

Abstract
Mechanosensitive channels are a class of ubiquitous membrane proteins gated by mechanical strain in the cellular membrane. MscS, the mechanosensitive channel of small conductance, is found in the inner membrane of Escherichia coli and its crystallographic structure in an open form has been recently solved. By means of molecular dynamics simulations we studied the stability of the channel conformation suggested by crystallography in a fully solvated lipid (POPC) bilayer, the combined system encompassing 224,340 atoms. When restraining the backbone of the protein, the channel remained in the open form and the simulation revealed intermittent permeation of water molecules through the channel. Abolishing the restraints under constant pressure conditions led to spontaneous closure of the transmembrane channel, whereas abolishing the restraints when surface tension (20 dyn/cm) was applied led to channel widening. The large balloon-shaped cytoplasmic domain of MscS exhibited spontaneous diffusion of ions through its side openings. Interaction between the transmembrane domain and the cytoplasmic domain of MscS was observed and involved formation of salt bridges between residues Asp62 and Arg128; this interaction may be essential for the gating of MscS. K+ and Cl− ions showed distinctively different distributions in and around the channel.
INTRODUCTION
The ability to perceive and react to different stimuli from the surrounding environment is essential for all living organisms. Mechanical forces produced by sound, gravity, or osmotic pressures, among other stimuli, are sensed by organisms through different mechanisms that involve mechanosensitive ion channels (Hamill and Martinac, 2001; Sukharev and Corey, 2004). These membrane proteins open and close upon application of mechanical stimuli, thus generating an ionic current that eventually becomes an electrical signal (mechanotransduction) or that simply helps, for instance, in the regulation of cell volume or the adaptation to osmotic pressures.
In bacteria, different mechanosensitive ion channels (MSCs) gated by mechanical strain in the cellular membrane have been identified (Martinac et al., 1987; Berrier et al., 1989; Sukharev et al., 1994). In particular, it has been shown that the mechanosensitive channel of large conductance, MscL, and the mechanosensitive channel of small conductance, MscS, both play an important role in osmoregulation and prevention of cell lysis in Escherichia coli (Levina et al., 1999; Blount and Moe, 1999; Sukharev, 2002).
MscL, with a conductance of ∼3.2 nS and an activation midpoint of 11 dyn/cm, has been extensively studied (see Hamill and Martinac, 2001; Sukharev and Corey, 2004, and references therein) and its crystallographic structure was the first one solved for a mechanosensitive ion channel (Chang et al., 1998). Gating mechanisms for this nonselective MSC have been explored and modeled using the available closed structure as a starting point (Sukharev et al., 2001; Betanzos et al., 2002; Perozo et al., 2002a,b; Gullingsrud et al., 2001; Gullingsrud and Schulten, 2003; Kong et al., 2002; Colombo et al., 2003; Markin and Sachs, 2004).
On the other hand, MscS, with a conductance of ∼1 nS and an activation midpoint of ∼5 dyn/cm (Sukharev, 2002; Bezanilla and Perozo, 2002), is now becoming the subject of intense scrutiny due to its voltage sensitivity and the recent release of its x-ray crystallographic structure at 3.9 Å resolution (Bass et al., 2002), the first structure available for a voltage-modulated ion channel.
The overall architecture of MscS is shown in Fig. 1. The protein is formed by seven identical subunits of 286 amino acids each; residues 27–280 are present in the crystal structure. The transmembrane domain has three α-helices per subunit as shown in Fig. 1 A. The first two transmembrane helices (TM1 and TM2) are tilted 27–35° with respect to the normal, and have charged residues (arginines) that may act as voltage sensors (Bass et al., 2002; Bezanilla and Perozo, 2002). The domain formed by these two transmembrane helices is similar to the controversial structure of the voltage sensor in the voltage-gated potassium channel KvAP (Jiang et al., 2003a,b; Perozo and Rees, 2003). The third transmembrane helix is divided by a pronounced kink into two subunits: TM3A and TM3B (Fig. 1 B). The division is observed at residue Gly113 and marks the end of the pore's cytoplasmic side. The transmembrane and highly hydrophobic pore, formed by TM3A, appears to be in an open state (Fig. 1 D) with a diameter of 6.5 Å in its narrowest section, located at the seven Leu105 side chains (Fig. 1 C). The second part of the third transmembrane helix, TM3B, is almost parallel to the membrane plane (Fig. 1 B), and the end of TM3B of one subunit slips below the loop between TM1 and TM2 of the next subunit (Fig. 1 D).
FIGURE 1.

Homoheptameric architecture of MscS. (A) Cartoon representation of the MscS crystal structure. Each subunit is presented in a different color. The suggested membrane position (Bass et al., 2002) is shown as a shaded bar. (B) Subunit B of the heptameric structure. This subunit is shown in cartoon and ribbons representation. The transmembrane domain is formed by three helices (TM1, TM2, and TM3A–TM3B) and a larger cytoplasmic domain. The TM3A–TM3B helix has a pronounced kink at residue Gly113, close to the narrowest part of the pore at the cytoplasmic side of the membrane. (C) Transmembrane helices of two monomers shown in surface and tube representations. Charged residues are shown in space-filling representation and colored blue (+) and red (−). Position of residues Leu105 and Leu109 (drawn in space-filling representation) is indicated by the arrow. (D) Top view of the transmembrane domain (residues 27–128). Pore residues (96–113) are shown in surface representation. Each subunit is labeled by a color and a letter. Note how the TM3B helix of one subunit is located just underneath the loop between TM1 and TM2 of the next subunit. (E) Surface representation of MscS. There are seven cytoplasmic openings located on the side and one at the bottom.
The cytoplasmic domain of MscS is formed by a large chamber (Fig. 1 E, Fig. 2) thought to act as a molecular filter or docking site for regulatory proteins (but otherwise of unknown function), with seven openings on the sides and one at the bottom. Polar and charged residues are abundant in the cytoplasmic domain of MscS.
FIGURE 2.

MscS in a membrane bilayer. (A) Stereo side view of MscS in a fully solvated membrane environment, 224,340 atoms. (B) Stereo view of MscS from its cytoplasmic side.
Researchers presently seek to understand the relationship between protein structure and function, the latter determined through different experimental techniques. Patch-clamp experiments not only have determined the conductance and midpoint activation values mentioned before, but also have revealed that MscS is reversibly inactivated at low pH (Cui et al., 1995; B. Martinac, personal communication). In addition, MscS shows slight anionic preference and is voltage-modulated (Martinac et al., 1987; Sukharev, 2002). The estimated energy of opening is ΔG = 11.4 ± 0.5 kT and the transition-related area change of the pore is ΔA = 8.4 ± 0.4 nm2, values that have been obtained using a two-state Boltzmann model and curvature data from video imaging (Sukharev, 2002).
Experiments have also shown that small deletions in the C-terminal region (bottom end of the cytoplasmic domain shown in Fig. 1 A, B, and E) failed to produce functional channels. However, the extracellular N-terminal segment, denoted at the top of Fig. 1 B, seems to tolerate significant (residues 8–21) deletions (Miller et al., 2003a). Moreover, cross-linking of C termini suggested that they move apart upon channel opening and are involved in channel activation (Koprowski and Kubalski, 2003). In addition, cross-linking of site-specific cysteine substitutions suggest that residues in position 58 (situated at the loop between helices TM1 and TM2, Fig. 1 B) can be located within 3 Å of each other compared to being 33 Å apart as in the crystal structure (Miller et al., 2003b). Altogether, these experimental results suggest that the protein is very flexible, its C-terminus is structurally highly relevant, and there are significant structural differences between the closed and open states.
Random mutagenesis experiments have identified some functionally relevant residues. The double mutation I48D/S49P (both residues located at the cytoplasmic end of TM1, Fig. 1 B) inactivates the channel, whereas T93R, L109S, A102P, V40K, and V40D have been identified as gain-of-function phenotypes (Miller et al., 2003a; Okada et al., 2002). It is not surprising that residues at positions 93, 102, and 109 affect the conduction properties of the channel, since they are located on the pore-lining helices (TM3A in Fig. 1 B). The role of positions 40, 48, and 49 is less clear (all located throughout TM1, Fig. 1 B).
Simulations of membrane proteins are becoming a standard tool to gain insight into the mechanisms of ion channels (Roux, 2002; Ash et al., 2004; Roux and Schulten, 2004). Although most of the simulations have focused on ion or water permeation, increasing attention has been paid to the channel gating, especially in MscL (Elmore and Dougherty, 2003; Gullingsrud and Schulten, 2003; Colombo et al., 2003) and in the KcsA potassium channel (Biggin and Sansom, 2002).
A molecular dynamics study of water permeation and ion conduction in MscS (Anishkin and Sukharev, 2004) suggested that the crystal structure of MscS does not represent an open state and that MscS gating involves a vapor-lock mechanism. It is rather unclear how the simulation conditions utilized (protein restrained and simulation of only part of the protein) along with possible distortions in the structure due to crystallization conditions affected these results.
In this report, we present a molecular dynamics exploration of the entire MscS in a fully solvated lipid bilayer, with and without restraints. We describe permeation properties of the restrained structure of MscS and the spontaneous closure of the MscS transmembrane pore after release of restraints. In addition, we describe the effect of surface tension on the transmembrane channel as well as features of the membrane-protein interaction and the conduction properties of MscS.
METHODS
In this section we consider the assembly of the system and the simulation conditions as well as the tools used to analyze our data and the equilibration phase of our simulation. A summary of the simulations carried out is provided at the end.
Assembly of protein, lipids, and solvent
Using VMD (Humphrey et al., 1996) and its Orient plug-in, the MscS crystal structure (Bass et al., 2002; PDB code 1MXM) was oriented by aligning the protein's axis of symmetry along the pore parallel to the z axis. By using the VMD Membrane plug-in, the molecule was then immersed in a previously equilibrated rectangular patch of POPC membrane bilayer with solvated lipid headgroups. The initial membrane position along the z axis was determined by matching positions of headgroups with the seven Tyr27 side chains of the MscS transmembrane domain. The resulting placement of MscS in the membrane was in agreement with the suggestion in Bass et al. (2002) (Fig. 1 A). Lipids within 0.8 Å of the protein and those located in the pore were removed. The final number of lipids was 299, 159 on the periplasmic side and 140 on the cytoplasmic side.
The membrane-protein complex was then solvated using the VMD plug-in Solvate. Water molecules located in the hydrophobic part of the membrane and inside the protein's transmembrane pore were eliminated. The VMD plug-in Autoionize was used to place ions randomly, corresponding to a salt strength of ∼200 mM KCl. The resulting system had zero charge and 52,015 water molecules. Residues Asp, Glu, Lys, and Arg were assumed to be charged throughout the protein. A snapshot of the constructed system of a total of 224,340 atoms is shown in Fig. 2. The initial size of the system was 130 × 130 × 172 Å3 and shrank to 108 × 111 × 178 Å3 during the equilibration.
Molecular dynamics
All simulations were performed using the parallel molecular dynamics program NAMD 2.5 (Kalé et al., 1999) and the CHARMM27 force field for lipids and proteins (MacKerell et al., 1998a,b, 1992, Schlenkrich et al., 1996) along with the TIP3P model for water (Jorgensen et al., 1983).
A cutoff of 12 Å (switching function starting at 10 Å) for van der Waals interactions was assumed. Periodic boundary conditions were used with a flexible cell. Distances between periodic images of the protein were >17 Å in all cases. The particle-mesh Ewald method was used to compute long-range electrostatic forces without cutoff. The density of grid points for particle-mesh Ewald was at least 1/Å3.
An integration time step of 1 fs was used, permitting a multiple time-stepping algorithm (Grubmüller et al., 1991; Schlick et al., 1999) to be employed in which interactions involving covalent bonds were computed every time step, short-range nonbonded interactions were computed every two time steps, and long-range electrostatic forces were computed every four time steps. The use of a multiple time-stepping algorithm may affect the membrane pressure profile (Gullingsrud and Schulten, 2004). However, it allowed us to improve the performance of the simulations by a factor of 2, and thus reach the multinanosecond timescale required to observe structural relaxation of MscS.
Langevin dynamics was utilized to keep a constant temperature of T = 300 K in all simulations. The Langevin damping coefficient was set to 1 ps−1 unless otherwise stated. This value was chosen so as to speed up conformational changes of the channel by reducing the solvent viscosity. Constant pressure simulations at 1 atm were conducted using the hybrid Nosé-Hoover Langevin piston method with a decay period of 200 fs and a damping timescale of 50 fs. When surface tension was applied, components of the pressure tensor were set using the formula (Feller and Pastor, 1999; Gullingsrud et al., 2001)
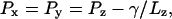 |
(1) |
where Px, Py, and Pz are the x, y, and z diagonal components of the pressure tensor, γ is the surface tension, and Lz is the size of the unit cell along the z axis, which is perpendicular to the plane of the membrane. The pressure along the z axis was kept at 1 atm and γ was set to 20 dyn/cm.
Analysis tools
To monitor the structural deformation of the protein, its root mean-square deviation (RMSD) was computed using VMD and coordinates saved every picosecond; the crystallographic structure served as the reference point, and only positions of protein backbone atoms were compared.
Pore radii were determined using HOLE (Smart et al., 1993). The HOLE program maximizes, using Monte Carlo simulated annealing, the radius of spheres located along and inside the pore defined by the van der Waals surface of the channel. The default all-atom set of van der Waals radii from AMBER (Weiner and Kollman, 1981) was used to allow comparisons with previously calculated radii profiles (Smart et al., 1997). Sample planes were spaced uniformly in the z direction with a 0.5 Å spacing. A note of caution should be added about the method utilized to measure the radius of the transmembrane pore as presented in Fig. 5. Our calculations give values that are smaller than stated in Bass et al. (2002) and subsequent publications, but are in agreement with values presented in Anishkin and Sukharev (2004). The disagreement could be due to the incorporation of hydrogen atoms and van der Waals radii of each atom in our calculations.
FIGURE 5.

Pore radius. (A, top) Pore of MscS in the crystal structure (excluding subunits C and D). The transmembrane domain is shown in transparent surface representation and colored by residue type (white, nonpolar residues; blue, basic residues; red, acidic residues; green, polar residues). Yellow spheres are drawn using the center and radii generated by HOLE. (A, bottom) Pore radius profile along the z axis for MscS with its backbone atoms restrained to the crystal structure (sim1) at t = 0 (black) and t = 4.45 ns (red). The shaded region marks the membrane boundaries. (B, top) Pore of MscS at the end of simulation sim2a (t = 8.45 ns) as in A. (B, bottom) Pore radius profile along the z axis of MscS at the end of simulations sim2a (t = 8.45 ns, green), sim2b (t = 9.05 ns, blue), and sim3a (t = 9.65 ns, yellow).
Conductances of the transmembrane pore of MscS were estimated using the Capsule module of HOLE (Smart et al., 1997). For this purpose, anisotropies of the channel were taken into account (spherocylinders instead of spheres were used for optimization and estimation of radii) and a uniform separation between sample planes of 0.25 Å was used. In addition, 10 times more steps as normal in the HOLE Monte Carlo optimization were performed. HOLE estimates conductances using a formula adapted from Hille's equation (for details, see Smart et al., 1997),
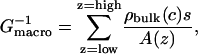 |
(2) |
where Gmacro is the macroscopically expected conductance, ρbulk is the resistivity of the permeant ions regarded as a function of the molar ion concentration c, s is the separation between the sample planes (s = 0.25 Å), A(z) is the area perpendicular to the channel direction vector, and the sum is performed over the sample planes along the z axis. HOLE also provides empirically corrected values of Gmacro using data from high resolution structures (“learning set”) with known conductances (Gfirst), the net overall charge of the channel (Gsecond), and its length. Corrections to Gmacro based on the channel length were not taken into account because the length of the MscS transmembrane channel was not well represented in the “learning set” used in HOLE. The resistivity of 1 M KCl was assumed to be 0.08 Ωm (Smart et al., 1997) along with a linear dependence of ρ on the ionic concentration. The final estimated conductance Gf was calculated as the average of Gfirst and Gsecond multiplied by 0.25 M, the molar concentration of KCl used in experiments (Martinac et al., 1987). The conductances summarized in Table 2 should be considered as estimates, correct only within a factor of 2.
TABLE 2.
Conductance of MscS
| Simulation (time) | Gmacro pS/M | Gfirst pS/M | Gsecond pS/M | Gf pS |
|---|---|---|---|---|
| sim1 (0 ns) | 2100 | 410 | 1000 | 176.25 |
| sim1 (4.45 ns) | 1900 | 360 | 850 | 151.25 |
| sim2a (8.45 ns) | 1500 | 270 | 680 | 118.75 |
| sim2b (9.05 ns) | 2900 | 580 | 1300 | 235.00 |
| sim3a (9.65 ns) | 2100 | 400 | 940 | 167.50 |
Values for different MscS states as determined by HOLE based on the simulated geometries of the MscS transmembrane pore (see Methods for definitions of Gmacro, Gfirst, and Gsecond). Gf was calculated as the average of the predicted conductances Gfirst and Gsecond multiplied by 0.25 M, the molar concentration of KCl used in experiments (Martinac et al., 1987). The large difference between values of Gfirst and Gsecond suggests an inaccuracy in the predicted conductances.
The area occupied by the transmembrane domain of MscS in the x,y plane was estimated using HOLE. In this case the HOLE program maximized the radius of spheres located along the pore defined by the lipid bilayer and occupied by the protein. The membrane pore radii were then averaged along the z axis. The obtained values (Table 1) should be considered as estimates that do not take into account the highly irregular shape of the protein.
TABLE 1.
Area of the periodic cell in the x,y plane for different simulations
| Simulation | Min. area (Å2) | Max. area (Å2) | Area at end of simulation (Å2) | Area occupied by protein (Å2) |
|---|---|---|---|---|
| sim1 | 12,023 | 12,316 | 12,052 | 992 |
| sim2a | 11,629 | 12,194 | 11,670 | 863 |
| sim2b | 12,079 | 12,491 | 12,322 | 1144 |
| sim3a | 11,640 | 12,078 | 11,978 | 840 |
The last column shows the estimated area occupied by the protein in the membrane calculated at the end of the respective simulation as indicated in Methods.
Conduction of water molecules was monitored by counting the number of water molecules present inside the pore (residues 96–113) every picosecond and counting full permeation events.
Averaged ion density maps were generated using VMD and trajectories of simulations sim2a and sim2b (see below) saved every picosecond. The cell size was set to the maximum value reached during the respective simulation. The grid resolution was set to 1 Å in each direction of the cell.
System equilibration
We equilibrated the protein-lipid-solvent system for 3 ns. This simulation step will be referred to below as sim0. The equilibration resulted in a close packing of lipids against the protein, preventing a leakage of water molecules through the membrane-protein interface from the cytoplasmic to the extracellular side.
Simulation sim0 was divided into three substeps. In the first step, “melting” of lipid tails was performed. For this purpose, all atoms of the simulated system were frozen except those of the lipid tails, and 5000 energy minimization steps and 0.5 ns of dynamics under NVT ensemble conditions were carried out. At this point a Langevin damping coefficient of 5 ps−1 was used. In all subsequent calculations the system was simulated as an NpT ensemble.
Subsequently, we carried out step two of simulation sim0. During this step the protein backbone atoms were harmonically restrained with a force constant of k = 1 (Kcal/mol)/Å2. The step began with an energy minimization of 1000 steps and continued then with 1.42 ns of dynamics. Initially, a Langevin damping coefficient of 5 ps−1 was used, and during the last 0.3 ns it was reduced to a value of 1 ps−1, a value that was also assumed in all subsequent simulations. At 1.42 ns, the packing of lipids surrounding the protein was inspected and some lipids reordered by means of interactive molecular dynamics (Stone et al., 2001; Grayson et al., 2003). The system was then minimized for 1000 steps and molecular dynamics resumed for 0.373 ns. To prevent water molecules from reaching the protein-lipid interface, forces were applied that directed them back to the nearest water-lipid interface. These forces were switched on for intervals of 50 ps every 300 ps of dynamics in the second step of simulation sim0 only.
In the third and final step of simulation sim0, 0.7 ns of NpT ensemble dynamics were carried out without forces applied to the water molecules. A satisfactory packing of lipids resulted and no leakage of water molecules through the membrane-protein interface was detected, as stated already above.
After simulation sim0, four simulations (sim1, sim2a, sim2b, and sim3a) were carried out for a total of 10.35 ns. A summary of these simulations is shown in Fig. 3. In these simulations all components, i.e., all 224,340 atoms including protein, membrane, water molecules, and ions, were free to move, except in sim1, where the protein backbone was restrained. Indeed, the entire MscS was free to move in sim2a, sim2b, and sim3a, namely, both transmembrane and cytoplasmic domains that dynamically influenced each other. Such influence had been suggested by experiments and appears to be relevant for MscS stability and function (Miller et al., 2003a,b; Koprowski and Kubalski, 2003).
FIGURE 3.

Summary of MscS simulations. Simulation sim0 consisted of 3 ns of equilibration with multiple substeps as described in the text. Permeation properties of MscS restrained to the crystal structure were studied in simulation sim1 (1.45 ns, protein backbone harmonically restrained). In simulation sim2a (4 ns), restraints were gradually suppressed leading to a nonsymmetrical closed state. The effect of surface tension on the open state was studied in simulation sim2b (3.7 ns). In simulation sim3a (1.2 ns), surface tension was applied to the closed state obtained in simulation sim2a; this resulted in a partially open state of MscS.
RESULTS AND DISCUSSION
The following sections describe results of our simulations of MscS with the protein backbone initially restrained and then released, as well as simulations of MscS under the influence of surface tension. The sections also discuss the conduction of ions through MscS, the behavior of its transmembrane helices, and the membrane surrounding it.
MscS with backbone restrained to the crystallographic structure
The first simulation (sim1) was designed to study MscS when restrained to the crystallographic structure. Indeed, in the case of this 1.45 ns simulation, the protein backbone was harmonically restrained with a force constant of k = 1 (Kcal/mol)/Å2 to the positions determined in the crystallographic structure. A snapshot of MscS at the end of this simulation is shown in Fig. 4, A and B. The pore was found to remain in the initially open state. This is concluded from the radius of the transmembrane pore measured as shown in Fig. 5 A. This figure presents the radius profile of the transmembrane pore of MscS. In the initial “crystal structure” the narrowest part of the transmembrane pore was found at the level of the seven Leu105 side chains (3.26 Å). During sim1, this part of the pore (neck) assumed a radius of 2.8 Å, a value that remained stable throughout sim1 (Supplementary Material, Fig. 20 A). In addition, a small displacement of the pore's neck toward the periplasm was observed (Fig. 5 A). Besides the channel opening (Fig. 5), we monitored in our simulations the pore constriction (Fig. 6), the RSMD values (Fig. 7), and water permeation (Fig. 8).
FIGURE 4.

Simulated conformations of MscS. (A) Side view of the final state (4.45 ns) of simulation sim1. The protein is shown in surface representation and the lipid bilayer in licorice representation. Transmembrane domains (residues 27–128) of subunits A and B (see Fig. 1) along with part of the membrane have been eliminated to reveal the pore's interior. Water molecules inside the pore are shown in space-filling representation. The water box is drawn in transparent blue. (B–E) Top views of final conformations of simulations sim1 (4.45 ns), sim2a (8.45 ns), sim2b (9.05 ns), and sim3a (9.65 ns), respectively. The transmembrane domain is shown in surface representation and the lipid bilayer in space-filling representation. The color code for each protein subunit is the same as in Fig. 1 D.
FIGURE 6.

(A) Detail of TM3A deformation upon closing of the pore. The backbone of subunit-C helix (residues 96–113) is shown in tube representation. Residues Gly101, Ala102, Leu105, and Ala106 are shown in licorice representation. Hydrogen bonds are shown in blue. (Left) Snapshot at the end of simulation sim1 (4.45 ns). (Right) Snapshot at t = 6.04 ns of simulation sim2a. (B) View of pore residues (96–113, TM3A) and surrounding lipids (shown in van der Waals representation with hydrogen atoms in white, carbon atoms in cyan, and oxygen atoms in red). Subunits are colored as in Fig. 1 D. Residues Leu105 of each subunit are shown in space-filling representation. Subunits A (light blue), C (red), and E (orange) show pronounced kinks and their respective Leu105 side chains block the pore. Subunit A is in close contact with a lipid, whereas the other subunits are not.
FIGURE 7.

RMSD with respect to the crystal structure. (A) RMSD of the whole protein backbone for simulations sim2a (red), sim2b (blue), and sim3a (green). The clearly discernible steps correspond to changes in harmonic restraints (see text). (B–D) RMSD per zone for simulations sim2a, sim2b, and sim3a, respectively. MscS zones are defined as follows: TM1–TM2 helices, residues 27–91 (black); TM3A–TM3B helix, residues 91–127 (red); pore (TM3A), residues 96–113 (green); and cytoplasmic domain, residues 128–280 (blue).
FIGURE 8.

Water permeation. Occupancy of the hydrophobic (transmembrane) pore of MscS by water molecules is shown as a function of time for simulations sim1 (A), sim2a (B), sim2b (C), and sim3a (D). The number of water molecules inside the pore was monitored every picosecond assuming the positions of Cα atoms of residues 96 and 113 as pore ends. Snapshots of the pore (shown in surface representation and colored by residue type as described in Fig. 5) with water molecules are shown for representative states.
During sim1, the transmembrane pore of MscS remained sufficiently open for permeation to occur, and, in fact, five full permeation events of water molecules were observed. As shown in Fig. 8 A, the pore transited from being completely filled with water, to being partially empty. This finding is in agreement with results presented in Anishkin and Sukharev (2004) and studies that showed liquid-vapor oscillations of water in hydrophobic nanopores (Beckstein and Sansom, 2003, 2004). Formation of single files of water molecules was observed inside the bottom opening of the cytoplasmic domain of MscS during sim1. However, the size of the bottom opening was greatly reduced.
Spontaneous closure of the MscS pore
In a second simulation, sim2a (4 ns), we explored the behavior of MscS without restraints, starting from the final state of sim1. However, we did not release the restraints entirely at one instant, but rather released them gradually over 0.9 ns, choosing force constants of k = 0.5, 0.1, and 0.01 (Kcal/mol)/Å2, in steps of 300 ps. Subsequently, we carried out 3.1 ns of free dynamics.
A top view of the final state of this simulation is shown in Fig. 4 C. One can immediately recognize a dramatic motion of the protein that led to a complete closure of the transmembrane pore. The pore radius at the end of the simulation was 1.27 Å in its narrowest section, i.e., at Leu105. However, the value of the radius fluctuated between 0.5 and 2.4 Å (Supplementary Material, Fig. 20 B). The closing of the transmembrane pore was accompanied by a displacement of the pore's neck by ∼5 Å toward the periplasmic side (see Fig. 5 B).
RMSD values of MscS during sim2a increased to a level of ∼4 Å, as seen in Fig. 7 A. The RMSD values were found to differ for different zones of the protein as shown in Fig. 7 B. The smallest RMSD value was found for the pore zone, whereas the largest values were found for the cytoplasmic domain and for helices TM1–TM2. Fig. 7 A for the overall RMSD value and Fig. 7 B for the zone RMSD values show a different time-dependence, in that the total RMSD value increased throughout sim2a, whereas the zone RMSD values reached stable plateaus in the 2–3 Å range. This suggests that the increase in the overall RMSD value is due to relative motions of protein domains (Supplementary Material, Fig. 15), and not due to intrinsic local changes of protein structure.
The sudden change in the RMSD values of the pore region in sim2a (Fig. 7 B, between 6.5 and 7.5 ns) is due to a deformation observed in the pore-lining α-helices and will be described in more detail below. The increase of the pore RMSD is trailing the increase of the RMSD values of TM1–TM2 and the cytoplasmic domain, suggesting a causal relationship.
A closer look at the dynamics of the pore in simulation sim2a revealed two interesting features. First, the bottom entrance of the transmembrane part of the pore, determined by the TM3B segment of TM3, moved up toward the periplasm (Supplementary Material, Fig. 19, sim2a), as already mentioned above. Second, the main feature of the pore closure was an asymmetric displacement of the seven Leu105 side chains along with the appearance of a kink in the α-helical structure of TM3A in subunits A, C, and E (Fig. 6 and Supplementary Material, Fig. 19, sim2a).
A key deformation causing the closing of the pore is shown in Fig. 6 A. The TM3A helix of subunit C buckled shoving Leu105 toward the pore center and moving residues Gly101, Ala102, and Ala106 along with it (Fig. 6). The motion involved breaking and transferring of α-helical hydrogen bonds. The helix TM3A is characterized by the sequence motif GxxGxxxGxxxxG, conserved in MscS homologs. This motif starts at residue Gly101 and ends at residue Gly113.
Two possible causes were identified for the deformation of pore lining structures: direct interaction with the lipid bilayer or tension transmitted through TM1 and TM2. As shown in Fig. 6 B, the deformation in subunit A (light blue) seems to be caused by the close proximity of lipids, whereas deformation in other subunits (C, red and E, orange) is not directly related to surrounding lipids (no contact is observed between lipids and TM3A in those cases). In both cases, the direct or indirect effect of the lipid bilayer tension on the overall structure of the protein could be overstated, since it is known that the force field parameters used in MD underestimate the correct area per lipid by ∼5% (Feller and Pastor, 1999; Gullingsrud and Schulten, 2004). This may explain the short timescale observed for the closure of the channel (Supplementary Material, Fig. 20 B).
The asymmetric movement of TM3A helices from different subunits seems to be compatible with the steric constraints experienced by seven helices that are in close proximity (Bezanilla and Perozo, 2002). However, distortions of helices resulted in a small lateral opening between TM3A helices of subunits A and B, compromising the integrity of the pore at the end of sim2a.
After release of harmonic restraints, the transmembrane zone of the pore remained empty and no conduction of water molecules could be discerned, as documented in Fig. 8 B. In addition, the distal opening of the cytoplasmic domain rapidly collapsed after release of restraints, and remained in a closed state during almost all of simulation sim2a (data not shown).
The closed state is favored under the chosen simulation conditions and should be the default state in a relaxed membrane (Bass et al., 2002). However, the spontaneous closure of the MscS transmembrane pore observed in simulation sim2a raised several questions that lead us to devise a third simulation, sim2b, in an attempt to keep the MscS pore open.
MscS assumes open state when surface tension is applied
The third simulation, sim2b (3.7 ns), applied surface tension to keep the MscS pore open, as was also done in experiments by Sukharev (2002). Coordinates of the system at 5.35 ns of sim2a were utilized as a starting point for sim2b (these coordinates correspond to the last step before complete release of restraints on the MscS backbone in sim2a, when the transmembrane pore is still in an open state; see Fig. 3). The simulation sim2b consisted of 0.4 ns of dynamics with the protein backbone harmonically restrained using a force constant of k = 0.01 (Kcal/mol)/Å2 and a surface tension of 20 dyn/cm. Then 3.3 ns of free dynamics were carried out with the same surface tension applied. A top view of the final state of sim2b is shown in Fig. 4 D. An increase in the transmembrane pore size can be easily recognized. Indeed, the pore radius at the end of sim2b is 3.75 Å at its narrowest part, at the level of residues Leu105 (Fig. 5 B). This value of the pore radius is even larger than the one observed in the original crystal structure (3.26 Å). Moreover, intermediate values of the pore radius at its narrowest section during sim2b were found to be ∼4 Å (Supplementary Material, Fig. 20 C). A minor displacement of the narrowest part of the pore toward the periplasm accompanied the increase in pore size, as shown in Fig. 5 B.
The surface tension applied in sim2b is approximately four times higher than what is needed to experimentally open MscS (Sukharev, 2002), and even higher than the rupture limit for a membrane. However, as mentioned before, the force-field parameters and simulation conditions (NpT) underestimate the experimental area per lipid (Feller and Pastor, 1999; Gullingsrud and Schulten, 2004), which may justify the large value used. Moreover, the high surface tension permitted us to induce the pore opening on the short timescale of molecular dynamics simulations. Simulations of MscL under surface tension have also utilized unphysiological tension values of 60 dyn/cm in vacuum (Gullingsrud et al., 2001) and 50–500 dyn/cm in a hydrated membrane environment (Colombo et al., 2003). In particular, opening of MscL in a hydrated membrane environment was observed only at 500 dyn/cm, which is more than an order-of-magnitude larger than the experimentally required magnitude. In none of our simulations did we observe membrane rupture.
The RMSD value for simulation sim2b is similar to that of sim2a, increasing to a level of ∼4 Å, as seen in Fig. 7 A. RMSD values for different zones of MscS reached rather stable plateaus at the end of sim2b, as shown in Fig. 7 C. However, RMSD values differ from each other for different zones of the protein. In particular, the RMSD value of the transmembrane helices TM1–TM2 is ∼1 Å larger than that of the pore, the cytoplasmic domain, and the transmembrane helix TM3A–TM3B. This can be explained as an effect of the applied surface tension and direct interaction of lipids with TM1 and TM2. As in sim2a, deformation of the TM3A–TM3B helix followed deformation of the TM1–TM2 transmembrane helices.
The in-plane protein areal expansion, ΔA, has been estimated from experiments to be 8.4 nm2 (Sukharev, 2002). According to our simulations (sim2b and sim2a), the area in the x,y plane of the periodic cell varies from 11,629 Å2 to 12,491 Å2 (see Table 1), which represents a change in total area of 8.62 nm2, close to the experimental value; this change in area observed between the closed state of sim2a and the open state of MscS of sim2b includes the expansion of the membrane bilayer itself. Therefore, we have estimated the area occupied by the protein itself (see Table 1). The change in area occupied by the protein observed between the final states of simulations sim2a and sim2b was 2.81 nm2. The inferred area per lipid is in good agreement with results presented in Gullingsrud and Schulten (2004).
Pore dynamics in sim2b showed a rather different behavior than in sim2a. Although a movement of the pore's bottom edge toward the periplasmic side was observed, the pore structure tilted due to movements of the cytoplasmic domain. These movements are directly linked to the pore through TM3B. In addition, and due to the applied surface tension, the pore remained open at all times (Supplementary Material, Figs. 19, sim2b, and 20 C). A further distortion of the α-helix in subunit G accompanied by the outward movement of its respective Leu105 side chain was observed (Supplementary Material, Fig. 19, sim2b.5 and sim2b.6) and is likely caused by direct interactions with the lipid bilayer (Fig. 6 B).
When surface tension was applied to keep the pore open, conduction of water molecules was enhanced and the transmembrane pore remained occupied by water molecules for almost the entire simulation sim2b (Fig. 8 C). On the other hand, the distal central opening of the cytoplasmic domain remained closed and conduction of water molecules through it was not observed.
MscS closed state under the influence of surface tension
The fourth and last simulation (sim3a) used as a starting point the last step of sim2a (see Fig. 3) and consisted of 1.2 ns of molecular dynamics with an applied surface tension of 20 dyn/cm. Our goal was to explore the effect of surface tension on the closed state of MscS obtained in sim2a.
The final state of sim3a is shown in Fig. 4 E. One can recognize that the surface tension led to a partial opening of the MscS transmembrane pore. As shown in the respective radius profile, the neck radius was 2.55 Å at the end of sim3a (Fig. 5); intermediate values for the neck radius of ∼2.8 Å were monitored (Supplementary Material, Fig. 20 D). The timescale of the MscS opening observed in sim3a is different and longer than the timescale of MscS closure observed in sim2a. Indeed, experiments suggest that the closure of MscS is faster than the opening (Sukharev, 2002).
Displacement of the pore's neck toward the cytoplasmic side along with a clear increase of radius in the periplasmic zone of the pore (Fig. 5 B) was also observed. The periplasmic opening seems to be caused by interaction of lipids with Tyr27 and with surrounding residues of some subunits. The effect of lipid-protein interaction on the periplasmic opening may be altered due to the presence of residues 1–26, missing in our model. Our finding is not in agreement (but not completely incompatible either) with the suggestion that the periplasmic side furnishes a pivotal point for the movement of TM1–TM2 in the gating mechanism (Bass et al., 2002; Bezanilla and Perozo, 2002). Since the behavior of different subunits is not conclusive, further simulations with smaller values of surface tension and longer simulation times are needed, along with a model for the periplasmic domain of MscS.
During sim3a, the RMSD value increased further as shown in Fig. 7 D. The RMSD values per zone also showed an increase, especially for the cytoplasmic domain and for the TM1–TM2 helices. It is important to note that the method used to apply surface tension induces a change in the area of the complete periodic cell. Of course, the water box was chosen big enough to absorb changes in MscS geometry; nevertheless, the generated flow of water molecules may exert a small perturbation on the cytoplasmic domain.
Finally, while trying to open the closed state in sim3a, conduction of water molecules was observed through the transmembrane segment of the MscS pore (Fig. 8 D). The shape of the pore permitted the formation of more than one file of water molecules. As in sim2a and sim2b, conduction of water molecules through the distal opening of the cytoplasmic domain was not observed.
Conduction of ions in MscS
Gating of MscS is monitored experimentally by ionic currents measured in patch-clamp experiments. Although presently molecular dynamics simulations cannot reach the timescale where natural conduction of ions arises, conductances can be estimated from the structure of the protein at different stages of the simulations using HOLE, as described in Methods. The resulting conductances of MscS are summarized in Table 2. The smallest value, 118.75 pS, is associated with the closed state at the end of sim2a (intermediates may have even smaller conductances); the largest value, 235 pS, is associated with the open state at the end of sim2b. The maximum value of the conductance predicted by HOLE is relatively small compared to the experimentally determined MscS conductance (∼1 nS). Thus, if our calculations are correct, it is likely that the open state of MscS corresponds actually to one of the intermediates arising in sim2b with a wider pore than that of the final state, or to an even wider state not observed on the timescale of our simulations. Clearly, further investigations are needed to address the conduction properties of MscS (see Roux et al., 2004, for a recent review on the subject of ion permeation). We note that overall ion conduction includes permeation of the cytoplasmic domain (Fig. 9).
FIGURE 9.

Ion diffusion across the cytoplasmic domain of MscS. (A) Stereo view of the diffusion of a Cl− ion from the bulk to the interior of the MscS cytoplasmic domain in simulation sim2a. (B) Stereo view of the diffusion of a K+ ion from inside of the MscS cytoplasmic domain toward the bulk in simulation sim2a. In both cases, the cytoplasmic domain of MscS (residues 128–280) is drawn in tube representation and colored as in Fig. 1 D. The ion pathway is formed by the superposition of sampled positions of the respective ion along simulation sim2a, which is shown in space-filling representation (sphere). The color of the ion indicates time, with red corresponding to initial stages of simulation sim2a and blue to final stages of simulation sim2a.
Although no spontaneous diffusion of ions through the transmembrane pore was observed (neither was this expected on the timescale of our simulations), an unusual concentration of Cl− ions was found in the periplasmic mouth of the channel (Fig. 10 A and Supplementary Material, Fig. 21 A). In fact, these ions were found close to the loops connecting helices TM2 and TM3A, where a small opening is formed by residues 83–98. The opening is present in all subunits of the crystal structure (see Fig. 1 C) and partially covered by residue Arg88 of the previous subunit. During simulations, the openings adopted different sizes in different subunits. A Cl− ion was found inside one of the openings at the end of sim3a. It is unclear if these openings can provide a pathway for Cl− ions into the hydrophobic crevices of the MscS transmembrane domain or if they just provide a binding site. It is also unclear how the mobility of TM1–TM2 and TM3A is affected by the presence of ions in this region. The gain-of-function mutation T93R hints at an important role of this zone in the conduction properties of MscS.
FIGURE 10.

Distribution of ions during simulation sim2a. (A) The average density of Cl− ions as defined by the scale bar is shown at a transversal slice crossing the center of the channel in the z direction. The approximate shapes of MscS and the lipid bilayer are depicted by a solid line. (B) The crystal structure of MscS is shown in cartoon representation. The approximate volumetric shape of MscS and the position of the lipid bilayer are depicted by a solid line. (C) The average density of K+ ions is shown as in A.
The distribution of ions inside the cytoplasmic domain of MscS (Fig. 10, A and C, and Supplementary Material, Fig. 21 A, C, D2, and D3) also showed a high concentration of Cl− close to the cytoplasmic mouth of the channel, contrasted by a weaker concentration of K+ ions found only in the zone distal from the membrane. Similarly, the distribution of positive and negative ions across the cytoplasmic domain of MscS showed a distinctively different pattern for the two types of ions (Supplementary Material, Fig. 21 D). The consistently large concentration of Cl− ions in the channel compared to K+ ions suggests faster conduction of Cl− than K+. We show in the Appendix for a one-dimensional channel model that the Ohmic resistance of a channel is related qualitatively to the ion concentration p0(x) (either Cl− or K+), according to
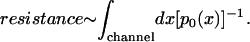 |
(3) |
Obviously, large p0(x) values reduce the resistance so that one expects Cl− ions to be conducted through the channel faster than K+ ions for which the p0(x) in the channel is small (see Fig. 10 and Supplementary Material, Fig. 21). Experiments show indeed that negative ions are conducted more readily by MscS than positive ions (Sukharev, 2002).
An exciting open problem is the role of the balloon-shaped cytoplasmic domain. In our simulations, spontaneous diffusion of ions through the cytoplasmic side openings of MscS was observed. For example, a Cl− ion transited from the bulk to the interior of the cytoplasmic domain in simulation sim2a. This event involved a series of interactions with conserved arginines (156, 184, 185, and 238) among other residues. The transit of a K+ ion from the inside of the cytoplasmic domain toward the bulk was also seen in sim2a. The positive ion interacted on its way with residues Glu134, Asp137, Asp159, and Glu187, among others. Of these, only Asp159 is conserved in more than 65% of the MscS homologs analyzed in Bass et al. (2002). The pathways of the ions are illustrated in Fig. 9.
Different degrees of openness were observed for the side openings of the cytoplasmic domain. In none of our simulations did the openings assume a completely closed state. Apparently, these side openings provide a more robust outlet and inlet than the distal opening of the cytoplasmic domain (Fig. 2).
Transmembrane helices of MscS and the gating mechanism
Upon insertion of the protein into the membrane bilayer, a strong interaction between the TM1–TM2 helices and the surrounding lipids became established. Indeed, inspection of the TM1–TM2 helices revealed an altered mobility after insertion. The tilt angle (with respect to the membrane normal) of helices TM1–TM2 was monitored during all simulations (data not shown) and revealed strong fluctuations. However, all subunits showed a decrease in their tilt angle of their TM1–TM2 domains (reductions of up to 8°) in sim2a, which seems to be in agreement with proposed movements associated with channel closure and the reorientation of α-helical transmembrane domains (Bass et al., 2002; Bezanilla and Perozo, 2002). The tilt angle of helices TM3A and TM3B (residues 96–113 and 114–127) increased (up to 45° for TM3A and 90° for TM3B in some cases) in sim2a and sim2b.
Due to strong fluctuations and the flexibility of the protein, a clear pattern could not be established for the overall motion of the transmembrane helices on the timescale of our simulations. The TM1–TM2 domains of some subunits moved according to the suggested gating mechanism in Bass et al. (2002), as shown in Fig. 11. However, a concerted movement of all subunits in the same direction was not seen. In fact, some subunits moved in one direction whereas the others moved in the opposite direction (Fig. 11), likely due to the overall displacement of the protein in an irregular environment. The movement of the transmembrane helices was found to be caused by the interaction of lipid headgroups with charged and polar residues of the TM1–TM2 helices (Arg46, Arg54, Arg59, Arg74, Lys60, and Tyr75). The rearrangement of the membrane (discussed below) lead to a close interaction of the charged and polar residues of each transmembrane subunit of MscS with at least one lipid headgroup. Based on these results we infer that these interactions explain the movement of the transmembrane helices and are a key element in the gating mechanism. Voltage sensitivity may arise from a rearrangement of side chains of these residues as well as movement of the membrane headgroups and changes of the number of ions present in the protein-water-lipid interface. Further simulations need to be performed to address this point.
FIGURE 11.

Movement of transmembrane helices. Transmembrane domains are shown in ribbon representation for subunit E (A) and subunit A (B). Red shows the state of helices in simulation sim2a at t = 8.45 ns and green shows the state of helices in simulation sim2b at t = 9.05 ns. The movement observed in subunit E is in agreement with the gating mechanism proposed in Bass et al. (2002). Subunits D and F showed similar behavior. The movement observed in subunit A (along with subunits B and G) is in the opposite direction.
In all of our simulations, the formation of up to four salt bridges (Figs. 12 and 13), with at least one appearing between residues Asp62 of one subunit and Arg128 of the subsequent subunit, was observed (an asterisk indicating the presence of the salt bridge has been placed in the label of different subunits in Supplementary Material, Fig. 19). Both residues are conserved in at least 65% of the sequences of 25 homologs of MscS in prokaryotes and archaea (Bass et al., 2002). Moreover, in four out of eight cases where the negatively charged residue Asp is not present in position 62, it is being replaced by residue Glu, also negatively charged. Similarly, in six out of nine cases where the positively charged residue Arg is not present in position 128, it is being replaced by Lys, also positively charged. These observations indicate a strong conservation of charge that will favor the formation of the respective salt bridge in similar structures. However, the specific role of these salt bridges in the gating mechanism is not clear. The movement of the TM1–TM2 domain could be limited by the presence of the salt bridge, reducing the sensitivity of the channel to membrane strain. On the other hand, the salt bridges provide direct links between the movement of the TM1–TM2 domains and the pore lining helices. Therefore, sensitivity of the channel to membrane strain could be increased due to their presence. Whether the protonation state for Asp62 and other residues changes throughout the opening or closing of the channel is a question that should be addressed in future studies of MscS gating. Preliminary simulations of MscS with all Asp and Glu residues protonated (low pH) have shown larger RMSD for the protein and inconclusive effects on gating of the pore.
FIGURE 12.

Contacts between the TM1–TM2 loop and the TM3B helix. (Top) Segments of helices TM1, TM2, and TM3 of subunit B are shown in blue along with helix TM3 of subunit A shown in light blue, all in tube representation. Residue Asp62 of subunit B and residue Arg128 of subunit A are shown in space-filling representation. The interaction of both residues is clearly discernible. (Bottom) Segments of helices TM1 and TM2 of subunit F (yellow), and helix TM3 along with part of the cytoplasmic domain of subunit E (orange) are shown in transparent surface and tube representation. The illustration shows how the TM1–TM2 loop closely interacts with the TM3B helix.
FIGURE 13.

Salt-bridge formation. Separation between the Cζ atom of residue Arg128 of one subunit and the Cγ atom of residue Asp62 of the subsequent subunit are shown for simulations sim1 (A) and sim2a (B–C). (The colors denote the subunits where the residue Arg128 is located: subunit A, black; subunit B, red; subunit C, green; subunit D, blue; subunit E, orange; subunit F, brown; and subunit G, gray.)
Membrane behavior
The surrounding membrane obviously plays an important role for the gating of MscS. Indeed, membrane lipids are responsible for the transmission of tension to the transmembrane domains of MscS. As sim1 proceeded, overall displacement by 2 Å of the protein toward the cytoplasm was observed (Supplementary Material, Fig. 15 and Fig. 18 A). After release of restraints in sim2a, a further displacement toward the periplasm by ∼2 Å of both the membrane and the protein occurred (Supplementary Material, Fig. 15 and Fig. 18 B). On the other hand, sim2b, in which a surface tension of 20 dyn/cm was applied, exhibited a fluctuating position of the protein (with respect to the membrane) that resulted in a small displacement of the protein toward the periplasm. In sim3a, the membrane and the protein moved together toward the cytoplasmic side. The relative motions described above suggest a faulty initial condition of the membrane, but may also be caused by the absence of the first 26 residues of MscS in our model.
The thickness of the membrane, computed as the average distance between phosphorus atoms of each layer, reached a value of ∼38 ± 1 Å in sim1, a value that remained without major variations in all subsequent simulations (in agreement with Gullingsrud and Schulten, 2004; data not shown). The lipid order parameter was computed and compared favorably to values presented in Gullingsrud and Schulten (2004), also showing good agreement with the experimentally determined values (Supplementary Material, Fig. 17).
Besides the relative movement of the protein and membrane under different external conditions, deformation of the membrane near the protein was observed in sim1 and subsequent simulations, as shown in Fig. 14. The arrangement of headgroups around the protein and its charged residues located in the protein-membrane interface revealed a dramatic thinning of the membrane bilayer close to the protein. As a result, side chains of charged residues reoriented and were not facing the hydrophobic zone of the membrane as suggested by the crystal structure, but rather interacted with membrane headgroups. This interaction is a key element in the gating mechanism and movement of transmembrane domains as discussed above.
FIGURE 14.

Stereo side views of part of the transmembrane domain. Phosphorus atoms of lipid bilayer headgroups are shown as tan spheres. The MscS transmembrane domain is shown in surface representation and colored by residue type (nonpolar residues, white; basic residues, blue; acidic residues, red; and polar residues, green). (Top) Initial conformation. (Bottom) Final conformation (both of simulation sim1). The deformation of the lipid bilayer close to the membrane-protein interface can be observed. One can discern a clear rearrangement of lipid headgroups around reoriented charged and polar residues (blue, red, and green) on the cytoplasmic side (bottom) of MscS.
CONCLUSIONS
Resolution of the structure of a protein has often proven to be a decisive step in the quest toward understanding function. However, a crystallographic or NMR structure depicts the protein in a single conformation that alone usually does not reveal the mechanism underlying the function. This is certainly true for a protein with a mechanical function like MscS, where function (mechanical and voltage-gating) implies significant movement. Crystallographic structures present an additional problem: the crystal environment may distort a protein from its conformation in the native environment. MscS, found as a closed membrane channel in a relaxed membrane, is seen in the crystal in an open channel form, suggesting a strong effect of the environment. Even when a resolved structure is close to that of the functional protein, the structure by itself may not reveal the mechanism without manipulation of the protein and monitoring of the response.
A solution to the problems described can be provided by molecular dynamics simulations, except that force fields may be error-prone and timescales covered are most often too brief. The shortcomings indicated can be overcome, e.g., when function may not depend on the accuracy of the force field and when external forces are applied that accelerate function without undue distortion. The latter path can be established only by trial and error. Unfortunately, MscS is a very large protein with over 2,000 amino acids. To study this protein in its lipid-membrane environment with a complex electrostatic force field requires one to simulate the dynamics of 200,000 atoms for 10 ns or longer, a considerable methodological challenge. The present study has met this challenge and described MscS accordingly in a series of scenarios that capture dynamics for MscS restrained to the crystal form, relaxation of the channel from the crystal form and its subsequent dynamics, as well as membrane-tension-induced channel behavior.
The simulations revealed key expected and unexpected characteristics of MscS that are likely essential for an eventual explanation of the mechanisms underlying the various functions of MscS. Being one of the earliest simulations of MscS, in fact the first that includes dynamics of the entire protein, the simulations mainly represent a fact-finding approach rather than one achieving an already reductionist picture of MscS. However, the behavior of MscS seen in the simulations proves to be thought-provoking and certainly will motivate further investigations.
The most dramatic feature observed in the simulations reported is the spontaneous closure of the MscS transmembrane pore when the crystallographic structure that exhibits an open pore was permitted to adjust itself to a membrane environment of 300 lipid and 52,000 water molecules. This is an extremely fortunate result since it is the first simulation of a spontaneous gating motion of a mechanosensitive channel. Undoubtedly, it will provide a sound basis for studying the mechanical coupling between protein and environment. The simulations above have taken the natural step of attempting to reopen the already closed channel and of attempting to prevent the closing by applying membrane tension, the latter proving highly successful. The atomic-level description permitted a detailed view of interactions and geometric transformations governing pore closing and opening. The role of charged and polar residues in the cytoplasmic side of the transmembrane domain was found to be extremely important. The membrane surrounding the protein adjusts and deforms, establishing close interactions with charged side chains, which were found to face lipid headgroups instead of the hydrophobic zone of the membrane as suggested by the crystallographic structure. Support for the suggested gating mechanism comes from the observation of TM1–TM2 helices moving in a hinge-like fashion, with the TM1–TM2 loop “swinging” and the TM2–TM3 loop acting as the hinge. Whether different units move in a concerted manner is not clear. However, the flexibility of these domains provides an efficient way for the protein to adapt to an irregular and changing membrane environment.
Beside its transmembrane pore, MscS sports a large, balloon-like chamber that acts most likely as a filter in front of the cytoplasmic entrance of the channel. This domain exhibited a dramatic role during the simulations. First, and most surprisingly, a central opening distal from the transmembrane channel closed quickly and remained closed during the simulations that kept the domain surrounded by its native aqueous environment. Second, the domain revealed, in keeping with its heptameric structure, seven lateral openings that experienced flowthrough of ions, the flow assisted through polar and charged amino acids. The openings remained large enough for ion passage throughout the simulations. Third and last, the arrangement of ions inside and around this cytoplasmic domain appeared to form a distinct pattern that should be relevant for its function.
The spatial rearrangements of MscS observed in our simulations suggest that this protein is very flexible, although most of its secondary structure elements remained intact (see Supplementary Material, Fig. 16). For example, the dynamics often broke the sevenfold symmetry of the crystallographic structure and results suggest that hypothetical mechanisms that assume symmetric reaction pathways for channel gating are ill-advised. What nature seems to hold in store is a protein that is very flexible and at the same time robust in regard to function. To achieve this with a multimeric system seems to be rather a miracle that definitely deserves much scrutiny.
Perhaps the most intriguing suggestion coming out of the simulations are interactions between the cytoplasmic domain and the transmembrane domain of MscS. Particularly interesting is the formation of salt bridges between the side chains located at the end of TM3B of one subunit and residues located in the loop between the TM1 and TM2 helices of the subsequent subunit. The connection between TM3B and the TM1–TM2 domains directly links the pore-lining helices to the movement of the transmembrane and cytoplasmic domains. It is unclear if this link immobilizes the TM1–TM2 domain or if it helps to open the pore, i.e., assists in gating.
We need to mention also key shortcomings of our simulations. First, missing residues in the crystal structure at the periplasmic zone may affect the movement of TM1–TM2 helices and the membrane position, whereas missing residues at the very end of the C-termini may stabilize the distal opening of the cytoplasmic domain. Second, the timescale of our simulations, the size of the membrane patch, and the symmetry of the periodic cell used limit the comparison of our results with experiments and add uncertainties. Third, changes in protonation states of Asp and Glu residues for different conformations of MscS were not yet considered.
Many questions still remain unanswered. Is the position of the membrane and its deformation near the protein an artifact of the initial conditions utilized? Is the small predicted conductance of the obtained open state a sign of an intermediate state? How is the pressure profile in the membrane modified by tension and deformation and how is it related to the interaction with charged and polar residues of the MscS transmembrane domain? Is this flexible protein affected directly by hydrostatic pressure and water flow? Why is there a concentration of negative ions in the periplasmic mouth of the channel and what is the relevance of the bimodal arrangement of ions inside the cytoplasmic domain of MscS? What is the effect of the membrane potential and how is it related to the protein-lipid interaction? What is the role of the observed salt bridges and how can the inactivation of the channel at low pH be explained?
A particular challenge to future work appears to be a re-engineering of MscS that eliminates much of the cytoplasmic domain. A small, re-engineered MscS may reveal much more readily its wide capacity of gating. Another challenge is to explain the role of the amazing cytoplasmic domain.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Justin Gullingsrud, Jordi Cohen, Trudy van der Straaten, Grischa Meyer, and members of the Theoretical and Computational Biophysics Group for helpful discussions, as well as our reviewers for their thoughtful suggestions. The molecular images in this article were created with the molecular graphics program VMD (Humphrey et al., 1996).
This work was supported by the National Institutes of Health (NIH P41 RR05969 and NIH 1 RO1 GM067887-01). The authors also acknowledge computer time provided by the National Science Foundation, National Resource Allocation Committee Grant MCA93S028.
APPENDIX
In the following we will determine the Ohmic resistance of ions in a membrane channel determined on the basis of the equilibrium probability distribution p0(r) as depicted in Fig. 10 and Supplementary Material, Fig. 21. Actually, we consider only a one-dimensional situation described by an equilibrium probability distribution p0(x). We also assume that in an unspecific, rather wide channel like MscS in the open state, conduction is a single particle property, e.g., ions are not conducted by a single-file multi-ion mechanism. A further analysis of ion conduction in membrane channels can be found in Cooper et al. (2004).
At times longer than 1 ps the motion of ions is described well by the Smoluchowski diffusion equation (Risken, 1989), that can be expressed in the form
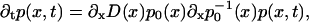 |
(4) |
where p(x,t) is the probability distribution of finding the ion at position x at time t, and where D(x) is the diffusion coefficient. Equation 4 needs to be complemented by boundary conditions. Actually, we are interested in the stationary behavior and expect in this case that for a small enough (compared to kBT) free energy difference, ΔG, applied along the channel, Ohm's law holds,
 |
(5) |
where R is the resistance.
Under stationary conditions, Eq. 4 reads
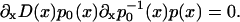 |
(6) |
In solving Eq. 6, we note immediately
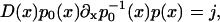 |
(7) |
where j is a constant, i.e., independent of x. From the theory of the Smoluchowski equation (Risken, 1989) one knows that j is, in fact, a current. To seek a solution that can be brought into the form of Ohm's law, i.e., Eq. 5, we postulate that at the entrance and exit of the channel at x = a and x = b, respectively, p(x) assumes the concentration values ca and cb. These conditions can be met quickly. We multiply Eq. 7 by [D(x) p0(x)]−1 and integrate, obtaining
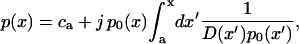 |
(8) |
where we have met already the condition p(a) = ca. In this equation the constant j is still unknown. Its value can be concluded, though, from the second boundary condition, i.e., from p(b) = cb. This condition reads, using Eq. 8,
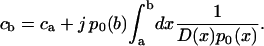 |
(9) |
We can now establish the connection to Ohm's law. For this purpose we recall p0(x) = Z−1 exp [ − βU(x)] where Z is the partition function and β = 1/kBT. One can then readily derive after a little algebra
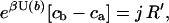 |
(10) |
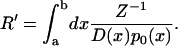 |
(11) |
The concentration of ions on the two sides of the channel is determined by the number of ions na, nb, and according to the Boltzmann distribution by the potential U(b) and U(a). Therefore it holds
 |
(12) |
Replacing accordingly ca, cb in Eq. 10, one obtains
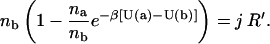 |
(13) |
Identifying log(na/nb) = ΔSab as the mixing entropy (Landau and Lifshitz, 1951) and defining ΔUab = U(a) − U(b), we can rewrite the last equation
 |
(14) |
We note that the exponent can be expressed in terms of the free energy difference ΔGab = ΔUab − kBT ΔSab, and in the case of ΔGab ≪ kBT, Eq. 14 can be written
 |
(15) |
where
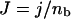 |
(16) |
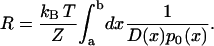 |
(17) |
Thereby, we have identified the resistance R of the channel, essentially expressed in terms of the equilibrium ion probability distribution p0(x). For an unspecific, rather wide channel, the unknown diffusion coefficient D(x) may be assumed to be constant and roughly independent of ion type, such that it matters much less in determining the ion conductivity. One can then assume that the resistance of an ion in an unspecific channel is governed by
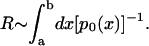 |
(18) |
References
- Anishkin, A., and S. Sukharev. 2004. Water dynamics and dewetting transitions in the small mechanosensitive channel MscS. Biophys. J. 86:2883–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash, W. L., M. R. Zlomislic, E. O. Oloo, and D. P. Tieleman. 2004. Computer simulations of membrane proteins. BBA Rev. Biomembr. In press. [DOI] [PubMed]
- Bass, R. B., P. Strop, M. Barclay, and D. C. Rees. 2002. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science. 298:1582–1587. [DOI] [PubMed] [Google Scholar]
- Beckstein, O., and M. S. P. Sansom. 2003. Liquid-vapor oscillations of water in hydrophobic nanopores. Proc. Natl. Acad. Sci. USA. 100:7063–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstein, O., and M. S. P. Sansom. 2004. The influence of geometry, surface character, and flexibility on the permeation of ions and water through biological pores. Phys. Biol. 1:42–52. [DOI] [PubMed] [Google Scholar]
- Berrier, C., A. Coulombe, C. Houssin, and A. Ghazi. 1989. A patch-clamp study of ion channels of inner and outer membranes and of contact zones of E. coli, fused into giant liposomes. FEBS Lett. 259:27–32. [DOI] [PubMed] [Google Scholar]
- Betanzos, M., C.-S. Chiang, H. R. Guy, and S. Sukharev. 2002. A large iris-like expansion of a mechanosensitive channel protein induced by membrane tension. Nat. Struct. Biol. 9:704–710. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F., and E. Perozo. 2002. Force and voltage sensors in one structure. Science. 298:1562–1563. [DOI] [PubMed] [Google Scholar]
- Biggin, P. C., and M. S. P. Sansom. 2002. Open-state models of a potassium channel. Biophys. J. 83:1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount, P., and P. C. Moe. 1999. Bacterial mechanosensitive channels: integrating physiology, structure and function. Trends Microbiol. 18:420–424. [DOI] [PubMed] [Google Scholar]
- Chang, G., R. H. Spencer, A. T. Lee, M. T. Barclay, and D. C. Rees. 1998. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- Colombo, G., S. J. Marrink, and A. E. Mark. 2003. Simulation of MscL gating in a bilayer under stress. Biophys. J. 84:2331–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, K., E. Jakobsson, and P. Wolynes. 2004. The theory of ion transport through membrane channels. Prog. Biophys. Mol. Biol. 46:51–96. [DOI] [PubMed] [Google Scholar]
- Cui, C., D. O. Smith, and J. Adler. 1995. Characterization of mechanosensitive channels in Escherichia coli cytoplasmic membrane by whole-cell patch clamp recording. J. Mol. Biol. 44:31–42. [DOI] [PubMed] [Google Scholar]
- Elmore, D. E., and D. A. Dougherty. 2003. Investigating lipid composition effects on the mechanosensitive channel of large conductance (MscL) using molecular dynamics simulations. Biophys. J. 85:1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, S. E., and R. W. Pastor. 1999. Constant surface tension simulations of lipid bilayers: the sensitivity of surface areas and compressibilities. J. Chem. Phys. 111:1281–1287. [Google Scholar]
- Grayson, P., E. Tajkhorshid, and K. Schulten. 2003. Mechanisms of selectivity in channels and enzymes studied with interactive molecular dynamics. Biophys. J. 85:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller, H., H. Heller, A. Windemuth, and K. Schulten. 1991. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Sim. 6:121–142. [Google Scholar]
- Gullingsrud, J., D. Kosztin, and K. Schulten. 2001. Structural determinants of MscL gating studied by molecular dynamics simulations. Biophys. J. 80:2074–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullingsrud, J., and K. Schulten. 2003. Gating of MscL studied by steered molecular dynamics. Biophys. J. 85:2087–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullingsrud, J., and K. Schulten. 2004. Lipid bilayer pressure profiles and mechanosensitive channel gating. Biophys. J. 86:3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O. P., and B. Martinac. 2001. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 81:685–740. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2003a. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003b. The principle of gating movement in a voltage-dependent K+ channel. Nature. 423:42–48. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- Kong, Y., Y. Shen, T. E. Warth, and J. Ma. 2002. Conformational pathways in the gating of Escherichia coli mechanosensitive channel. Proc. Natl. Acad. Sci. USA. 99:5999–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprowski, P., and A. Kubalski. 2003. C-termini of the Escherichia coli mechanosensitive ion channel (MscS) move apart upon the channel opening. J. Biol. Chem. 278:11237–11245. [DOI] [PubMed] [Google Scholar]
- Landau, L. D., and E. M. Lifshitz. 1951. Statistical Physics, 3rd Ed. Butterworth-Heinemann, Oxford, UK.
- Levina, N., S. Totemeyer, N. R. Stokes, P. Louis, M. A. Jones, and I. Booth. 1999. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. EMBO J. 18:1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, Jr., A. D., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998a. All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- MacKerell, Jr., A. D., B. Brooks, C. L. Brooks III, L. Nilsson, B. Roux, Y. Won, and M. Karplus. 1998b. CHARMM: the energy function and its parameterization with an overview of the program. In The Encyclopedia of Computational Chemistry. P. Schleyer et al., editors. John Wiley & Sons, Chichester, UK. 271–277.
- MacKerell, Jr., A. D., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1992. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB J. 6:A143–A143. [Google Scholar]
- Markin, V. S., and F. Sachs. 2004. Thermodynamics of mechanosensitivity. Phys. Biol. 1:110–124. [DOI] [PubMed] [Google Scholar]
- Martinac, B., M. Buechner, A. H. Delcour, J. Adler, and C. Kung. 1987. Pressure-sensitive ion channel in Escherichia coli. Proc. Natl. Acad. Sci. USA. 84:2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S., W. Bartlett, S. Chandrasekaran, S. Simpson, M. Edwards, and I. R. Booth. 2003a. Domain organization of the MscS mechanosensitive channel of Escherichia coli. EMBO J. 22:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S., M. D. Edwards, C. Ozdemir, and I. R. Booth. 2003b. The closed structure of the MscS mechanosensitive channel. J. Biol. Chem. 278:32246–32250. [DOI] [PubMed] [Google Scholar]
- Okada, K., P. C. Moe, and P. Blount. 2002. Functional design of bacterial mechanosensitive channels. J. Biol. Chem. 277:27682–27688. [DOI] [PubMed] [Google Scholar]
- Perozo, E., D. M. Cortes, P. Sompornpisut, A. Kloda, and B. Martinac. 2002a. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418:942–948. [DOI] [PubMed] [Google Scholar]
- Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002b. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- Perozo, E., and D. C. Rees. 2003. Structure and mechanism in prokaryotic mechanosensitive channels. Curr. Op. Struct. Biol. 13:432–442. [DOI] [PubMed] [Google Scholar]
- Risken, H. 1989. The Fokker-Planck Equation: Methods of Solution and Applications, 2nd Ed. Springer, New York.
- Roux, B. 2002. Theoretical and computational models of ion channels. Curr. Op. Struct. Biol. 12:182–189. [DOI] [PubMed] [Google Scholar]
- Roux, B., T. Allen, S. Berneche, and W. Im. 2004. Theoretical and computational models of biological ion channels. Quar. Rev. Biophys. 37:15–103. [DOI] [PubMed] [Google Scholar]
- Roux, B., and K. Schulten. 2004. Computational studies of membrane channels. Structure. 12:1343–1351. [DOI] [PubMed] [Google Scholar]
- Schlenkrich, M., J. Brickmann, A. D. MacKerell, Jr., and M. Karplus. 1996. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhauser, Boston, MA. 31–81.
- Schlick, T., R. Skeel, A. Brünger, L. Kalé, J. A. Board, Jr., J. Hermans, and K. Schulten. 1999. Algorithmic challenges in computational molecular biophysics. J. Comput. Phys. 151:9–48. [Google Scholar]
- Smart, O., J. Goodfellow, and B. Wallace. 1993. The pore dimensions of gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart, O. S., J. Breed, G. R. Smith, and M. S. P. Sansom. 1997. A novel method for structure-based prediction of ion channel conductance properties. Biophys. J. 72:1109–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, J., J. Gullingsrud, P. Grayson, and K. Schulten. 2001. A system for interactive molecular dynamics simulation. In 2001 ACM Symposium on Interactive 3D Graphics. J.F. Hughes and C.H. Séquin, editors. New ACM SIGGRAPH, New York. 191–194.
- Sukharev, S. 2002. Purification of the small mechanosensitive channel of Escherichia coli (MscS): the subunit structure, conduction, and gating characteristics in liposomes. Biophys. J. 83:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukharev, S., M. Betanzos, C.-S. Chiang, and H. R. Guy. 2001. The gating mechanism of the large mechanosensitive channel MscL. Nature. 409:720–724. [DOI] [PubMed] [Google Scholar]
- Sukharev, S., and D. Corey. 2004. Mechanosensitive channels: multiplicity of families and gating paradigms. Sci. STKE. 219:re4. [DOI] [PubMed]
- Sukharev, S. I., P. Blount, B. Martinac, F. R. Blattner, and C. Kung. 1994. A large-conductance mechanosensitive channel in E. coli encoded by MscL alone. Nature. 368:265–268. [DOI] [PubMed] [Google Scholar]
- Weiner, P. K., and P. A. Kollman. 1981. AMBER: assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 2:287–303. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.