

Abstract
Treatment of met-myoglobin (FeIII) with H2O2 gives rise to ferryl myoglobin, which is closely related to compound II in peroxidases. Experimental studies have given conflicting results for this species. In particular, crystallographic and extended x-ray absorption fine-structure data have shown either a short (∼170 pm) or a longer (∼190 pm) Fe–O bond, indicating either a double or a single bond. We here present a combined experimental and theoretical investigation of this species. In particular, we use quantum refinement to re-refine a crystal structure with a long bond, using 12 possible states of the active site. The states differ in the formal oxidation state of the iron ion and in the protonation of the oxygen ligand (O2−, OH−, or H2O) and the distal histidine residue (with a proton on Nδ1, Nε2, or on both atoms). Quantum refinement is essentially standard crystallographic refinement, where the molecular-mechanics potential, normally used to supplement the experimental data, is replaced by a quantum chemical calculation. Thereby, we obtain an accurate description of the active site in all the different protonation and oxidation states, and we can determine which of the 12 structures fit the experimental data best by comparing the crystallographic R-factors, electron-density maps, strain energies, and deviation from the ideal structure. The results indicate that FeIII OH− and FeIV OH− fit the experimental data almost equally well. These two states are appreciably better than the standard model of compound II, FeIV O2−. Combined with the available spectroscopic data, this indicates that compound II in myoglobin is protonated and is best described as FeIV OH−. It accepts a hydrogen bond from the distal His, which may be protonated at low pH.
INTRODUCTION
Myoglobin is the major heme-containing protein of cardiac and skeletal muscle and serves as an oxygen buffer in respiring tissues. The oxygen affinity of myoglobin lies between that of hemoglobin, which releases oxygen in passing through respiring tissues, and cytochrome c oxidase, which uses molecular oxygen in oxidative respiration. It is the high myoglobin content that gives continually respiring tissues their red color.
Myoglobin has been studied intensively for decades (Philips, 2001) and it was the first protein for which the three-dimensional structure was solved (Kendrew et al., 1960). The structure showed a globular protein, built up mainly from α-helices. The active site of myoglobin is situated in a hydrophobic pocket near the center of the molecule and consists of a prosthetic heme group (iron protoporphyrin IX) with a FeII ion in the center of a porphyrin ring, coordinated by the four nitrogen atoms of the pyrrole rings. The ion is also bound to the Nε2 atom of the conserved proximal histidine (His) residue. The sixth coordination site, completing the octahedral coordination around the iron ion, is empty or occupied by O2. A nearby His residue (the distal His) forms a hydrogen bond to the bound O2 molecule, thereby stabilizing the binding and favoring O2 compared to other competing diatomic gases, e.g., CO and NO (Spiro and Kozlowski, 2001). Mutation of the proximal His ligand or surrounding hydrophobic side chains in general leads to dissociation of the heme group (Hargrove et al., 1996).
Heme groups are also present in many other proteins. They show an impressively varied chemistry, with functions ranging from electron transfer (cytochromes) to binding and transport of small molecules and catalysis of a great wealth of reactions (e.g., peroxidases, oxidases, and catalases; see da Silvia and Williams, 1994; Kaim and Schwederski, 1994; Smith and Veitch, 1998; Matsunaga and Shir, 2004). All these proteins bind heme, but the surrounding protein and the axial ligand vary extensively between different groups of proteins and are considered to tune the properties of the heme group (Poulos, 1996).
Much effort has been put into investigations of the reaction cycles of these heme proteins. The reaction cycle of peroxidases (Gajhede, 2001; and see Fig. 1, this article) starts with the resting FeIII state, which is either six-coordinate with a water molecule (1) or five-coordinate with an empty coordination site (2). This state binds H2O2 (3), which is then deprotonated to a hydroperoxide intermediate (4). When this intermediate is re-protonated on the distal oxygen atom, the O–O bond breaks automatically in a heterolytic manner, resulting in water and compound I (5; formally FeV O2−, but one of the reducing equivalents is located in the porphyrin ring or on a nearby protein residue). Compound I is extremely reactive and can abstract an electron from almost any substrate (the reduction potential is close to 1 V; Gajhede, 2001). This results in a substrate radical and compound II (FeIV O2−; 6). Compound II is also reactive with a similar reduction potential and can abstract an electron from another substrate molecule, thereby returning to the resting state after the uptake of two protons.
FIGURE 1.
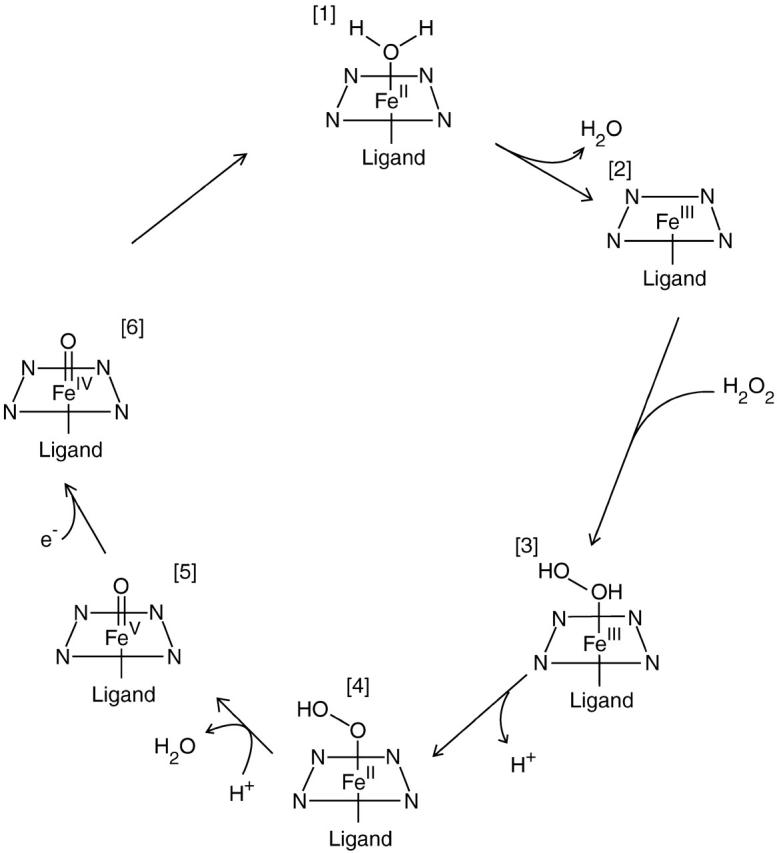
The proposed reaction cycle for peroxidases (Gajhede, 2001). Compounds I and II are complexes 5 and 6.
The normal function of myoglobin is to bind and store O2. However, it has been shown that it may also support a peroxidase-like activity, i.e., a H2O2-dependent oxidation of many substrates (Giulivi and Cadenas, 1994). During the reversible O2 binding, oxymyoglobin auto-oxidizes at a relatively slow rate (Tajima and Shikama, 1987) to yield met-myoglobin (i.e., FeIII) and a superoxide radical (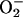 ). The latter disproportionates to H2O2 and O2. H2O2 is a reactive species and can cause potential damage. The peroxidase activity of myoglobin may take care of this problem (Giulivi and Cadenas, 1994). The reaction product of H2O2 with met-myoglobin is considered to be compound I (Keilin and Hartree, 1950). This species is labile and rapidly takes up an electron by a poorly known mechanism to form compound II (ferryl myoglobin), which is quite stable (Giulivi and Cadenas, 1994). It has also been suggested that the O–O bond may break homolytically, giving rise to compound II and a hydroxyl radical (neutral OH˙). Native myoglobins possess a lower peroxidase activity than peroxidases, which has been attributed to the absence of a distal arginine residue and the failure of the distal histidine to function as an acid-base catalyst (Matsui et al., 1999). By mutations that place the histidine in a similar distance to the heme as in the active site of peroxidases, compound I of myoglobin has been observed, probably owing to an increased rate of heterolysis (Ozaki et al., 1997; Matsui et al., 1999). However, compound I has also been observed for native myoglobin through stopped-flow rapid-scan spectrophotometry (Egawa et al., 2000).
). The latter disproportionates to H2O2 and O2. H2O2 is a reactive species and can cause potential damage. The peroxidase activity of myoglobin may take care of this problem (Giulivi and Cadenas, 1994). The reaction product of H2O2 with met-myoglobin is considered to be compound I (Keilin and Hartree, 1950). This species is labile and rapidly takes up an electron by a poorly known mechanism to form compound II (ferryl myoglobin), which is quite stable (Giulivi and Cadenas, 1994). It has also been suggested that the O–O bond may break homolytically, giving rise to compound II and a hydroxyl radical (neutral OH˙). Native myoglobins possess a lower peroxidase activity than peroxidases, which has been attributed to the absence of a distal arginine residue and the failure of the distal histidine to function as an acid-base catalyst (Matsui et al., 1999). By mutations that place the histidine in a similar distance to the heme as in the active site of peroxidases, compound I of myoglobin has been observed, probably owing to an increased rate of heterolysis (Ozaki et al., 1997; Matsui et al., 1999). However, compound I has also been observed for native myoglobin through stopped-flow rapid-scan spectrophotometry (Egawa et al., 2000).
Earlier crystallographic and extended x-ray absorption fine-structure (EXAFS) investigations have indicated that the Fe–O bond length in compound II in myoglobin, horseradish peroxidase (HRP), and catalase is 164–171 pm, i.e., close to what has been observed for compound I (Chance et al., 1986a; Penner-Hahn et al., 1986; Gouet et al., 1996), thereby indicating that it is a FeIV O2− species. However, in three recent crystal structures of compound II in myoglobin, HRP, and catalase, appreciably longer Fe–O bonds were observed, 184–192 pm (Hersleth et al., 2002; Berglund et al., 2002; Murshudov et al., 2002). Such a long Fe–O bond was also observed in a recent crystal structure of compound I in cytochrome c peroxidase (187 pm), which is electronically equivalent to compound II of HRP, because the other oxidizing equivalent is on a tryptophane residue (Bonagura et al., 2003). A similar bond length (193 pm) has also been reported in an EXAFS study of compound II in HRP (Chance et al., 1984). This was interpreted as a protonation of O2− to OH− or even as FeIII OH˙. However, this interpretation was recently questioned in another EXAFS study of chloroperoxidase and HRP, which gave a long Fe–O bond only for the former enzyme (Green et al., 2004). Considering the importance of compound II in the catalytic cycle of many different types of enzymes, it is highly interesting to settle the actual character of compound II. In this article we therefore use our recently developed quantum-refinement method (Ryde et al., 2002) to re-refine the crystal structure of compound II in myoglobin using various oxidation and protonation states of the active site, to see which fit the experimental data best.
METHODS
Quantum refinement
Quantum refinement (Ryde et al., 2002; Ryde and Nilsson, 2003b) is essentially standard crystallographic refinement supplemented by quantum chemical calculations for a small part of the protein. Crystallographic refinement programs change the protein model (coordinates, occupancies, B-factors, etc.) to improve the fit of the observed and calculated structure-factor amplitudes (usually estimated as the residual disagreement, the R-factor). Owing to the limited resolution normally obtained for biomolecules, the experimental data are supplemented by some chemical information, usually in the form of a molecular-mechanics (MM) force field (Kleywegt and Jones, 1998). Then, the refinement takes the form of a minimization or simulated annealing calculation by molecular dynamics using an energy function of the form
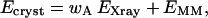 |
(1) |
where EXray is a penalty function that describes how well the model agrees with the experimental data (we have used a maximum-likelihood refinement target using amplitudes, MLF; see Pannu and Read, 1996; Adams et al., 1997). EMM is an MM energy function with bond, angle, dihedral, and nonbonded terms, and wA is a weight factor, which is necessary because EMM is in energy units whereas EXray is in arbitrary units. It determines the relative importance of the crystallographic raw data and the MM force field for the final structure.
Quantum chemistry can be introduced in this function by replacing the MM potential for a small (but interesting) part of the protein (system 1) by a quantum mechanics (QM) calculation, yielding a QM energy for system 1, EQM1. To avoid double counting we must then subtract the MM energy of system 1, EMM1,
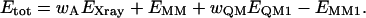 |
(2) |
Thereby, we introduce an accurate energy function for the system of interest. Such an energy function is implemented in the software ComQum-X (Ryde et al., 2002), which is a combination of the software TURBOMOLE (Ahlrichs et al., 2000) and the crystallography and NMR system (CNS) (Brünger et al., 2000). The factor wQM in Eq. 2 is another weight, which is needed because the CNS MM force field is based on a statistical analysis of crystal structures (Engh and Huber, 1991). Therefore, the force constants are not energy-derived, as is the QM term, but they are in arbitrary statistical units. Experience has shown that the CNS force constants are typically three times larger than energy-based force constants (Engh and Huber, 1991), and wQM = 3 has therefore been used throughout this work (Ryde et al., 2002).
Special attention is needed if there is a covalent bond between the QM system and the surrounding proteins. This is a well-known problem in the popular combined QM and MM methods (QM/MM; Monard and Merz, 1999; Mulholland, 2001; Ryde, 2003) (ComQum-X can also be seen as a QM/MM method with restraints to crystallographic raw data), and a simple and robust solution (Nicoll et al., 2001) is to truncate the QM system with hydrogen atoms, the positions of which are linearly related to the corresponding carbon atom in the protein (Ryde et al., 2002). Of course, EMM1 is also calculated with these hydrogen atoms, so that artifacts introduced by the hydrogen truncation may cancel out. Following crystallographic custom, protons and electrostatic interactions are ignored in the refinement, except of course in the quantum chemical calculations.
ComQum-X has been tested by re-refining the structure of N-methylmesoporphyrin bound to ferrochelatase (Ryde et al., 2002). The results showed that we may improve the structure locally in terms of the Rfree factor (the Rfree factor is an R-factor calculated for a set of randomly chosen intensities, typically ∼5%, which are set aside from the beginning and not used during the refinement; Brünger, 1993). Moreover, we have shown (Ryde and Nilsson, 2003a) that refinement with ComQum-X of a medium-resolution (170 pm) crystal structure of cytochrome c553 brings the geometry of the heme group and its ligands closer to that observed in an atomic-resolution structure (97 pm) of the same protein (Benini et al., 2000). For example, the errors in the Fe-ligand distances are reduced from 3–9, 12, and 32 pm to 1, 0, and 2 pm (for the porphyrin, histidine, and methionine ligands, respectively). We have also shown that ComQum-X can be used to deduce the protonation state of metal-bound solvent molecules in protein structures (Ryde and Nilsson, 2003b; Nilsson and Ryde, 2004). We used the catalytic zinc ion in alcohol dehydrogenase as a test case, because the pKa of the zinc-bound water molecule is known from kinetic measurements, and we were able to reproduce the correct protonation state of both an alkoxide ion and a water molecule.
Computational details
The quantum-refinement calculations and vacuum geometry optimizations were performed with the density functional Becke-Perdew86 method (Becke, 1988; Perdew, 1986). They were sped up by expanding the Coulomb interactions in an auxiliary basis set, the resolution-of-identity approximation (Eichkorn et al., 1995, 1997). These calculations employed the DZpdf basis set for iron (Schäfer et al., 1992; Sigfridsson et al., 2001) and the 6-31G* basis set for all other atoms, except the O-ligand, for which a set of diffuse functions (6-31+G*) was also included (Hehre et al., 1986).
Most of the calculations were run on the model Fe(Por)(Im)(H2O/OH−/O2−) (Im), where Por is porphine, i.e., the porphyrin ring without side chains (our previous calculations have shown that the porphyrin side chains have a minor effect on the geometry of the heme group; Sigfridsson and Ryde, 2003) and Im is imidazole, the standard model of His residues. Thus, the model includes both the proximal His ligand and the distal His residue. However, some vacuum calculations were run on a smaller complex without the model of the distal His residue.
All quantum-refinement calculations are based on the crystal structure of compound II in myoglobin at 135-pm resolution (collected at 100 K, pH 5.2; Hersleth et al., 2002). Coordinates, occupancies, B-factors, and structure factors of the structure were obtained from the Brookhaven Protein Data Bank files 1GJN. From these files we also obtained the space group, unit-cell parameters, resolution limits, R-factors, and the test set used for the evaluation of the Rfree factor.
The full protein was used in all calculations, including all crystal water molecules in the Protein Data Bank file. The full geometry of the proteins was optimized until the change in Etot was below 10−6 Hartree (2.6 J/mol) and the norm of the Cartesian gradients was below 10−3 a.u. In each cycle of the geometry optimization, the surrounding protein was allowed to relax by one cycle of crystallographic minimization and one cycle of individual B-factor refinement. However, the new coordinates and B-factors were accepted only if the Rfree factor was reduced. For the protein, we used the standard CNS force field (protein_rep.param, water_rep.param, and ion.param) and for the heme group, the parameter file from the original refinement (these parameters are of little importance, because the heme group is treated by quantum chemistry in the refinement). For the other program parameters, we used data from the Protein Data Bank files or the default choices.
The wA factor in Eq. 2, poses a special problem, because it is the only undetermined parameter in ComQum-X. In standard crystallographic refinements, it is determined so that the MM and crystallographic forces have a similar magnitude (Brünger and Rice, 1997). However, there is no warranty that this is the optimum choice, especially not if a part of the protein is supplemented by a QM energy function. Therefore, we have determined the optimum choice of wA for each system by optimizing the Rfree factor. This was done by refining each structure at (at least) five different values of wA, 0.006, 0.06, 0.25 (the default choice of CNS), 1, and 6. The Rfree factor was always lowest for wA = 0.25 or 1 and increased monotonously for higher and lower values of wA.
Relative energies of the various protein complexes were estimated by solving the Poisson-Boltzmann equation numerically using MEAD 2.2 software (Bashford, 1997; Tishmack et al., 1997). Charges for the quantum system were fitted to the electrostatic potential using the Merz-Kollman scheme (Besler et al., 1990), as implemented in Gaussian-98 software (Frisch et al., 1998). In the charge calculation, the wavefunction was perturbed by protein and solvent point charges, scaled by a factor of 4. For the rest of the protein (and side chains of the heme group), partial charges from the CHARMM param27 force field (MacKerell et al., 1998) were used. Optimized radii were used for all protein atoms in the MEAD calculations (Nina and Roux, 1997) and for the atoms that change protonation in the various calculations (the iron oxygen ligand and the distal imidazole group), a radii of 120, 155, and 150 PM were used for the H, N, and O atoms, respectively (Ullmann et al., 2002). A unity dielectric constant (ε = 1) was used for the quantum system, whereas ε = 4 in the remaining protein, and ε = 80 in the surroundings (water). These values have previously been shown to provide reasonable agreement with experimental results (Ullmann et al., 2002; Bashford et al., 1993; T. Rasmussen, K. Nilsson, and U. Ryde, unpublished).
The MEAD calculations were based on the quantum-refined geometries. All water molecules were removed. The MEAD calculations were performed with 1513 grid points, with a coarse grid with a spacing of 100 pm, centered on the geometric center of the protein, and a dense grid with a spacing of 20 pm, centered on the iron ion. The solvent energies are the average of seven calculations in which the grid origin was moved 10 pm in positive and negative directions along each Cartesian axis. The maximum difference among the four calculations is 2 kJ/mol.
The reported energies are the sum of the QM energies of the quantum system and the MEAD solvation energies of the QM system in the protein and the surrounding solvent. The intrinsic and solvation energy of the protein was assumed to be constant for the various states. The QM energies were calculated with the B3LYP function, as implemented in Gaussian-98 (Frisch et al., 1998; Hertwig and Koch, 1997), because this functional is known to give more accurate energies, especially for transition metal systems (Bauschlicher, 1995; Siegbahn and Blomberg, 2000; the same functional was used for the charge calculations). The wavefunction was perturbed by the protein and solvent point charges, scaled by a factor of 4, but the energy was calculated without these charges in a single SCF iteration.
RESULTS AND DISCUSSION
Vacuum calculations
As mentioned above, compound II is normally assumed to be FeIV O2− (which is often written as Fe=O, but we prefer to explicitly show the formal oxidation and protonation states), but the FeIV OH− and FeIII OH˙ forms have also been suggested (Hersleth et al., 2002). The aim of the present investigation is to determine the actual protonation and oxidation state of compound II in myoglobin, observed in the crystal structure of Hersleth et al. (2002). Therefore, we have studied three formal oxidation states for iron, FeIII, FeIV, and FeV, and three protonation states for water, H2O, OH−, and O2−. Based on the expected protonation states for the various ions, we decided to study the following six combinations: FeIII H2O, FeIII OH−, FeIII O2−, FeIV OH−, FeIV O2−, and FeV O2−.
To get a first feeling of the optimum structures of these complexes and also to get some insight in their most stable spin states (some have not been observed experimentally), we optimized the structures of the three reasonable spin states (high-, intermediate-, and low-spin), using the Fe(Por)(Im)(H2O/OH−/O2−) model (Fig. 2). The results of these calculations are shown in Table 1.
FIGURE 2.

The small Fe(Por)(Im)(OH−) model of the heme active site used in some of the vacuum calculations.
TABLE 1.
Vacuum structures of the Fe(Por)(His)(O/OH/H2O) model and relative energies of the various spin states (ΔE)
| Ox. state | # Unpaired spin | Distance to Fe (pm)
|
ΔE kJ/mol | |||
|---|---|---|---|---|---|---|
| Ligand | NPor | NHis | O | |||
| III | H2O | 5 | 205–208 | 215 | 240 | |
| III | OH− | 1 | 201–202 | 206 | 182 | 0.0 |
| III | 3 | 202–203 | 261 | 191 | 75.6 | |
| III | 5 | 208–210 | 256 | 188 | 106.2 | |
| III | O2−* | 1 | 203–204 | 219 | 166 | 0.0 |
| III | 3 | 203–204 | 218 | 165 | 5.8 | |
| IV | OH− | 0† | 196–205 | 204 | 179 | 24.2 |
| IV | 2 | 196–203 | 205 | 178 | 0.0 | |
| IV | 4 | 196–213 | 218 | 183 | 101.2 | |
| IV | H2O | 0† | 200 | 194 | 205 | 3.1 |
| IV | 0 | 194–204 | 194 | 204 | 38.2 | |
| IV | 2 | 198–199 | 194 | 205 | 0.0 | |
| IV | 4 | 198 | 218 | 241 | 22.2 | |
| IV | O2− | 0† | 202 | 217 | 165 | 32.0 |
| IV | 2 | 202 | 217 | 165 | 0.0 | |
| IV | 4 | 209 | 217 | 164 | 90.5 | |
| V | OH− | 1 | 201–202 | 205 | 181 | 0.0 |
| V | 3 | 202–203 | 262 | 191 | 76.1 | |
| V | 5 | 208–210 | 257 | 188 | 106.6 | |
| V | O2− | 1 | 197–206 | 213 | 163 | 5.5 |
| V | 3 | 199 | 213 | 165 | 0.0 | |
| V | 5 | 199 | 214 | 165 | 181.5 | |
| Crystal | 197–205 | 214 | 192 | |||
The His ligand dissociates in the sextet state.
Open-shell singlet.
FeIII H2O (met-myoglobin) is experimentally known to be in the high-spin (sextet) state (Hersleth et al., 2002). The FeIII OH− model is most stable in the low-spin (doublet) state (Sitter et al., 1988), as can be seen in Table 1, whereas the FeIII O2− model has nearly degenerate double and quartet states (the former is 6 kJ/mol more stable). For the FeIV models, the triplet (intermediate-spin) state was found to be lowest in energy. Again, this is in accordance with experimental data for compound II in myoglobin, as well as in peroxidases (Foote et al., 1989). For the compound I model (FeV O2−), the doublet and quartet states are almost degenerate, but in our calculations, the quartet is slightly lower in energy. In the peroxidases, the doublet state is observed experimentally, with a J-coupling constant of only 0–10 cm−1 (Green, 2001) to the radical on heme or on an amino acid. Since the axial ligation differs between myoglobin and peroxidases (the latter has a carboxylate group hydrogen-bonded to the His ligand, giving it some imidazolate character), and since the two states have very similar geometries, we decided to run all FeV O2− calculations with the quartet state. In the following, we will only discuss these spin states.
The geometries of the various complexes are also shown in Table 1. For the most stable spin states, we get a Fe–O distance of 240 pm for FeIII H2O, 182 pm for FeIII OH−, 166 pm for FeIII O2−, 178 pm for FeIV OH−, 165 pm for FeIV O2−, and 163–165 pm for FeV O2− Thus, based on these data, neither of the models give a Fe–O distance close to that observed in the crystal structure of compound II in myoglobin (192 pm), but that of FeIII OH− is closest to it.
Interestingly, the crystal structure shows a long Fe–NHis bond length of 214 pm. This is appreciably longer than in the OH− models (204–206 pm), but similar to what is found for the H2O and O2− complexes (213–219 pm). Thus, these small models give little explanation to the observed crystal structure.
The results of these vacuum calculations are similar to those obtained in earlier studies of similar sites (Ghosh et al., 1994; Antony et al., 1997; Kuramochi et al., 1997; Wirstam et al., 1999; Harris, 2001; Harris and Loew, 2001; Rovira and Fita, 2003; Rydberg et al., 2004). In particular, a recent study of compound II in catalase and peroxidase gave Fe–O distances of 177 pm when protonated and 168–169 pm when unprotonated (Rovira and Fita, 2003).
As mentioned above, the distal His forms a hydrogen bond to the oxygen ligand in myoglobin via the Nε2 atom. It is conceivable that such a hydrogen bond may modify the Fe–O distance. Therefore, we added a model of the distal His to the quantum system, yielding the Fe(Por)(Im)(H2O/OH−/O2−)(Im) model (Fig. 3). Unfortunately, the protonation state of a His residue is not clear from crystal structures; it can be protonated either on the Nδ1 atom, the Nε2 atom, or on both atoms. We will call these three possibilities HID, HIE, and HIP. Clearly, if there is a water ligand, the distal His residue must be of the HID type, to accept a hydrogen bond (Fig. 3 a), whereas if the ligand is O2−, it must be either HIE or HIP, to donate a hydrogen bond. However, for the OH− ligand, all three protonation states are possible. This gives us 12 different models of the active site, which were all optimized and are described in Table 2. For the FeIV H2O–HIE state, the proton moved from H2O to HIE, forming the FeIV OH− HIP complex. Likewise, the FeIII O2− HIP complex reorganized to the FeIII OH− HIE complex.
FIGURE 3.

The Fe(Por)(Im)(OH−)(Im) model of the active heme site used in both the vacuum and quantum-refinement calculations. The three figures illustrate the three possible protonation states of the distal His model—HID, HIE, and HIP.
TABLE 2.
Vacuum structures of the Fe(Por)(Im)(O/OH/H2O)(Im) model
| Distance to Fe (PM)
|
||||||
|---|---|---|---|---|---|---|
| Ox. state | Ligand | His | NPor | NHis | O | O–N/H |
| III | H2O | HID | 205–209 | 222 | 219 | 267/104 |
| III | OH− | HID | 201–202 | 207 | 180 | 292/100 |
| III | HIE | 201–202 | 206 | 184 | 282/178 | |
| III* | HIP | 201–202 | 201 | 188 | 255/148 | |
| III | O2− | HIE | 201–203 | 219 | 170 | 271/165 |
| IV* | H2O | HID | 196–202 | 197 | 194 | 253/105 |
| IV | OH− | HID | 201–202 | 209 | 171 | 253/111 |
| IV | HIE | 198–202 | 205 | 182 | 284/180 | |
| IV | HIP | 195–203 | 201 | 186 | 268/161 | |
| IV | Arg | 201–204 | 200 | 189 | 269/163 | |
| IV | O2− | HIE | 202–203 | 217 | 166 | 286/182 |
| IV | HIP | 202–203 | 213 | 168 | 255/142 | |
| IV | Arg | 202 | 212 | 167 | 277/172 | |
| V | O2− | HIE | 200–201 | 211 | 166 | 282/179 |
| V | HIP | 200 | 209 | 167 | 270/164 | |
| Crystal | 197–205 | 214 | 192 | 270/− | ||
Fixed N–H distance.
From this table, it can be seen that the hydrogen bond has a quite strong influence on the Fe–O distance. Thus, if the O-ligand is a donator (the HID structures), the Fe–O distances decrease by 2–9 pm, whereas when it is an acceptor, the Fe–O distance increases by 1–8 pm, more for OH− than O2− and also more for HIP than HIE. Therefore, the Fe–O bonds in the FeIV OH− HIP (186 pm) and FeIII OH− HIE structures (184 pm) are only 6–8 pm shorter than in the crystal structure.
However, the hydrogen bond to the distal His residue also affects the (quite flexible) distance to the proximal His residue. The HIP models give the shortest bonds and the HID models the longest with a variation of up to 8 pm. As with the small model, it is in general the models with the shortest Fe–O bond that give a Fe–NHis bond length most similar to the crystal structure. Yet, the FeIV OH−/HID model also gives a quite reasonable Fe–NHis bond length (209 pm).
This indicates that there actually may be a reasonable interpretation of the crystal structure, although the direct comparison with vacuum structures cannot unambiguously point out the best candidate because different models give the best results for the Fe–O and Fe–NHis bonds. If we simply take the sum of the absolute deviations of these two bonds, the FeIV OH− HIP (16 pm) and FeIII OH− HIE (17 pm) models give the best results. However, such a treatment does not take into account the other bond lengths (and angles) in the structure, nor does it take into account the relative strength of the bonds (a Fe–O2− bond is much stiffer than a Fe–H2O bond). All this is considered in the quantum refinement procedure, which can be seen as a quantum chemical method with restraints to the crystallographic raw data. Thus, it also disregards possible errors in the crystallographic coordinates (which are not the raw data but the result of an involved process of interpretation) and it considers how well the positions of the various atoms are determined (giving a stronger restraint to a well-determined atom than to a more poorly determined one), thereby obtaining an optimum compromise between crystallography and quantum chemistry.
Finally, we also tested if the distal residues in heme peroxidases have a similar effect. Thus, we added CH3NHC(NH2)2 as a model of the distal arginine (Arg) residue in these enzymes. To ensure that this model forms a hydrogen bond to the oxo group only with the inner nitrogen atom, we also added a water molecule, bridging the oxo group and the terminal nitrogen atom of the Arg model. The results of these optimizations (for FeIV OH− and FeIV O2−) are also shown in Table 2 (Arg entries). It can be seen that the geometries are similar to those obtained with the His models, especially the charged HIP model; the Fe–O bond is 167 pm for the FeIV O2− structure, but 189 pm in the FeIV OH− structure. The latter estimate is close to the distance observed in the recent crystal structures of compound II in HRP and compound I in cytochrome c peroxidase (184–187 PM) (Berglund et al., 2002; Bonagura et al., 2003). This shows that myoglobin and peroxidase models behave in a similar manner.
Quantum refinement
We re-refined the crystal structure of compound II in myoglobin (Hersleth et al., 2002) using the same 12 models of the heme site as in the vacuum calculations. All 12 models converged to distinct structures, but two states, which we also tried, FeIV H2O HID and FeIII O2− HIP, converged to FeIV OH− HIP and FeIII OH− HID by an internal proton transfer. All structures were optimized for a range of values for the wA factor, to find the optimum choice. For all models, either wA = 1 or the default wA = 0.25 gave the minimum in Rfree factor and these results are presented in Table 3. It should be noted that most of the entries in this table are comparable only if the same value of wA has been used.
TABLE 3.
ComQum-X results for the active site in myoglobin compound II (Hersleth et al., 2002)
| Distance to Fe (PM)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ox. state | Ligand | His | wA | ΔEQM1 kJ/mol | NPor | NHis | O | O–N/H | Rfree | R |
| III | H2O | HID | 0.25 | 41.9 | 202–209 | 212 | 206 | 259/107 | 0.20594 | 0.18058 |
| III | 1.0 | 56.2 | 199–206 | 210 | 200 | 262/107 | 0.20572 | 0.18020 | ||
| III | OH− | HID | 0.25 | 22.8 | 199–204 | 209 | 182 | 277/100 | 0.20560 | 0.18041 |
| III | 1.0 | 34.9 | 197–203 | 208 | 185 | 272/100 | 0.20561 | 0.18020 | ||
| III | HIE | 0.25 | 33.4 | 198–204 | 207 | 185 | 273/171 | 0.20565 | 0.18048 | |
| III | 1.0 | 34.6 | 196–203 | 208 | 188 | 271/168 | 0.20568 | 0.18020 | ||
| III | HIP | 0.25 | 41.9 | 199–203 | 205 | 191 | 259/143 | 0.20594 | 0.18047 | |
| III | 1.0 | 44.8 | 197–203 | 207 | 192 | 264/150 | 0.20577 | 0.18018 | ||
| III | O2− | HIE | 0.25 | 34.1 | 199–204 | 211 | 174 | 273/167 | 0.20587 | 0.18068 |
| III | 1.0 | 44.0 | 197–203 | 209 | 182 | 271/163 | 0.20579 | 0.18027 | ||
| IV | OH− | HID | 0.25 | 40.9 | 198–204 | 209 | 177 | 271/105 | 0.20573 | 0.18055 |
| IV | 1.0 | 47.9 | 197–203 | 209 | 183 | 271/104 | 0.20569 | 0.18024 | ||
| IV | HIE | 0.25 | 32.8 | 197–204 | 207 | 184 | 273/170 | 0.20567 | 0.18050 | |
| IV | 1.0 | 34.4 | 196–203 | 208 | 187 | 270/168 | 0.20570 | 0.18027 | ||
| IV | HIP | 0.25 | 44.9 | 196–205 | 206 | 186 | 270/164 | 0.20564 | 0.18047 | |
| IV | 1.0 | 45.9 | 195–204 | 207 | 189 | 270/163 | 0.20565 | 0.18020 | ||
| IV | O2− | HIE | 0.25 | 33.3 | 200–205 | 211 | 169 | 280/177 | 0.20587 | 0.18075 |
| IV | 1.0 | 46.2 | 197–204 | 210 | 176 | 276/173 | 0.20579 | 0.18037 | ||
| IV | HIP | 0.25 | 129.3 | 200–204 | 211 | 171 | 270/160 | 0.20572 | 0.18068 | |
| IV | 1.0 | 152.2 | 197–204 | 210 | 179 | 271/161 | 0.20567 | 0.18032 | ||
| V | O2− | HIE | 0.25 | 41.4 | 200–204 | 210 | 169 | 277/174 | 0.20590 | 0.18075 |
| V | 1.0 | 51.5 | 197–204 | 209 | 176 | 274/170 | 0.20583 | 0.18037 | ||
| V | HIP | 0.25 | 125.1 | 200–204 | 210 | 170 | 275/167 | 0.20570 | 0.18062 | |
| V | 1.0 | 146.5 | 197–203 | 209 | 178 | 274/167 | 0.20570 | 0.18033 | ||
| Crystal | 197–205 | 214 | 192 | 270/− | 0.20593 | 0.18010 | ||||
Fe–ligand distances, strain energies (ΔEQM1), and R-factors are tabulated for the various oxidation and protonation states.
We will use six different criteria to evaluate which of the structures fit the crystal data best. These are the R and Rfree factors, strain energies, the divergence of the Fe–O and Fe–NHis distances from those observed in vacuum, and electron-density maps. Strain energies (ΔEQM1 in Table 3) are calculated as the difference in QM energy of the active site optimized in vacuum or in the protein, and therefore measures how much the structure has to be distorted to fit into the experimental electron density.
All calculations except one give improved structures in terms of the Rfree factor. However, the reduction (and variation between the various structures) is very small, from 0.2056 to 0.2059. This is quite normal for the quantum refinement (Ryde and Nilsson, 2003b) and reflects that the Rfree factor is a global property of the whole crystal structure, and we make only small changes for 57 atoms (out of 1412) in the active site. Our previous calculations (Ryde and Nilsson, 2003b) have shown that even if the variation in the Rfree factor is small, it is reproducible and makes sense (e.g., Rfree shows a nice minimum when the wA factor is varied). Therefore, we have some confidence to compare Rfree factors obtained for the various models.
From Table 3 it can be seen that the lowest Rfree factors are obtained for FeIII OH− (especially with HID and HIE) and FeIV OH− (especially with HIP), whereas FeIII H2O, FeIII O2−, FeIV O2−, and FeV O2− give high values.
The standard R-factor shows a similar variation, but it is always slightly higher than in the original crystal structure (0.1802–0.1807, compared to 0.1801). This actually also flags an improvement, because both the Rfree factor and the difference between Rfree and R should be minimized. The R-factor is smallest for FeIII OH− HID and FeIII H2O HID, whereas the O2− models give the largest values. The R-factor is always lower for wA = 1 than for wA = 0.25, reflecting the stronger restraints toward the crystallographic data in the former calculations.
The strain energy (ΔEQM1) is lowest (23–35 kJ/mol) for the FeIII OH− HID/HIE and FeIV OH− HIE structures. Most of the other structures have a strain energy of 40–50 kJ/mol, but the two FeIV/FeV O2− HIP structures have an appreciably higher strain energy (125–152 kJ/mol).
The Fe–O distances show a quite appreciable variation for the various models, 169–206 pm. This reflects that this bond is quite stiff in the quantum chemical calculations, especially for O2− and OH−. It is notable that all structures, except those with FeIII H2O, give Fe–O bonds shorter than those in the original crystal structure, although the difference for FeIII OH− HIP is not large. The stiffness of this bond is also reflected in the small deviation between the ComQum-X and vacuum distances for the various models. It varies between 1 and 12 pm (19 pm for FeIII H2O) and is smallest for FeIII OH− HIE and FeIV OH− HIP. As usual, the O2− structures (but also the FeIV OH− HID structure) give the worst results.
The Fe–NHis distance shows an even smaller variation, 205–212 pm, reflecting that it is quite flexible in the quantum chemical calculations. It is notable that it is always shorter than in the original crystal structure, indicating that this distance was somewhat too long. Interestingly, the smallest deviation from the vacuum structures is obtained for the FeV O2− and the FeIV OH− HID structures, i.e., those that gave the worst Fe–O bonds and strain energies. This reflects the inconsistency between the Fe–O and Fe–NHis bonds, observed already for the vacuum structures.
If we consider all of these results, it is clear that the normal interpretation of compound II, FeIV O2− fits the crystal data worst. It is also clear that FeIII O2− and FeV O2− are poor interpretations of the crystal structure. On the other hand, FeIII OH− HID and HIE seem to give the best results, both being always among the best structures with all criteria. The FeIV OH− HIE and HIP structures also seem to give a reasonable fit to the crystallographic data.
These conclusions are confirmed by an inspection of the electron-density maps. Fig. 4 shows a comparison of the original crystal structure and the re-refined structure with the FeIV OH− HIE model. Only small differences between the two structures can be seen. This figure also shows the fo–fc difference map of the original crystal structure at the ±3σ level, which has quite a few features of both positive and negative residual density. However, if these are compared with the difference maps of two ComQum-X structures (the FeIII OH− HIE and FeIV OH− HIE models), shown in Fig. 5, a strong improvement (less residual density) can be seen for both structures. On the other hand, it is very hard to distinguish between the two ComQum-X structures because they show similar features of almost equal volumes.
FIGURE 4.

A comparison of the original crystal structure (magenta) and the re-refined structure of FeIV OH− HIE. Also included is the fo−fc difference map of the original crystal structure at the ±3.0σ level (cyan and yellow).
FIGURE 5.
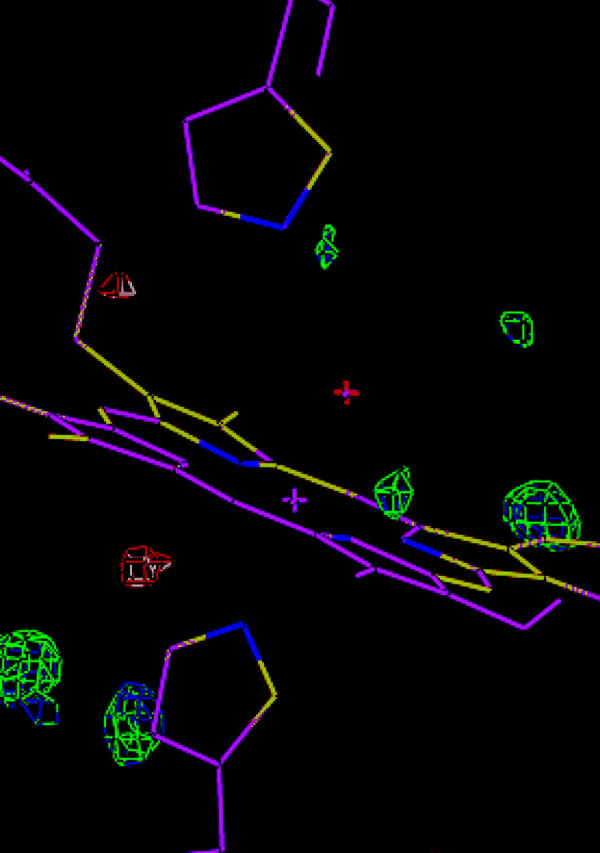
A comparison of the ComQum-X structures of the FeIII OH− HIE (magenta) and FeIV OH− HIE models. Also included are the fo−fc difference maps of the two structures at the ±3.0σ level (blue and white for FeIII and green and red for FeIV).
Concluding remarks
We have studied compound II in myoglobin by re-refining a 135-pm resolution crystal structure using 12 different protonation and oxidation states of the iron ion, the O-ligand, and the distal His residue. The results clearly show that the crystal structure does not contain a FeIV O2− complex. Instead, it seems to contain a FeIII OH− or FeIV OH− complex. However, none of the tested models fit perfectly into the electron density. Instead, there is an inconsistency between the observed Fe–O and Fe–NHis distances, which are not reproduced by any of the tested vacuum structures. This may indicate that the crystal structure is actually a mixture of at least two different oxidation or protonation states. Alternatively, the surrounding of the proximal His ligand in myoglobin may stabilize an elongated Fe–NHis bond length (Chance et al., 1986a).
The basis of our quantum refinement method is of course that our quantum chemical calculations give accurate results. Previous investigations and calibrations have shown that this is the case for metal complexes studied with the present density-functional method and basis set (Ryde and Nilsson, 2003b). In particular, the method reproduces Fe–NPor, Fe–NHis, and Fe–SMet bond lengths in cytochrome models within 3, 2, and 3 pm, respectively, and reproduce Fe–ligand bond lengths in a 95-pm crystal structure of cytochrome c553 to within 3 pm (Ryde and Nilsson, 2003a).
In the present investigation, we see that our optimized Fe–O distance for compound I (FeV O2−), 165–167 pm, is in excellent accordance with experimental (crystallographic and EXAFS) data, 165–172 pm, for cytochrome P450, catalase, cytochrome c peroxidase, and HRP (Schlichting et al., 2000; Chance et al., 1986b; Gouet et al., 1996; Green et al., 2004). Moreover, our results reproduce the expectation that deprotonated compound II (FeIV O2−) should have a similar Fe–O distance (165–168 pm; it is 164–171 pm in a crystal structure of catalase and EXAFS structures of myoglobin and HRP; Chance et al., 1986a; Penner-Hahn et al., 1986; Gouet et al., 1996). Therefore, we have some confidence that our calculated structures can be trusted to within 3–5 pm.
Some of the systems studied have the same atoms and the same number of electrons. Therefore, their energies are comparable. It can be of some interest to compare these energies to get a feeling of the intrinsic stabilities and how they are changed in the protein. First, the HIE form of FeIII (i.e., where OH− is the acceptor of a hydrogen bond from the distal His) is 28 kJ/mol more stable than the HID form in vacuum. In the protein this has increased to 95 kJ/mol (for the structures obtained with wA = 0.25). For FeIV, the HID form is most stable both in vacuum (by 46 kJ/mol) and in the protein (by 5 kJ/mol). Finally, we can also compare FeIII H2O HID with FeIII OH− HIP. It turns out that the latter form is most stable, both in vacuum and in the protein (by 83 and 23 kJ/mol, respectively).
In the original article of the crystal structure (Hersleth et al., 2002), it was suggested that the compound II structure represents a FeIII OH˙ state or possibly FeIV OH−. This conclusion was based on the long Fe–O distance and the short hydrogen bond to the distal His residue (N–O distance of 270 pm). In our vacuum calculations, the N–O hydrogen-bond length varies quite appreciably, e.g., from 252 to 293 pm in vacuum. All distances for HIE are ∼285 pm, whereas those of HID and HIP vary much more. The length of the H-acceptor interaction is 142–182 pm, indicating a quite strong interaction. In the ComQum-X structures, the variation is smaller, 259–280 pm—i.e., quite well centered around the value in the original structure. Thus, our results give no evidence that the oxidation and protonation state of the active site can be determined from the hydrogen-bond length.
The suggestion that the species observed is FeIII OH˙ also needs some comments. This state involves the same nuclei and the same number of electrons as the FeIV OH− state. The only difference is the transfer of an electron from FeIII to OH˙. Thus, they are two electronic states of the same complex. In our calculations, one of them should therefore come out as the ground state, whereas the other is an excited state. To show the electronic structure of the various states, we have calculated the spin density of the different groups in the various complexes in Table 4. It can be seen that the FeIV OH− complexes show a spin population of 1.39–1.55 on iron. This is intermediate between what is expected for FeIII and FeIV. The spin population on OH− varies with the distal His model. In the HIE and HIP complexes, there is 0.13–0.17 e on OH−, indicating almost a pure OH− ion. The rest of the spin is located either in the porphyrin ring or on the distal His model. However, in the HID complex there are 0.46 unpaired electrons on the oxygen ligands, indicating that its electronic structure is almost between OH− and OH˙. These trends remain also in the ComQum-X structures, even if the length of the hydrogen bond is almost identical in these. We have made many attempts to find a pure wavefunction that corresponds to the FeIII OH˙ description, but without any success. Thus, it is hard to discern between the FeIII OH˙ and FeIV OH− descriptions and it may actually be that the electron density is so delocalized that the truth is in between these two descriptions (as was seen for the HID complex). Moreover, it can be expected that the character varies significantly with small changes in the surroundings. A conclusive description of the actual electronic structure of the states can only be obtained with more advanced, multiconfigurational methods.
TABLE 4.
Spin densities of the Fe(Por)(Im)(H2O/OH−/O2−)(Im) vacuum models
| Spin density on
|
|||||||
|---|---|---|---|---|---|---|---|
| Ox. state | Ligand | His | Fe | O-ligand | Por | Improx | Imdist |
| III | H2O | 4.13 | 0.03 | 0.72 | 0.11 | ||
| III | HID | 4.16 | 0.07 | 0.66 | 0.10 | 0.01 | |
| III | OH− | 0.87 | 0.21 | −0.07 | 0.00 | ||
| III | HID | 0.84 | 0.24 | −0.08 | −0.01 | 0.00 | |
| III | HIE | 0.92 | 0.14 | −0.06 | −0.01 | 0.00 | |
| III | HIP | 0.97 | 0.08 | −0.05 | 0.00 | 0.00 | |
| III | O2− | 1.12 | 0.79 | −0.90 | −0.01 | ||
| III | HIE | 1.05 | 0.63 | −0.67 | −0.02 | 0.00 | |
| IV | H2O | 1.07 | 0.01 | 0.92 | 0.01 | ||
| IV | HID | 1.21 | 0.03 | 0.75 | 0.00 | 0.00 | |
| IV | OH− | 1.53 | 0.26 | 0.21 | 0.00 | ||
| IV | HID | 1.55 | 0.45 | −0.02 | −0.01 | 0.03 | |
| IV | HIE | 1.43 | 0.17 | 0.12 | −0.01 | 0.29 | |
| IV | HIP | 1.39 | 0.13 | 0.48 | 0.00 | 0.00 | |
| IV | O2− | 1.17 | 0.89 | −0.05 | −0.01 | ||
| IV | HIE | 1.25 | 0.81 | −0.05 | −0.01 | 0.00 | |
| IV | HIP | 1.37 | 0.65 | −0.06 | 0.02 | 0.02 | |
| V | OH− | 0.87 | 0.21 | −0.07 | −0.01 | ||
| V | O2− | 1.43 | 0.94 | 0.63 | 0.00 | ||
| V | HIE | 1.38 | 0.84 | 0.44 | 0.00 | 0.33 | |
| V | HIP | 1.16 | 0.82 | 0.99 | 0.05 | −0.01 | |
The fact that the crystal should contain compound II and not compound I or FeIII is based on an investigation of the electron paramagnetic resonance spectrum, which shows that the FeIII signal disappears when treating the crystals with H2O2 (Hersleth et al., 2002). It is replaced by a compound I signal, but it amounts to <2% of the original signal, indicating that the majority of the sites are in a different state with an even number of electrons. The conclusion that it is compound II is also supported by the change in color of the crystal (Hersleth et al., 2002). It has been checked by microspectrophotometry that the oxidation state of myoglobin compound II crystals does not change significantly during data collection at SNBL/BM1 at the European Synchrotron Radiation Facility in Grenoble, France.
The experimental evidence for compound II in heme proteins is somewhat messy. As mentioned before, a crystal structure of compound II in catalase (Gouet et al., 1996) gives a short Fe–O bond of 171 pm, indicating the FeIV O2− state. However, the present crystal structure, two recent structures of compound II in HRP and catalase, as well as the crystal structure of compound I in cytochrome c peroxidase (with the extra oxidizing equivalent on a tryptophane radical), indicate a longer bond, 184–192 pm (Berglund et al., 2002; Hersleth et al., 2002; Murshudov et al., 2002; Bonagura et al., 2003). These long bonds were interpreted as a single Fe–O bond caused by the protonation of the oxygen atom. Likewise, there are three EXAFS studies that indicate a short Fe–O bond of compound II in myoglobin and HRP (164–170 pm; Chance et al., 1986a; Penner-Hahn et al., 1986; Green et al., 2004), but there is also another study on HRP that gave a long Fe–O bond (193 pm; Chance et al., 1984). The two spectra from HRP are clearly different, showing that the difference is not caused by errors in the fitting procedure (Penner-Hahn et al., 1986). Thus, at least two forms of compound II seem to exist. A long EXAFS Fe–O bond (182 pm) was also found for compound II in chloroperoxidase (with an axial cysteine Fe ligand), but the same authors obtained a short bond for HRP (170 pm) and they question the long bonds observed in the other enzymes (Green et al., 2004). Thus, the nature of compound II in heme proteins is still controversial.
The situation is even more complicated by the observation of two pH-dependent states of compound II in peroxidases by resonance Raman and variable-temperature magnetic circular dichroism (Paeng and Kincaid, 1988; Sitter et al., 1985; Andersson and Dawson, 1991; Foote et al., 1989). For HRP, the transition between the two states is around pH 8.5 and the alkaline form has a higher frequency of the Fe–O bond (788 compared to 776 cm−1; Sitter et al., 1988). Both pH states are suggested to be FeIV O2−, but at high pH, there should be no hydrogen bond to the distal His residue (the Fe–O frequency is insensitive to deuterium exchange; Sitter et al., 1988). For myoglobin, a similar transition is observed around pH 3.5 and the form at normal pH is most similar to the alkaline form of HRP compound II (Sitter et al., 1985; Foote et al., 1989). The low-pH form is highly unstable and has been interpreted as having a protonated distal His residue that swings out into the solvent, making the heme site more exposed (Foote et al., 1989). Thus, these two pH forms cannot explain the differences in the Fe–O distance, observed by EXAFS and crystallography (concerning the protonation of the iron-bound water molecule).
This is in accordance with the resonance Raman spectrum of our myoglobin crystals and solution (T. Uchida, H.-P. Hersleth, T. Kitagawa, K. K. Andersson, unpublished data): It shows an appreciably lower Fe–O frequency (below 700 cm−1), in agreement with the long Fe–O bond.
If we take all this experimental data together with our results and trust that the crystal is not reduced during data collection, the most probable interpretation of compound II in our crystals of myoglobin seems to be the FeIV OH− state in the HIE or HIP forms, in which the distal His donates a hydrogen bond to the iron-bound OH− group (these two states also fit the crystal structure best). It is possible that the distal His group protonates in the physiological pH range, which could explain the variation observed in the O–N distance between the O-ligand and the distal His group in crystal structures collected at pH 5.2 (270 pm), 6.8 (288 pm), and 8.7 (294 pm; H.-P. Hersleth, C. H. Görbitz, K. K. Andersson, unpublished data; Hersleth et al., 2001). These structures show no significant changes in the other structural parameters. This interpretation is in accordance with a 16-pm increase in the O–N distance for the FeIV OH− HIE/HIP models in Table 2.
Supplementary Material
Acknowledgments
We are grateful for help from the team at the Swiss-Norwegian Beam Line/BM1 at the European Synchrotron Radiation Facility, Grenoble, France.
This investigation has been supported by grants from the Crafoord Foundation and the Swedish Research Council. It has also been supported by computer resources of Lunarc at Lund University.
References
- Adams, P. D., N. S. Pannu, R. J. Read, and A. T. Brunger. 1997. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc. Natl. Acad. Sci. USA. 94:5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlrichs, R., M. Bär, H.-P. Baron, R. Bauernschmitt, S. Böcker, M. Ehrig, K. Eichkorn, S. Elliott, F. Haase, M. Häser, H. Horn, C. Huber, C. Kölmel, M. Kollwitz, C. Ochsenfeld, H. Öhm, A. Schäfer, U. Schneider, O. Treutler, M. von Arnim, F. Weigend, P. Weis, and H. Weiss. 2000. TURBOMOLE. Universität Karlsruhe, Germany.
- Andersson, L. A., and J. H. Dawson. 1991. EXAFS spectroscopy of heme-containing oxygenases and peroxidases. Struct. Bond. 74:1–40. [Google Scholar]
- Antony, J., M. Grodzicki, and A. X. Trautwein. 1997. Local density functional study of oxoironIV porphyrin complexes and their one-electron oxidized derivatives. Axial ligand effects. J. Phys. Chem. A. 101:2692–2701. [Google Scholar]
- Bashford, D. 1997. An object-oriented programming suite for electrostatic effects in biological molecules. In Scientific Computing in Object-Oriented Parallel Environments, Lecture Notes in Computer Science, Vol. 134. Y. Ishikawa, R.R. Oldehoeft, J.V.W. Reynders, and M. Tholburn, editors. Springer, Berlin, Germany. 233–240.
- Bashford, D., D. A. Case, C. Dalvit, L. Tennant, and P. E. Wright. 1993. Electrostatic calculations of side-chain pKa values in myoglobin and comparison with NMR data for histidines. Biochemistry. 32:8045–8056. [DOI] [PubMed] [Google Scholar]
- Bauschlicher, C. W. 1995. A comparison of the accuracy of different functionals. Chem. Phys. Lett. 246:40–44. [Google Scholar]
- Becke, A. 1988. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 38:3098–3100. [DOI] [PubMed] [Google Scholar]
- Benini, S., A. González, W. R. Rypniewski, K. S. Wilson, J. J. Van Beeumen, and S. Ciurli. 2000. Crystal structure of oxidized Bacillus pasteurii cytochrome c553 at 0.97-Å resolution. Biochemistry. 39:13115–13126. [DOI] [PubMed] [Google Scholar]
- Berglund, G. I., G. H. Carlsson, A. T. Smith, H. Szörke, A. Henriksen, and J. Hajdu. 2002. The catalytic pathway of horseradish peroxidase at high resolution. Nature. 417:463–468. [DOI] [PubMed] [Google Scholar]
- Besler, B. H., K. M. Merz, and P. A. Kollman. 1990. Atomic charges derived from semiempirical methods. J. Comput. Chem. 11:431–439. [Google Scholar]
- Bonagura, C. A., B. Bhaskar, H. Shimizu, H. Li, M. Sundaramoorthy, D. E. McRee, D. B. Goodin, and T. L. Poulos. 2003. High-resolution crystal structures and spectroscopy of native and compound I cytochrome c peroxidase. Biochemistry. 42:5600–5608. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T. 1993. Assessment of phase accuracy by cross validation: the free R-value. Methods and applications. Acta Crystallogr. D49:24–36. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., and L. M. Rice. 1997. Crystallographic refinement by simulated annealing. Methods and applications. Meth. Enzymol. 277:243–269. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., P. D. Adams, G. M. Clore, W. L. Delano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, M. Niges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 2000. Crystallography and NMR System CNS, Ver. 1.0. Yale University, New Haven, CT. [DOI] [PubMed]
- Chance, M., L. Powers, Y. Chiang, T. Poulos, G. R. Schonbaum, I. Yamazaki, and K. G. Paul. 1984. X-ray absorption studies of intermediates in peroxidase activity. Arch. Biochem. Biophys. 235:596–611. [DOI] [PubMed] [Google Scholar]
- Chance, M., L. Powers, C. Kumar, and B. Chance. 1986a. X-ray absorption studies of myoglobin peroxide reveal functional differences between globins and heme enzymes. Biochemistry. 25:1259–1265. [DOI] [PubMed] [Google Scholar]
- Chance, M., L. Powers, T. Poulos, and B. Chance. 1986b. Cytochrome c peroxidase compound ES is identical with horseradish peroxidase compound I in iron-ligand distances. Biochemistry. 25:1266–1270. [DOI] [PubMed] [Google Scholar]
- da Silvia, J. J. R. F., and P. J. Williams. 1994. The Biological Chemistry of the Elements. Clarendon Press, Oxford, UK.
- Egawa, T., H. Shimada, and Y. Ishimura. 2000. Formation of compound I in the reaction of native myoglobin with hydrogen peroxide. J. Biol. Chem. 275:34858–34866. [DOI] [PubMed] [Google Scholar]
- Eichkorn, K., O. Treutler, H. Öhm, M. Häser, and R. Ahlrichs. 1995. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 240:283–290. [Google Scholar]
- Eichkorn, K., F. Weigend, O. Treutler, and R. Ahlrichs. 1997. Auxiliary basis sets for main row and transition metals and their use to approximate Coulomb potentials. Theor. Chim. Acta. 97:119–124. [Google Scholar]
- Engh, R. A., and R. Huber. 1991. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Crystallogr. A47:392–400. [Google Scholar]
- Foote, N., P. M. A. Gadsby, C. Greenwood, and A. J. Thompson. 1989. pH-dependent forms of the ferryl heme in myoglobin peroxide analysed by variable-temperature magnetic circular dichroism. J. Biol. Chem. 261:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle, and J. A. Pople. 1998. GAUSSIAN 98, Rev. A.5. Gaussian Inc., Pittsburgh, PA.
- Gajhede, M. 2001. Horseradish peroxidase. In Handbook of Metalloproteins, Vol. 1. A. Messerschmidt, R. Huber, T. Poulos, and K. Wieghardt, editors. J. Wiley & Sons, Chichester, UK. 195–209.
- Ghosh, A., J. Almlöf, and L. Que. 1994. Density functional theoretical study of oxo(porphyrinato)ironIV complexes, models of peroxidase compound I and II. J. Phys. Chem. 98:5576–5579. [Google Scholar]
- Giulivi, C., and E. Cadenas. 1994. Ferrylmyoglobin: formation and chemical reactivity toward electron-donating compounds. Methods Enzymol. 233:189–202. [DOI] [PubMed] [Google Scholar]
- Gouet, P., H.-M. Jouve, P. A. Williams, I. Andersson, P. Adreoletti, L. Nussaume, and J. Hajdu. 1996. Ferryl intermediates of catalase captured by time-resolved Weissenberg crystallography and UV-VIS spectroscopy. Nat. Struct. Biol. 3:951–956. [DOI] [PubMed] [Google Scholar]
- Green, M. T. 2001. The structure and spin coupling of catalase compound I: a study of noncovalent effects. J. Am. Chem. Soc. 123:9218–9219. [DOI] [PubMed] [Google Scholar]
- Green, M. T., J. H. Dawson, and H. B. Gray. 2004. Oxoiron(IV) in chloroperoxidase compound II is basic: implications for P450 chemistry. Science. 304:1653–1656. [DOI] [PubMed] [Google Scholar]
- Hargrove, M. S., A. J. Wilkinson, and J. S. Olson. 1996. Structural factors governing hemin dissociation from met-myoglobin. Biochemistry. 35:11300–11309. [DOI] [PubMed] [Google Scholar]
- Harris, D. L. 2001. High-valent intermediates of heme proteins and model compounds. Curr. Opin. Chem. Biol. 5:724–735. [DOI] [PubMed] [Google Scholar]
- Harris, D. L., and G. H. Loew. 2001. Proximal ligand effects on electronic structure and spectra of compound I of peroxidases. J. Porph. Phtalocyanins. 5:334–344. [Google Scholar]
- Hehre, W. J., L. Radom, P. v. R. Schleyer, and J. A. Pople. 1986. Ab Initio Molecular Orbital Theory. Wiley-Interscience, New York.
- Hersleth, H. P., B. Dalhus, C. H. Göbitz, and K. K. Andersson. 2002. An iron hydroxide moiety in the 1.35 Å resolution structure of hydrogen peroxide derived myoglobin compound II at pH 5.2. J. Biol. Inorg. Chem. 7:299–304. [DOI] [PubMed] [Google Scholar]
- Hersleth, H. P., B. Dalhus, C. H. Görbitz, and K. K. Andersson. 2001. Compound II in peroxidases: new resonance forms suggested by pH-dependent structures of myoglobin intermediates formed by oxidation with peroxides. J. Inorg. Biochem. 86:260. (Abstr.) [Google Scholar]
- Hertwig, R. H., and W. Koch. 1997. On the parameterization of the local correlation functional. What is Becke-3-Lyp? Chem. Phys. Lett. 268:345–351. [Google Scholar]
- Kaim, W., and B. Schwederski. 1994. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life. John Wiley & Sons, Chichester, UK.
- Keilin, D., and E. F. Hartree. 1950. Effect of drying upon the absorption spectra of hemoglobin and its derivatives. Nature. 166:513–518. [DOI] [PubMed] [Google Scholar]
- Kendrew, J. C., R. E. Dickerson, B. E. Strandberg, R. G. Hart, D. R. Davies, D. C. Phillips, and V. C. Shore. 1960. Structure of myoglobin—three-dimensional Fourier synthesis at 2-Å resolution. Nature. 185:422–427. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J., and T. A. Jones. 1998. Databases in protein crystallography. Acta Crystallogr. D54:1119–1131. [DOI] [PubMed] [Google Scholar]
- Kuramochi, H., L. Noodleman, and D. A. Case. 1997. Density functional study on the electronic structures of model peroxidase compounds I and II. J. Am. Chem. Soc. 119:11442–11451. [Google Scholar]
- MacKerell, A. D., D. M. Bashford, R. L. D. Bellott, Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics. Studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Matsui, T., S. Ozaki, E. Liong, G. N. Phillips, and Y. Watanabe. 1999. Effect of the location of distal histidine in the reaction of myoglobin with hydrogen peroxide. J. Biol. Chem. 274:2838–2844. [DOI] [PubMed] [Google Scholar]
- Matsunaga, I., and Y. Shir. 2004. Peroxide-utilizing biocatalysts: structural and functional diversity of heme-containing enzymes. Curr. Opin. Chem. Biol. 8:127–132. [DOI] [PubMed] [Google Scholar]
- Monard, G., and K. M. Merz. 1999. Combined quantum mechanical/molecular mechanical methodologies applied to biomolecular systems. Acc. Chem. Res. 32:904–911. [Google Scholar]
- Mulholland, A. J. 2001. The QM/MM approach to enzymatic reactions. In Theoretical Biochemistry—Processes and Properties of Biological Systems, Theoretical and Computational Chemistry, Vol. 9. L.A. Eriksson, editor. Elsevier Science, Amsterdam, The Netherlands.
- Murshudov, G. N., A. I. Grebenko, J. A. Brannigan, A. A. Antson, V. V. Barynin, G. G. Dodson, Z. Dauter, K. S. Wilson, and W. R. Melik-Adamyan. 2002. The structures of Micrococcus lysodeikticus catalase, its ferryl intermediate (compound II) and NADPH complex. Acta Crystallogr. D58:1972–1982. [DOI] [PubMed] [Google Scholar]
- Nicoll, R. M., S. A. Hindle, G. MacKenzie, I. H. Hillier, and N. A. Burton. 2001. Quantum mechanical/molecular mechanical methods and the study of kinetic isotope effects: modelling the covalent junction region and application to the enzyme xylose isomerase. Theor. Chim. Acta. 106:105–112. [Google Scholar]
- Nilsson, K., and U. Ryde. 2004. Protonation status of protein ligands can be determined by quantum refinement. J. Inorg. Biochem. 98:1539–1546. [DOI] [PubMed] [Google Scholar]
- Nina, M., and B. Roux. 1997. Atomic radii for continuum electrostatics calculations based on molecular dynamics free energy simulations. J. Phys. Chem. B. 101:5239–5248. [Google Scholar]
- Ozaki, S., T. Matsui, and Y. Watanabe. 1997. Conversion of myoglobin into a peroxygenase: a catalytic intermediate of sulfoxidation and epoxidation by the F43H/H64L mutant. J. Am. Chem. Soc. 119:6666–6667. [Google Scholar]
- Paeng, K. J., and J. R. Kincaid. 1988. The resonance Raman spectrum of horseradish peroxidase compound I. J. Am. Chem. Soc. 110:7913–7915. [Google Scholar]
- Pannu, N. S., and R. J. Read. 1996. Improved structure refinement through maximum likelihood. Acta Crystallogr. A52:659–668. [Google Scholar]
- Penner-Hahn, J. E., K. S. Eble, T. J. McMurry, M. Renner, A. L. Balch, J. T. Groves, and K. O. Hodgsen. 1986. Structural characterization of horseradish peroxidase using EXAFS spectroscopy. Evidence for Fe=O ligation in compound I and II. J. Am. Chem. Soc. 108:7819–7825. [DOI] [PubMed] [Google Scholar]
- Perdew, J. P. 1986. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B. 33:8822–8824. [DOI] [PubMed] [Google Scholar]
- Philips, G. N. 2001. Myoglobin. In Handbook of Metalloproteins, Vol. 1. A. Messerschmidt, R. Huber, T. Poulos, and K. Wieghardt, editors. Wiley & Sons, Chichester, UK. 5–15.
- Poulos, T. L. 1996. The role of the proximal ligand in heme enzymes. J. Biol. Inorg. Chem. 1:356–359. [Google Scholar]
- Rovira, C., and I. Fita. 2003. The proximal hydrogen-bonded residue controls the stability of the compound II intermediate of peroxidases and catalases. J. Phys. Chem. B. 107:5300–5305. [Google Scholar]
- Rydberg, P., E. Sigfridsson, and U. Ryde. 2004. On the role of the axial ligand in heme proteins—a theoretical study. J. Biol. Inorg. Chem. 9:203–223. [DOI] [PubMed] [Google Scholar]
- Ryde, U. 2003. Combined quantum and molecular mechanics calculations on metalloproteins. Curr. Opin. Chem. Biol. 7:136–142. [DOI] [PubMed] [Google Scholar]
- Ryde, U., and K. Nilsson. 2003a. Quantum chemistry can improve protein crystal structures locally. J. Am. Chem. Soc. 125:14232–14233. [DOI] [PubMed] [Google Scholar]
- Ryde, U., and K. Nilsson. 2003b. Quantum refinement—a combination of quantum chemistry and protein crystallography. J. Mol. Struct. 632:259–275. [Google Scholar]
- Ryde, U., L. Olsen, and K. Nilsson. 2002. Quantum chemical geometry optimisations in proteins using crystallographic raw data. J. Comput. Chem. 23:1058–1070. [DOI] [PubMed] [Google Scholar]
- Schäfer, A., H. Horn, and R. Ahlrichs. 1992. Fully optimized contracted Gaussian basis set for atoms Li to Kr. J. Chem. Phys. 97:2571–2577. [Google Scholar]
- Schlichting, I., J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko, and S. G. Sligar. 2000. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 287:1615–1622. [DOI] [PubMed] [Google Scholar]
- Siegbahn, P. E. M., and M. R. A. Blomberg. 2000. Transition-metal systems in biochemistry studied by high-accuracy quantum chemical methods. Chem. Rev. 100:421–437. [DOI] [PubMed] [Google Scholar]
- Sigfridsson, E., M. H. M. Olsson, and U. Ryde. 2001. A comparison of the inner-sphere reorganisation energies of cytochromes, iron-sulphur clusters, and blue copper proteins. J. Phys. Chem. B. 105:5546–5552. [Google Scholar]
- Sigfridsson, E., and U. Ryde. 2003. The importance of porphyrin distortions for the ferrochelatase reaction. J. Biol. Inorg. Chem. 8:273–282. [DOI] [PubMed] [Google Scholar]
- Sitter, A. J., C. M. Reczek, and J. Terner. 1985. Heme-linked ionization of horseradish peroxidase compound II monitored by the resonance Raman Fe(IV)=O stretching vibration. J. Biol. Chem. 260:7515–7522. [PubMed] [Google Scholar]
- Sitter, A. J., J. R. Shifflett, and J. Terner. 1988. Resonance Raman spectroscopic evidence for heme iron-hydroxide ligation in peroxidase alkaline forms. J. Biol. Chem. 263:13032–13038. [PubMed] [Google Scholar]
- Smith, A. T., and N. C. Veitch. 1998. Substrate binding and catalysis in heme peroxidases. Curr. Opin. Chem. Biol. 2:269–278. [DOI] [PubMed] [Google Scholar]
- Spiro, T. G., and P. M. Kozlowski. 2001. Is the CO adduct of myoglobin bent, and does it matter? Acc. Chem. Res. 34:137–144. [DOI] [PubMed] [Google Scholar]
- Tajima, G., and K. Shikama. 1987. Autoxidation of oxymyoglobin. an overall stoichiometry including subsequent side reactions. J. Biol. Chem. 262:12603–12606. [PubMed] [Google Scholar]
- Tishmack, P. A., D. Bashford, E. Harms, and R. I. Van Etten. 1997. Use of 1H NMR spectroscopy and computer simulations to analyze histidine pKa changes in a protein tyrosine phosphatase: experimental and theoretical determination of electrostatic properties in a small protein. Biochemistry. 36:11984–11994. [DOI] [PubMed] [Google Scholar]
- Ullmann, G. M., L. Noodleman, and D. A. Case. 2002. Density functional calculation of pKa values and redox potentials in the bovine Rieske iron-sulfur protein. J. Biol. Inorg. Chem. 7:632–639. [DOI] [PubMed] [Google Scholar]
- Wirstam, M., M. R. A. Blomberg, and P. E. M. Siegbahn. 1999. Reaction mechanism of compound I formation in heme peroxidases: a density functional theory study. J. Am. Chem. Soc. 121:10178–10185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.