for 30 min before centrifuging a third time as above. The pellets were resuspended in the same buffers to 5–6 mgChl mL−1 or more. EPR samples for both X-band and Q-band were prepared from each anion-treated PSII sample.
For PSII samples treated with ethanol, 95% ethanol (ACS certified) was added to the concentrated PSII-enriched membrane fragments (without other subsequent treatments) to 5% vol/vol (∼0.9 M) before EPR sample preparation.
Preparation of EPR samples
Q-band EPR samples of PSII-enriched membrane fragments were prepared in 2 mm outer diameter clear fused quartz tubes (Wilmad Glass, Buena, NJ). Samples were loaded into the tubes at a concentration of 5 mgChl mL−1 or less using a 25 μL Hamilton syringe, then centrifuged in a Sorvall high-speed centrifuge SS-34 rotor at a speed of 10,000–15,000 rpm for 10–30 min. Supernatant was removed and additional sample was loaded into the same tube and centrifuged in this way two to three times until the sample of ∼3 cm height at an average concentration of 20–30 mgChl mL−1 was obtained. Samples were then dark adapted on ice for 1–2 h and frozen in liquid nitrogen just before use.
X-band EPR samples of PSII-enriched membrane fragments were prepared in 4-mm outer diameter clear fused quartz tubes (Wilmad Glass) at a concentration of 5–10 mgChl mL−1 and a sample height of ∼3 cm. For samples that had been anion-treated, precision bored tubes were used to facilitate signal intensity comparisons. Samples were dark adapted on ice for 3–4 h, frozen, and stored in liquid nitrogen until use.
After collection of spectra of dark-adapted samples, the S2 state was achieved by illuminating EPR samples in a dry ice/methanol bath (195 K) for 8 min using a Dolan-Jenner model 180 Fiber-lite illumination system similar to that described for O2 evolution assays, or using a pair of Dolan-Jenner model 190 Fiber-lite illuminators. The dual light beams were each passed through ∼6 cm of 5 mM CuCl2 solution in cylindrical glass containers before reaching the sample.
EPR spectroscopy
X-band EPR spectroscopy was carried out using a Bruker Instruments (Billerica, MA) EMX 6/1 EPR spectrometer equipped with either a standard ER4102ST cavity (9.5 GHz) or a dual mode ER4116DM cavity operating in perpendicular mode (9.7 GHz). Temperature was controlled with an Oxford Instruments (Eynsham, UK) ESR 900 liquid helium cryostat. Signals were observed at 10.0 K using EPR settings that included microwave power of 20 mW, modulation frequency of 100 kHz, and modulation amplitude of 20 G for the standard cavity or 18 G for the dual mode cavity. Frequency was monitored using an EMX 048T frequency meter.
Q-band (34 GHz) EPR spectroscopy was carried out using a Bruker Instruments EMX EPR spectrometer equipped with a 10-inch magnet and an ER 5106 QT low-temperature Q-band resonator. Temperature was controlled with an Oxford Instruments ER 4112HV-Q liquid helium cryostat. S2 state signals were observed at 8.0 K using EPR settings of 5.1–5.3 mW, modulation frequency of 100 kHz, and modulation amplitude of 20–24 G. In general, Q-band EPR spectroscopy is insensitive compared to X-band and a spectrum of PSII at Q-band was collected over a period that was several times longer than the analogous spectrum at X-band.
Because the Q-band EPR spectrometer was not equipped with an external frequency meter, calibration of the Q-band g-factors was carried out using two signals that were naturally present in the PSII samples: the dark stable radical from tyrosine D and the small background signal from Mn2+ that is apparently nonspecifically associated with PSII (this is enhanced at Q-band relative to X-band). In addition to the spectra presented, dark-adapted and illuminated samples were examined at 1 μW, 5 G modulation amplitude, and 8.0 K to view these signals. The g-factor of the baseline crossover point of the Tyr D signal was taken to be gy = 2.004 (Brok et al., 1985). The g-factor of the Mn2+ signal (found by averaging the field positions of the four outer hyperfine peak positions) was taken to be g = 2.002; although the exact nature of the Mn2+ species is not known, this value was taken as a consensus value based on that of a variety of Mn2+ species (de Wijn and van Balderen, 1967; Meirovitch et al., 1978; Pilbrow, 1990). These signals, taken as internal standards, allowed the determination of sample g-factors to three significant digits (±0.01).
Conversion of spectra from Bruker binary to ASCII format was accomplished using EWPlot (Scientific Software Services, Plymouth, MI) written by Philip D. Morse of Illinois State University. Each difference spectrum was found by subtracting the spectrum of the dark-adapted sample from that of the sample after illumination.
The intensities of signals were assessed by measuring the peak-to-trough heights in baseline-corrected spectra (linear correction only) and by double integration of the signals, where possible. The use of signal height as a measure of relative intensity is valid as long as the shapes of the signals do not change. The intensities of X-band g = 4.1 signals and Q-band multiline signals were the easiest to assess by double integration, since these generally integrated as complete signals. Integration of the X-band multiline signal was somewhat less reliable as indicated by a lopsided appearance of the integral, probably due to other underlying light-induced peaks. For height measurements of the multiline signal the heights of the second, third, and fourth main peaks from the left of the signal center were averaged. Except where otherwise noted, errors for the signal heights were estimated by propagation of the errors estimated in EPR spectral noise, tube volume, and sample concentration.
EPR simulation
Calculations of transition energies and transition probabilities were carried out using an updated version of a Fortran program originally written by W. Richard Dunham as described previously (Haddy et al., 1992). The program diagonalizes a spin Hamiltonian matrix that includes a Zeeman term at a given magnetic field strength, ZFS parameters D, and rhombicity λ = E/D, and assuming a true g-factor of 2.00 for all principal axes.
RESULTS
Light-induced signals at low field
PSII-enriched membrane fragments were examined using Q-band EPR spectroscopy with particular interest in the low-field region since this was the region in which the signal corresponding to the X-band g = 4.1 signal was expected to appear. Two light-induced signals were detected in PSII samples after illumination at 195 K with positions corresponding to g-factors in the range of g = 3–5 (Fig. 1 A). One signal was observed at about g = 4.6 and the other at about g = 3.1. Both were broad, with peak-to-trough widths of 500 ± 100 G and with no additional observable structure. The two low-field signals were comparable in size to the various iron signals observed in the dark. The presence of these signals corresponded to the S2 state, as confirmed by observation of a multiline signal at g = 2 in the same sample. In addition, a corresponding X-band sample prepared from the same sample stock and illuminated in the same way showed a g = 4.1 signal (Fig. 1 B) as well as a multiline signal centered at ∼3400 G (not shown).
FIGURE 1.

Production of low-field S2 state signals from PSII-enriched membrane fragments at Q-band (A) and X-band (B): (a) illuminated for 8 min at 195 K (dry ice/methanol); (b) dark-adapted; and (c) illuminated minus dark-adapted difference spectrum (spectrum a minus spectrum b). PSII membrane fragments were prepared in 0.4 M sucrose, 20 mM Mes, pH 6.3, and 20 mM NaCl. Q-band spectra (A) represent a sum of 32 scans taken at 8 K using 34 GHz microwave frequency, 5 mW microwave power, and modulation amplitude of 24 G. X-band spectra (B) represent a sum of six scans taken at 10 K using 9.65 GHz microwave frequency, 20 mW microwave power, and modulation amplitude of 18 G. The X-band difference spectrum c is plotted using 2× the intensity scale as in spectra a and b.
Background signals
As at X-band, the EPR spectra of dark-adapted PSII-enriched membrane fragments at Q-band showed several signals as background signals that completely subtracted away in the illuminate minus dark-adapted difference spectra. These signals were characterized to aid recognition of light-induced signals. Three signals with peak positions at g = 5.5–5.6, g = 6.0, and g = 6.5 were observed (Fig. 1 A, curve b), which were attributed to axial and near axial Fe3+ signals that are also observed at X-band. Because higher frequency bands resolve differences in g-factors, the Q-band signals in this region were better resolved than the corresponding X-band signals, which overlapped greatly and showed apparent peaks at g = 5.6, g = 6.0, g = 6.2, and g = 6.7 (Fig. 1 B, curve b).
The dark-stable rhombic Fe3+ signal of PSII samples was found at Q-band to appear around g = 4.3, similar to the X-band signal, but with one significant and interesting difference. The X-band powder pattern generally shows a central sharp line at g = 4.31–4.32 with the high- and low-field powder lines appearing as broad features at g = 4.7–4.8 and g = 4.1 (Fig. 1 B, curve b). At Q-band the signal was similar, but the central line was split into two sharp lines at g = 4.33 and g = 4.24, with the broad features appearing at about g = 4.1 and g = 4.5 (Fig. 1 A, curve b). This extra central line must be due to the resolution of one of the off-principal axis lines (extra powder lines), which in general become evident in regions where the operating frequency approaches the magnitude of the zero field splitting (Aasa, 1970).
Cl−-containing samples
The low-field Q-band signals were examined for five Cl−-containing samples, which were all control samples for various experiments. Three of these, which had undergone slightly different treatments, are shown in the figures: one was washed three times with Cl−-containing buffer (Fig. 1 A), one had been Cl−-depleted before Cl− addition (Fig. 2 A), and the other was not treated further after PSII preparation (Fig. 3 B). The two low-field signals were found to have g-factors of 4.55 ± 0.02 and 3.15 ± 0.01 (average of five samples, including data not shown). The width of the signal at g = 3.1 was 450–550 G, whereas that of the signal at g = 4.6 was generally 500 G or more, with a broadening of the “peak” portion of the signal making this assessment difficult in some cases (such as that in Fig. 1 A).
FIGURE 2.

Effect of Cl− depletion and replacement on the S2 state EPR signals from Photosystem II at low-field Q-band (A) and at X-band (B): (a) 20 mM NaCl; (b) Cl−-depleted; (c) 20 mM NaN3; and (d) 20 mM NaF. Spectra shown are difference spectra of illuminated minus dark-adapted samples. Samples of PSII-enriched membrane fragments were depleted of Cl− as described in 20 mM Mes, pH 6.3, then treated with no anion, 20 mM NaCl, NaN3, or NaF, as indicated. Sample illumination and EPR conditions at Q-band and at X-band were as described in the legend to Fig. 1, except that 48 scans were summed for the Q-band spectra.
FIGURE 3.

Effect of ethanol on the EPR signals from the S2 state of PSII-enriched membrane fragments, including the multiline signal at Q-band (A), the lower field signals at Q-band (B), and both signals at X-band (C): (top) +5% ethanol; and (bottom) no addition. All spectra shown are difference spectra of illuminated minus dark-adapted samples. The samples were illuminated for 8 min at 195 K (dry ice/methanol). PSII samples were prepared in 0.4 M sucrose, 20 mM Mes, pH 6.3, 20 mM NaCl, with or without the addition of 5% ethanol by volume, as indicated. The same samples were used for the spectra in A and B. The samples shown in C were the same as, but different fractions of, those shown in A and B. EPR conditions at Q-band and at X-band were as described in the legend to Fig. 1, except that 24 scans were summed for the Q-band multiline spectra.
In general, the relative sizes of the two signals were found to not correlate well for the various samples containing Cl−. The height of the g = 3.1 signal relative to that of the g = 4.6 signal was 68%, 37%, and 87% for the three signals shown in Figs. 1 A, 2 A, and 3 B, respectively; the two other signals not presented here showed g = 3.1 signals with heights of >100% and <20% relative to the g = 4.6 signal.
Anion-treated PSII
The effects of chloride depletion followed by addition of inhibitory anions on the Q-band signals at g = 4.6 and 3.1 were investigated. Fluoride and azide are competitive inhibitors of chloride-activated O2 evolution (Haddy et al., 1999; Sandusky and Yocum, 1984, 1986) and each causes a narrowing of the X-band g = 4.1 signal. Fluoride also causes a significant increase in the integrated signal intensity at X-band, so fluoride-treated PSII samples were initially used in this study at Q-band to facilitate identification of a signal associated with the X-band g = 4.1 signal.
The effects of Cl− depletion and addition of 20 mM Cl−, F−, and 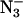 on the two light-induced signals at low field are shown in Fig. 2 A. The samples in which Cl− had been removed or replaced with another anion showed slight shifts in the positions of the two low-field Q-band signals. These samples had either been Cl−-depleted before F− or
on the two light-induced signals at low field are shown in Fig. 2 A. The samples in which Cl− had been removed or replaced with another anion showed slight shifts in the positions of the two low-field Q-band signals. These samples had either been Cl−-depleted before F− or 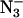 addition, or the Cl− had been replaced by successive washes with F− or
addition, or the Cl− had been replaced by successive washes with F− or 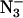 -containing buffer. Based on averages of six samples (three with F−, two with
-containing buffer. Based on averages of six samples (three with F−, two with 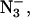 and one Cl−-depleted, including data not shown), the signals appeared at positions of g = 4.60 ± 0.02, an increase in g-factor of 0.05 over the Cl−-containing samples, and g = 3.09 ± 0.02, a decrease in g-factor of 0.06 compared to the Cl−-containing samples. Although these shifts are small, they appear to be significant.
and one Cl−-depleted, including data not shown), the signals appeared at positions of g = 4.60 ± 0.02, an increase in g-factor of 0.05 over the Cl−-containing samples, and g = 3.09 ± 0.02, a decrease in g-factor of 0.06 compared to the Cl−-containing samples. Although these shifts are small, they appear to be significant.
The F−-treated samples also showed a Q-band signal at g = 4.1 that appeared as a shoulder on the g = 4.6 signal. This feature was evident in several F−-treated samples, leading to the conclusion that the shoulder at g = 4.1 was not an artifact due to baseline irregularities. It is not clear whether the apparent lack of this shoulder in other samples was because of their lower resolution/higher broadening or because it was truly absent.
Aliquots of the same samples were examined at X-band (Fig. 2 B and Table 1) so that the trend in signal intensities at the two bands could be compared. At X-band, the signal at g = 4.1 can be quantified by integration because of its symmetrical nature (consistent with the interpretation that all principal axis resonances are contained therein) as long as the baseline is of good quality. This is the preferred method of measurement because the signal width is affected by the anion treatment. In this experiment and in others, the linewidth of the g = 4.1 signal at X-band narrowed from 345–350 G in Cl−-containing samples to 300–308 G in both the azide and F−-treated samples, a decrease of 10–15%. Only the F−-treated sample showed a clear increase in the integrated signal intensity of ∼50%, which corresponded to a doubling of signal height.
TABLE 1.
Relative signal heights/intensities for low-field S2 state signals at Q-band and X-band
| Sample | Cl− | No anion | 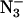 |
F− |
|---|---|---|---|---|
| Q-band signal at g = 4.6 | ||||
| g-factor | 4.55 | 4.62 | 4.61 | 4.61 |
| Height/rel. units | 100 | 84.9 | 70.8 | 179 |
| Width/G | 550–700 | 400–450 | 400–450 | 380–420 |
| Q-band signal at g = 3.1 | ||||
| g-factor | 3.16 | 3.11 | 3.09 | 3.08 |
| Height/rel. units | 37.4 | 66.7 | 88.0 | 215 |
| Width/G | 400–450 | 400–450 | 500–550 | 380–420 |
| X-band signal at g = 4.1 | ||||
| g-factor | 4.06 | 4.13 | 4.17 | 4.20 |
| Intensity/rel. units | 100 | 107 | 106 | 156 |
| Height/rel. units | 100 | 109 | 154 | 215 |
| Width/G | 340–370 | 330–360 | 290–310 | 300–315 |
| O2 evolution activity | ||||
| Relative to Cl− | 100 | 36.1 | ∼0 | 10.9 |
Samples, which correspond to those presented in Fig. 2, were Cl−-depleted and subsequently treated with 20 mM sodium salt of the anion indicated. Signal heights were measured from peak to trough; intensities at X-band were also measured by double integration. Each measurement was scaled to account for the number of scans and the sample concentration and normalized to the height of the g = 4.6 signal for the Cl−-containing sample at Q-band or the height/intensity of the g = 4.1 signal for the Cl−-containing sample at X-band. Measurements at X-band were averaged from two aliquots; measurements at Q-band were from single aliquots of the same samples. Error is estimated to be 10–15% for Q-band signals and 10% for X-band signals. The O2 evolution activity of the Cl−-containing sample was 653 μmolO2 mgChl−1 h−1.
For the Q-band signals at g = 3.1 and g = 4.6 the conditions required for signal measurement by integration were generally not met. It was evident that the shapes of the two signals were not symmetrical, strongly suggesting that each signal represented an incomplete portion of a powder pattern. As a result, the signal sizes could be compared based on height only. Signal widths were generally in the range of 450–550 G, although in the case of the F−-treated samples it was evident that both Q-band signals were at least 10% narrower than those from other samples, as at X-band.
The sizes of the Q-band signals at g = 3.1 and g = 4.6 correlated fairly well with each other in samples that contained F−, 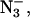 or remained Cl−-depleted, as is evident in the signal height measurements (Table 1). An increase in the heights of the two signals in the F−-treated sample relative to the others was particularly notable. However because of the narrowing of the signals in F−-containing samples, the increase was probably not as great as the height measurement suggests. This effect is probably similar to that shown at X-band, where the F−-containing sample was seen to increase to 215% in height but only 156% in intensity relative to the Cl−-containing sample. The signal intensities of the Cl−-containing sample did not seem to fit in with the trend shown by the others, since the intensity of the g = 3.1 signal was much lower than that of the g = 4.6 signal and g = 3.1 signals from the other samples. This seemed to be a reflection of the general inconsistent appearance of the signal at g = 3.1 for Cl−-containing samples as mentioned above.
or remained Cl−-depleted, as is evident in the signal height measurements (Table 1). An increase in the heights of the two signals in the F−-treated sample relative to the others was particularly notable. However because of the narrowing of the signals in F−-containing samples, the increase was probably not as great as the height measurement suggests. This effect is probably similar to that shown at X-band, where the F−-containing sample was seen to increase to 215% in height but only 156% in intensity relative to the Cl−-containing sample. The signal intensities of the Cl−-containing sample did not seem to fit in with the trend shown by the others, since the intensity of the g = 3.1 signal was much lower than that of the g = 4.6 signal and g = 3.1 signals from the other samples. This seemed to be a reflection of the general inconsistent appearance of the signal at g = 3.1 for Cl−-containing samples as mentioned above.
In summary the effects of the anions were similar at both bands (Table 1). Height and intensity comparisons indicated that the signal enhancement due to F− was similar at both bands, although somewhat more pronounced at Q-band. Only the Q-band signal at g = 3.1 from the Cl−-containing sample showed departure from the correlation, since it did not show a height comparable to its X-band counterpart.
Effect of ethanol
The effect of 5% ethanol on the Q-band multiline and low-field signals from the S2 state was examined. The X-band signal at g = 4.1 is suppressed by the presence of ethanol or methanol, with a concurrent increase in intensity and/or linewidth narrowing of the multiline signal, a well known observation typified in Fig. 3 C. At Q-band, the multiline signal was also observed to increase in intensity in response to the addition of 5% ethanol (Fig. 3 A), as observed for the corresponding signal at X-band. The presence of ethanol caused an increase in the integrated intensity of the Q-band multiline signal of ∼3.2-fold and an increase in line height of ∼2.0-fold. The X-band multiline signal for the same sample showed an increase in integrated intensity in the presence of ethanol of ∼1.9-fold and an increase in line height of ∼1.4-fold (Fig. 3 C). The difference in the multiline signal intensities for the two bands is probably attributable to partial signal saturation in each case. A more significant observation is the difference in intensities between the two quantification methods, which probably indicates that the individual linewidths were greater in the absence of ethanol, although overlap obscures this from direct observation.
Examination of the lower-field light-induced signals at Q-band (Fig. 3 B) revealed that the height of the signal at g = 4.6 was not greatly affected by the presence of ethanol, since it showed less than 10% difference in height compared with the control. The signal at g = 3.1, on the other hand, was essentially removed by the presence of ethanol, decreasing in height to only 20% or less than that of the signal in the absence of ethanol. In the corresponding X-band samples (Fig. 3 C), the loss in the signal at g = 4.1 was also relatively complete, with only ∼10% remaining as measured by either height or intensity. The feature appearing at ∼8500 G in Fig. 3 B in the ethanol-containing sample was not clearly reproducible and has uncertain significance at this time. The average g-factor of the light-induced signal observed in the two ethanol-containing samples studied was g = 4.53, which is similar to that of the Cl−-containing samples studied.
Theoretical interpretation
The Q-band resonances at g = 3.1 and g = 4.6 can both be explained by a model for an S = 5/2 system similar to that proposed in the earlier study carried out at P-band (Haddy et al., 1992). For this case, the unusual g-factors observed at Q-band arise because the system is in a range where the transition frequency is similar to the zero-field splitting. In this range, g-factors are likely to be more dependent on the operating frequency and transitions that are not normally observed may become observable.
The signal at g = 3.1 corresponds very well to the transition within the middle Kramers doublet, levels 3–4, for a system in this rapidly changing frequency region. The parameters found in the previous P-band study of the g = 4.1 signal, D = 0.43 cm−1 and λ = 0.25, predicted a g-factor of 3.07 at 34 GHz, which matches the observed signal fairly well. A slight adjustment of the value of D to 0.455 cm−1 using the same λ = 0.25 reproduces the observed g-factor of g = 3.15. The dependence of g-factor on operating frequency for this value of λ is shown in the Aasa diagram of Fig. 4, in which the x axis can be viewed as a relative field scale, adjusted for frequency such that g-factors for different frequencies are correlated. Given the proposed value of D, at X-band frequency all three principal axis transitions are observed and the signal has a symmetrical appearance, although the principal g-factors are somewhat different. As the frequency increases, two of the principal axis resonances (labeled y and z axes) shift out of range of observation and the third (x axis) shifts toward g = 2.00. As a result, the P-band (15 GHz) spectrum shows increased g-anisotropy and the Q-band (34 GHz) spectrum shows only the gx resonance. The shape of the Q-band signal is not symmetrical, probably because it does not represent a normal distribution of sampling about the x axis.
FIGURE 4.

Aasa diagram showing the dependence of transition field position on frequency for the level 3-to-4 transition of an S = 5/2 system with rhombicity λ = 0.25. Principal axis transitions are shown corresponding to the z axis (solid line), x axis (dash-dotted line), and y axis (dotted line). The diagram shows hν/D on the y axis, where hν is the transition or operating frequency. The relative field position is given as g/gobs on the x axis, where g is the true g-factor, which has been set to 2.00 for this calculation. Horizontal dashed lines indicate the positions of hν/D at 34 GHz (long-dash line) and 9.5 GHz (short-dash line) for D = 0.455 cm−1.
The source of the signal at g = 4.6 is less clear, but there are two possible sources for a resonance at this position arising from the same S = 5/2 spin system. One possibility is that it represents a remnant of the z-axis transition of the middle Kramers doublet in the form of an off-principal axis line associated with the y-z plane. It is known that an off-principal axis line would continue to appear around g = 4.6 at hν/D values higher than that at which the z-axis transition disappears. The off-principal axis lines are not shown in Fig. 4, but they are well described by Aasa (1970). Further refinement of the parameters may reveal conditions for which this transition would appear along with the x-axis transition at g = 3.0.
Another possible source of the signal at g = 4.6 is two unusual transitions that become observable in the region where hν ∼ D, between levels 2 and 4 and between levels 3 and 5 (Fig. 5), each of which is associated with only a single principal axis. As shown by the diagram, transitions between these levels are not possible below 25–30 GHz for the proposed ZFS values, but where they appear on the diagrams they occur with significant transition probabilities. In particular, at 34 GHz the relative transition probabilities for both transitions are found to be around 1.8, which compares well with the relative transition probability of 1.7 for the x-axis Kramers doublet transition. In this interpretation, the main signal at g = 4.6 would be due to the level 3-to-5 transition, whereas the shoulder at g = 4.1 that becomes resolved in the F−-treated samples would be accounted for by the level 2-to-4 transition.
FIGURE 5.

Aasa diagram showing the dependence of transition field position on frequency for the level 2-to-4 (A) and the level 3-to-5 (B) transitions of an S = 5/2 system with rhombicity λ = 0.25. Principal axis transitions are shown for the z axis (solid line) and y axis (dotted line). No principal x axis transition appears for either case. Other details are as given in the legend to Fig. 4.
Spectral simulations at Q-band and at P-band (15.5 GHz) using the ZFS parameters of D = 0.455 cm−1 and λ = 0.25 are shown in Figs. 6, A and B, next to an experimental Q-band spectrum from Fig. 3 B and a P-band spectrum reproduced from the earlier study (Haddy et al., 1992). One of the Q-band simulations includes only the middle Kramers doublet transition (Fig. 6 A, curve c) and the other simulation includes all possible transitions (Fig. 6 A, curve b), accounting for transition probabilities. The P-band simulation (Fig. 6 B) shows only the middle Kramers doublet transition, but a simulation including all possible transitions did not reveal others with significant probability.
FIGURE 6.

Simulation of the Q-band (34.0 GHz) signal at g = 3.1 (A) and the P-band (15.5 GHz) signal at g = 4 (B), using preliminary parameters for an S = 5/2 system in which D = 0.455 cm−1 and λ = 0.25. For A: (curve a) experimental spectrum for the Cl−-containing control, reproduced from Fig. 3 B; (curve b) simulation including all possible transitions scaled according to the relative transition probabilities; (curve c) simulation including only the transition for the middle Kramers doublet (levels 3–4). Both simulations included a total of 2500 summation angles and a linewidth of 450 G. For B: (curve a) experimental spectrum reproduced from Haddy et al. (1992); (curve b) simulation including the transition for the middle Kramers doublet, using 2500 summation angles and a linewidth of 250 G.
Possible sources of the signal at g = 4.6 from other spin systems are considered in Discussion.
DISCUSSION
In this article we present evidence in support of a near rhombic S = 5/2 origin of the X-band g = 4.1 signal from the S2 state of the Mn cluster of PSII. At Q-band frequency (34 GHz), low-field light-induced signals appeared at g = 3.1 and g = 4.6. The Q-band signal at g = 3.1 showed an increase in intensity in response to the presence of the chloride-competitive anion F−, as does the X-band signal at g = 4.1. In addition, the Q-band signal at g = 3.1 was not observed in the presence of ethanol, consistent with its identity as the X-band g = 4.1 signal. The signal at g = 3.1 (g = 3.15 in control PSII samples) matches well the position expected for the middle Kramers doublet (levels 3–4) using the zero-field splitting parameters of D = 0.455 cm−1 and λ = 0.25. These parameters are fairly close to the parameters of D = 0.43 cm−1 and λ = 0.25 that were previously determined in the study at P-band frequency (15.5 GHz) (Haddy et al., 1992). The unusual position of this peak is characteristic of the case for a spin S = 5/2 system in which the transition frequency is similar to the ZFS, hν/D ∼ 1.
The interpretation of the Q-band signal at g = 4.6 is less clear. The signal appeared to correlate with the signal at g = 3.1 in response to chloride depletion and anion replacement (Fig. 2 A), but it was observed in the presence of ethanol, unlike the signal at g = 3.1 (Fig. 3 B). The signal at g = 4.6 can be accounted for by the transition between levels 3 and 5 of the S = 5/2 spin manifold and a second feature observed in the presence of F− at g = 4.1 is consistent with a level 2-to-4 transition. However because of the lack of complete correlation with the behavior of the X-band g = 4.1 signal, careful consideration of the possible sources for this signal is necessary and will be discussed below.
Likelihood that the g = 4.6 signal arises from the same S = 5/2 source
The observation of a signal from the 3-to-5 and/or the 2-to-4 transition from an S = 5/2 system would be unusual, but expected in this region of hν/D. As noted above, such a transition is not possible below a certain frequency, which corresponds to 25–30 GHz for the S = 5/2 system described here. At Q-band, transition probability calculations show these transitions would be comparable to the Kramers doublet transition. Thus the only reason these signals would not be observed would be linewidth broadening.
Ethanol and methanol reduce the intensity of the g = 4.1 signal and increase the intensity of the multiline signal. They also modify the shape of the multiline signal in a manner that appears to increase the resolution of the hyperfine structure. Methanol and other small alcohols including ethanol have been shown to bind at the manganese cluster using ESEEM measurements (Force et al., 1998); however it was also found that direct binding was not the sole cause of the interconversion. Recent studies have shown that the loss of g = 4.1 signal intensity in the presence of methanol correlates closely with the appearance of multiline signal intensity and that this change occurs with an apparent binding constant around 50–65 mM (Deak et al., 1999; Force et al., 1998). This is consistent with the idea that the S = 5/2 signal converts to the S = 1/2 multiline signal form under the influence of ethanol. Given these considerations, it does not appear likely that the g = 4.6 signal could be from the same S = 5/2 spin manifold as the X-band g = 4.1 signal, even though a signal at this position is possible or even expected. It is worth noting, however, that only two PSII samples containing ethanol were studied at Q-band, so the observations will need to be confirmed. In the meantime, additional sources for a signal near g = 4.6 are considered below.
Other possible sources of the signal at g = 4.6
Several other light-induced signals that have been previously identified at X-band were considered as possible sources of the Q-band signal at g = 4.6. The acceptor-side iron can show low-field signals in the oxidized Fe(III)QA state (Aasa et al., 1989; Diner and Petrouleas, 1987; Petrouleas and Diner, 1986). At X-band these appear at g = 5.6 and g = 8 and arise from the middle and the lowest Kramers doublets, respectively, of the S = 5/2 system in an axial environment. By studying the temperature dependence of the signals, Aasa and coworkers found that the ZFS parameters for the system are D = 1.0 ± 0.3 cm−1 and λ = E/D = 0.10 ± 0.01 (Aasa et al., 1989). Using these parameters, the g-factors expected for these signals at 34 GHz were calculated to be g = 5.6 and g = 7.8. Even using the lowest possible value of D = 0.7 cm−1, the positions of the signals would shift by only a small amount and are therefore unable to account for the observed Q-band signal at g = 4.6. Further, the conditions of the experiments carried out here would not be expected to result in the oxidized acceptor side Fe(III) signals, since this signal is usually generated by the presence of an acceptor-side oxidizing agent (quinones or ferricyanide), which was not part of the sample preparation protocol here.
The signals at g = 10 and g = 6, which appear as a result of IR illumination at 65 K of samples containing the S2 state multiline signal (Boussac et al., 1998a,b), were considered as possible sources of the Q-band signal at g = 4.6. These signals were attributed to an S = 5/2 spin system in an axial environment, with D ≤ 1.0 cm−1 and E/D = −0.05 (Boussac et al., 1998b). It is conceivable that at higher frequencies the signal at g = 10 could shift to a lower g-factor, such as g = 4.6, accounting for the data observed here; a signal such as this would be in a frequency-dependent region and likely show broadening. (The signal at g = 6 would disappear at higher frequencies rather than shift position.) However, the conditions of the experiments performed here again do not correspond well to the conditions used for observation of the g = 6 and 10 signals; in addition, the g = 6 and 10 signals were not observed at X-band.
A signal that warrants careful attention as a possible candidate is that observed at about g = 4.7 from the state designated 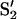 and that appears as a result of decay of the S3 state at 77 K or after NIR illumination of the S3 state at 50 K (Ioannidis and Petrouleas, 2000, 2002; Nugent et al., 1997; Sanakis et al., 2001). This or a similar signal has also been observed in PSII samples at elevated pH (pH 8.1) after white-light illumination of the S1 state at 243 K (Ioannidis and Petrouleas, 2002). The conditions used for the observation of the
and that appears as a result of decay of the S3 state at 77 K or after NIR illumination of the S3 state at 50 K (Ioannidis and Petrouleas, 2000, 2002; Nugent et al., 1997; Sanakis et al., 2001). This or a similar signal has also been observed in PSII samples at elevated pH (pH 8.1) after white-light illumination of the S1 state at 243 K (Ioannidis and Petrouleas, 2002). The conditions used for the observation of the  signal do not match those used in this study, since there was no preoxidation of the acceptor-side iron and the temperature was not kept low enough for the
signal do not match those used in this study, since there was no preoxidation of the acceptor-side iron and the temperature was not kept low enough for the  signal to remain stable. It is more likely that the high-pH version of the
signal to remain stable. It is more likely that the high-pH version of the  signal, which is stable at higher temperatures, was present in a fraction of centers. However, given the pH of the samples used here (pH 6.3), this possibility seems low. Furthermore, the possibility is essentially negated if the observation that 4% ethanol prevents formation of the g = 4.7 signal in S3 state samples incubated at 77 K (Nugent et al., 1997) is relevant for the high-pH version of the signal as well. Nevertheless, this remains a possibility to be explored in future studies.
signal, which is stable at higher temperatures, was present in a fraction of centers. However, given the pH of the samples used here (pH 6.3), this possibility seems low. Furthermore, the possibility is essentially negated if the observation that 4% ethanol prevents formation of the g = 4.7 signal in S3 state samples incubated at 77 K (Nugent et al., 1997) is relevant for the high-pH version of the signal as well. Nevertheless, this remains a possibility to be explored in future studies.
Comparison with studies by other researchers
The observation of the Q-band signal at g = 3.1 confirms that the S2 state signal from the Mn cluster known as the g = 4.1 signal originates from an S = 5/2 spin system, and in particular represents a transition within the middle Kramers doublet. Other studies that also support an S = 5/2 spin state origin include a pulsed EPR study, in which the ZFS parameters were found to be D > 0.25 cm−1 and E/D ∼ 1/3 (Astashkin et al., 1994) and a SQUID magnetization study of the IR-induced form of the g = 4 signal, in which the best-fit parameters were found to be D = 1.7 cm−1 or −1.05 cm−1 and E/D = 0.25 (Horner et al., 1998). In the pulsed EPR study, the rhombicity E/D was found to be somewhat larger than in this study. Those measurements could have been complicated by a background contribution from rhombic Fe3+, which would explain a higher value of E/D. In the SQUID study the rhombicity E/D was found to be similar to that reported here, but the absolute values of D were much higher (error estimates were not reported). It may be significant that the signal was produced by IR illumination, because although the g = 4.1 signal produced this way is essentially the same as that produced in sucrose solution by 200 K illumination (Boussac and Rutherford, 2000), it is possible that there actually is a difference in the values of D for the two signals (but E/D would have to be the same to produce the same g-factors at X-band). This possibility would need to be tested in future studies.
The finding of an S = 5/2 center as the origin of the g = 4.1 signal is not in agreement with studies carried out by Smith and coworkers, who have argued in favor of an S = 3/2 origin of the S2 state g = 4.1 signal. Based on a Q-band spectrum from sucrose-containing PSII membrane fragments with features at g = 4.35 and g = 4.14, the S = 3/2 zero-field splitting parameters were suggested to be D > 5 cm−1 and E/D ∼ 0.017 (Smith et al., 1993). The parameters were later modified to D > 1 cm−1 and E/D ≤ 0.02 for the same sample system, based on the same Q-band spectrum and a temperature-dependence study (Smith and Pace, 1996); at the same time, the signal in PSII samples containing ethylene glycol, was correlated with an S = 3/2 system with D < 1 cm−1 (Smith and Pace, 1996). A later analysis of the same data from sucrose-containing samples included the 15.5 GHz data found in our previous study. This extensive theoretical analysis (Åhrling et al., 1998) hypothesized that the two g = 4.1 signals arose from two dimers of the Mn cluster, both with S = 3/2, one from a radical-bridged inner dimer in the ground state and the other from an outer dimer in an excited state. The radical-bridged inner dimer signal was thought to be that observed in sucrose-containing samples (similar to the conditions used for both this present and our previous studies) and the multifrequency data were fitted to parameters D = 1.1 cm−1, E/D = 0.037, with true g-factors of g⊥ = 2.15, g‖ = 2.0 (Åhrling et al., 1998).
The major difference between the Smith and Pace studies and the experiments described here is that two different signals have been identified as the Q-band signal associated with the X-band g = 4.1 signal. The data presented by Smith and coworkers identified a signal at g = 4.1–4.3 with the S2 state (Smith et al., 1993; Smith and Pace, 1996), whereas we did not observe a light-induced signal of that description. Instead we observed light-induced signals at g = 3.1 and g = 4.6, whereas dark-stable background signals at g = 4.3 and g = 4.2 were identified as arising from the rhombic iron of PSII. Q-band is known to be problematic in terms of cavity tuning and sensitivity. Even using the state-of-the-art equipment available to us the subtraction of spectra taken before and after illumination was occasionally troublesome because of the difficulty in reproducing the same cavity tuning after illumination. Thus it would not be unusual to mistakenly identify a portion of the rhombic iron signal, which has an unusual appearance at Q-band, with a light-induced signal particularly if it is expected in this region. An inconsistency with the Q-band data identified as the S2 state g = 4.1 signal in the study by Smith and coworkers may be hinted at by the results of the careful theoretical study that was based on the same Q-band data. To fit the data from three bands (X-, P-, and Q-bands) for the sucrose-containing PSII samples the value of g⊥ ∼ 2.15 was invoked for one of the true g-factors (Åhrling et al., 1998), which is an unusually high value for manganese-containing compounds.
Relationship of our results with the P-band study
In the P-band (15.5 GHz) study, the signal at g = 4 from the Cl−-containing control PSII samples showed resolution of two g-factors and was fitted to the middle Kramers doublet of an S = 5/2 system with zero-field splitting parameters D = 0.43 cm−1 and λ = 0.25 (Haddy et al., 1992). These parameters led to a prediction that at 34 GHz, a signal corresponding to a single principal axis resonance would appear at g = 3.07. This study at Q-band revealed a signal at g = 3.15 for Cl−-containing samples, which can lead to a refinement of the ZFS values. Preliminary simulations using D = 0.455 cm−1 and λ = 0.25 have reproduced fairly well the g-factors and signal shapes observed at Q-band and P-band (Fig. 6, A and B). These values are considered to be a starting point for future simulations because the parameters were not well refined by optimization and the simulation did not include the underlying Mn nuclear hyperfine coupling that is expected to be present. Using the higher D value of this study, all three principal axis g-factors (g = 4.67, 3.88, and 3.24) appear in the new P-band simulation, whereas only two of the three principal axis g-factors appeared in the g = 4 region in the earlier P-band simulation. The original conclusion that two principal axes represented the observed spectrum was dependent on the accuracy of the baseline. It may also have been influenced by the use of direction cosines to calculate g-factors from principal axis values in the original P-band simulation, which would tend to distort the results somewhat in frequency-dependent regions. The simulations shown in Fig. 6, A and B, employed a matrix diagonalization procedure to compute g-factors for all directions used in the summation and so would not be subject to this source of inaccuracy.
One discrepancy between this study and the original P-band study concerns the signal from F−-containing PSII samples. At P-band, the signal from these samples did not show g-factor resolution, but rather appeared more like its X-band counterpart, and was fitted to an S = 5/2 system with ZFS parameters D = 0.53 cm−1 and λ = 0.27 (Haddy et al., 1992). These parameters led to a prediction that at 34 GHz a signal corresponding to a single principal axis resonance would be observed at g = 3.47, a difference of 0.4 g-factor lower field compared to the prediction for the Cl−-containing sample. This prediction resulted primarily from the difference in the zero-field splitting parameter D. The Q-band results presented here do not reveal two widely separated signals for the two types of samples. Rather, only a small difference was observed between Cl−-containing control samples (g = 3.15) and samples that were treated with F− or 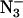 or Cl−-depleted (g = 3.09). The similarity in the g-factors for the two types of samples suggests that the parameter D was similar. If true, then the narrower overall appearance of the spectrum from the F−-treated sample at P-band may be attributed mainly to a difference in the rhombicity parameter. This direction can be pursued in future analyses of the multifrequency data available.
or Cl−-depleted (g = 3.09). The similarity in the g-factors for the two types of samples suggests that the parameter D was similar. If true, then the narrower overall appearance of the spectrum from the F−-treated sample at P-band may be attributed mainly to a difference in the rhombicity parameter. This direction can be pursued in future analyses of the multifrequency data available.
Acknowledgments
We thank James Bautista, Pam Dolan, Robielyn Ilagan and other members of the Frank Laboratory for assistance using the Q-band EPR spectrometer. We also thank W. Richard Dunham for providing the original version of the simulation program.
This work was supported by grants from the National Science Foundation (MCB-0111356 and MCB-0314380), National Institutes of Health (GM32715 and GM30353), United States Department of Agriculture (523130), and the University of Connecticut Research Foundation.
References
- Aasa, R. 1970. Powder line shapes in the electron paramagnetic resonance spectra of high-spin ferric complexes. J. Chem. Phys. 52:3919–3930. [Google Scholar]
- Aasa, R., L.-E. Andréasson, S. Styring, and T. Vänngård. 1989. The nature of the Fe(III) EPR signal from the acceptor-side iron in photosystem II. FEBS Lett. 243:156–160. [Google Scholar]
- Åhrling, K. A., P. J. Smith, and R. J. Pace. 1998. Nature of the Mn centers in Photosystem II. Modeling and behavior of the g = 4 resonances and related signals. J. Am. Chem. Soc. 120:13202–13214. [Google Scholar]
- Astashkin, A. V., Y. Kodera, and A. Kawamori. 1994. Pulsed EPR study of manganese g = 4.1 signal in plant Photosystem II. J. Magn. Reson. B. 105:113–119. [Google Scholar]
- Berthold, D. A., G. T. Babcock, and C. F. Yocum. 1981. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 134:231–234. [Google Scholar]
- Boussac, A., J.-J. Girerd, and A. W. Rutherford. 1996. Conversion of the spin state of the manganese complex in Photosystem II induced by near-infrared light. Biochemistry. 35:6984–6989. [DOI] [PubMed] [Google Scholar]
- Boussac, A., H. Kuhl, S. Un, M. Rögner, and A. W. Rutherford. 1998a. Effect of near-infrared light on the S2-state of the manganese complex of Photosystem II from Synechococcus elongatus. Biochemistry. 37:8995–9000. [DOI] [PubMed] [Google Scholar]
- Boussac, A., S. Un, O. Horner, and A. W. Rutherford. 1998b. High-spin states (S ≥ 5/2) of the Photosystem II manganese complex. Biochemistry. 37:4001–4007. [DOI] [PubMed] [Google Scholar]
- Boussac, A., and A. W. Rutherford. 2000. Comparative study of the g=4.1 EPR signals in the S2 state of photosystem II. Biochim. Biophys. Acta. 1457:145–156. [DOI] [PubMed] [Google Scholar]
- Britt, R. D. 1996. Oxygen evolution. In Oxygenic Photosynthesis: The Light Reactions. D. R. Ort and C. F. Yocum, editors. Kluwer Academic Publishers, Dordrecht, the Netherlands. 137–164.
- Brok, M., F. C. R. Ebskamp, and A. J. Hoff. 1985. The structure of the secondary donor of Photosystem II investigated by EPR at 9 and 35 GHz. Biochim. Biophys. Acta. 809:421–428. [Google Scholar]
- Casey, J. L., and K. Sauer. 1984. EPR detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim. Biophys. Acta. 767:21–28. [Google Scholar]
- Deak, Z., S. Peterson, P. Geijer, K. A. Åhrling, and S. Styring. 1999. Methanol modification of the electron paramagnetic resonance signals from the S0 and S2 states of the water-oxidizing complex of Photosystem II. Biochim. Biophys. Acta. 1412:240–249. [DOI] [PubMed] [Google Scholar]
- Debus, R. J. 1992. The manganese and calcium ions of photosynthetic oxygen evolution. Biochim. Biophys. Acta. 1102:269–352. [DOI] [PubMed] [Google Scholar]
- de Paula, J. C., W. F. Beck, A.-F. Miller, R. B. Wilson, and G. W. Brudvig. 1987. Studies of the manganese site of Photosystem II by electron spin resonance spectroscopy. J. Chem. Soc., Faraday Trans. 1. 83:3635–3651. [Google Scholar]
- de Paula, J. C., J. B. Innes, and G. W. Brudvig. 1985. Electron transfer in Photosystem II at cryogenic temperatures. Biochemistry. 24:8114–8120. [DOI] [PubMed] [Google Scholar]
- de Wijn, H. W., and R. F. van Balderen. 1967. Electron spin resonance of manganese in borate glasses. J. Chem. Phys. 46:1381–1387. [Google Scholar]
- Diner, B. A., and V. Petrouleas. 1987. Light-induced oxidation of the acceptor-side Fe(II) of Photosystem II by exogenous quinones acting through the QB binding site. II. Blockage by inhibitors and their effects on the Fe(III) EPR spectra. Biochim. Biophys. Acta. 893:138–148. [Google Scholar]
- Force, D. A., D. W. Randall, G. A. Lorigan, K. L. Clemens, and R. D. Britt. 1998. ESEEM studies of alcohol binding to the manganese cluster of the oxygen evolving complex of Photosystem II. J. Am. Chem. Soc. 120:13321–13333. [Google Scholar]
- Ford, R. C., and M. C. W. Evans. 1983. Isolation of a photosystem 2 preparation from higher plants with highly enriched oxygen evolution activity. FEBS Lett. 160:159–164. [Google Scholar]
- Franzén, L.-G., Ö. Hansson, and L.-E. Andréasson. 1985. The roles of the extrinsic subunits in Photosystem II as revealed by EPR. Biochim. Biophys. Acta. 808:171–179. [Google Scholar]
- Haddy, A., W. R. Dunham, R. H. Sands, and R. Aasa. 1992. Multifrequency EPR investigations into the origin of the S2-state signal at g = 4 of the O2-evolving complex. Biochim. Biophys. Acta. 1099:25–34. [PubMed] [Google Scholar]
- Haddy, A., J. A. Hatchell, R. A. Kimel, and R. Thomas. 1999. Azide as a competitor of chloride in oxygen evolution by Photosystem II. Biochemistry. 38:6104–6110. [DOI] [PubMed] [Google Scholar]
- Hansson, Ö., R. Aasa, and T. Vänngård. 1987. The origin of the multiline and g = 4.1 electron paramagnetic resonance signals from the oxygen-evolving system of Photosystem II. Biophys. J. 51:825–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner, O., E. Riviere, G. Blondin, S. Un, A. W. Rutherford, J.-J. Girerd, and A. Boussac. 1998. SQUID magnetization study of the infrared-induced spin transition in the S2 state of Photosystem II: Spin value associated with the g = 4.1 signal. J. Am. Chem. Soc. 120:7924–7928. [Google Scholar]
- Ioannidis, N., and V. Petrouleas. 2000. Electron paramagnetic resonance signals from the S3 state of the oxygen-evolving complex. A broadened radical signal induced by low-temperature near-infrared light illumination. Biochemistry. 39:5246–5254. [DOI] [PubMed] [Google Scholar]
- Ioannidis, N., and V. Petrouleas. 2002. Decay products of the S3 state of the oxygen-evolving complex of Photosystem II at cryogenic temperatures. Pathways to the formation of the S = 7/2 S2 state configuration. Biochemistry. 41:9580–9588. [DOI] [PubMed] [Google Scholar]
- Kim, D. H., R. D. Britt, M. P. Klein, and K. Sauer. 1990. The g = 4.1 EPR signal of the S2 state of the photosynthetic oxygen-evolving complex arises from a multinuclear Mn cluster. J. Am. Chem. Soc. 112:9389–9391. [Google Scholar]
- Kim, D. H., R. D. Britt, M. P. Klein, and K. Sauer. 1992. The manganese site of the photosynthetic oxygen-evolving complex probed by EPR spectroscopy of oriented Photosystem II membranes: The g = 4 and g = 2 multiline signals. Biochemistry. 31:541–547. [DOI] [PubMed] [Google Scholar]
- Lindberg, K., and L.-E. Andréasson. 1996. A one-site, two-state model for the binding of anions in photosystem II. Biochemistry. 35:14259–14267. [DOI] [PubMed] [Google Scholar]
- Lindberg, K., T. Vänngård, and L.-E. Andréasson. 1993. Studies of the slowly exchanging chloride in photosystem II of higher plants. Photosynth. Res. 38:401–408. [DOI] [PubMed] [Google Scholar]
- Meirovitch, E., H. Brumberger, and H. Lis. 1978. Mn2+-electron spin resonance spectra of several lectins. Biophys. Chem. 8:215–219. [DOI] [PubMed] [Google Scholar]
- Nugent, J. H. A., S. Turconi, and M. C. W. Evans. 1997. EPR Investigation of water oxidizing Photosystem II: Detection of new EPR signals at cryogenic temperatures. Biochemistry. 36:7086–7096. [DOI] [PubMed] [Google Scholar]
- Ono, T., J.-L. Zimmermann, Y. Inoue, and A. W. Rutherford. 1986. EPR evidence for a modified S-state transition in chloride-depleted Photosystem II. Biochim. Biophys. Acta. 851:193–201. [Google Scholar]
- Ono, T.-A., H. Nakayama, H. Gleiter, Y. Inoue, and A. Kawamori. 1987. Modification of the properties of S2 state in photosynthetic O2-evolving center by replacement of chloride with other anions. Arch. Biochem. Biophys. 256:618–624. [DOI] [PubMed] [Google Scholar]
- Petrouleas, V., and B. A. Diner. 1986. Identification of Q400, a high potential electron acceptor of Photosystem II, with the iron of the quinone-iron acceptor complex. Biochim. Biophys. Acta. 849:264–275. [Google Scholar]
- Pilbrow, J. R. 1990. Transition Ion Electron Paramagnetic Resonance. Oxford University Press, Oxford, U.K.
- Sanakis, Y., N. Ioannidis, G. Sioros, and V. Petrouleas. 2001. A novel S = 7/2 configuration of the Mn cluster of Photosystem II. J. Am. Chem. Soc. 123:10766–10767. [DOI] [PubMed] [Google Scholar]
- Sandusky, P. O., and C. F. Yocum. 1984. The chloride requirement for photosynthetic oxygen evolution: analysis of the effects of chloride and other anions on amine inhibition of the oxygen-evolving complex. Biochim. Biophys. Acta. 766:603–611. [Google Scholar]
- Sandusky, P. O., and C. F. Yocum. 1986. The chloride requirement for photosynthetic oxygen evolution: factors affecting nucleophilic displacement of chloride from the oxygen-evolving complex. Biochim. Biophys. Acta. 849:85–93. [Google Scholar]
- Smith, P. J., K. A. Åhrling, and R. J. Pace. 1993. Nature of the S2 state electron paramagnetic resonance signals from the oxygen-evolving complex of Photosystem II: Q-band and oriented X-band studies. J. Chem. Soc., Faraday Trans. 1. 89:2863–2868. [Google Scholar]
- Smith, R. J., and R. J. Pace. 1996. Evidence for two forms of the g = 4.1 signal in the S2 state of photosystem II. Two magnetically isolated manganese dimers. Biochim. Biophys. Acta. 1275:213–220. [Google Scholar]
- Zimmermann, J.-L., and A. W. Rutherford. 1986. Electron paramagnetic resonance properties of the S2 state of the oxygen-evolving complex of Photosystem II. Biochemistry. 25:4609–4615. [DOI] [PubMed] [Google Scholar]