

Abstract
Structural information for mammalian DNA pol-β combined with molecular and essential dynamics studies have provided atomistically detailed views of functionally important conformational rearrangements that occur during DNA repair and replication. This conformational closing before the chemical reaction is explored in this work as a function of the bound substrate. Anchors for our study are available in crystallographic structures of the DNA pol-β in “open” (polymerase bound to gapped DNA) and “closed” (polymerase bound to gapped DNA and substrate, dCTP) forms; these different states have long been used to deduce that a large-scale conformational change may help the polymerase choose the correct nucleotide, and hence monitor DNA synthesis fidelity, through an “induced-fit” mechanism. However, the existence of open states with bound substrate and closed states without substrates suggest that substrate-induced conformational closing may be more subtle. Our dynamics simulations of two pol-β/DNA systems (with/without substrates at the active site) reveal the large-scale closing motions of the thumb and 8-kDa subdomains in the presence of the correct substrate—leading to nearly perfect rearrangement of residues in the active site for the subsequent chemical step of nucleotidyl transfer—in contrast to an opening trend when the substrate is absent, leading to complete disassembly of the active site residues. These studies thus provide in silico evidence for the substrate-induced conformational rearrangements, as widely assumed based on a variety of crystallographic open and closed complexes. Further details gleaned from essential dynamics analyses clarify functionally relevant global motions of the polymerase-β/DNA complex as required to prepare the system for the chemical reaction of nucleotide extension.
INTRODUCTION
The genome template in every cell of the human body is continuously being contaminated by environmental chemicals, harmful radiation, and reactive oxygen species (Ames et al., 1990; Lindhal and Wood, 1999). Cells have therefore devised various efficient ways to repair the DNA damage to help maintain genome integrity. The base excision repair pathway is used by cells to repair damage due to oxidation, alkylation, and ultraviolet radiation (Seeberg et al., 1995; Wilson, 1998). DNA pol-β is the enzyme in this repair pathway that fills small nucleotide gaps with relatively high accuracy (“fidelity”). It selects from the pool of four (dATP, dCTP, dGTP, and dTTP) the single dNTP (2′-deoxyribonucleoside 5′-triphosphate) complementary to the template residue. The base substitution error frequency for pol-β is one per 103 bases, not including extrinsic correction machinery (Wilson, 1998).
Pol-β has been crystallized (Sawaya et al., 1997) in complexes representing three intermediates of the opening/closing transition: “open binary complex”, containing pol-β bound to a DNA substrate with single nucleotide gap; “closed ternary complex”, containing pol-β·gap·ddCTP (pol-β bound to gapped DNA as well as 2′,3′-dideoxyribocytidine 5′-triphosphate (ddCTP)); and “open binary product complex”, pol-β·nick (pol-β bound to nicked DNA). Fig. 1, b and c, illustrate the significant conformational difference between these open and closed forms. Structurally, pol-β is composed of only two domains, an N-terminal 8-kDa region that exhibits deoxyribosephosphate lyase activity, and a 31-kDa C-terminal domain that possesses nucleotidyl transfer activity. The 31-kDa domain resembles all structurally characterized polymerases to date, containing finger, palm, and thumb subdomains (Joyce and Steitz, 1994). Studies on pol-β, therefore, can serve as a model for other DNA polymerases. The relatively small size of pol-β (335 protein residues and 16 DNA basepairs) also renders it attractive for computational studies.
FIGURE 1.

General pathway for nucleotide insertion by DNA pol-β (a) and corresponding crystal open (b) and closed (c) conformations of pol-β/DNA complex. E, DNA polymerase; dNTP, 2′-deoxyribonucleoside 5′-triphosphate; PPi, pyrophosphate; DNAn/DNAn+1, DNA before/after nucleotide incorporation to DNA primer. T6 is the template residue (G) corresponding to the incoming dCTP.
Based on kinetic and structural studies of several polymerases (Ahn et al., 1997, 1998; Vande Berg et al., 2001; Shah et al., 2001; Suo and Johnson, 1998; Kraynov et al., 1997; Zhong et al., 1997; Dahlberg and Benkovic, 1991; Kuchta et al., 1987; Wong et al., 1991; Patel et al., 1991; Frey et al., 1995; Capson et al., 1992) complexed with primer/template DNA, a common nucleotide insertion pathway has been characterized for some polymerases (e.g., HIV-1 reverse transcriptase, phage T4 DNA polymerase, Escherichia coli DNA polymerase I Klenow fragment) that undergo transitions between open and closed forms, like pol-β (Fig. 1 a). At step 1, after DNA binding, the DNA polymerase incorporates a 2′-deoxyribonucleoside 5′-triphosphate (dNTP) to form an open substrate complex; this complex is assumed to undergo conformational change to align catalytic groups and form a closed ternary complex at step 2; the nucleotidyl transfer reaction then follows: 3′-OH of the primer strand attacks the Pα of the dNTP to extend the primer strand and form the ternary product complex (step 3); this complex then likely undergoes a reverse conformational change (step 4) back to the open enzyme form. This transition is followed by dissociation of pyrophosphate (PPi) (step 5), after which the DNA synthesis/repair cycle can begin anew.
The conformational rearrangements involved in steps 2 and 4 are believed to be key for monitoring DNA synthesis fidelity (Sawaya et al., 1997). Binding of the correct nucleotide is thought to induce the first conformational change (step 2) whereas binding of an incorrect nucleotide may alter or inhibit the conformational transition. This “induced-fit” mechanism was thus proposed to explain the polymerase fidelity in selection of correct dNTP (Sawaya et al., 1997). This mechanism suggests that the conformational changes triggered by binding of the correct nucleotide will align the catalytic groups as needed for catalysis, whereas the incorrect substrate will interfere with this process. However, no direct experiments have shown this effectively.
Although crystal structures of many DNA polymerases have revealed either open binary conformations without dNTP or closed ternary conformations with dNTP, there are notable exceptions. For example, addition of a dNTP to a crystal of an open binary DNA complex of Klentaq (A-family) results in an open ternary complex (Li et al., 1998); open rather than closed complexes, as expected, were also observed in a ternary pol-β R283A mutant (Beard et al., 1996). Conversely, closed rather than open complexes were captured in crystals of B-family polymerases without substrate (Rodriguez et al., 2000) and binary complexes of DNA pol-λ (Garcia-Diaz et al., 1998). Furthermore, subdomain conformations of Bacillus Pol I (A-family) are known to be sensitive to specific crystal lattice contacts (Johnson et al., 2003). These variations in outcomes as a function of the active site context are likely because subdomain rearrangements are subject to potential distortions because of crystal packing forces (Arnold et al., 1995), crystallization conditions (e.g., necessity for extra moeity to stall chemical reaction), or factors other than presence/absence of substrate. Thus, dynamics simulations can be useful to pinpoint the nature of conformational changes in pol-β and their relation to the presence or absence of a substrate in the active site.
Broadly speaking, there is great interest in understanding polymerase mechanisms because of the importance of DNA polymerases in maintaining genome integrity and the relation of their malfunctioning to many diseases (e.g., cancer, premature aging). Still, detailed mechanisms of the conformational changes induced by binding of the correct dNTP and how these cycles differ for polymerases in diverse classes remain unknown. In this work, we analyze dynamics simulations to delineate the structural and dynamical changes that occur during the conformational change before the nucleotidyl transfer reaction (step 2, Fig. 1 a), when the enzyme active site is occupied by the correct substrate on one hand, or no substrate at all. (Modeling studies of incorrect substrates have been reported by Florián et al., 2003 and Yang et al., 2002b and are under study by R. Radhakrishnan and T. Schlick, unpublished data). We seek here to identify the rate-limiting conformational motions and mechanistic details associated with specific residues in the active site of pol-β to complement structural and kinetic data. Our use of principal component analysis (also called essential dynamics) aids visualization and quantification of the conformational changes that are difficult to recognize by visual inspection of dynamics trajectories because of the rapid thermal motions of atoms and voluminous number of conformations.
Significantly, we observe contrasting global subdomain motions as well as alignments of the catalytic residues in the presence versus absence of substrate in the pol-β binding site. The correct incoming substrate triggers a thumb subdomain “closing” motion with the alignment of the active site protein residues that favors the subsequent chemical step. The absence of the substrate leads to a thumb “opening” motion and is accompanied by local rearrangements near the active site that disfavor the chemical step. These results highlight the delicate and sophisticated network of global and local conformational changes that play an important role in recognizing the incoming substrate and hence polymerase fidelity.
COMPUTATIONAL METHODOLOGY
Model building
Two initial models, pol-β/DNA with substrate and without substrate, in an intermediate form between crystal open (1BPX) and closed (1BPY) states before nucleotidyl transfer reaction (prechemistry) were constructed. Such intermediate states are intended to hasten the capturing of subdomain motions: all simulations from the crystal open state (1BPX) with substrate and crystal closed state (1BPY) without substrate do not reveal any significant global motion within 5 ns (K. Arora, unpublished data). Indeed, the intermediate structure of pol-β has been successful in capturing the polymerase opening (after the chemical reaction of nucleotidyl transfer) in the polymerase kinetic cycle (Fig. 1 a) (Yang et al., 2002a,b); the intermediate forms used in our studies also resemble a crystal intermediate (J. M. Krahn, W. A. Beard, and S. H. Wilson, unpublished data) and are found on the closing pathway delineated by the stochastic pathway approach (K. Arora and T. Schlick, unpublished data). Here we investigate the enzyme dynamics before the chemical reaction of nucleotidyl transfer (prechemistry) in the presence of the substrate and without substrate in the active site to test the proposed “induced-fit” hypothesis (Li et al., 1998; Doublié and Ellenberger, 1998; Kiefer et al., 1998; Koshland, 1994; Beard and Wilson, 1998; Doublié et al., 1999).
For the first model with substrate, the intermediate pol-β complexed with DNA primer/template was constructed as an average of the crystallographic open, binary gapped complex (1BPX) and closed, ternary complex (1BPY) from the PBD/RCSB resource (Berman et al., 2000). The incoming nucleotide (dCTP) along with catalytic and nucleotide binding Mg2+ ions is placed in the active site as in the ternary crystal closed complex. The hydroxyl group was added to the 3′ terminus of the primer DNA strand. The missing protein residues 1–9 in the crystal structure were placed by using the INSIGHT II package, version 2000. CHARMM's subroutine HBUILD (Brünger and Karplus, 1988) was employed to add all hydrogen atoms to the crystallographic heavy atoms. The second model of the intermediate pol-β without substrate was constructed similarly, but without the incoming basepair (dCTP), catalytic magnesium ion, and nucleotide binding Mg2+ ion in the active site. In the intermediate state, the thumb subdomain is partially closed (Fig. 2 a1), i.e., the thumb root-mean-square deviation (RMSD) is 2.5 Å compared to crystal open and 3.5 Å compared to crystal closed complex. Cubic periodic domains for both initial models were constructed using our program PBCAID (Qian et al., 2001). To neutralize the system at an ionic strength of 150 mM, water molecules (TIP3 model) with minimal electrostatic potential at the oxygen atoms were replaced by Na+, and those with maximal electrostatic potential were replaced with Cl−. All Na+ and Cl− ions were placed >8 Å away from any protein or DNA atom and from each other. The electrostatic potential for all bulk oxygen atoms was calculated using the DelPhi package (Klapper et al., 1986). In total, both neutral systems consist of ∼40,000 atoms including water of solvation (11,249 water molecules).
FIGURE 2.
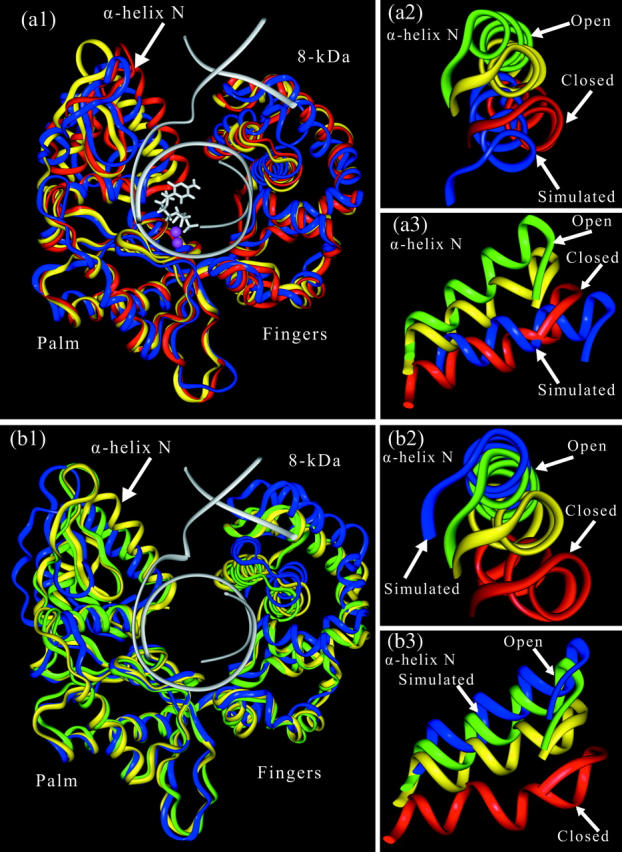
Cα traces of superimposed pol-β/DNA complex with dCTP (a1) and without dCTP (b1) for the intermediate starting structure (yellow), crystal closed (red), and crystal open (green) and the trajectory final structures (blue). Notable are the residue motions in the thumb subdomain and the 8-kDa domain. The positions of α-helix N in the simulated systems are compared to the crystal structures and shown from two points of view in pabels a2 and a3, and panels b2 and b3.
Minimization, equilibration, and dynamics protocol
Energy minimizations, equilibrations, and dynamics simulations for both systems were performed using the program CHARMM (Chemistry Department, Harvard University, Cambridge, MA) (Brooks et al., 1983) with the all-atom version C26a2 force field (MacKerell and Banavali, 2000). First, each system was minimized with fixed positions of all protein and DNA heavy atoms using the method of steepest descent for 10,000 steps; this is followed by adapted basis Newton-Raphson minimization (Brooks et al., 1983; Schlick, 1992) for 20,000 steps. The 30 ps of equilibriation at 300 K was followed by further minimization using steepest descent and adapted basis Newton-Raphson until the gradient of RMSD was 10−6 kcal/mol Å. Finally, the protein/DNA/counterions/water coordinates were reequilibrated for 30 ps at 300 K by the Langevin multiple time-step method LN (See Yang et al., 2002a) for a thorough examination of the stability and reliability of the integrator for large macromolecular systems in terms of thermodynamic, structural, and dynamic properties compared to single-time-step Langevin as well as Newtonian (Velocity Verlet) propagators (Yang et al., 2002a). Our triple-time-step protocol uses an inner time step Δτ = 1 fs for updating local bonded interactions; medium time step Δtm = 2 fs to update nonbonded interactions within 7 Å; and outer time step of Δt = 120 fs for calculating remaining terms; this yields a speed-up factor of 4 over single-time-step Langevin dynamics at Δτ = 1 fs. The SHAKE algorithm was employed to constrain the bonds involving hydrogen atoms. Electrostatic and van der Waals interactions were smoothed to zero at 12 Å with a shift function and a switch function, respectively. The Langevin collision parameter of γ = 10 ps−1 was chosen to couple the system to a 300°C heat bath. The total simulation length for each system is ∼10 ns.
Geometric characteristics
The radius of gyration, Rg, and root-mean-square deviations of the Cα atomic positions in the dynamic trajectories with respect to the crystal, closed ternary complex and the open binary gapped complex, drms, were monitored as a function of time. The radius of gyration is defined as:
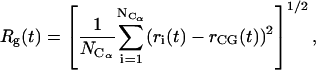 |
(1) |
where ri(t) and rCG(t) are the coordinates of atom i and of the center of gravity of the polymerase, respectively, at time t. 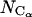 is the total number of the Cα atoms involved. The RMSD is defined as:
is the total number of the Cα atoms involved. The RMSD is defined as:
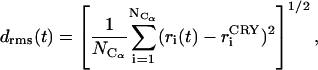 |
(2) |
where  is the coordinate of atom i in the crystal structure, after a least-square fit superimposition with dynamic structures at time t.
is the coordinate of atom i in the crystal structure, after a least-square fit superimposition with dynamic structures at time t.
The RMSD measure is not always effective in describing the trends in domain motions because different direction of motion can be realized. Therefore, we have devised a scheme to represent the RMSD data more effectively such that both the magnitude and direction of domain motions can be appreciated. Namely, we project the RMSD of the simulated structure on the line joining the geometric centers of α-helix N in the crystal open and closed conformation (see Fig. S1 in online Supplementary Material); the crystal open and closed conformations form two vertices of the triangle, and a simulated structure forms the third. The length of each side of the triangle is given by the RMSD of the thumb's α-helix N when the two structures at the end of the line are superimposed with respect to pol-β's palm subdomain. The α-helix N RMSD between crystal open and closed conformation is fixed (6.96 Å). The shift distance (h) describes the displacement of the α-helix N of the simulated structure in the direction perpendicular to the line joining the geometric centers of α-helix N in the crystal open and closed conformation. When h is constant, the RMSD values alone are good indicators of the motion toward one of the crystal states; when h is not constant, RMSDs may be misleading. The variables L1 and L2 thus can describe the projected RMSD of the simulated structure with respect to the crystal open and closed conformations, respectively.
Motion analysis by essential dynamics
To better analyze the global motions of pol-β, the dynamics trajectories were analyzed according to the principal component (PCA) or essential dynamics (ED) method (Amadei et al., 1993). This method aims to describe the overall dynamics of systems with a few collective, “essential” degrees of freedom in which anharmonic motion occurs. These motions comprise most of the positional fluctuations and are often functionally relevant. The remaining degrees of freedom represent not necessarily independent harmonic motions orthogonal to “essential” subspace that collectively describe much smaller positional fluctuations. The large number of biological systems that have been studied by this approach indicates its utility for analysis of domain motions (van Aalten et al., 1995, 1997; deGroot et al., 1996, 2001; Weber et al., 1998; Stella et al., 1999; Arcangeli et al., 2000a,b, 2001). To describe their collective motions, the covariance matrix C of atomic fluctuations along the dynamics trajectory (Amadei et al., 1993) is constructed. The matrix elements are given by
 |
(3) |
where Xk is the coordinate vector at kth configuration (snapshot), and 〈X〉 is the average structure over the entire dynamics simulation:  Diagonalization of covariance matrix produces the eigenvectors and eigenvalues as entries of Λ from the spectral decomposition:
Diagonalization of covariance matrix produces the eigenvectors and eigenvalues as entries of Λ from the spectral decomposition:
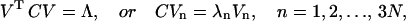 |
(4) |
where Λ is the diagonal matrix with eigenvalues λi: Λ = diag(λ1, λ2, …, λ3N). Each eigenvector Vn defines the direction of motion of N atoms as an oscillation about the average structure 〈X〉. The normalized magnitude of corresponding eigenvalue is a measure of the amplitudes of motion along the eigenvector Vn. If eigenvalues are arranged in order of decreasing value, the first few describe the largest positional fluctuations. Here we have applied this analysis only to Cα atoms of the polymerase primarily to produce a manageable (diagonalizable) covariance matrix. This is reasonable because it has been systematically shown that there is a great similarity between the motions along the first few eigenvectors of Cα matrix and those along the first few eigenvectors derived from the all-atom matrix (Amadei et al., 1993). Of course, the raw data (configuration series) come from dynamics trajectories that allow all degrees of freedom to vary. A total of 335 atoms were included in the mode analysis, resulting in 1005 PCs (3 × 335). Snapshots were sampled from 10 ns of each trajectory at frequency of Δt = 100 ps.
RESULTS AND DISCUSSION
Polymerase-β/DNA complex with bound correct incoming substrate (dCTP)
Subdomain motions
From the intermediate structure between the crystal open and closed conformations, we followed the motion of the polymerase subdomains over the 10-ns trajectory. The thumb moves toward the crystal closed conformation, although the structure after 10 ns is not superimposable with the crystal conformation (see Fig. 2 a1). The motion of the thumb subdomain is strikingly visible from the position of α-helix N in the thumb subdomain. Fig. 2, a2 and a3, highlights the closing motion and the final position of the α-helix N of the thumb subdomain by the end of the 10-ns trajectory. A closing motion away from the open conformation is evident. To assess the extent of motion quantitatively, we monitored the root-mean-square deviation of the α-helix N Cα atoms throughout the trajectory with respect to the starting and crystal structures, and the radius of gyration, as shown in supplemental Fig. S3 a and in Fig. 4 a. The RMSD with respect to the starting structure suggests a movement >4 Å for α-helix N. However, the total RMSD of α-helix N with respect to the crystal closed conformation shows an unexpected increasing trend. This is because although the thumb moves toward the closed conformation as is clear from Fig. 2, it does so at some angle to the closed conformation. Our modified scheme dissecting h, L1, and L2 allows us to quantify this axis shift h and to represent global motions more effectively (Fig. 3 a). This modified scheme is important because the shift h rises to ∼7 Å in the simulation with substrate (see supplemental Fig. S2).
FIGURE 4.

Radius of gyration (Rg) for all Cα atoms (a) simulated closing of pol-β with substrate (b) simulated opening of pol-β without substrate in the binding site.
FIGURE 3.

Evolution of the root-mean-square deviations (RMSD) of the Cα residues in α-helix N of the thumb subdomain in the simulated structure with respect to the crystal open (green) and crystal closed structures (red); (a) simulated closing of pol-β with substrate and (b) simulated opening of pol-β without substrate in the binding site after removing the shift distances.
Motion of key residues in the active site
The thumb motion is accompanied by conformational changes of key protein residues in the substrate (dCTP) binding site. According to the current induced-fit hypothesis, the large conformational closing of the thumb directs the system to the subsequent step of nucleotidyl transfer along the polymerase kinetic pathway. From structural analyses of pol-β in the crystal open and closed conformations, several key residues have been implicated to act in concert with the thumb's movement (Sawaya et al., 1997). In our simulation with substrate, we follow the dynamics of these key protein residues in addition to large global conformational changes. Essentially, we observe the rotation of Asp-192 to ligand with the catalytic Mg2+, rotation of Arg-258 to bond with Tyr-296, and the flip of Phe-272 that positions itself between Arg-258 and Asp-192 to prevent the bond reformation. These local conformational changes in the substrate binding site can be visualized by superimposing the pol-β structure after 10 ns with the crystal closed and starting structures (Fig. 5 a). These residues in the simulated structure overlap well with the crystal closed form, suggesting the approach of a stable closed conformation. In Fig. S4 (see Supplementary Material), the time evolution of crucial interatomic distances between key residues shows the bond breaking between Arg-258 and Asp-192 and bond formation between Arg-258 and Tyr-296 as discussed above.
FIGURE 5.

Positions of key residues Tyr-296, Arg-258, Asp-192, and Phe-272 in the 10-ns simulated (blue), crystal closed (red), crystal open (green), and starting intermediate (yellow) structures; (a) trajectory with substrate and (b) without substrate.
Interestingly, we observe the rotation of Arg-258 in our 10-ns trajectories. Because the full Arg-258 rotation has been suggested to be a slow conformational change—from studies of pol-β's opening after chemistry (Yang et al., 2002a) and transition path sampling before chemistry (Radhakrishnan and Schlick, 2004)—Arg-258's barrier may exist before reaching our intermediate structure. Indeed in that starting structure, Arg-258 was already significantly rotated toward the final closed conformation as in the ternary closed crystal complex. Further experiments and computational studies are underway to assess the barrier region associated with Arg-258 rotation.
Nucleotidyl transfer geometry and the magnesium coordination sphere
According to the general kinetic pathway of pol-β, the nucleotidyl transfer reaction that follows subdomain closing involves the attack on the Pα of the substrate by 3′-hydroxy of the primer, extending the DNA primer strand by one base. This reaction is favored when the Pα distance is 3 Å from the 3′-hydroxy of the primer. The 3′-OH is absent in the closed crystal complex; we thus modeled it based on coordinates of C3′ in the starting structure. In the initial modeled structure, the Pα–O3′ distance was close to 3 Å; this distance evolves after 10 ns to ∼4.2 Å. This value is larger than required for the chemical reaction (see Fig. 6) and therefore we further examined this behavior. Our 1-ns control simulation from the crystal closed complex, resulted in a somewhat larger Pα–O3′ distance as well (K. Arora, unpublished data). Thus, we believe that the larger Pα–O3′ may reflect the force-field parameters and/or the fact that the nucleotidyl transfer step has a higher energy barrier and is rate limiting compared to the conformational change. The latter is supported by the suggested alternate mechanism for fidelity, where the rate-limiting step for nucleotide transfer is determined by stabilization of transition state during chemical step (Showalter and Tsai, 2002). To determine whether conformational or chemical steps are rate limiting, mixed quantum and molecular mechanics simulations are needed (e.g., R. Radhakrishnan and T. Schlick, unpublished data).
FIGURE 6.

Coordination sphere of catalytic (Mg2+) and nucleotide (Mg2+) binding magnesium ions in the pol-β/DNA complex with bound dCTP substrate after 10 ns. All the distances within 2 Å are depicted by white dotted lines. WAT2 is the crystallographically observed water. The dCTP:Pα-P10:O3′ distance crucial for nucleotidyl transfer reaction is shown in a green dotted line.
The magnesium ions are required for the nucleotidyl transfer reaction according to the two-metal-ion catalytic mechanism (Beese and Steitz, 1991). Therefore, we monitored the coordination and motion of both Mg2+ (catalytic and substrate binding) ions throughout the simulation. The observed distances between the Mg2+ ions and the ligands are listed in Table 1 and Fig. 6. We find that the coordination sphere of two Mg2+ ions is close to that observed in the crystal structure with a few noted exceptions. Specifically, the O1A oxygen atom from the substrates Pα, a bridging ligand coordinated to both magnesium ions in the crystal structure, is coordinated only to the catalytic magnesium. Lacking the coordination with the O1A oxygen atom, the nucleotide binding ion coordinates with a water molecule (WAT3 in Fig. 6) not observed in the crystal structure. We also find the nucleotide binding magnesium to coordinate with two water molecules instead of one as observed in the crystal structure. Interestingly one water molecule (WAT1) becomes bound to a catalytic magnesium ion (see Fig. 6), a possibility predicted by crystallographic structures (Sawaya et al., 1997). We also note the larger distance between the catalytic ion and the modeled O3′, not present in the crystal structure; this O3′ might be expected to coordinate with the catalytic ion to complete the coordination sphere (see Fig. 6). These details may reflect imperfections of the force field, which is not parameterized to account for ligand to metal charge transfer and polarization effects. Still, the studies here shed light on the nature of subtle local conformational changes in the catalytic region on the substrate binding, like the rotations of Asp-192 and Asp-256, with all conserved aspartates (190, 192, 256) tightly coordinating the Mg2+ ions. The other interatomic distances between substrate and magnesium ions crucial for catalysis are maintained throughout the simulation and are close to those observed in crystal closed conformation (see Table 1).
TABLE 1.
Active site interatomic distances (in Å) for the simulated pol-β with substrate and closed crystal form (1BPY)
| Distance | 1BPY (x-ray) | Pol-β/DNA/dCTP (10 ns) |
|---|---|---|
| Mg2+(A)-Asp-190:OD2 | 2.2 | 1.78 |
| Mg2+(A)-Asp-192:OD1 | 2.2 | 1.84 |
| Mg2+(A)-Asp-256:OD2 | 2.6 | 1.83 |
| Mg2+(A)-dCTP:O1A | 1.9 | 1.84 |
| Mg2+(A)-WAT1 | n/a | 2.11 |
| Mg2+(B)-Asp-190:OD1 | 2.7 | 1.78 |
| Mg2+(B)-Asp-192:OD2 | 2.0 | 1.80 |
| Mg2+(B)-dCTP:O2B | 2.1 | 1.83 |
| Mg2+(B)-dCTP:O2G | 2.0 | 1.89 |
| Mg2+(B)-WAT2 | 2.0 | 2.02 |
| Mg2+(B)-WAT3 | n/a | 2.00 |
| P10:O3′-dCTP:Pα | n/a | 4.20 |
Mg2+(A), catalytic magnesium; Mg2+(B), nucleotide binding magnesium; dCTP, 2′ -deoxyadenosine 5′ -triphosphate; P10, primer nucleotide; n/a, absent in the crystal structure.
Polymerase-β/DNA complex without substrate
To test the “induced-fit” mechanism, we performed an analogous simulation of our enzyme/DNA system without the dCTP substrate and the Mg2+ ions in the binding site. If the induced-fit mechanism were to hold, we expect an opening motion of the thumb subdomain to occur rather than the closing thought to be triggered by the substrate in the binding site.
Subdomain motions
The superimposition of the pol-β structure after 10 ns with the crystal open and starting intermediate structures (superimposed according to Cα atoms in the palm subdomain) depicts the large thumb subdomain movement toward the crystal open form (see Fig. 2 b1). Also shown in the Fig. 2, b2 and b3, is the α-helix N position compared to the starting, crystal open and closed structures. The overall opening trend is visible. The α-helix N in the simulated state is close to the crystal open conformation (RMSD = 1.4 Å). The time evolution of the thumb subdomain motion and overall dynamics of polymerase was followed by computing the RMSD and radius of gyration (Rg), throughout the simulation (see Figs. 3 b and 4 b). Again, the overall opening motion of the enzyme and the significant motion of the α-helix N are evident. In contrast to the closing motion, the shift h of the α-helix N is almost constant and small in this case (1–2 Å), and thus the total RMSD is a good measure of approach to the open state (supplemental Fig. S2). This observation of overall opening supports the induced-fit mechanism for maintaining polymerase fidelity.
We also note the relaxation of the thumb in either the closed or open conformation depending on whether there is substrate or no substrate in the polymerase binding site, within a few nanoseconds. This observation ties well with the recent experimental studies that suggest that subdomain motions are relatively fast and occur on the timescale of the order of a few nanoseconds (Vande Berg et al., 2001; Kim et al., 2003). However, any estimate of the timescale warrants caution because our starting point is an intermediate structure designed to accelerate the conformational change event. The complete time of subdomain motions is likely slower than a few nanoseconds.
Motion of key active site residues
In sharp contrast to the pol-β simulation with substrate in the binding site, the 10-ns trajectory of pol-β without dCTP and Mg2+ ions in the substrate binding site reveals that residues in the active site flip or rotate to a conformation that resembles the open crystal structure. We observe the flip of Asp-192, rotation of Arg-258 toward Asp-192 to engage in forming salt bridge, breaking of the Tyr-296 and Arg-258 bond, and the flip of Phe-272 to allow for the rotation of Arg-258. These changes are easily visualized by superimposition of the simulated structure after 10 ns with the crystal open and the intermediate starting structure (see Fig. 5 b). Crucial interatomic distances between these key residues are also shown in supplemental Fig. S4.
To clearly show the final position of the α-helix N with respect to the crystal open and closed conformation, the time evolution of the shift distance (h) (see supplemental Fig. S1) was plotted for both trajectories of pol-β with and without substrate (see supplemental Fig. S2). For the trajectory with substrate in the active site, the shift distance (h) increases after 3 ns, suggesting the drift of α-helix N away from the line joining the geometric centers of α-helix N in the crystal open and closed structures. Although the α-helix N moves toward the closed conformation, it does so at some angle. On the other hand, the shift distance remains almost constant for the opening motion without substrate suggesting better overlap of the simulated structure with the crystal open form. Beside these subdomain motions, approach to open or closed conformations, gains support by the associated rearrangement of local side chains in the pol-β active site, motions crucial for subsequent catalysis.
The results here highlight how the presence of the correct substrate or the absence of substrate in the pol-β binding site invokes contrasting response of protein residues in the microenvironment of the binding site. The binding site is thus extremely sensitive to the presence of the substrate. The thumb subdomain of the polymerase also shows the contrasting motions: closing in the presence of the substrate and opening without substrate, although not perfectly superimposable with the crystal conformation because of limited sampling. The two Mg2+ ions (catalytic and nucleotide binding) are also crucial for maintaining the tight active site geometry. The importance of Mg2+ in maintaining polymerase fidelity has been highlighted by several experimental studies like Beese and Steitz (1991) and Steitz (1993). Although here we modeled catalytic Mg2+ and nucleotide binding Mg2+ in the polymerase active site as in crystal closed form, we cannot distinguish their individual roles on polymerase dynamics; this is addressed elsewhere (Yang et al., 2004).
ESSENTIAL DYNAMICS ANALYSIS
Essential dynamics analysis was performed on the two dynamics trajectories analyzed above (pol-β/DNA with and without substrate) to separate large-scale correlated motions from small-scale random harmonic vibrations. Note that although principal component analysis is not appropriate for predicting long-time dynamics of proteins (Balsera et al., 1996; Clarage et al., 1995) due to sampling errors, it is reasonable when large-scale motions are observed during the simulation length (Balsera et al., 1996). The two covariance matrices were diagonalized as detailed under methodology, and the resulting eigenvalues for the first 50 modes are shown in supplemental Fig. S5. There are only a few dominant eigenvectors with large eigenvalues, suggesting that the essential subspace that encapsulates the large-scale protein motions is small relative to the total number of degrees of freedom. Specifically, the top 10 eigenvalues describe 80% of the overall motion magnitude for both systems. Figs. 7, a–c, and 8, a–c, illustrate the contribution of each Cα atom to the motion along the top five principal component (PCs), trajectories along the PC axes, and the probability distribution of fluctuations from corresponding PCs for our two DNA/pol-β systems.
FIGURE 7.

Results of the essential dynamics analysis of pol-β with substrate. (a) Contribution of each Cα atom to the motions along the first five normalized eigenvectors. (b) Time evolution of projection of these eigenvectors on the dynamics trajectory. (c) Corresponding probability distributions together with fitted Gaussian distributions.
FIGURE 8.

Results of the essential dynamics analysis of pol-β without substrate. (a) Contribution of each Cα atom to the motions along the first five normalized eigenvectors. (b) Time evolution of projection of these eigenvectors on the dynamics trajectory. (c) Corresponding probability distributions together with fitted Gaussian distributions.
The projections of the trajectory amplitudes on the first few eigenvectors for both systems suggest that a significant component of the motion in the protein is concentrated in the thumb (residues 252–335) and 8-kDa (residues 1–90) regions. The thumb motion is particularly evident in PCs 1 and 2 with substrate and PCs 1 and 3 without substrate. Besides the thumb and 8-kDa regions, there is persistent motion in both cases in three loop regions corresponding to residue numbers 242–250, 200–210, and 302–310 (see PC 2). PCs 3, 4, and 5 single out motions pertaining exclusively to the loop of residues 300–310 in the thumb subdomain of pol-β with substrate. From the probability distributions of displacements along the corresponding eigenvectors, we see that PC 1 corresponds to slow global motions with probability distributions far from Gaussian (Figs. 7 c and 8 c). PC 1 of pol-β with substrate has three distinct peaks, suggesting structural transitions among different conformational states during closing; this suggests that closure occurs via a series of metastable states separated by low energy barriers, corroborating transition path sampling studies of the system (Radhakrishnan and Schlick, 2004). The other PCs represent oscillations and lower amplitude motions. The comparison between the sampled and ideal Gaussian distributions yields a correlation coefficient, which can be plotted as the function of the eigenvector number as shown in supplemental Fig. S6. The correlation coefficients between the actual distribution (by projecting the eigenvectors on the trajectories) and fitted Gaussians is between 0.46 and 0.99. Beyond PC 3, correlation coefficients are higher than 0.95, indicating a reduced dimension essential space in which the most functionally relevant motions of protein reside.
Specific motions corresponding to PCs can also be visualized by projecting all trajectory frames onto a specific eigenvector and the new trajectory; a visual inspection reveals the motion in the direction defined by the eigenvector. A rendering of the motion along the first three eigenvectors for both cases is shown in Fig. 9, a and b. Motions are evident along the thumb and 8-kDa domain regions, as well as solvent-exposed loops of the protein: L1 (residues 200–210), L2 (residues 242–250), and L3 (residues 302–310). The palm and finger subdomains are almost superimposable and show the least movement. Subtle differences also exist between the two cases. The thumb subdomain explores different conformational spaces in the case with and without substrate (closing versus opening trends), as evident from PC 2 and other PCs. Another striking difference is the occurrence of larger 8-kDa domain motion when the substrate is present in the binding site (especially PC 1); this larger 8-kDa motion is correlated with the thumb subdomain closing movement. Protein residues in the thumb and 8-kDa domains come to spatial proximity and engage in bond formation that finally locks the molecule in the closed conformation. When the substrate is absent, the thumb undergoes an opening motion to move farther away from the 8-kDa domain, thus invoking smaller movements of 8-kDa domain.
FIGURE 9.

Twenty-five frames taken at equally spaced intervals from the motions along the first three eigenvectors of (a) pol-β/DNA with dCTP and (b) pol-β/DNA without substrate in the binding site. Frames correspond to displacements between the minimum and maximum displacement corresponding to eigenvalues. Different subdomains of polymerase are color coded: thumb (yellow), palm (green), fingers (light blue), and 8-kDa (mauve). The loop regions showing persistent movement are labeled as L1 (residues 200–210), L2 (residues 242–250), and L3 (residues 302–310).
CONCLUDING REMARKS
We have performed nanosecond-range molecular dynamics simulations of pol-β with and without substrate to investigate the induced-fit hypothesis of pol-β's large subdomain rearrangement. This investigation is warranted because correlation between the polymerase's subdomain conformation and the presence/absence of an incoming nucleotide is not obvious from static crystal structures. In the presence of substrate, we observe systematic rearrangement of residues in the microenvironment of the substrate, stabilizing the closed state for subsequent nucleotidyl transfer reaction. Without the nucleotide substrate, an opening motion is accompanied with a rearrangement of the residues in the binding site that may disfavor chemistry. Although our trajectories start from intermediate structures of pol-β (between crystallographic “open” (1BPX) and “closed” (1BPY) complexes), this construction facilitates capturing motions that appear intrinsic to the enzyme. This intermediate is likely close to a structure along the conformation transition pathway; indeed it resembles the crystal structure of intermediate complex (thumb RMSD 1.3 Å) of pol-β with a mismatch (J. M. Krahn, W. A. Beard, and S. H. Wilson, unpublished data) and corresponds to the intermediate (thumb RMSD 1.4 Å) determined by our stochastic path simulations of pol-β (K. Arora and T. Schlick, unpublished data).
Our studies support the substrate-induced conformational change in pol-β and are consistent with the crystal structures of pol-β in open and closed forms that reveal a movement of the main-chain atoms of the thumb subdomain up to 7 Å upon nucleotide binding. The complementary principal component analysis helps to extract salient features from these trajectories. Interestingly, the first few principal components in both cases (with and without substrate) correspond to similar modes, with most of the motion concentrated in the residues of the thumb and 8-kDa subdomains. These results further support the functional importance of the thumb subdomain motion that polymerases employ in monitoring synthesis fidelity. Natural extensions of this work involve the study of polymerase dynamics in the presence of different incoming substrates, as done for the chemical reaction (Florián et al., 2003). Studies of mismatches were also performed for the opening after chemistry (Yang et al., 2002b) and are underway by transition path sampling for a G:A mismatch (R. Radhakrishnan and T. Schlick, unpublished data).
In addition, other studies that can generate complete reaction pathways—by Elber's stochastic path approach (Elber et al., 2003) and Chandler's transition path sampling (Bolhuis et al., 2000)—will undoubtedly shed further insights on polymerase mechanisms at atomic resolution.
DNA POLYMERASE β MOVIES
Movies from our 10-ns dynamics simulations of DNA pol-β with and without substrate can be viewed to better comprehend the motion described here. The animations show the large global motions as well as accompanying local conformational changes in the active site. We have also prepared movies of the top two eigenvectors from our essential dynamics analysis that smoothly depict the backbone dynamics of the pol-β. These products can be viewed on http://monod.biomath.nyu.edu/index/papdir/pap_2_107.html.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Drs. Samuel Wilson and Suse Broyde for helpful advice and insights. We are indebted to Dr. William Beard for his insightful comment on the induced-fit hypothesis and to Linjing Yang and Ravi Radhakrishnan for many fruitful discussions. Molecular images were generated using visual molecular dynamics (Humphrey et al., 1996) and INSIGHT II.
The work was supported by National Science Foundation grant ASC-9318159 and National Institutes of Health grant R01 GM55164. We also acknowledge the American Chemical Society Petroleum Research Fund for support (or partial support) of this research (award PRF39115-AC4 to T. Schlick).
Abbreviations used: pol-β, DNA polymerase-β; dNTP, 2′-deoxyribonucleoside 5′-triphosphate; ddCTP, 2′, 3′-dideoxyribocytidine 5′-triphosphate; dCTP, 2′-deoxyribocytidine 5′-triphosphate; ED, essential dynamics; MD, molecular dynamics; PCA, principal component analysis.
References
- Ahn, J., V. S. Kraynov, X. Zhong, B. G. Werneburg, and M.-D. Tsai. 1998. DNA polymerase β: effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem. J. 331:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, J., B. G. Werneburg, and M.-D. Tsai. 1997. DNA polymerase β: structure-fidelity relationship from pre-steady-state kinetic analyses of all possible correct and incorrect base pairs for wild type and R283A mutant. Biochemistry. 36:1100–1107. [DOI] [PubMed] [Google Scholar]
- Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- Ames, B. N., M. K. Shigenaga, and T. M. Hagen. 1990. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA. 90:7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangeli, C., A. R. Bizzari, and S. Cannistraro. 2000a. Concerted motions in copper plastocyanin and azurin: an essential dynamics study. Biophys. Chem. 90:45–56. [DOI] [PubMed] [Google Scholar]
- Arcangeli, C., A. R. Bizzari, and S. Cannistraro. 2000b. Molecular dynamics simulations and essential dynamics study of mutated plastocyanin: structural, dynamical and functional effects of a disulfide bridge insertion at the protein surface. Biophys. Chem. 92:183–189. [DOI] [PubMed] [Google Scholar]
- Arcangeli, C., A. R. Bizzari, and S. Cannistraro. 2001. Dynamic simulations of 13 TATA variants refine kinetic hypotheses of sequence/activity relationships. Biophys. Chem. 308:681–703. [DOI] [PubMed] [Google Scholar]
- Arnold, E., J. Ding, S. H. Hughes, and Z. Hostomsky. 1995. Structures of DNA and RNA polymerases and their interactions with nucleic acid substrates. Curr. Opin. Struct. Biol. 5:27–38. [DOI] [PubMed] [Google Scholar]
- Balsera, M. A., W. Wriggers, Y. Oono, and K. Schulten. 1996. Principal component analysis and long time protein dynamics. J. Phys. Chem. 100:2567–2572. [Google Scholar]
- Beard, W. A., W. P. Osheroff, R. Prasad, M. R. Sawaya, M. Jaju, T. G. Wood, J. Kraut, T. A. Kunkel, and S. H. Wilson. 1996. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β. J. Biol. Chem. 271:12141–12144. [DOI] [PubMed] [Google Scholar]
- Beard, W. A., and S. H. Wilson. 1998. Structural insights into DNA polymerase β fidelity: hold it tight if you want it right. Chem. Biol. 5:R7–R13. [DOI] [PubMed] [Google Scholar]
- Beese, L. S., and T. A. Steitz. 1991. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 9:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The protein data bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis, P. G., C. Dellago, P. L. Geisseler, and D. Chandler. 2000. Transition path sampling: throwing ropes over mountains in the dark. Annu. Rev. Phys. Chem. 12:A147–A152. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Brünger, A. T., and M. Karplus. 1988. Polar hydrogen positions in proteins: empirical energy placement and neutron diffraction comparison. Proteins. 4:148–156. [DOI] [PubMed] [Google Scholar]
- Capson, T. L., J. A. Peliska, B. F. Kaboord, M. W. Frey, C. Lively, M. Dahlberg, and S. J. Kovic. 1992. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 31:10984–10994. [DOI] [PubMed] [Google Scholar]
- Clarage, J. B., T. Romo, B. K. Andrews, B. M. Pettitt, and G. N. Phillips, Jr. 1995. A sampling problem in the molecular dynamics simulations of macromolecules. Proc. Natl. Acad. Sci. USA. 92:3288–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg, M. E., and S. J. Benkovic. 1991. Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry. 30:4835–4843. [DOI] [PubMed] [Google Scholar]
- deGroot, B. L., X. Daura, A. E. Mark, and H. Grubmüller. 2001. Essential dynamics of reversible peptide folding: memory-free conformational dynamics governed by internal hydrogen bonds. J. Mol. Biol. 309:299–313. [DOI] [PubMed] [Google Scholar]
- deGroot, B. L., D. M. F. van Aalten, A. Amadei, and H. J. C. Berendsen. 1996. The consistency of large concerted motions in proteins in molecular dynamics simulations. Biophys. J. 71:1707–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié, S., and T. Ellenberger. 1998. The mechanism of action of T7 DNA polymerase. Curr. Opin. Struct. Biol. 8:704–712. [DOI] [PubMed] [Google Scholar]
- Doublié, S., M. R. Sawaya, and T. Ellenberger. 1999. An open and closed case for all polymerases. Structure. 7:R31–R35. [DOI] [PubMed] [Google Scholar]
- Elber, R., A. Cárdenas, A. Ghosh, and H. Stern. 2003. Bridging the gap between long time trajectories and reaction pathways. Adv. Chem. Phys. 126:93–129. [Google Scholar]
- Florián, J., M. F. Goodman, and A. Warshel. 2003. Computer simulations of the chemical catalysis of DNA polymerases: discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 125:8163–8177. [DOI] [PubMed] [Google Scholar]
- Frey, M. W., L. C. Sowers, D. P. Millar, and S. J. Benkovic. 1995. The nucleotide analog 2-aminopurine as a spectroscopic probe of nucleotide incorporation by the Klenow fragment of Escherichia coli polymerase I and bacteriophage T4 DNA polymerase. Biochemistry. 34:9185–9192. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz, M., K. Bebenek, J. M. Krahn, L. Blanco, T. A. Kunkel, and L. C. Pedersen. 1998. A structural solution for the DNA polymerase λ-dependent repair of DNA gaps with minimal homology. Mol. Cell. 13:561–572. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Johnson, S. J., J. S. Taylor, and L. S. Beese. 2003. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc. Natl. Acad. Sci. USA. 100:3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce, C. M., and T. A. Steitz. 1994. Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 63:777–822. [DOI] [PubMed] [Google Scholar]
- Kiefer, J. R., C. Mao, J. C. Braman, and L. S. Beese. 1998. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 391:302–305. [DOI] [PubMed] [Google Scholar]
- Kim, S. J., W. A. Beard, J. H. Harvey, D. D. Shock, and J. R. Knutson. 2003. Rapid segmental and subdomain motions of DNA polymerase β. J. Biol. Chem. 278:5072–5081. [DOI] [PubMed] [Google Scholar]
- Klapper, I., R. Hagstrom, R. Fine, K. Sharp, and B. Honig. 1986. Focusing of electric fields in the active site of Cu-Zn superoxide dismutase: effects of ion strength and amino-acid modification. Proteins. 1:47–59. [DOI] [PubMed] [Google Scholar]
- Koshland, D. E. 1994. The key-lock theory and the induced fit theory. Angew Chem. Int. Ed. Engl. 33:2375–2378. [Google Scholar]
- Kraynov, V. S., B. G. Werneburg, X. Zhong, H. Lee, J. Ahn, and M.-D. Tsai. 1997. DNA polymerase β: analysis of the contributions of tyrosine-271 and asparagine-279 to substrate specificity and fidelity of DNA replication by pre-steady-state kinetics. Biochem. J. 323:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchta, R. D., V. Mizrahi, P. A. Benkovic, K. A. Johnson, and S. J. Benkovic. 1987. Kinetic mechanism of DNA polymerase I (Klenow). Biochemistry. 26:8410–8417. [DOI] [PubMed] [Google Scholar]
- Li, Y., S. Korolev, and G. Waksman. 1998. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 17:7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhal, T., and R. D. Wood. 1999. Quality control by DNA repair. Science. 286:1897–1905. [DOI] [PubMed] [Google Scholar]
- MacKerell, A. D., Jr., and N. K. Banavali. 2000. All-atom empirical force field for nucleic acids. II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 21:105–120. [Google Scholar]
- Patel, S. S., I. Wong, and K. A. Johnson. 1991. Pre-steady-state kinetic analysis of processive DNA replication inducing complete characterization of an exonuclease-deficient mutant. Biochemistry. 30:511–525. [DOI] [PubMed] [Google Scholar]
- Qian, X., D. Strahs, and T. Schlick. 2001. A new program for optimizing periodic boundary models of solvated biomolecules (PBCAID). J. Comput. Chem. 22:1843–1850. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan, R., and T. Schlick. 2004. Orchestration of cooperative events in DNA synthesis and repair mechanism unravelled by transition path sampling of DNA polymerase β's closing. Proc. Natl. Acad. Sci. USA. 101:5970–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, A. C., H. W. Park, C. Mao, and L. S. Beese. 2000. Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon thermococcus sp. 9 degrees n-7. J. Mol. Biol. 299:447–462. [DOI] [PubMed] [Google Scholar]
- Sawaya, M. R., R. Parsad, S. H. Wilson, J. Kraut, and H. Pelletier. 1997. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 36:11205–11215. [DOI] [PubMed] [Google Scholar]
- Schlick, T. (1992). Optimization methods in computational chemistry. In Reviews in Computational Chemistry, Vol. III. K. B. Lipkowitz and D. B. Boyd, editors. VCH Publishers, New York, NY. 1–71.
- Seeberg, E., L. Eide, and M. Bjoras. 1995. The base excision repair pathway. Trends Biochem. Sci. 20:391–397. [DOI] [PubMed] [Google Scholar]
- Shah, A. M., S. X. Li, K. S. Anderson, and J. B. Sweasy. 2001. Y265H mutator mutant of DNA polymerase β. Proper teometric alignment is critical for fidelity. J. Biol. Chem. 276:10824–10831. [DOI] [PubMed] [Google Scholar]
- Showalter, A. K., and M.-D. Tsai. 2002. A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry. 41:10571–10576. [DOI] [PubMed] [Google Scholar]
- Steitz, T. A. 1993. DNA- and RNA-dependent DNA polymerases. Curr. Opin. Struct. Biol. 3:31–38. [Google Scholar]
- Stella, L., E. E. Iorio, M. Nicotra, and G. Ricci. 1999. Molecular dynamics simulations of human glutathione transferase p1–1: conformational fluctuations of the apo-structure. Proteins. 37:10–19. [DOI] [PubMed] [Google Scholar]
- Suo, Z., and K. A. Johnson. 1998. Selective inhibition of HIV-1 reverse transcriptase by an antiviral inhibitor, (R)-9-(2-phosphonylmethoxypropyl)adenine. J. Biol. Chem. 273:27250–27258. [DOI] [PubMed] [Google Scholar]
- van Aalten, D. M. F., B. L. deGroot, J. B. C. Findlay, H. J. C. Berendsen, and A. Amadei. 1997. A comparison of techniques for calculating protein essential dynamics. J. Comput. Chem. 18:169–181. [Google Scholar]
- van Aalten, D. M. F., J. B. C. Findlay, A. Amadei, and H. J. C. Berendensen. 1995. Essential dynamics of the cellular retinol-binding protein-evidence for ligand induced conformational changes. Protein Eng. 8:1129–1135. [DOI] [PubMed] [Google Scholar]
- Vande Berg, B. J., W. A. Beard, and S. H. Wilson. 2001. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase β. J. Biol. Chem. 276:3408–3416. [DOI] [PubMed] [Google Scholar]
- Weber, W., H. Demirdjian, R. D. Lins, J. M. Briggs, R. Ferreira, and J. A. McCammon. 1998. Brownian dynamics and essential dynamics studies of HIV-1 integrase catalytic domain. J. Biomol. Struct. Dyn. 16:733–745. [DOI] [PubMed] [Google Scholar]
- Wilson, S. H. 1998. Mammalian base excision repair and DNA polymerase β. Mutat. Res. 407:203–215. [DOI] [PubMed] [Google Scholar]
- Wong, I., S. S. Patel, and K. A. Johnson. 1991. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry. 30:526–537. [DOI] [PubMed] [Google Scholar]
- Yang, L., K. Arora, W. A. Beard, S. H. Wilson, and T. Schlick. 2004. The critical role of magnesium ions in DNA polymerase β's closing and active site assembly. J. Am. Chem. Soc. 126:8441–8453. [DOI] [PubMed] [Google Scholar]
- Yang, L., W. A. Beard, S. H. Wilson, S. Broyde, and T. Schlick. 2002a. Polymerase β simulations reveal that Arg258 rotation is a slow step rather than large subdomain motion per se. J. Mol. Biol. 317:651–671. [DOI] [PubMed] [Google Scholar]
- Yang, L., W. A. Beard, S. H. Wilson, B. Roux, S. Broyde, and T. Schlick. 2002b. Local deformations revealed by dynamics simulations of DNA polymerase β with DNA mismatches at the primer terminus. J. Mol. Biol. 321:459–478. [DOI] [PubMed] [Google Scholar]
- Zhong, X., S. S. Patel, B. G. Werneburg, and M. D. Tsai. 1997. DNA polymerase β: multiple conformational changes in the mechanism of catalysis. Biochemistry. 36:11891–11900. [DOI] [PubMed] [Google Scholar]