

Abstract
Reverse iontophoresis uses a small low electric current to noninvasively extract blood analytes, e.g., glucose, across the skin. The simultaneous quantification of the analyte extracted and of an additional endogenous substance of fixed and known concentration in the body permits the blood level of interest to be found without the need for an invasive calibration procedure. The transport phenomena underlying this approach, applied to glucose monitoring, has been investigated in vitro, using Na+ and neutral model solutes as endogenous “internal standards” (specifically, urea, glycerol, mannitol, and sucrose). The cathodal extracted fluxes of glucose under conditions of modified skin permselectivity were related to those of the different, potential internal standards. Flux ratios depended upon the iontophoretic conditions and the size of the neutral internal standards, whereas high variability was observed with Na+. Constant flux ratios were obtained with mannitol, glycerol, urea, and sucrose for which the mechanism of electrotransport was identical to that of glucose. The advantage of using a neutral internal standard, however, must be weighed against the need to identify and validate the marker under physiological conditions and the additional analytical chemistry necessary for the practical quantification of this substance.
INTRODUCTION
Human skin acts as a biological barrier, which renders transdermal drug delivery a significant challenge despite the skin's easy accessibility and large available surface. Iontophoresis enhances the delivery of charged and uncharged polar species via application of a low electric current (Kalia et al., 2004). The symmetry of iontophoresis makes the technique equally interesting, furthermore, for the clinical monitoring of drugs (Delgado-Charro and Guy, 2003; Leboulanger et al., 2004a,b,c) and biomarkers such as glucose (Glikfeld et al., 1989; Potts et al., 2002, Rao et al., 1995; Tamada et al.,1995; Tierney et al., 2001).
The total flux of a solute (Ji) during iontophoresis is the sum of electromigration (JEM), convective flow (JEO), and passive diffusion (Jp):
 |
(1) |
Electromigration is the movement of small ions across the skin under the direct influence of the electric field. Electron fluxes are transformed into ionic fluxes via the electrode reactions; and ionic transport proceeds through the skin to maintain electroneutrality. The total charge transported depends on the strength of the electric field and the duration of application. Iontophoresis sets in motion a number of ions across the skin, and all of them compete to carry a fraction of the current. The contribution of each ion to charge transport is called its transport number, the overall sum of which equals 1. According to Faraday's law, the electromigration flux of each ion in the iontophoretic circuit is given by
 |
(2) |
where ti is the transport number, zi is the valence of the ith ion, F is Faraday's constant, and I is the total current. The transport number depends on the ion's relative mobility (ui) and charge zi and upon its relative concentration ci:
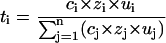 |
(3) |
Given that sodium and chloride ions are the principal extracellular electrolytes present in the body at high concentrations, they will invariably carry a major part of the current during iontophoresis in vivo.
Electroosmosis is the principal transport mechanism of uncharged molecules and of high molecular weight cations (Pikal, 1992). The skin is negatively charged at physiological pH, and acts therefore as a permselective membrane to cations. This preferential passage of counterions induces an electroosmotic solvent flow that carries neutral molecules in the anode-to-cathode direction. The volume flow JV (volume × time−1 × area−1) is proportional to the potential gradient established by the electric field (Pikal, 1992)
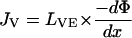 |
(4) |
where LVE is the electroosmotic flow coefficient describing the direction and the magnitude of the volume flow resulting from the driving force, −dΦ/dx. The electroosmotic flux contribution to the transport of a solute s present in the anodal compartment at molar concentration cs is then
 |
(5) |
Electroosmosis assists the transport of high molecular weight cations and retards the passage of anions at pH 7. It can be modified by altering the permselectivity of the membrane and by manipulation of the formulation in the electrode chambers, that is by changing the value of LVE (Santi and Guy, 1996a).
Passive diffusion of the solute j may be expressed as
 |
(6) |
where Dj is the aqueous diffusivity of the solute, and Δcj/h represents its concentration gradient across the skin.
The contributions of electromigration, electroosmosis, and passive diffusion to the total iontophoretic flux depend on the structure and physicochemical properties of the species being transported. For small ions such as Na+ or Cl−, electromigration dominates; on the other hand, neutral solutes are transported by electroosmosis and (usually) to a much lesser extent by passive diffusion.
The reverse iontophoretic extraction of glucose across the skin is primarily electroosmotic and has been successfully used to monitor glycemia in diabetics (GlucoWatch Biographer, Cygnus, Redwood City, CA) (Potts et al., 2002; Tamada et al., 1995; Tierney et al., 2001). The commercially available wrist-worn device tracks glucose continuously for up to 13 h, making six measurements per hour. However, because the glucose extraction efficiency varies significantly within and between patients, the device must be calibrated against a conventional fingertip blood glucose reading before each use. This essential step has been perceived as a disadvantage despite the fact that the GlucoWatch provides tremendously more information to the diabetic than one or two “finger-sticks” per day.
The long-term objective of the research presented here is to refine the iontophoresis technology so as to avoid the invasive calibration step. The use of an internal standard has been proposed as a strategy to attain this goal. As iontophoresis is nonspecific, many ions and small uncharged species (in addition to the analyte of interest) are moved across the skin. Instead of detecting uniquely the single target substance extracted by iontophoresis and calibrating its transdermal measurement via a blood assay, we propose to monitor the extraction of two species simultaneously: the compound of interest, the temporal change in the concentration of which is of clinical importance (i.e., glucose), and a second analyte, the physiological concentration of which is known and essentially fixed. If the iontophoretic transport of the analyte (A) and the latter internal standard (IS) are independent of one another, then their fluxes (J) out of the skin should obey the relationship
 |
(7) |
where [A] and [IS] are the blood concentrations of the two substances, and K is a constant. Previous work (Sieg et al., 2003) established the validity of this equation in vitro, in a series of experiments in which glucose and sodium concentrations were varied over their clinically relevant ranges. However, subsequent in vivo studies (Sieg et al., 2004) found much more variability in glucose extraction, both within and between subjects (Sieg et al., 2004). As a consequence, the “constant” K was only truly constant for about two-thirds of the population.
A first objective of the work described here, therefore, is to understand the in vitro-in vivo differences that have been observed. Further in vitro experiments have been performed under conditions known to modify the skin's net charge (i.e., its permselectivity). Specifically, by manipulating the pH on either side of the barrier (Santi and Guy, 1996a; Marro et al., 2001), it was possible to investigate whether the iontophoretic extraction of Na+ and glucose varied in parallel and to assess the impact of these changes on the constancy of the parameter K in Eq. 7. A second objective was to examine different, small, neutral solutes as alternative internal standards for glucose extraction. In this case, both analyte and internal standards were moved across the skin by electroosmosis. So as to evaluate the robustness of this approach, electroosmosis was modified to different degrees using different background electrolytes (Santi and Guy, 1996b).
MATERIALS AND METHODS
Chemicals
d-glucose, Tris base (α,α,α-Tris-(hydroxymethyl)-methylamine), sodium chloride, potassium chloride, d-mannitol, urea, glycerol, d-sucrose, calcium chloride, EDTA, sodium hydroxide, and hydrochloric acid were analytical grade and purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). d-[1-14C]-mannitol, [14C]-urea, [14C(U)]-glycerol, [14C(U)]-sucrose (specific activities 51.5, 54.3, 142.7, and 401.0 mCi/mmol, respectively) were obtained from PerkinElmer Life Sciences, Rungis, France), and d-[6-3H]-glucose (specific activity 35.0 mCi/mmol) was purchased from Amersham Pharmacia Biotech (Orsay, France). Deionized water (resistivity >18.2 MOhm/cm2) was used to prepare all solutions.
Skin preparation
Porcine ears were obtained <2 h after slaughter of the animal (Société d'Exploitation d'Abbatage, Annecy, France) and cleaned under running cold water. The whole skin was removed carefully from the outer region of the ear and separated from the underlying cartilage with a scalpel. The tissue was then dermatomed to a thickness of 750 μm (Zimmer Air Dermatome, Dover, Ohio) and cut into small squares (∼9 cm2), which were wrapped individually in Parafilm and maintained at −20°C for no longer than 2 weeks before use.
Reverse iontophoresis
Side-by-side diffusion cells (transport area = 0.78 cm2) with three compartments representing the anodal, subdermal, and cathodal chambers (Leboulanger et al., 2004b) were used in the iontophoresis experiments. Volumes were 1.5 ml for the electrode chamber and 3.5 ml for the subdermal compartment.
A piece of excised skin was clamped between the central compartment and each electrode chamber, with the dermal surface facing the central compartment, and the cell was assembled as shown in Fig. 1. The background buffer, 10 mM Tris, was used for all experiments at pH 6.3, 7.4, or 8.5. All chambers were initially filled with this buffer for a 1-h equilibration period. After replacing the buffer by the appropriate electrode and subdermal solutions, constant current (0.5 mA/cm2) was applied for 6 h via Ag/AgCl electrodes connected to a constant current power supply (KEPCO APH-1000DM, KEPCO Inc., Flushing, NY). After each hour, the current was interrupted, and the entire contents of anodal and cathodal chamber were withdrawn and replaced by fresh receiver solution. All experiments were performed in quadruplicate, using skin samples from four different pigs. The fluxes shown correspond to the 5–6th h of iontophoresis; steady values were typically attained after 3 h of current passage.
FIGURE 1.

Schematic view of the setup for the reverse iontophoresis experiments.
Glucose and sodium extraction at different pH
In these experiments, the subdermal (donor) solution comprised 10 mM glucose, 133 mM NaCl, and 4 mM KCl in Tris buffer at pH 6.3, 7.4, or 8.5, respectively. The anodal compartment contained 100 mM NaCl in Tris buffer, the cathodal compartment Tris buffer alone at the same pH as the donor solution. A control experiment with pH 7.4 in the donor and pH 8.5 in the electrode chambers was also performed.
Simultaneous extraction of neutral compounds
To study the simultaneous extraction of glucose and model neutral compounds, the subdermal solution contained 133 mM NaCl, 4mM KCl, 5 mM glucose, and either urea, glycerol, mannitol, or sucrose, (again, at a concentration of 5 mM); further, the solution was spiked with ∼0.2 μCi/ml of the corresponding 14C-isotope and ∼0.5 μCi/ml of 3H-glucose. The pH of the subdermal solution was 7.4 for all experiments except for the measurements at pH 6.3 when this lower pH was also maintained in the central compartment (see Results and Discussion). In all experiments, the anodal and cathodal solutions were identical and comprised 10 mM Tris buffer at pH 6.3 or pH 8.5 together with additional electrolytes as described below. Passive controls, following the same experimental procedure without current application, were performed for the experiments with urea and glycerol.
Sample analysis
Samples from the first series of measurements were quantified by high-performance ion chromatography using a Dionex IC 600 system (Dionex, Sunnyvale, CA). Glucose was assayed by anion separation with pulsed amperometric detection; Na+ was quantified by cation separation with suppressed conductivity detection (Sieg et al., 2004).
For the experiments using radioactivity, samples from the electrode chambers were mixed with 5 ml scintillation cocktail (Ultima Gold XR, PerkinElmer Life Sciences) and then analyzed by liquid scintillation counting (LS 6500, Beckmann Instruments France SA, Gagny, France) for 3H-glucose and the 14C-isotope from the second neutral compound.
Statistical analysis
Iontophoretic fluxes and values of K were determined as the mean ± SD and were compared with a one-way ANOVA followed by Tukey's Multiple Comparison Test. Excel MS Windows 97 and GraphPad Prism 3.02 Software (GraphPad Software, San Diego, CA) were used for data analysis.
RESULTS AND DISCUSSION
As earlier explained, at conditions close to physiological pH, electroosmosis is predominantly in the anode-to-cathode direction. All solutes investigated were much more efficiently extracted, therefore, at the cathode, and attention is focused specifically on these data alone.
pH dependence of glucose and Na+ transport
By modification of the pH, we aimed to gradually change the skin's permselectivity and hence to modify the extraction of glucose by electroosmosis. If Na+ electromigration was affected in a similar manner, then we would expect the extraction constant K (Eq. 7) to remain essentially constant.
Although the pH and composition of the subdermal milieu in vivo cannot be manipulated, it is possible to perform rather specific modifications in the in vitro experimental setup. To avoid the additional complication of a pH gradient across the skin, therefore, it was decided initially to vary the pH on both sides of the skin in systematic steps of 1.1 pH units between 8.5 and 6.3. In this way, the skin retained its cation permselectivity (the isoelectric point of porcine ear skin having been shown to be ∼4.4 (Marro et al., 2001)), and the variability in electroosmotic behavior observed with a significant pH gradient across the skin (Kim et al., 1993) was precluded.
The skin's permselectivity depends strongly on the pH of the surrounding media (Marro et al., 2001; Phipps and Gyory, 1992). With a change in the fixed charge on the membrane, the preferential passage of cations and the extent of electroosmotic flow can be modified. Glucose and Na+ fluxes after 6 h of iontophoresis at different pHs are shown in Table 1. Although no differences were observed when the pH was changed from 7.4 to 8.5, both glucose and sodium electrotransport were reduced significantly when the pH was decreased to 6.3. However, the amplitude of this modification was not the same for the two solutes. Whereas sodium fluxes decreased to ∼75% of its value at pH 7.4 (that is, from 10.7 (±0.7) to 8.0 (±0.3) μmol × cm−2 × h−1), electroosmotic transport of the glucose decreased by one half (from 54.3 (±5.5) to 27.4 (±8.6) nmol × cm−2 × h−1). Given that the subdermal glucose and Na+ concentrations were kept constant, the proportionality constant K (calculated from Eq. 7) fell significantly from 0.068 ± 0.007 to 0.045 ± 0.014. Note that the values for Na+ extraction are 2–3 orders of magnitude higher than those for glucose; in other words, sodium electromigration is much more efficient than glucose extraction by electroosmosis at concentrations close to physiological conditions. This difference is reflected in the value of the constant K, which is <0.1.
TABLE 1.
Simultaneous glucose and Na+ extraction fluxes and the deduced sodium transport number ( ), together with calculated proportionality constant K (Eq. 7), as a function of pH
), together with calculated proportionality constant K (Eq. 7), as a function of pH
| pH subdermal/electrodes | Glucose flux (nmol × cm−2 × h−1) | Na+ flux (μmol × cm−2 × h−1) | 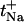 |
K |
|---|---|---|---|---|
| 6.3/6.3 | 27.4 ± 8.6 | 8.0 ± 0.3 | 0.43 ± 0.01 | 0.045 ± 0.014 |
| 7.4/7.4 | 54.3 ± 5.5 | 10.7 ± 0.7 | 0.57 ± 0.02 | 0.068 ± 0.007 |
| 8.5/8.5 | 56.8 ± 4.0 | 11.5 ± 1.2 | 0.62 ± 0.07 | 0.066 ± 0.005 |
| 7.4/8.5 | 56.4 ± 9.4 | 11.7 ± 0.7 | 0.62 ± 0.04 | 0.064 ± 0.007 |
This differential modification of electromigration and electroosmosis across the skin has been alluded to previously (Marro et al., 2001; Phipps and Gyory, 1992). The transport number of Na+ in iontophoresis under normal physiological conditions (∼0.6 at pH 7.4 (Phipps and Gyory, 1992)) reflects the net negative charge on the skin and is significantly higher than in simple aqueous solution (∼0.4) (Burnette and Ongpipattanakul, 1987). Similarly, because of this cationic permselectivity, electroosmosis proceeds in the anode-to-cathode direction, achieving ∼1 μl × cm−2 × h−1 at neutral pH (Burnette and Ongpipattanakul, 1987; Marro et al., 2001). However, if the pH of the solution bathing the skin is lowered to 4, while the Na+ transport number decreases by ∼40%, electroosmotic flow is completely attenuated from the anode and now occurs in the opposite, cathode-to-anode, direction (Marro et al., 2001). That is, alteration of the net charge on the skin has a far more dramatic impact on convective flow than on electromigration. For reverse iontophoresis, it is clear that the high subdermal concentration of NaCl guarantees that Na+ will dominate the carrying of current toward the cathode, regardless of the level of negative charge of the skin; on the other hand, changes in the latter parameter will result in significant changes in electroosmosis.
In previous experiments in vivo in man, Na+ transport numbers from 0.48 to 0.64 were determined at pH 8.5 (I = 0.6 mA) (Sieg et al., 2004). Normalized glucose fluxes ranged from 0.25 to 6.25 μl × cm−2 × h−1, and the inferred constant K varied between 0.01 and 0.13. Compared to the results from this investigation, the Na+ transport numbers were in good agreement: average values in vitro and in vivo 0.60 vs. 0.55, respectively). Notably, this overlap was found despite the facts that i), the in vitro subdermal solution did not contain all physiologically relevant ions, and ii), porcine rather than human skin was used. The normalized glucose flux at pH 8.5 in vitro was 5.7 μl × cm−2 × h−1 (I = 0.4 mA), and is comparable to the upper value observed in vivo. It is important to emphasize that the in vivo experiments were performed with a slightly higher current, and that the up to 10-fold difference in glucose fluxes was not observed in vitro. To correct for these important interindividual differences that are not mirrored by the Na+ extraction flux, attention was next focused on the identification of a potential neutral internal standard transported by the same mechanism as glucose, namely electroosmosis.
Simultaneous extraction of neutral solutes and glucose
In examining the transport behavior of the neutral internal standard candidates relative to that of glucose, it was particularly interesting to determine the flux ratios under different conditions of skin permselectivity. In addition to exogenous factors, such as the composition of the electrode formulations and the current applied, iontophoretic extraction in vivo depends on a number of biological factors (including net charge on the skin, local blood flow, etc.) that are difficult to control and modulate in vitro. Nevertheless, the electrical properties of the skin can be modified in vitro by changing the pH, as before, and by the use of different background electrolytes, and this was the strategy adopted here.
The candidates for the internal standard were chosen based upon their molecular weight (MW, ranging from 60 to 342), biological relevance (urea and glycerol are present at sufficiently high concentrations in the blood to be extracted by reverse iontophoresis in amounts sufficient for relatively straightforward analysis), and similar physicochemical properties (glycerol, mannitol, and sucrose having multiple-OH functions like glucose). The cathodally extracted fluxes of glucose and the model solutes at 6 h are shown in Table 2. For urea and glycerol, passive diffusion contributed significantly to the overall transport, and the uniquely electroosmotic contribution (JEO) was therefore calculated after subtracting passive transport from total measured flux. If extraction into a solution at pH 8.5 in the presence of a relatively low level of background electrolyte is considered as a reference value, addition of 10 mM EDTA significantly increased electroosmosis. On the other hand, supplementing the formulation with 100 mM CaCl2 decreased convective transport by 50%. A comparable reduction was induced by simply lowering the pH of the cathode solution to 6.3, an effect already observed in the first part of this study. Calcium has been shown to shield the skin's net negative charge (presumably via an interaction with carboxylic acid groups in the skin (Phipps and Gyory, 1992)). Conversely, EDTA has been suggested to increase the skin's permselectivity by complexing endogenous divalent cations, such as Ca2+, thus enhancing electroosmosis toward the cathode (Santi and Guy, 1996b). It is noted that, for all solutes (except urea at pH 6.3), convective flow was essentially the same at each set of experimental conditions studied. Although there is evidence of molecular hindrance (i.e., a sieving effect as the convective volume flow passes through narrow channels) under certain iontophoretic conditions (Ruddy and Hadzija,1992), the results described here are consistent with the hypothesis that electrotransport via electroosmosis is relatively constant for low molecular weight solutes (Pikal, 1992).
TABLE 2.
Cathodal extraction fluxes (nmol × h−1 × cm−2) at 6 h for glucose and the neutral internal standard candidates
| MW | pH 8.5 + 30 mM NaCl | pH 8.5 + 30 mM NaCl + 10 mM EDTA | pH 8.5 + 100 mM CaCl2* | pH 6.3 + 30 mM NaCl* | n | |
|---|---|---|---|---|---|---|
| Urea | 60 | 41.4 ± 7.9† | 52.0 ± 6.6† | 26.5 ± 10.5† | 27.2 ± 7.9† | 4 |
| JEO | 33.2 ± 3.6 | 46.5 ± 3.9 | 20.1 ± 6.2 | 25.5 ± 6.8† | 4 | |
| Glycerol | 92 | 36.7 ± 8.5† | 49.6 ± 9.2 | 21.7 ± 11.8 | 16.1 ± 6.5 | 4 |
| JEO | 33.9 ± 7.9 | 47.4 ± 8.4 | 18.7 ± 9.4 | 14.2 ± 5.4 | 4 | |
| Mannitol | 182 | 28.2 ± 2.6 | 41.5 ± 4.7 | 14.7 ± 2.6 | 11.8 ± 4.4 | 4 |
| Glucose | 180 | 26.2 ± 3.7 | 39.6 ± 5.0 | 15.2 ± 5.5 | 12.4 ± 4.1 | 16 |
| Sucrose | 342 | 27.2 ± 5.0 | 40.8 ± 6.0 | 16.0 ± 5.8 | 12.6 ± 6.7 | 4 |
The electroosmotic contributions (corrected for passive diffusion) for urea and glycerol fluxes are given as JEO.
The extraction fluxes at pH 8.5 + 100 mM CaCl2 were not significantly different, for any solute, from those at pH 6.3 + 30 mM NaCl.
These values are significantly different (p < 0.01) from all others in the corresponding columns.
In the second part of the study, the subdermal concentration of all the neutral solutes considered was 5 mM. Under these conditions, according to Eq. 7, the flux ratio (glucose/internal standard) must equal the constant K, which represents, therefore, the relative efficiency of glucose transport to that of the candidate internal standard. Thus, if K is greater than unity, glucose transport is higher than that of the standard, whereas K < 1 indicates that the standard is more efficiently extracted than glucose. Modification of skin permselectivity had a significant impact on K (Table 3, Fig. 2), the values of which increased gently with increasing molecular weight. Not surprisingly, mannitol, as an isomer of glucose, was reverse iontophoretically extracted at the same rate as glucose, yielding K values close to unity under all experimental conditions. This behavior is entirely consistent with an earlier in vitro study (Sieg et al., 2003). Urea (MW 60) transport was significantly higher than that of glucose, and K values, which differed significantly between the iontophoretic conditions, were consistently <1. Glycerol (MW 92) showed behavior intermediate between mannitol and urea. Finally, sucrose (having the highest MW, nearly double that of glucose) yielded values of K sensitive to the composition of the cathode formulation. At pH 8.5, in the presence or not of EDTA, K was close to 1; on the other hand, at high calcium concentration and at lower pH, K equaled 1.2.
TABLE 3.
Calibration constant K (glucose/internal standard) (n = 4)
| Internal standard | MW | pH 8.5 + 30 mM NaCl | pH 8.5 + 30 mM NaCl + 10 mM EDTA | pH 8.5 + 100 mM CaCl2* | pH 6.3 + 30 mM NaCl* |
|---|---|---|---|---|---|
| Na+ | 23 | 0.064 ± 0.007 | - | - | 0.045 ± 0.014 |
| Urea | 60 | 0.61 ± 0.04 | 0.71 ± 0.05 | 0.51 ± 0.11 | 0.47 ± 0.03 |
| Glycerol | 92 | 0.75 ± 0.05 | 0.79 ± 0.05 | 0.71 ± 0.06 | 0.74 ± 0.03 |
| Mannitol | 182 | 0.92 ± 0.01 | 0.92 ± 0.02 | 0.95 ± 0.04 | 0.95 ± 0.03 |
| Sucrose | 342 | 0.98 ± 0.02 | 1.00 ± 0.06 | 1.19 ± 0.12 | 1.20 ± 0.15 |
The extraction fluxes at pH 8.5 + 100 mM CaCl2 were not significantly different, for any solute, from those at pH 6.3 + 30 mM NaCl.
FIGURE 2.

Values of the calibration constant K (mean ± SD; n = 4) for each internal standard candidate at each set of experimental conditions studied (see Table 3).
Although the values of K determined in this series of experiments, over all solutes and all cathodal solution conditions, differed by no more than a factor of 2, (i.e., ranging from ∼0.6 at the lowest for urea up to ∼1.2 at the highest for sucrose), the trend in Fig. 2, with respect to molecular weight, deserves further discussion. The K values reported were determined from Eq. 7 using the absolute fluxes measured at 6 h. Although these fluxes have no electromigration contribution (Eq. 1), they do include passive diffusion as well as electroosmosis. As mentioned above, the passive transport of urea and glycerol did contribute significantly to the total and it might be anticipated, therefore, that this phenomenon would explain, at least in part, why K deviates from unity. That is, when glucose and the internal standard are present subdermally at equal concentrations
 |
(8) |
where JEO and Jp refer to the electroosmotic and passive fluxes during iontophoresis of the solutes concerned: i.e., glucose (Glu) and the internal standard (IS). It follows, therefore, that if the JEO are identical, K will not equal 1 if the passive contributions for glucose and the internal standard are different, and if they make up a measurable component of the experimentally measured fluxes.
Although this issue is of mechanistic interest, and relevant to previous work that has focused on the relative contributions of electromigration and electroosmosis to the iontophoresis of cationic drugs (Guy et al., 2000; Yoshida and Roberts, 1993), it must nevertheless be emphasized that this phenomenon elicited only a modest (no more than two-fold) effect on the absolute value of the calibration constant K. Of greater practical importance is whether, for any particular candidate internal standard, the value of K remains fixed despite changes in the skin's permselectivity. Such was clearly not the case when Na+ was examined for this role in vivo (Sieg et al., 2004). For mannitol and glycerol, deviations of K were small. However, these solutes are either present physiologically at levels too low (mannitol, ∼34 ± 18 μM (Lentner, 1984)) or are subject to significant systemic variation (unbound glycerol, ∼120 ± 65 μM (Lentner, 1984)) to be useful internal standards for glucose monitoring in vivo. Sucrose and urea showed slightly greater deviations in K. Again, sucrose (plasma concentration ∼1.8 ± 1.2 μM (Lentner, 1984)) is not a practical option for the same reason as mannitol. Urea outperformed Na+, both in terms of absolute divergence of the observed K, and in terms of variability. The fact that K for urea is >10 times the corresponding Na+ value confirms that its extraction is primarily achieved by the same mechanism as glucose. In addition, blood concentrations of urea are relatively stable, sufficiently high (4–8 mM (Bankir and Trinh-Trang-Tan, 2000)) and in rapid equilibrium with those in the extracellular fluid (the tissue compartment sampled by reverse iontophoresis) to offer a pragmatic option for a useful internal standard (Rosdahl et al., 1998). Finally, it is important to point out that the variability observed (coefficients of variation typically <10%) in the values of K obtained with urea as an internal standard would be acceptable in clinical practice. In a recent study, in which lithium serum levels in bipolar patients were successfully predicted using sodium as an internal standard, the variability in K was on the order of no more than 7% (Leboulanger et al., 2004c).
Of course, it remains to be seen whether the conclusions from this work are directly applicable in vivo, in human subjects. It should be recalled that the skin model used, although considered extremely useful and faithful, was from pig rather than man. In addition, the subdermal milieu in vivo is much more complex than that employed in the in vitro reported here. Equally, although it has been possible to deliberately modify skin permselectivity in these experiments by modulating the cathodal formulation, there are undoubtedly other physiological and/or environmental factors that can also play a role in the real world and these issues can only be elucidated and examined in vivo (for example, although the intraindividual variation in the blood concentration of urea over a 24-h period is quite small, intersubject variability is greater, which means that it may be necessary to verify periodically a “personal” K for each diabetic (Bankir and Trinh-Trang-Tan, 2000)). Such is clearly the next logical step in this research.
Acknowledgments
This work was supported by the United States National Institutes of Health (EB 001420) and United States Army Medical Research Acquisition Activity grant DAMD17-02-1-0712 (Fort Detrick, MD). The information presented does not necessarily reflect the position or the policy of the United States government, and no official endorsement should be inferred.
References
- Bankir, L., and M. M. Trinh-Trang-Tan. 2000. Urea and the kidney. In The Kidney. B. M. Brenner, editor. W. B. Saunders, Philadelphia, PA. 637–679.
- Burnette, R. R., and B. Ongpipattanakul. 1987. Characterization of the permselective properties of excised human skin during iontophoresis. J. Pharm. Sci. 76:765–773. [DOI] [PubMed] [Google Scholar]
- Delgado-Charro, M. B., and R. H. Guy. 2003. Transdermal reverse iontophoresis of valproate: a non-invasive method for therapeutic drug monitoring. Pharm. Res. 20:1508–1513. [DOI] [PubMed] [Google Scholar]
- Glikfeld, P., R. S. Hinz, and R. H. Guy. 1989. Noninvasive sampling of biological fluids by iontophoresis. Pharm. Res. 6:988–990. [DOI] [PubMed] [Google Scholar]
- Guy, R. H., Y. N. Kalia, M. B. Delgado-Charro, V. Merino, A. Lopez, and D. Marro. 2000. Iontophoresis: electrorepulsion and electroosmosis. J. Control. Release. 64:129–132. [DOI] [PubMed] [Google Scholar]
- Kalia, Y. N., A. Naik, J. Garrison, and R. H. Guy. 2004. Iontophoretic drug delivery. Adv. Drug Deliv. Rev. 56:619–658. [DOI] [PubMed] [Google Scholar]
- Kim, A., P. G. Green, G. Rao, and R. H. Guy. 1993. Convective solvent flow across the skin during iontophoresis. Pharm. Res. 10:1315–1320. [DOI] [PubMed] [Google Scholar]
- Leboulanger, B., R. H. Guy, and M. B. Delgado-Charro. 2004a. Reverse iontophoresis for non-invasive transdermal monitoring. Physiol. Meas. 25:R35–R50. [DOI] [PubMed] [Google Scholar]
- Leboulanger, B., R. H. Guy, and M. B. Delgado-Charro. 2004b. Non-invasive monitoring of phenytoin by reverse iontophoresis. Eur. J. Pharm. Sci. 22:427–433. [DOI] [PubMed] [Google Scholar]
- Leboulanger, B., J. M. Aubry, G. Bondolfi, R. H. Guy, and M. B. Delgado-Charro. 2004c. Non-invasive lithium monitoring by reverse iontophoresis in vivo. 10.1373/Clin. Chem. 2004.034249. [DOI] [PubMed]
- Lentner, C. 1984. Geigy Scientific Tables, Vol. 3. Ciba-Geigy, Basel, Switzerland.
- Marro, D., R. H. Guy, and M. B. Delgado-Charro. 2001. Characterization of the iontophoretic permselectivity properties of human and pig skin. J. Control. Release. 70:213–217. [DOI] [PubMed] [Google Scholar]
- Phipps, J. B., and J. R. Gyory. 1992. Transdermal ion migration. Adv. Drug Deliv. Rev. 9:137–176. [Google Scholar]
- Pikal, M. J. 1992. The role of electroosmotic flow in transdermal iontophoresis. Adv. Drug Deliv. Rev. 9:201–237. [DOI] [PubMed] [Google Scholar]
- Potts, R. O., J. A. Tamada, and M. J. Tierney. 2002. Glucose monitoring by reverse iontophoresis. Diabetes Metab. Res. Rev. 18:S49–S53. [DOI] [PubMed] [Google Scholar]
- Rao, G., R. H. Guy, P. Glikfeld, W. R. LaCourse, L. Leung, J. A. Tamada, R. O. Potts, and N. T. Azimi. 1995. Reverse iontophoresis: noninvasive glucose monitoring in vivo in humans. Pharm. Res. 12:1869–1873. [DOI] [PubMed] [Google Scholar]
- Rosdahl, H., K. Hamrin, U. Ungerstedt, and J. Henrikson. 1998. Metabolite levels in human skeletal muscle and adipose tissue studied with microdyalisis at low perfusion flow. Am. J. Physiol. 274:E939–E945. [DOI] [PubMed] [Google Scholar]
- Ruddy, S. B., and B. W. Hadzija. 1992. Iontophoretic permeability of polyethylene glycols through hairless rat skin: application of hydrodynamic theory for hindered transport through liquid-filled pores. Drug Des. Discov. 8:207–224. [PubMed] [Google Scholar]
- Santi, P., and R. H. Guy. 1996a. Reverse iontophoresis—parameters determining electroosmotic flow: I. pH and ionic strength. J. Control. Release. 38:159–165. [Google Scholar]
- Santi, P., and R. H. Guy. 1996b. Reverse iontophoresis—parameters determining electroosmotic flow: II. Electrode chamber formulation. J. Control. Release. 42:29–36. [Google Scholar]
- Sieg, A., R. H. Guy, and M. B. Delgado-Charro. 2003. Reverse iontophoresis for noninvasive glucose monitoring: the internal standard concept. J. Pharm. Sci. 92:2295–2302. [DOI] [PubMed] [Google Scholar]
- Sieg, A., R. H. Guy, and M. B. Delgado-Charro. 2004. Noninvasive glucose monitoring by reverse iontophoresis in vivo: application of the internal standard concept. Clin. Chem. 50:1383–1390. [DOI] [PubMed] [Google Scholar]
- Tamada, J. A., N. J. V. Bohannon, and R. O. Potts. 1995. Measurement of glucose in diabetics subjects using non invasive transdermal extraction. Nat. Med. 1:1198–1201. [DOI] [PubMed] [Google Scholar]
- Tierney, M. J., J. A. Tamada, R. O. Potts, L. Jovanovic, S. Garg, and Cygnus Research Team. 2001. Clinical evaluation of the GlucoWatch biographer: a continual, non-invasive glucose monitor for patients with diabetes. Biosens. Bioelectron. 16:621–629. [DOI] [PubMed] [Google Scholar]
- Yoshida, N. H., and M. S. Roberts. 1993. Solute molecular size and transdermal iontophoresis across excised human skin. J. Control. Release. 25:177–195. [Google Scholar]