

Abstract
Gramicidin A was studied by continuous wave electron spin resonance (CW-ESR) and by double-quantum coherence electron spin resonance (DQC-ESR) in several lipid membranes (using samples that were macroscopically aligned by isopotential spin-dry ultracentrifugation) and vesicles. As a reporter group, the nitroxide spin-label was attached at the C-terminus yielding the spin-labeled product (GAsl). ESR spectra of aligned membranes containing GAsl show strong orientation dependence. In DPPC and DSPC membranes at room temperature the spectral shape is consistent with high ordering, which, in conjunction with the observed high polarity of the environment of the nitroxide, is interpreted in terms of the nitroxide moiety being close to the membrane surface. In contrast, spectra of GAsl in DMPC membranes indicate deeper embedding and tilt of the NO group. The GAsl spectrum in the DPPC membrane at 35°C (the gel to Pβ phase transition) exhibits sharp changes, and above this temperature becomes similar to that of DMPC. The dipolar spectrum from DQC-ESR clearly indicates the presence of pairs in DMPC membranes. This is not the case for DPPC, rapidly frozen from the gel phase; however, there are hints of aggregation. The interspin distance in the pairs is 30.9 Å, in good agreement with estimates for the head-to-head GAsl dimer (the channel-forming conformation), which matches the hydrophobic thickness of the DMPC bilayer. Both DPPC and DSPC, apparently as a result of hydrophobic mismatch between the dimer length and bilayer thickness, do not favor the channel formation in the gel phase. In the Pβ and Lα phases of DPPC (above 35°C) the channel dimer forms, as evidenced by the DQC-ESR dipolar spectrum after rapid freezing. It is associated with a lateral expansion of lipid molecules and a concomitant decrease in bilayer thickness, which reduces the hydrophobic mismatch. A comparison with studies of dimer formation by other physical techniques indicates the desirability of using low concentrations of GA (∼0.4–1 mol %) accessible to the ESR methods employed in the study, since this yields non-interacting dimer channels.
INTRODUCTION
Gramicidins, a family of linear pentadecapeptide antibiotics, are produced by the bacterium Bacillus brevis, which is found in soil. In fact, gramicidin is one of the first known antibiotics. In 1939, Hotchkiss and Dubois (1940) isolated the substance tyrothricin and later showed that it was composed of two substances, gramicidin and tyrocidine. These were the first antibiotics to be manufactured commercially, well before penicillin.
Gramicidins are composed of alternating D- and L-amino acids and form β-type helices with a hydrogen-bonding pattern of the backbone similar to that in β-sheets. The sequence of the major B. brevis product, valine gramicidin A (GA), is HCO-L-Val1-Gly2-L-Ala3-D-Leu4-L-Ala5-D-Val6-L-Val7-D-Val8-L-Trp9-D-Leu10-L-Trp11-D-Leu12-L-Trp13-D-Leu14-L-Trp15-NHCH2CH2OH. The single-stranded helices may form head-to-head dimers (Urry et al., 1971; Urry, 1971; Veatch et al., 1974). In organic solvents GA is conformationally polymorphic and forms single- or double-stranded helices (Wallace, 1998). The double-stranded helices are either parallel or antiparallel and exist in a variety of conformations of different lengths and numbers of residues per turn. The equilibrium between a double-helical conformation and GA monomers in organic solvents was extensively studied with a variety of techniques (Killian, 1992).
The primary interest of gramicidin lies in the ability of the peptide to form ion channels in lipid membranes (Andersen, 1984). The channels specifically conduct monovalent cations (alkali metals, H+, Tl+, Ag+, and 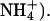 Based on accumulated evidence, the head-to-head dimer has been shown to be the predominant ion-conducting channel form in membranes (Ketchem et al., 1997; Kovacs et al., 1999; but see also Burkhart et al., 1998). However, a number of studies exhibited that two (or more) dimer structures are present in membranes in equilibrium. The equilibrium can be shifted from one conformation to the other by changing the length of the alkyl chains of the bilayer (Mobashery et al., 1997), the chemical nature of the lipid (Sychev et al., 1993), or the presence of ions (Doyle and Wallace, 1998). A temperature dependence of the equilibrium between double helices and head-to-head structures was demonstrated by Fourier-transform infrared (FTIR) spectroscopy for DMPC, DPPC, and DSPC (Wallace et al., 1981; Fahsel et al., 2002; Zein and Winter, 2000). Also, the conformation of GA in hydrated membranes may depend on solvent history before introducing it into the membrane (Bouchard and Auger, 1993), and the transition to the thermodynamically stable state may be slow (Cotten et al., 1997). Cross and co-workers have given a prescription for how to obtain the GA membrane samples in the thermodynamically stable conductive form (LoGrasso et al., 1988).
Based on accumulated evidence, the head-to-head dimer has been shown to be the predominant ion-conducting channel form in membranes (Ketchem et al., 1997; Kovacs et al., 1999; but see also Burkhart et al., 1998). However, a number of studies exhibited that two (or more) dimer structures are present in membranes in equilibrium. The equilibrium can be shifted from one conformation to the other by changing the length of the alkyl chains of the bilayer (Mobashery et al., 1997), the chemical nature of the lipid (Sychev et al., 1993), or the presence of ions (Doyle and Wallace, 1998). A temperature dependence of the equilibrium between double helices and head-to-head structures was demonstrated by Fourier-transform infrared (FTIR) spectroscopy for DMPC, DPPC, and DSPC (Wallace et al., 1981; Fahsel et al., 2002; Zein and Winter, 2000). Also, the conformation of GA in hydrated membranes may depend on solvent history before introducing it into the membrane (Bouchard and Auger, 1993), and the transition to the thermodynamically stable state may be slow (Cotten et al., 1997). Cross and co-workers have given a prescription for how to obtain the GA membrane samples in the thermodynamically stable conductive form (LoGrasso et al., 1988).
Spin-label electron spin resonance (ESR) spectroscopy is a proven and valuable approach in membrane biophysics. The method has been successfully applied to GA/phospholipid mixtures. Previous work utilizing spin-labeled lipids showed that gramicidin channels indeed alter acyl-chain behavior (Tanaka and Freed, 1985; Ge and Freed, 1993). ESR spectroscopy, strengthened by the 2D-ELDOR technique, has provided a reliable way to clearly distinguish the bulk lipids and boundary lipids and to observe GA aggregation and formation of the HII phase (Ge and Freed, 1999; Costa-Filho et al., 2003). However, the spin-labeled lipids are less useful for studying the behavior and conformation of the GA molecules in the channel. Furthermore, the lipid probes are useful only if a significant fraction of lipid exists as boundary lipid, which requires GA concentrations >5% mol. In that case a signal from the reporter spin-label molecules may be detectable on the background of the signal coming from the unperturbed lipid fraction. On the other hand, much lower GA concentrations are needed to observe non-interacting single channels and to relate the results to energetic measurements or molecular dynamics simulations. The measurements of transmembrane electrical potential, for example, are done at nanomolar concentrations of GA (Schönknecht et al., 1992) to ensure that each channel becomes an isolated entity. A theoretical analysis shows (Nielsen et al., 1998) that in addition to 20 molecules of the boundary, the channel-induced bilayer perturbation involves 50–100 molecules. Consequently, molar ratios GA/lipid below 2 mol % should suffice for the purpose of single channel observation. The challenge for ESR spectroscopy was to find a spin-label that is sensitive to channel formation at these concentrations of GA.
Clearly, a spin-label directly attached to the GA molecule is very likely to detect conformational changes and aggregation of the GA molecules. We have therefore spin-labeled GA (GAsl) with the label attached at the C-terminus, which is located at each end of the head-to-head dimer (compare to Fig. 1). It seems clear from this figure that the spin-labeling should not affect the formation of head-to-head (N-terminus to N-terminus) dimers. We estimated the distance between the two nitroxide fragments in GAsl, based on the known structure for the GA channel (Ketchem et al., 1997) and molecular modeling. We found 30.2 Å between the oxygens and 29.5 Å between the nitrogen atoms of the NO groups at the different C-termini of the GAsl dimer (Fig. 1). The expected distance between two nitroxide moieties in the dimer(s) is in the range that is very suitable for double-quantum coherence (DQC)-ESR distance measurements, which provide very accurate distances (Borbat and Freed, 2000; Borbat et al., 2002).
FIGURE 1.
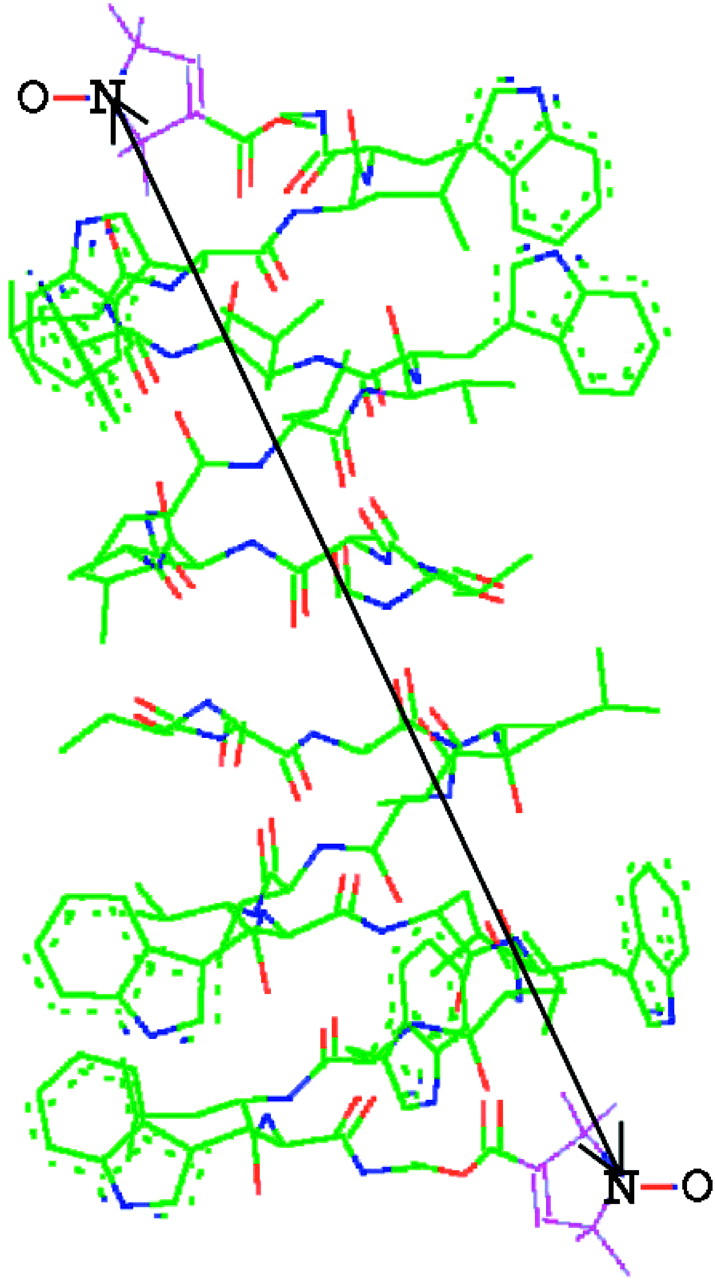
GAsl in the head-to-head conformation. The arrow indicates a distance of 29.5 Å between the nitroxide nitrogen atoms at the spin-labeling sites on the different C-termini of the dimer obtained from the structure for the GA channel given by Ketchem et al. (1997) and molecular modeling of the nitroxide tethers (see text).
In this study we examine how the behavior of spin-labeled GA molecules in aligned membranes and vesicles depends on the composition and phase of the lipid membranes. We utilize continuous-wave and double-quantum coherence (CW and DQC) spectroscopy, and we discuss the results in terms of matching/mismatch between the length of the GA channel and the hydrophobic thickness of the membrane. Our results are compared with those obtained by previous techniques, and the ability of the ESR methods to study low concentrations (<2 mol %) is pointed out.
EXPERIMENTAL
Materials
DMPC, DPPC, and DSPC were obtained from Avanti Polar Lipids (Birmingham, AL). The spin-label 2,2,5,5-tetramethyl-3-pyrrolin-1-oxyl-3-carboxylic acid was purchased from Sigma (St. Louis, MO), and the GA, 4-dimethylamino pyridine, and N,N′–dicyclohexylcarbodiimide (DCC) were purchased from Fluka (Sigma-Aldrich, St. Louis, MO).
Synthesis of GAsl
In the spin-labeling we used a modification of the mild one-pot esterification method by Hassner and Alexanian (1978), which targets the free hydroxyl at the C-terminus of the GA molecule. The molecular mass of the product, as determined by mass spectroscopy, corresponded exactly to the expected spin-labeled compound; see Scheme 1 below.
SCHEME 1.

To remove any traces of water, first 0.53 g of GA was dissolved in 25 ml C2H5OH. The GA solution was dried in a rotor evaporator and then incubated under vacuum overnight in a reagent flask. Dry reagents were then added to the reagent flask: 80 mg 2,2,5,5-tetramethyl-3-pyrrolin-1-oxyl-3-carboxylic acid, 90 mg DCC, and 6 mg of 4-dimethylamino pyridine as a catalyst. Seven milliliters of anhydrous dimethylformamide was added, and the resulting solution was allowed to stir for 24 h under argon.
The thin-layer chromatography spots for GA and product tend to streak in the 90:10 CHCl3/methanol system. Also, no spot of free spin-label is seen on the thin-layer chromatography plate. Apparently, the label entirely reacts with DCC to form a yellow product that moves faster in the system, compared to the control spot for free label.
After dimethylformamide was removed by a high vacuum line overnight, the reaction mixture was dissolved in 95:5 CHCl3/methanol and applied to a column. The column was packed with 230–400 mesh silica gel 60. Chromatography was carried out under nitrogen pressure in 95:5 CHCl3/methanol. The fractions were collected, and ESR spectra of each fraction were recorded. The faster moving yellowish fraction shows a strong ESR signal with a relatively fast rotational correlation time τR ∼ 10−10 s.
Although some GAsl (τR = 3 × 10−10 s) is apparently present in slower moving fractions, the main portion stayed at the start line and was eluted by a 70:30 CHCl3/methanol mixture; 387 mg of dry, slightly yellowish powder was collected. Mass spectroscopy showed that the product was a mixture of labeled and unlabeled GA with the extent of labeling ∼40–45%. No noticeable amounts of other products were detected.
Since the concentration of radical pairs observed by DQC is proportional to the square of the extent of labeling, 80 mg of the product was reintroduced into the same reaction with 13 mg of the spin-label, 14.6 mg DCC, and ∼1.2 mg 4-dimethylamino pyridine. The yield was 62 mg of ∼80% labeled product.
The spin-labeled gramicidin A (GAsl) is a white or slightly yellowish powder, insoluble in water and soluble in organic solvents. It may be easily introduced into membranes and shows well-resolved slow-motional ESR spectra. The spin-labeled product is stable in CHCl3/MeOH solutions and in membranes at neutral pH, although it slowly releases free spin-label at pH ≥8.
Sample preparation
Preparation of vesicle samples
Measured amounts of lipid and GA were dissolved in chloroform/methanol 3:1 v/v. We used 4 mg lipid samples with 0.4–2% mol. GAsl for CW-ESR and 15 mg samples containing 0.12% GAsl for DQC-ESR. The solvent was evaporated by nitrogen gas flow. To ensure complete removal of the solvent the sample was evacuated 10–12 h (see further description below about the choice of solvent and the removal procedure). The samples were hydrated with excess water (90 μL for 4 mg and 200 μL for 15 mg samples) for 30 min, above the chain-melting temperature, with several vortex mixing cycles. Then the suspensions were collected in 1.6-mm diameter glass capillaries (CW) or 2.5-mm Suprasil tubes (DQC) and pelleted by using a microhematocrit centrifuge. The supernatant was removed and the capillaries were sealed.
In some cases, measurements at 77 K were necessary to characterize a conformation of GAsl, which is stable in DPPC membranes above 35°C. Below this temperature the conformation transforms into a different one. Although the full conversion at room temperature takes hours, to trap the maximum fraction of the metastable state the samples were first quenched in freezing pentane (143 K) and then transferred to liquid nitrogen for the measurements. Control experiments on other samples showed that the freezing rate itself does not affect their CW-ESR lineshape at 77 K.
Choice of the cosolubilization solvent
To observe possible differences between single- and double-helical forms of GAsl; we prepared DMPC and DPPC membranes containing GAsl using different cosolubilization solvents. It was reported previously (Bouchard and Auger, 1993) that incorporation in DMPC membranes from trifluoroethanol (TFE) produces only the head-to-head single-helical form, but incorporation from chloroform/methanol (CM) causes a significant admixture of the double-helical form. If the vesicular samples are frozen in liquid nitrogen immediately after preparation to preserve the state of GAsl dimers, we expected different outer hyperfine splittings in the ESR spectrum from the use of TFE versus CM. Such a difference should emerge from the different distances that exist between the C-termini, which results in their being at different locations in the membrane, with different polarities, which affect the nitroxides (see Results). However, the values of the outer splitting for both TFE and CM were equal.
We did see conspicuous differences in the spectra (but not in the outer splitting values) if both samples were just evacuated overnight. The difference is much greater for DPPC. It may be ascribed to intermolecular dipole-dipole broadening between nitroxides at 77 K for the TFE reconstitution. For these samples we also see a slightly increased mobility of the spin-label over the whole temperature range 5–55°C, with the effect being more pronounced around the main transition point. Longer periods of evacuation (24–36 h) do change the TFE-derived spectra, but not CHCl3/methanol ones, and eventually the former do become identical to the latter. None of the samples used in our CW and DQC experiments showed any conspicuous features of dipole-dipole broadening, and they were virtually identical to control “magnetically diluted” samples, which were prepared with a 1:5 mixture of GAsl and unlabeled GA instead of pure GAsl.
There are two conclusions that can be drawn from these results. First, it shows that the vesicular samples are obtained even at short incubation times in a thermodynamically equilibrated state, independent of their solvent history. Second, it shows that the use of TFE, the solvent, which is reported to favor formation of head-to-head dimers in solution (Bouchard and Auger, 1993), is not necessary in our case with GAsl. What is more, it is hard to remove TFE from the membrane, making TFE less convenient than CHCl3/methanol. Traces of TFE in the DPPC membrane are apparently not uniformly dissolved in the gel phase, but form some additional fluid areas. It would appear that GAsl tends to concentrate in such areas yielding an increase in intermolecular dipole-dipole broadening.
Preparation of aligned membranes
Isopotential spin-dry ultracentrifugation (ISDU)-aligned lipid membranes were prepared as described by Ge et al. (1994). It involves sedimentation of the membrane fragments (in the gel phase) with simultaneous evaporation of the water phase in a vacuum ultracentrifuge. Dry lipid/GAsl mixtures, typically 9 mg per centrifuge bucket (volume 0.9 ml), which yield a final aligned sample with area ∼1.1 cm2, were first prepared as described for vesicles. Then 1 ml of deionized water per 10 mg of lipid was added. The mixture was sonicated 12 min above the chain-melting temperature at a frequency of 20 kHz, and an incident power of 20 W/cm2. For the ISDU procedure, a Beckman L8M-70 centrifuge (Beckman Coulter, Fullerton, CA) with standard SW27 rotors at 15,000 rpm was used. The procedure took 64 h at 8°C for DMPC and 24 h for DPPC and DSPC at 20°C. After ultracentrifugation, the dry membranes were checked for their alignment using a polarizing microscope (Nicon, Instrument Division, Garden City, NJ). Only samples that exhibited the distinctive interference pattern indicative of an aligned membrane were used in the ESR experiments. The samples were then hydrated either in 100% relative humidity or in excess water. In the latter case, an ∼3.5-mg sample was sealed in a flat glass cell with 10 mg water. The cell (0.3-mm clearance) was used directly in the ESR measurements.
Calorimetry
A differential scanning calorimeter, DSC-4 Perkin Elmer (PerkinElmer, Wellesley, MA), was used to check the main transition and pretransition temperatures in vesicles and aligned membrane samples. For samples containing 0.4% GAsl the values obtained of 41.4°C (main transition) and 34°C (pretransition) in DPPC and 23.7°C and ∼15°C, respectively, for DMPC are indicative of fully hydrated samples, and they do not depend on the hydration procedure. In general, the pretransition peaks are sharper in the case of vesicles than in aligned membranes. An increase in the concentration of GAsl to 2% increases the main transition temperatures by ∼2°C and broadens the DSC peaks.
CW-ESR spectroscopy
ESR spectra were obtained on a Bruker EMX spectrometer (Bruker, Billerica, MA) at a frequency of 9.55 GHz under standard conditions. The field sweeps were calibrated with a Bruker ER 035 Gaussmeter. The microwave frequency was monitored with a frequency counter.
DQC-ESR measurements
The distances between nitroxide spin-labels were determined using DQC-ESR (Borbat and Freed, 1999, 2000; Borbat et al., 2002). In DQC-ESR we used the six-pulse sequence (see Fig. 2 a) designed to extract weak dipolar couplings from ESR spectra dominated by hyperfine and g-tensor interactions. The method cancels out the larger effects of these hyperfine and g-tensor interactions, which are refocused by this pulse sequence. Thus the time dependence of the signal is just due to the dipolar coupling(s). The sequence utilizes short and intense pulses with a large B1 microwave field to irradiate and detect the signal from a large fraction of the spins in the sample and to enhance the effects of the dipolar coupling.
FIGURE 2.
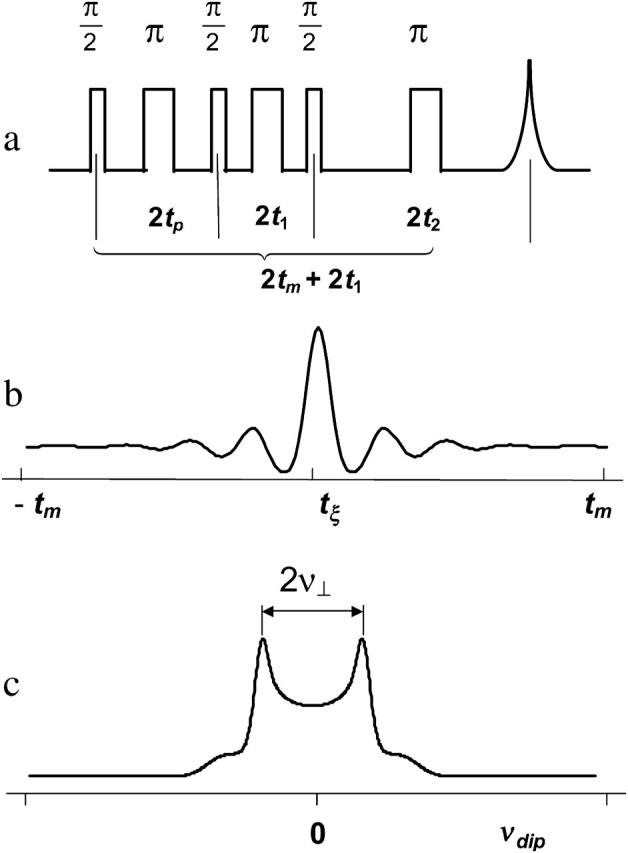
(a) The six-pulse DQC-ESR sequence designed to measure distances between electron spins in solids. The amplitude of the echo formed at 2(tp + t1 + t2) is recorded as a function of tξ ≡ 2tp−tm, with t1 and tm ≡ tp + t2 being kept constant. The phase cycling leaves only the part of the echo modulated by the dipolar coupling a. For the dipolar coupling in DQC, 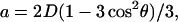 wherein
wherein 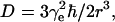 r is the distance between electron spins, and θ is the angle between the vector connecting the electron spins on the nitroxides and the magnetic field, B0. (b) An illustrative temporal envelope of the signal recorded with the six-pulse sequence for a nitroxide biradical in an isotropic medium. (c) The dipolar spectrum, produced by Fourier transformation of b, has the shape of a Pake doublet with an inner splitting of 2ν⊥ = 4D/3.
r is the distance between electron spins, and θ is the angle between the vector connecting the electron spins on the nitroxides and the magnetic field, B0. (b) An illustrative temporal envelope of the signal recorded with the six-pulse sequence for a nitroxide biradical in an isotropic medium. (c) The dipolar spectrum, produced by Fourier transformation of b, has the shape of a Pake doublet with an inner splitting of 2ν⊥ = 4D/3.
The six-pulse DQC signal is recorded versus tξ ≡ tp−t2 (see Fig. 2 b), and in the limit of nonselective pulses it takes on the simple form of 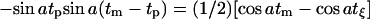 (Borbat and Freed, 2000).
(Borbat and Freed, 2000).
Here, 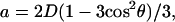 where
where 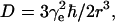 r is the distance between two electron spins, and θ is the angle between the vector connecting the electron spins and the magnetic field, B0. In general, this expression should be averaged over all possible values of a, i.e., all possible θ and r. The signal is symmetric with respect to tξ = 0 and is referred to as a “zero dead-time” signal, since the initial part (tξ = 0) of the signal is not lost. The Fourier transformation of the temporal envelope of the DQC signal versus tξ for two spins (see Fig. 2 b) yields the dipolar spectrum, which, in disordered solids for not very broad distributions over r, has the shape of the Pake doublet with a splitting 2ν⊥ of 4D/3 (see Fig. 2 c).
r is the distance between two electron spins, and θ is the angle between the vector connecting the electron spins and the magnetic field, B0. In general, this expression should be averaged over all possible values of a, i.e., all possible θ and r. The signal is symmetric with respect to tξ = 0 and is referred to as a “zero dead-time” signal, since the initial part (tξ = 0) of the signal is not lost. The Fourier transformation of the temporal envelope of the DQC signal versus tξ for two spins (see Fig. 2 b) yields the dipolar spectrum, which, in disordered solids for not very broad distributions over r, has the shape of the Pake doublet with a splitting 2ν⊥ of 4D/3 (see Fig. 2 c).
DQC signals were recorded at 17.3 GHz using π/2 pulses of 3.2 ns, corresponding to B1 ≈ 28 G. To better enable low-temperature experiments at this frequency, the previously described FT-ESR spectrometer (Borbat et al., 1997, 2001) was equipped with an Oxford Instruments helium-flow cryostat, model CF 935 (Oxford Instruments, Eynsham, UK). The temperature was set to 77 K, and DQC signals were recorded using four 40-min scans. They were analyzed as previously described (Borbat and Freed, 1999, 2000; Borbat et al., 2002).
Spectral analysis
Nonlinear least-squares analyses of the CW-ESR spectra based on the stochastic Liouville equation (Meirovitch et al., 1982; Schneider and Freed, 1989) were performed using the fitting program by Budil et al. (1996). The magnetic A-tensor and g-tensor components needed for simulations were obtained from fitting rigid limit spectra recorded at 77 K. The value of the Azz component was taken as half of the outer splitting at the rigid limit. Initial values for (Axx+ Ayy) were then estimated from the isotropic splittings aiso = (1/3) (Axx+ Ayy + Azz) at/above room temperature in low-viscosity solvents (toluene, ethanol, and ethanol/water) which at 77 K showed Azz values close to that value in the specific membrane. The g-tensor values were initially taken as gxx, gyy, and gzz = 2.0090, 2.0061, and 2.0022, according to the values for similar pyrroline radicals (Lebedev et al., 1992). Our high-field ESR measurements at 170 GHz for GAsl in DMPC vesicles yielded the same values (unpublished results); the gxx component was varied slightly depending on the polarity (concomitant with Azz at 77 K).
The motional parameters for the gel phase are found to be well into the ESR slow-motion regime (R⊥ ∼ 106 s−1, R‖ < 5 × 106 s−1), and the spectra are, in general, close to the rigid limit. Thus the utilization of ISDU-aligned membranes provides particularly valuable information on the orientation of the nitroxide moiety relative to the membrane normal (Budil et al., 1996). Such information is difficult to obtain from vesicles in this motional range. The ESR spectrum in vesicles is well described by the microscopic-order and macroscopic-disorder model (Meirovitch et al., 1984; Budil et al., 1996). Because in this case all orientations of the membrane normal relative to the magnetic field are averaged, the orientation of the nitroxide moiety manifests itself only as a result of anisotropic molecular motion around key axes. As one approaches the rigid limit, the vesicular spectrum converges to a “powder” spectrum, which is not sensitive to any properties of molecular structure (except the magnetic tensors of the nitroxide).
In the Pβ phase of DMPC and especially DPPC we see an increase in the rate of molecular motion. The separation between outer spectral extremes decreases, but the spectra still retain some features of rigid limit spectra. These spectra can be approximately simulated with the fast internal motion model (Liang and Freed, 1999; Hubbell and McConnell, 1971) where the internal dynamics of the C-terminus leads to partial averaging of the magnetic tensors, and the global motion is described in terms of a diffusion tensor.
The least-squares fits for the aligned samples were performed simultaneously for the 0, 45, and 90° orientations using the same set of parameters (Budil et al., 1996).
RESULTS AND DISCUSSION
The spectra from the aligned membranes of DPPC, DSPC, and DMPC containing GAsl show strong orientation-dependence (see Figs. 3–7).
FIGURE 3.

Experimental (solid line) and simulated (dots) ESR spectra of GAsl in aligned DPPC (a) and DSPC (b) membrane for three orientations of the local director relative to the membrane normal (0°, 45°, and 90°). T = 22°C, corresponding to gel phase. Molar ratio GAsl/lipid = 0.6%.
FIGURE 4.

Experimental (solid line) and simulated (dots) ESR spectra of GAsl in aligned DMPC membrane for three orientations of the local director relative to the membrane normal (0°, 45°, and 90°). T = 2°C. Molar ratio GAsl/DMPC = 0.6%.
FIGURE 5.

Temperature dependence of ESR spectra of GAsl/DMPC (0.6 mol %) in aligned membrane. Experimental (solid line) spectra in 0° (a–d) or 90° (e) orientations. Dotted lines are simulations for d and e.
FIGURE 6.

ESR spectra of GAsl/DPPC (0.6 mol %), aligned membrane, 0° orientation, with temperature dependence.
FIGURE 7.

Experimental (solid line) and simulated (dots) ESR spectra of GAsl in aligned DPPC membrane for three orientations of the local director relative to the membrane normal (0°, 45°, and 90°). T = 36°C, corresponding to Pβ phase. Molar ratio GAsl/DPPC = 0.6%.
Aligned membranes of DPPC and DSPC, the gel phase
Fig. 3 a displays the spectrum of GAsl in aligned DPPC at 22°C. The dashed line shows a one-component fit with ordering and diffusion parameters given in Table 1. The fit is acceptable, although (see below) there are indications that the spectrum contains a component that corresponds to another form of GAsl in the membrane. The spectrum may also have a small admixture of a disordered signal, since we cannot completely rule out the presence of small, less-oriented regions in the sample, or small regions that lost their alignment upon hydration. The lineshape at room temperature does not depend on the GAsl concentration in the range of 0.4–2%. It does vary slightly from sample to sample and can change slowly upon storage. In the latter case the peak, marked by an arrow in Fig. 3 a, can grow in amplitude and become sharper. This can be well simulated with a slight increase in the order parameter S. Alternatively it could indicate an admixture with a broader and less-ordered second component, which disappears during storage (i.e., a hysteresis effect, see below), and this would also lead to a sharpening of the peak marked in Fig. 3 a. This uncertainty does not affect the main conclusions that we obtain from the simulations: at room temperature GAsl in DPPC shows a well-aligned spectrum corresponding to Z ordering (i.e., the Z axis of the magnetic tensors preferentially aligns parallel to the membrane normal) with an order parameter S ∼ 0.55. This ordering (see also below) implies an alignment of GAsl in the membrane sketched in Fig. 8 a.
TABLE 1.
Molecular motion, potential, and diffusion-tilt parameters of GAsl in different lipid environments used in the best fits to experimental spectra
| Lipid system | Diffusion-tilt angle, β | C20 | C40 | Cone angle, ϕ | R⊥ | R‖ |
|---|---|---|---|---|---|---|
| DMPC, 2°C | 44° | 4 | −4 | 39° | 7.9 × 105 | 2.0 × 106 |
| DMPC, 21.5°C | 38° | 2 | −0.8 | 22° | 3.2 × 106 | 2.0 × 107 |
| DPPC, 22°C | 0° | 2.2 | 0 | 0° | 1.0 × 106 | 2.0 × 106 |
| DPPC, 36°C | 45° | 1.6 | −0.5 | 17° | 7.9 × 106 | 4.0 × 107 |
| DMPC, 55°C, vesicles | — | 1.16 | 0 | — | 7.9 × 107 | 6.3 × 108 |
The results are shown for aligned samples, except the last row, which is given for vesicles.
FIGURE 8.
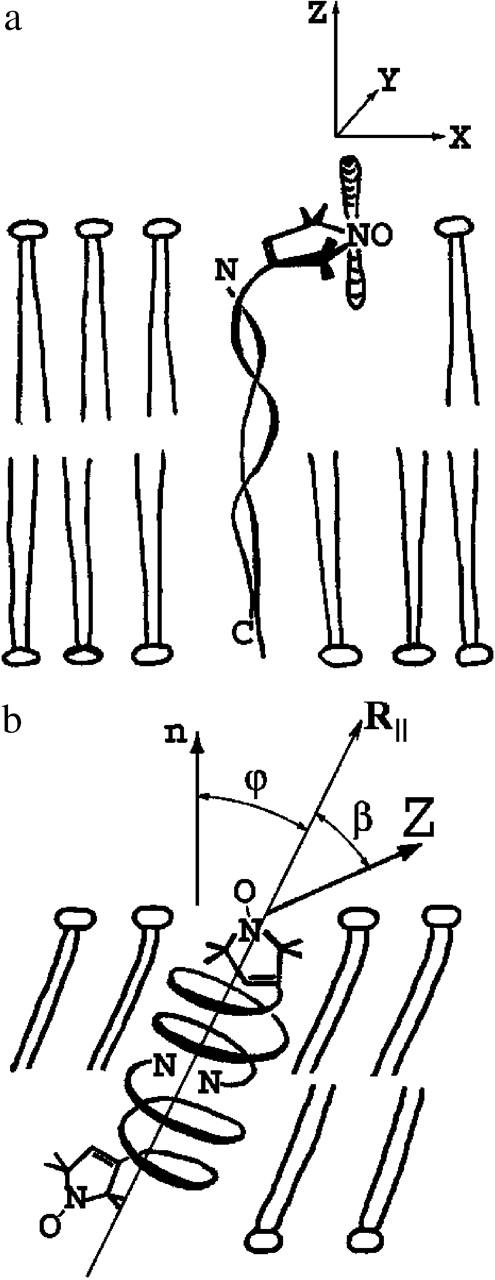
Schematic drawings of the conformations of GAsl in DMPC (a) and DPPC (b). For angles β and ϕ, see text.
As seen in Fig. 3 b, in aligned DSPC membranes GAsl exhibits an ESR spectrum that is practically indistinguishable from DPPC (except for the small sharp peaks from small amounts of free spin-label).
Aligned membranes of DMPC in the gel phase
In aligned DMPC membranes the spectrum of GAsl looks less ordered (see Fig. 4) than in DPPC or DSPC membranes (see Fig. 3). That is, the spectra display less dramatic variation with orientation, although they do still exhibit strong orientation dependence. The lines are sharp, however, and they have very specific features that can only be simulated by an orienting potential corresponding to the nitroxides being tilted relative to the membrane normal, i.e., a conical distribution. To consider how this is implemented, we first consider the restoring potential relative to the membrane normal (Schneider and Freed, 1989),
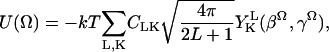 |
(1) |
where Ω = (0, βΩ, γΩ) represents the angles relating the diffusion (and ordering) tensors relative to the membrane normal. The values 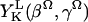 are spherical harmonics, and CLK represents the expansion coefficients. Usually one retains only lowest order (i.e., L = 2 and 4) terms.
are spherical harmonics, and CLK represents the expansion coefficients. Usually one retains only lowest order (i.e., L = 2 and 4) terms.
For K = 0, which implies axially symmetric ordering of the nitroxide, we can then write
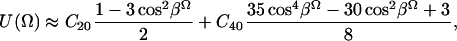 |
(2) |
and the potential, U(Ω) is determined with just one angle βΩ. For convenience below, we will let βΩ ≡ ϕ. This means that the most favored direction of the diffusion axis in the Lβ phase forms a cone relative to the membrane normal and may suggest (although not necessarily) a tilt of GAsl molecules in the membrane. The cone angle corresponding to the minimum in U(Ω) obtained from differentiating U(Ω) is
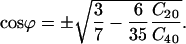 |
(3) |
For the ratio C20/C40 = −1 obtained from the simulations, this yields ϕ = 39° (see Fig. 8 b).
We also found it necessary to introduce a substantial diffusion-tilt angle (β = 44°) for the nitroxide moiety (compare to Table 1). Here β is the angle between the main diffusion axis of the molecule and the magnetic Z axis (i.e., the gzz or Azz component of the corresponding tensor). This means that the most favored direction of the nitroxide magnetic tensors is tilted relative to the principal diffusion and ordering axis of the nitroxide moiety (see Fig. 8 b).
Aligned DMPC membranes in the Pβ phase
With an increase in temperature we see a gradual change in the lineshape for the aligned DMPC membranes (Fig. 5). At 21.5°C, in the Pβ phase, near the main transition point (23.7°C), the deep splitting of the central component almost disappears, and the spectra for all orientations look less resolved. This corresponds to an increase in molecular motional parameters R⊥ and R‖ (Table 1) and also a decrease in the cone angle ϕ and the diffusion-tilt angle β. Changes of this sort may be expected at the transition to the rippled Pβ phase, which is thinner and less ordered than the Lβ phase.
Aligned DPPC membranes in the Pβ phase
With an increase in temperature from 2°C to ∼34°C (the pretransition temperature), the spectrum in the DPPC membrane goes through a slow and systematic change (see Fig. 6 for the spectra ranging from 24° to 36°C). At ∼35°C, however, an abrupt change occurs. The sharp central peak disappears and the spectrum in the aligned DPPC membrane becomes similar to the spectrum for aligned DMPC in the Pβ phase (Figs. 5 and 6).
The transition at 35–36°C is reversible, but has a hysteresis. The hysteresis time apparently depends on the extent of hydration and tends to decrease with the number of heating/cooling cycles. It varies from days for some samples that were hydrated in 100% relative humidity at room temperature to ∼1 h for the samples that were hydrated and held in excess water. For both samples the DSC shows a sharp peak at the main transition point at 41.4°C and an obvious pretransition range at 34–36°C. This suggests that both samples are thermodynamically “fully hydrated.” We consider the difference in the hysteresis time between the samples held at 100% humidity and in excess water as a “hydration paradox” effect (Rand and Parsegian, 1989).
Simulation of the spectra of GAsl in the DPPC membrane at 36°C shows a substantially tilted position of the spin-label (Fig. 7, Table 1) and noticeable mobility, just like DMPC in the Pβ phase.
Polarity: further evidence of different conformations
The polarity of the local environment is another important parameter widely used to characterize the position of the nitroxide moiety and the whole spin-labeled molecule in the membrane. Fig. 9 a shows ESR spectra of 0.4% GAsl in DMPC and DPPC vesicles at 77 K. At these conditions, all molecular motions that could affect the value of the outer splitting 2Azz are frozen, and any differences have to be attributed entirely to different polarity. The parameter 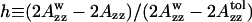 (see Lassman et al., 1973) was used to characterize the local environment's hydrophobicity for the nitroxide moiety, where 2Azz,
(see Lassman et al., 1973) was used to characterize the local environment's hydrophobicity for the nitroxide moiety, where 2Azz, 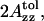 and
and 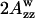 are the values of the outer splitting at 77 K in the lipid phase, in toluene and in water/glycerol mixture (1:1), respectively. As seen from Table 2, the local environment of the NO group in DMPC vesicles is much more hydrophobic than in DPPC or DSPC, when they are frozen from room temperature.
are the values of the outer splitting at 77 K in the lipid phase, in toluene and in water/glycerol mixture (1:1), respectively. As seen from Table 2, the local environment of the NO group in DMPC vesicles is much more hydrophobic than in DPPC or DSPC, when they are frozen from room temperature.
FIGURE 9.

GAsl in DPPC (solid line) and DMPC (dashes) at 77 K (a) and 10°C (b). Molar ratio GAsl/lipid = 0.6%.
TABLE 2.
Values of the outer splitting 2Azz and the hydrophobicity parameter, 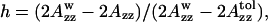 for GAsl in different membranes and solvents at 77 K
for GAsl in different membranes and solvents at 77 K
| 2Azz,G | h | |
|---|---|---|
| GAsl in toluene | 66.9 | 1 |
| GAsl in ethanol | 68.4 | 0.69 |
| Free spin-label in glycerol/water 1:1 | 71.7 | 0 |
| GAsl in DMPC membrane | 67.5 | 0.87 |
| GAsl in DPPC membrane frozen from 20°C to 77K | 70.6 | 0.23 |
| GAsl in DPPC membrane quenched from 40°C | 68.7 | 0.63 |
| GAsl in DSPC membrane | 70.4 | 0.27 |
2Azz, 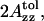 and
and 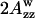 are the values of outer splitting at 77 K in the lipid phase, toluene, and water/glycerol mixture 1:1, respectively.
are the values of outer splitting at 77 K in the lipid phase, toluene, and water/glycerol mixture 1:1, respectively.
Fig. 9 b shows ESR spectra of 0.4% GAsl in DMPC and DPPC vesicles at 10°C. Although similar in shape, the spectra differ in the values of outer splitting 2A′zz. The value for DPPC is ∼2.6 G larger than for DPPC. Since in this very slow-motional range (see above) most details of molecular structure seen in aligned membranes, such as angles β and ϕ, are not manifested in vesicles, we attribute the difference to different polarity of the nitroxide environment.
As seen from the ESR spectra in the aligned membrane, GAsl in DPPC takes on different conformations above and below 35°C. The hysteresis in the heating/cooling cycle allows us to lock in (at least partially) the high-temperature conformation and to check the polarity of its environment at 77 K. The polarity (see Table 2) drops sharply at the Lβ–Pβ transition point. This means that at this transition point the spin-label goes deeper into the membrane with an accompanying change in orientation.
GAsl takes on the head-to-head conformation in the gel phase only in DMPC, not DPPC
The concept of hydrophobic mismatch gives a plausible explanation for these experimental results. Hydrophobic mismatch plays an important role in the interaction between proteins and lipids (Dumas et al., 1999). The difference between the hydrophobic length of protein and membrane is known to affect lateral segregation/aggregation of proteins in membranes and protein backbone conformation and activity (Marsh, 1995; Killian, 1998; Froud et al., 1986; Johannson et al., 1981). The hydrophobic membrane thickness in the Lα phase can be approximately estimated as 1.75(n − 1) Å, where n is the number of carbon atoms (Lewis and Engelman, 1983). In the gel phase the value is ∼30% larger (Sperotto and Mouritsen, 1988). More specifically, for the gel phase, x-ray diffraction gives a hydrocarbon thickness of 30.3 Å in DMPC (Tristram-Nagle et al., 2002) and 34.4 Å in DPPC (Nagle and Tristram-Nagle, 2000) membranes. For GA, it was found (Martinac and Hamill, 2002) that small changes in the phospholipid acyl-chain length in the Lα phase of unsaturated lipids, Di-C18:1-PC to Di-C20:1-PC, can switch GA from a stretch-activated to a stretch-inactivated channel. That corresponds to an increase in hydrocarbon thickness from 27.1 to ∼31.2 Å (Nagle and Tristram-Nagle, 2000). It has also been shown (Elliott et al., 1983) that the gramicidin channel loses its conductivity at the monoacylglycerol-squalene bilayer hydrophobic thickness ∼29–30 Å. For unsaturated phospatidylcholines a dramatic change in conductivity occurs between Di-C20:1-PC to Di-C22:1-PC, within the ∼31–35 Å range (Mobashery et al., 1997). We observe, for approximately the same range of hydrophobic membrane thicknesses, a switch for GAsl in the Lβ phase from head-to-head dimers in DMPC to another conformation in DPPC. We justify our assignment of the head-to-head conformation below (see next subsection).
The length of the head-to-head form of GA is known to be 26 Å (Wallace, 1998). In the gel phase this distance almost matches the hydrophobic thickness of the DMPC membrane, but in the case of DPPC, and especially DSPC, the membrane is substantially thicker. Due to the mismatch, the gel phase of DPPC (34.4 Å) apparently does not favor dimer formation, at least in its channel form. Our results given above indicate that the nitroxide moiety “pops up” to the polar region at the membrane surface and takes on an unconstrained (i.e., Z-aligned) conformation. With further increase in the membrane thickness and hydrophobic mismatch, formation of the head-to-head conformation in the DSPC membrane becomes even less likely than in DPPC. The similar, nearly identical spectra of GAsl in the two membranes are consistent with these spectra representing a single state of the GAsl molecules, rather than a superposition of different dimer (or monomer) forms.
However, in DMPC, GAsl most likely forms head-to-head dimers. In that case our results suggest that the conjoining N-termini pull the opposite ends of each GAsl molecule deeper into the bilayer, causing a decrease in polarity and a tilt of the nitroxide ring. In the Pβ phase of DPPC (above 35°C) GAsl starts to form channels, which may be associated with a lateral expansion of lipid molecules and a concomitant decrease in bilayer thickness and hydrophobic mismatch.
Good orientation-dependence for GAsl in DMPC membranes with low water content versus complete loss of macroscopic alignment in DPPC upon drying (results not shown) gives another hint that the behavior of GAsl in the two lipids is substantially different.
GAsl in the Lα phase
In the Pβ phase (see above) of DPPC we observe formation of GAsl dimers. The head-to-head dimer is apparently the major form of GAsl also in the Lα phase. Near the main transition point for both DMPC and DPPC we observe a substantial change in the spectrum that corresponds to a transition to a faster motional regime. Since our ISDU samples, especially in the DMPC membranes, lose their macroscopic alignment at the main transition point, we confined the comparative study in the Lα phase to vesicles. The phase transition manifests itself in a second mobile component that emerges at a temperature slightly below the main transition point (compare to Fig. 10 a for DMPC). Above the main transition point the more mobile component grows, at the expense of the less mobile one (marked with an arrow in Fig. 10 a). The slower component, however, does not disappear completely and can be observed over the whole temperature range studied up to 60°C. As seen in Fig. 10 b, at 55°C the spectra in DMPC and DPPC are almost identical and the simulation (Fig. 10 c) gives coefficients of rotational diffusion R⊥ = 8 × 107, R‖∼109, in the intermediate motional regime for the main (faster motional) component.
FIGURE 10.

Temperature dependence of ESR spectra of GAsl (0.6 mol %) in (a) DMPC vesicles around and above the main transition; (b) DMPC vesicles (solid line) and DPPC vesicles (dots) at 55°, corresponding to Lα phase; and (c) simulation for DMPC at 55°C (dashes).
Direct observation of dimer formation by DQC
Results of our DQC experiments are strongly supportive of the above analysis of CW-ESR spectra. The DQC-ESR dipolar spectrum for 0.12 mol % GAsl in DMPC is shown in Fig. 11 a. DQC clearly shows the presence of dimers. The interspin distance is 30.9 ± 0.5 Å, in good agreement with our estimates for the head-to-head dimer (see below). Our DQC results for DMPC also rule out any significant lateral aggregation and formation of GAsl hexamers as described in Mou et al. (1996), which would yield a much less resolved spectrum or a loss of signal. Thus we conclude that GAsl at our experimental conditions exists mainly as isolated head-to-head dimers.
FIGURE 11.

DQC-ESR spectra of GAsl dimers in (a) DMPC vesicles and (b) DPPC vesicles for a sample rapidly frozen to 77 K from 313 K. Molar ratio GAsl/lipid = 0.12% in both cases. The splitting in the dipolar spectrum a corresponds to an average distance of 30.9 Å.
Unlike the results for DMPC, the DQC data for DPPC membranes do not yield a resolved dipolar spectrum (Pake doublet), if the samples are frozen from the gel phase (22°C) to 77 K. However, if a DPPC sample is quickly frozen from the Pβ or Lα phase, then DQC shows a strong signal from radical pairs, similar to the signal in DMPC (Fig. 11 b). The interspin distance in these pairs is 31.4 ± 0.5 Å. (More details on the DQC results are given in the Appendix.)
The most detailed structure of the GA channel in the DMPC membrane was obtained by NMR (Ketchem et al., 1997). Our initial estimate for the distance between the nitroxide fragments in the GAsl dimer is based on this structure and the assumption of a free unconstrained conformation of the spin-label fragment. Taking into account the distribution of the free electron density along the NO-bond we expect an interspin distance ∼30 Å. A slightly different actual distance of 30.9 Å may be explained by a tilt of the nitroxide tethers toward the membrane surface, consistent with our analysis of the CW-ESR spectra. If we assume that nitroxide tethers have some tendency to stretch toward the membrane surface, one should also expect a slight increase in the interspin distance for the head-to-head dimer of GAsl with an increase in the membrane thickness, in good accord with our observations for DPPC samples, quenched from the Pβ phase.
In the Lβ phase of DPPC, GAsl is not grouped in isolated pairs with a well-defined interspin distance
The weakness of the DQC signal from pairs in DPPC samples frozen from the Lβ phase indicates that GAsl is not grouped into isolated pairs with a relatively narrow distribution of interspin distance. However, it does not necessarily imply a uniform distribution of monomers. A double-stranded dimer conformation(s) with a range of interspin distances is one possibility. This seems likely, given that the length of some double-stranded structures reported in organic solvents matches well with the hydrophobic thickness for the DPPC gel phase (Wallace, 1998). There is a general consensus in favor of the presence of an antiparallel double β5.6 helical dimer (Langs, 1988) in membranes in equilibrium with the head-to-head form (Sychev et al., 1993; Zein and Winter, 2000; Burkhart et al., 1998). A high-resolution NMR structure for the conformer in benzene/ethanol was solved by Pascal and Cross (1993). The total dimer length is 36–37 Å, which for the case of GAsl would give an interspin distance ∼40–41 Å, well within the range of DQC.
A plausible explanation for the absence of a strong signal from isolated pairs in the Lβ phase of DPPC may emerge from the indication of aggregation obtained by DQC and the primary ESE decay (see Appendix). The dipolar spectrum will be unresolved and weakened by multiple interactions between electron spins at various distances possible in the aggregate. Mou et al. (1996), on the basis of atomic-force microscope images in supported saturated phosphatidylcholines in the gel phase, inferred a basic aggregation unit, most probably a hexamer of GA, which can be observed even at low GA concentrations. The assumption that the double-stranded dimers in the membrane form aggregates, whereas head-to-head dimers exist mainly as separate entities, can also resolve the controversy between the data of atomic-force microscopy in the gel phase and the data of other methods, obtained mainly for the fluid phase (Tank et al., 1982; Veatch et al., 1975; Macdonald and Seelig, 1988). The role of tryptophan residues may provide a key to the difference in the aggregation behavior of the conformers. It is well established that tryptophan residues disrupt the structure of the hydrophobic part of the bilayer, and tend to be excluded from the bilayer (Yau et al., 1998). The preference of tryptophan for membrane interfaces is an important factor in the stability of the head-to-head dimer (Cotten et al., 1997; Roux, 1998). An important feature of the antiparallel double β5.6 helical structure is a uniform distribution of Trp residues along the dimer axis (Pascal and Cross, 1993). Their submerged position should cause an energy penalty, which can be partially reduced by aggregation of several dimers. Killian et al. (1987) showed an important role of tryptophans in GA aggregation and HII phase formation at higher GA concentrations.
Although there is strong evidence of double-helical dimers in membranes in the literature, we should also consider the possibility of a free monomer state existing in the gel phase of DPPC membrane (see Appendix). In consideration of this latter possibility we note that dimerization constants for GA were measured in DOPC (Bamberg and Läuger, 1973) and glyceroester membranes (Veatch et al., 1975), thylakoid membranes (Schönknecht et al., 1992), and 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) (Rokitskaya et al., 1996). The dimerization constant for DOPC in the Lα phase at 25°C was reported in the range 2 × 1013 to 1014 mol−1 cm2 (Bamberg and Läuger, 1973; Veatch et al., 1975), depending on the method used. For a surface area per DOPC molecule of 0.82 nm2 and GA concentration 1 mol % it yields that 4.9–10.5% of the GA molecules present as monomers. Rokitskaya et al. (1996) give the values of the dissociation and association rate constants in DPhPC, from which we get dimerization constants of 9.6 × 1013 mol−1 cm2 and 1.87 × 1013 mol−1 cm2 at 26 and 18°C, respectively. Taking a surface area per molecule 0.59 nm2 like in fluid DPPC (Marsh, 1990) we get, for 1% GA, 4.2% monomers at 26°C and 9.3% at 18°C. DPhPC is in the Lα phase at these temperatures. The decrease in the association is apparently concomitant with a decrease in the bilayer fluidity at 18°C. Such a decrease is much more pronounced for a lipid phase that undergoes a phase transition. In the gel phase we may expect a greater fraction of GAsl monomers, with the nitroxide moiety located at the membrane surface.
Benefits obtained by utilization of Gasl: comparison with other methods
Spin-labeled GA gave us an opportunity to carry out an ESR study of gramicidin-lipid systems at much lower molar ratios of GA/lipid than have been used before in ESR (Tanaka and Freed, 1985; Ge and Freed 1993, 1999; Costa-Filho et al., 2003), or in FTIR, circular dichroism (CD), or x-ray (Sychev et al., 1993; Zein and Winter, 2000). The ratios suffice for the purpose of studying single non-interacting channels and the results can be directly related to energetic measurements (Nielsen et al., 1998) or molecular dynamics simulations (Woolf and Roux, 1996; Chiu et al., 1999).
In DMPC, over the whole temperature range, or in DPPC above the Lβ–Pβ phase transition point (35°C), GAsl forms head-to-head dimers. However, in the Lβ phase in DPPC we find a different conformation. Qualitatively similar results were obtained for gramicidin in DMPC, DSPC, and DPPC by FTIR spectroscopy (Zein and Winter, 2000). They observed that in the DMPC membrane, no significant change in the conformational behavior in DMPC/GA mixtures (5 mol %) occurs within the temperature range 10–35°C; the equilibrium of the gramicidin species seems to be in favor of head-to-head dimers. In DPPC/GA mixtures (5 mol %) at a temperature slightly below the main transition, they observe significant structural rearrangements: the fractional intensity of head-to-head dimers increases with a concomitant decrease in the fractional intensities attributed to other species. Wallace et al. (1981) obtained similar results earlier with the CD method. Whereas CD indicates the same GA conformation for both Lβ and Lα phases of DMPC, a clear change in the CD pattern was observed upon going from DMPC to DPPC and DSPC in the gel phase. As in our case, the head-to-head conformer becomes the main component in all fluid phases (Zein and Winter, 2000).
Although qualitatively similar, there are some differences between our ESR/DQC-ESR data and the FTIR data of Zein and Winter (2000). In their case, there is a substantial fraction of the head-to-head conformation below the main phase transition of DPPC. Although no quantitative numbers are available on absolute fractions, it is clearly seen that below the Lα–Lβ transition the fraction of head-to-head dimers does not disappear but reduces approximately by one-half. Our data show much greater change; only a small fraction of head-to-head dimers may be present in the Lβ phase according to DQC. The absence of a well-resolved Pake doublet detected by DQC-ESR in DPPC vesicles frozen from the Lβ phase means that the concentration of radical pairs in the sample is at least a factor-of-10 lower than in the sample which is quenched from the Pβ phase. The CW-ESR spectra in these phases can be reasonably well simulated with a single (at least 90%) component. We also observe a much sharper change (within an ∼2°C range at the Lβ–Pβ transition), compared to a gradual increase through a 20°C range in the fractional intensity of the head-to-head dimer observed by Zein and Winter (2000).
The most plausible explanation for the discrepancy is the high molar fraction of GA used in Zein and Winter (2000). In the case of the low concentration of ∼0.4% GAsl that we use, the phase behavior of the mixture is approximately the same as in pure lipids. A 5%-GA fraction causes substantial change in the phase behavior. Thus, Zein and Winter (2000), by DSC, observe a broad gel-fluid coexistence region, instead of a sharp (within 0.3°C) main transition and a well-defined pretransition. At 5 mol % of gramicidin, one-half of the lipid molecules are in direct contact with the channel surface (Nielsen et al., 1998), and there is no unperturbed lipid present, nor any well-defined Lα, Pβ, or Lβ phase, but just fluid-like and gel-like phases at temperatures above and below the broad gel-fluid coexistence region. For example, at gramicidin concentrations above 4% we could not obtain well-aligned membrane samples by ISDU. The viscosity in the lipid bilayer increases with an increase in GA concentration, and that should also cause kinetic effects, such as hysteresis and retaining thermodynamically unstable states. (See, also in this connection, the effects of “solvent history,” observed at GA concentration ∼10%; Bouchard and Auger, 1993.) Hence, at higher concentrations we would expect significantly modified behavior, largely due to cooperative effects of the gramicidin molecules in the membrane, compared to what we observe in our low concentration experiments.
CONCLUSIONS
We obtained a spin-labeled analog of gramicidin A, GAsl, which allowed us to study, by CW-ESR and DQC-ESR, GA channels under sufficiently dilute conditions for non-interacting single channels in several lipid membranes. This included both macroscopically aligned (by ISDU) membrane multilayers and vesicles. The labeling position at the C-terminus of the GA molecule makes GAsl a very convenient compound with which to study head-to-head dimer formation (i.e., N-terminus to N-terminus).
The use of well-aligned membranes substantially increased the resolution of the CW spectra compared to vesicles and yielded (after detailed spectral analysis) information on the preferred orientation of the nitroxide moieties relative to the membrane normal, which would be difficult to obtain from vesicles. This allowed us to assign the spectrum of GAsl in the DMPC membrane to the head-to-head conformation. It was found that in DPPC and DSPC, GAsl exists in a different conformation. Further evidence of head-to-head dimer formation in DMPC (but not DPPC or DSPC) was provided by the polarity of the local environment of the nitroxide moiety in the lipids.
Direct evidence of head-to-head dimer formation has been obtained by DQC-ESR. This method clearly indicates the presence of pairs in the DMPC membrane with an interspin distance of 30.9 Å. No such pairs are detected in the samples frozen from the Lβ phase of DPPC.
Given that the length of the head-to-head dimer of GAsl matches the hydrophobic thickness of the DMPC membrane, but that it is substantially shorter than the thickness of DPPC and DSPC, these results are consistent with the concept of hydrophobic mismatch.
Above the Lβ–Pβ phase transition point in DPPC (above 35°C), we do observe formation of head-to-head dimers by both CW-ESR and DQC-ESR. This most likely occurs because the lateral expansion of lipid molecules and concomitant decrease in bilayer thickness reduce the hydrophobic mismatch in the Pβ phase compared to the Lβ phase. Hysteresis at the heating/cooling cycle allowed us to trap the Pβ phase conformation and to detect, by DQC-ESR, radical pairs with an interspin distance of 31.4 Å.
Acknowledgments
We thank Pieter C. Dorrestein for carrying out mass-spectroscopic measurements and providing help in the experiments.
This work was supported by National Institutes of Health grants GM 25852 and RR16292, and National Science Foundation-Chemistry grant CHE-0098022.
APPENDIX: FURTHER ANALYSIS OF DQC-ESR SPECTRA
Here we compare the experimental time-domain DQC-ESR and spin-echo ESR signals from GAsl in DPPC, from samples rapidly frozen from the Lβ and Lα phases. We show in Fig. 12 a the time-domain DQC signal (we collect typically one-half of the signal versus tξ with some extra points at ∼0), from a sample prepared by rapid freeze from the Lα phase. This signal has a background due to intermolecular dipolar couplings, which was removed as described previously (Borbat et al., 2002). Subsequent Fourier transformation yields a dipolar spectrum with a splitting corresponding to ∼31.4 Å (see Fig. 11 b). It is not as well resolved as that from DMPC dimers (see Fig. 11 a), and it indicates that there is also a substantial population of pairs with larger distances, possibly double-helix dimers. Fig. 12 b is the DQC signal from the sample prepared by freezing DPPC in the Lβ phase. It is significantly weaker and with no hint of an oscillation. The latter suggests the existence of a broad distribution in distances of the order 30–40 Å. This is supported by the primary echo decay signals obtained for the two cases. The logarithm of the primary echo decay for the sample, rapidly quenched from the Lα phase (not shown), is linear, but with small oscillations that correspond to that in the DQC signal. However, the logarithm of the primary echo decay from the sample frozen from the Lβ phase (not shown) decays during the first 0.5 μs by an order-of-magnitude more rapidly than from the Lα phase for a π/2 pulse with a width of 3 ns, whereas it becomes more linear for longer π/2 pulses (not shown). Such observations could be attributed to a large fraction of antiparallel double-helical dimers with a broad range of distances e.g., 33–40 Å, and/or it may indicate aggregation of GAsl at these conditions. The faster decay of the initial part of the primary echo decay and the weaker background signal in the DQC from the sample frozen from the Lβ phase suggests the presence of a significant fraction of GAsl in aggregates, which would yield a rapid decay as a result of the intermolecular dipolar interactions and thus would not be visible in DQC, where the echo occurs after a time lapse of 2tm + 2t1 (see Fig.2 a).
FIGURE 12.

Time-domain DQC-ESR signals from GAsl/DPPC (0.12 mol %) samples. (a) A sample rapidly frozen from the Lα phase; (b) a sample frozen from the Lβ phase. Note that the smaller amplitude in case b is due to the property of DQC that it senses only the dipolar-modulated part of the spin-echo. There are fewer detectable nitroxide pairs in b as compared to a. The background signal is caused by intermolecular dipolar couplings (Borbat et al., 2002). A smaller background signal in b may be caused by a significant fraction of GA existing in the Lβ phase in the form of small aggregates, leading to a rapid decay of the echo on the timescale required to acquire the signal from GAsl pairs.
References
- Andersen, O. S. 1984. Gramicidin channels. Annu. Rev. Physiol. 46:531–548. [DOI] [PubMed] [Google Scholar]
- Bamberg, E., and P. Läuger. 1973. Channel formation kinetics of gramicidin A in lipid bilayer membranes. J. Membr. Biol. 11:177–194. [DOI] [PubMed] [Google Scholar]
- Borbat, P. P., R. Crepeau, and J. H. Freed. 1997. Multifrequency two-dimensional Fourier transform ESR: an X\Ku-band spectrometer. J. Magn. Reson. 127:155–167. [DOI] [PubMed] [Google Scholar]
- Borbat, P. P., and J. H. Freed. 1999. Multiple-quantum ESR and distance measurements. Chem. Phys. Lett. 313:145–154. [Google Scholar]
- Borbat, P. P., and J. H. Freed. 2000. Double quantum ESR and distance measurements. In Biological Magnetic Resonance, Vol. 19. Distance Measurements in Biological Systems by EPR. L.J. Berliner, G.R. Eaton, and S.S. Eaton, editors. Kluwer Academic/Plenum Publishers, New York. 383–459.
- Borbat, P. P., A. J. Costa-Filho, K. A. Earle, J. K. Moscicki, and J. H. Freed. 2001. Electron spin resonance in studies of membranes and proteins. Science. 291:266–269. [DOI] [PubMed] [Google Scholar]
- Borbat, P. P., H. S. Mchaourab, and J. H. Freed. 2002. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J. Am. Chem. Soc. 124:5304–5314. [DOI] [PubMed] [Google Scholar]
- Bouchard, M., and M. Auger. 1993. Solvent history dependence of gramicidin-lipid interactions: a Raman and infrared spectroscopic study. Biophys. J. 65:2484–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budil, D. E., S. Lee, S. Saxena, and J. H. Freed. 1996. Non-linear least squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J. Magn. Reson. A. 120:155–189. [Google Scholar]
- Burkhart, B. M., N. Li, D. A. Langs, W. A. Pangborn, and W. L. Duax. 1998. The conducting form of gramicidin A is a right-handed double-stranded double helix. Proc. Natl. Acad. Sci. USA. 95:12950–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., S. Subramaniam, and E. Jakobsson. 1999. Simulation study of gramicidin/lipid bilayer system in excess water and lipid. I. Structure of the molecular complex. Biophys. J. 76:1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Filho, A. J., R. H. Crepeau, P. P. Borbat, M. Ge, and J. H. Freed. 2003. Lipid-gramicidin interactions: dynamic structure of the boundary lipid by 2D-ELDOR. Biophys. J. 84:3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten, M., F. Xu, and T. A. Cross. 1997. Protein stability and conformational rearrangements in lipid bilayers: linear gramicidin, a model system. Biophys. J. 73:614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, D. A., and B. A. Wallace. 1998. Shifting the equilibrium mixture gramicidin double helices toward a single conformation with multivalent cationic salts. Biophys. J. 75:635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas, F., M. C. Lebrun, and J.-F. Tocanne. 1999. Is the protein/lipid hydrophobic matching principle relevant to membrane organization and functions? FEBS Lett. 458:271–277. [DOI] [PubMed] [Google Scholar]
- Elliott, J. R., D. Needham, J. P. Dilger, and D. A. Haydon. 1983. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Biophys. Acta. 735:95–103. [DOI] [PubMed] [Google Scholar]
- Fahsel, S., E. M. Pospiech, M. Zein, T. L. Hazlet, E. Gratton, and R. Winter. 2002. Modulation of concentration fluctuations in phase-separated lipid membranes by polypeptide insertion. Biophys. J. 83:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froud, R. J., C. R. A. Earl, J. M. East, and A. G. Lee. 1986. Effects of lipid fatty acyl chain structure on the activity of the (Ca2+-Mg2+)-ATPase. Biochim. Biophys. Acta. 860:354–375. [DOI] [PubMed] [Google Scholar]
- Ge, M., and J. H. Freed. 1993. An electron spin resonance study of interactions between gramicidin A′ and phosphatidylcholine bilayers. Biophys. J. 65:2106–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, M., and J. H. Freed. 1999. Electron-spin resonance study of aggregation of gramicidin in dipalmitoylphosphatidylcholine bilayers and hydrophobic mismatch. Biophys. J. 76:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, M., D. E. Budil, and J. H. Freed. 1994. ESR studies of spin-labeled membranes aligned by isopotential spin-dry ultracentrifugation: lipid-protein interactions. Biophys. J. 67:2326–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassner, A., and V. Alexanian. 1978. Direct room temperature esterification of carboxylic acids. Tetrahedr. Lett. 19:4475–4478. [Google Scholar]
- Hotchkiss, R. D., and R. J. Dubois. 1940. Fractionation of bactericidal agent from cultures of a soil bacillus. J. Biol. Chem. 132:791–792. [Google Scholar]
- Hubbell, W., and H. McConnell. 1971. Molecular motion in spin-labeled phospholipids and membranes. J. Am. Chem. Soc. 93:314–326. [DOI] [PubMed] [Google Scholar]
- Johannson, A., G. A. Smith, and J. C. Metcalfe. 1981. The effect of bilayer thickness on the activity of the (Na+-K+)-ATPase. Biochim. Biophys. Acta. 641:416–421. [DOI] [PubMed] [Google Scholar]
- Ketchem, R. R., B. Roux, and T. A. Cross. 1997. High-resolution polypeptide structure in a lamellar phase lipid environment from solid-state NMR-derived orientational constants. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- Killian, J. A. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–416. [DOI] [PubMed] [Google Scholar]
- Killian, J. A. 1992. Gramicidin and gramicidin-lipid interactions. Biochim. Biophys. Acta. 1113:391–425. [DOI] [PubMed] [Google Scholar]
- Killian, J. A., K. N. J. Burger, and B. de Kruijff. 1987. Phase separation and hexagonal HII phase formation by gramicidins A, B and C in dioleoylphosphatidylcholine model membranes. A study on the role of the tryptophan residues. Biochim. Biophys. Acta. 897:269–284. [DOI] [PubMed] [Google Scholar]
- Kovacs, F., J. Quine, and T. A. Cross. 1999. Validation of the single-stranded channel conformation of gramicidin A by solid-state NMR. Proc. Natl. Acad. Sci. USA. 96:7910–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langs, D. A. 1998. Three-dimensional structure at 0.86 Å of the uncomplexed form of the transmembrane ion channel peptide gramicidin A. Science. 241:188–191. [DOI] [PubMed] [Google Scholar]
- Lassman, G., B. Ebert, A. N. Kuznetsov, and W. Damerau. 1973. Characterization of hydrophobic regions in proteins by spin-labeling technique. Biochim. Biophys. Acta. 310:298–304. [DOI] [PubMed] [Google Scholar]
- Lebedev, Y. S., O. Y. Grinberg, A. A. Dubinsky, and O. G. Poluektov. 1992. Investigation of spin-labels and probes by millimeter band EPR. In Bioactive Spin-Labels. R.I. Zhdanov, editor. Springer Verlag, New York.
- Lewis, B. A., and D. M. Engelman. 1983. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 166:203–210. [DOI] [PubMed] [Google Scholar]
- Liang, Z., and J. H. Freed. 1999. An assessment of the applicability of multifrequency ESR to study the complex dynamics of biomolecules. J. Phys. Chem. B. 103:6384–6396. [Google Scholar]
- LoGrasso, P. V., F. Moll III, and T. A. Cross. 1988. Solvent history dependence of gramicidin A conformations in hydrated lipid bilayers. Biophys. J. 54:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, P. M., and J. Seelig. 1988. Dynamic properties of gramicidin A in phospholipid membranes. Biochemistry. 27:2357–2364. [DOI] [PubMed] [Google Scholar]
- Marsh, D. 1990. Handbook of Lipid Bilayers. CRC Press, Boca Raton, FL.
- Marsh, D. 1995. Lipid-protein interactions and heterogeneous lipid distribution in membranes. Mol. Membr. Biol. 12:244–249. [DOI] [PubMed] [Google Scholar]
- Martinac, B., and O. Hamill. 2002. Gramicidin A channels switch between stretch activation and stretch inactivation depending on bilayer thickness. Proc. Natl. Acad. Sci. USA. 99:4308–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirovitch, E., A. Nayeem, and J. H. Freed. 1984. Analysis of protein-lipid interactions based on model simulations of electron spin resonance spectra. J. Phys. Chem. 88:3454–3465. [Google Scholar]
- Meirovitch, E., D. Igner, E. Igner, G. Moro, and J. H. Freed. 1982. Electron-spin relaxation and ordering in smectic and supercooled nematic liquid crystals. J. Chem. Phys. 77:3915–3938. [Google Scholar]
- Mobashery, N., C. Nielsen, and O. S. Andersen. 1997. The conformational preference of gramicidin channels is a function of lipid bilayer thickness. FEBS Lett. 412:15–20. [DOI] [PubMed] [Google Scholar]
- Mou, J., D. M. Czajkowsky, and Z. Shao. 1996. Gramicidin A aggregation in supported gel state phosphatidylcholine bilayers. Biochemistry. 35:3222–3226. [DOI] [PubMed] [Google Scholar]
- Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, C. M., M. Goulian, and O. S. Andersen. 1998. Energetics of inclusion-induced bilayer deformation. Biophys. J. 74:1966–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal, S. M. and T. A. Cross. 1993. High resolution structure and dynamic implications for a double-helical gramiciden A conformer. J. of Biomolec. NMR. 3:495–513. [DOI] [PubMed] [Google Scholar]
- Rand, R. P., and V. A. Parsegian. 1989. Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta. 988:351–376. [Google Scholar]
- Rokitskaya, T. I., Y. N. Antonenko, and E. A. Kotova. 1996. Photodynamic inactivation of gramicidin channels: a flash-photolysis study. Biochim. Biophys. Acta. 1275:221–226. [DOI] [PubMed] [Google Scholar]
- Roux, B. 1998. Environment of a membrane protein. In The Encyclopedia of Computational Chemistry. P.v.R. Scleyer, N.L. Allinger, T. Clark, J. Gasteiger, P.A. Kollman, H.F. Schaefer III, and P.R. Schreiner, editors. John Wiley & Sons, Chichester, UK.
- Schneider, D. J., and J. H. Freed. 1989. Calculating slow motional magnetic resonance spectra: a user's guide. In Biological Magnetic Resonance, Vol. 8, Spin-labeling, Theory and Application. L.J. Berliner and J. Reuben, editors. Plenum Press, New York. 1–76.
- Schönknecht, G., G. Althoff, and W. Junge. 1992. Dimerization constant and single-channel conductance of gramicidin in thylakoid membranes. J. Membr. Biol. 126:265–275. [DOI] [PubMed] [Google Scholar]
- Sperotto, M. M., and O. G. Mouritsen. 1988. Dependence of lipid membrane phase transition temperature on the mismatch of protein and lipid hydrophobic thickness. Eur. Biophys. J. 16:1–10. [Google Scholar]
- Sychev, S. V., L. I. Barsukov, and V. T. Ivanov. 1993. The double ππ5.6 helix of gramicidin A predominates in unsaturated lipid membranes. Eur. Biophys. J. 22:279–288. [DOI] [PubMed] [Google Scholar]
- Tanaka, H., and J. H. Freed. 1985. Electron spin resonance studies of lipid-gramicidin interactions utilizing oriented multibilayers. J. Phys. Chem. 89:350–360. [Google Scholar]
- Tank, D. W., E. S. Wu, P. R. Meers, and W. W. Webb. 1982. Lateral diffusion of gramicidin C in phospholipid multibilayers. Effects of cholesterol and high gramicidin concentration. Biophys. J. 40:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristram-Nagle, S., Y. Liu, J. Legleiter, and J. F. Nagle. 2002. Structure of gel phase DMPC determined by x-ray diffraction. Biophys. J. 83:3324–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urry, D. W. 1971. The gramicidin A transmembrane channel: a proposed π(L,D) helix. Proc. Natl. Acad. Sci. USA. 68:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urry, D. W., M. C. Goodall, J. D. Glickson, and D. F. Mayers. 1971. The gramicidin A transmembrane channel: characteristics of head-to-head dimerized π(L,D) helices. Proc. Natl. Acad. Sci. USA. 68:1907–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch, W. R., R. Mathies, M. Eisenberg, and L. Stryer. 1975. Simultaneous fluorescence and conductance studies of planar bilayer membranes containing highly active and fluorescent analogs of gramicidin A. J. Mol. Biol. 99:75–92. [DOI] [PubMed] [Google Scholar]
- Veatch, W. R., E. T. Fossel, and E. R. Blout. 1974. The conformation of gramicidin A. Biochemistry. 13:5249–5256. [DOI] [PubMed] [Google Scholar]
- Wallace, B. A. 1998. Recent advances in the high resolution structures of bacterial channels: gramicidin A. J. Struct. Biol. 121:123–141. [DOI] [PubMed] [Google Scholar]
- Wallace, B. A., W. R. Veatch, and E. R. Blout. 1981. Conformation of gramicidin A in phospholipid vesicles: circular dichroism studies of effects of ion binding, chemical modification, and lipid structure. Biochemistry. 20:5754–5760. [DOI] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1996. Structure, energetics and dynamics of lipid-protein interaction: a molecular dynamics study of the gramicidin A channel in DMPC bilayer. Proteins Struct. Funct. Genet. 24:92–114. [DOI] [PubMed] [Google Scholar]
- Yau, W.-M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- Zein, M., and R. Winter. 2000. Effect of temperature, pressure and lipid acyl chain length on the structure and phase behavior of phospholipid-gramicidin bilayers. Phys. Chem. Chem. Phys. 2:4545–4551. [Google Scholar]