

Abstract
Near-field scanning optical microscopy (NSOM) has been used to study the nanoscale distribution of voltage-gated L-type Ca2+ ion channels, which play an important role in cardiac function. NSOM fluorescence imaging of immunostained cardiac myocytes (H9C2 cells) demonstrates that the ion channel is localized in small clusters with an average diameter of 100 nm. The clusters are randomly distributed throughout the cell membrane, with some larger fluorescent patches that high-resolution images show to consist of many small closely-spaced clusters. We have imaged unstained cells to assess the contribution of topography-induced artifacts and find that the topography-induced signal is <10% of the NSOM fluorescence intensity. We have also examined the dependence of the NSOM signal intensity on the tip-sample separation to assess the contributions from fluorophores that are significantly below the cell surface. This indicates that chromophores >∼200 nm below the probe will have negligible contributions to the observed signal. The ability to quantitatively measure small clusters of ion channels will facilitate future studies that examine changes in protein localization in stimulated cells and during cardiac development. Our work illustrates the potential of NSOM for studying membrane domains and protein localization/colocalization on a length scale which exceeds that available with optical microscopy.
INTRODUCTION
The current renaissance in optical imaging technologies enables the study of biologically relevant molecules with resolution that is capable of detecting macromolecular complexes and signaling domains (Tsien, 2003). One approach, near-field scanning optical microscopy (NSOM), utilizes a subwavelength-aperture light source for simultaneous mapping of topographic and optical properties of a sample surface with a lateral spatial resolution beyond the classical diffraction limit (Pohl et al., 1984; Lewis et al., 1984). This method combines advantages of both atomic force microscopy and optical imaging, with typical optical resolution of 40–100 nm. In this context it is worth noting that other high-resolution fluorescence methods, such as 4 Pi detection, have yet to demonstrate such resolution for biological samples (Hell, 2003). Although most biological applications of NSOM have focused on imaging individual macromolecules (Dunn, 1999; Zenobi and Deckert, 2000), the possibility of cellular imaging is particularly exciting (Edidin 2001; de Lange et al., 2001; Lewis et al., 2003). The surface sensitivity and high spatial resolution give NSOM significant potential for studies of the lateral organization of membrane domains and protein clusters and complexes.
Several studies of NSOM imaging of cell surfaces have been reported. For example, NSOM has been used to examine human red blood cells invaded by the malaria parasite Plasmodium falciparum (Enderle et al., 1997). Selective immunofluorescence labeling of parasite and erythrocyte proteins allowed simultaneous dual-color excitation, detection, and colocalization studies of the proteins on the cell membrane with a resolution of ∼100 nm. Simultaneous topographic and fluorescence NSOM imaging of recombinant Escherichia coli-expressing green fluorescent protein showed different green fluorescent protein levels for individual bacteria (Tamiya et al., 1997). In another study NSOM was used to demonstrate that erbB2, a member of the epidermal growth factor receptor tyrosine kinase family, was concentrated in irregular membrane patches with a mean diameter of ∼0.5 μm (Nagy et al., 1999). The cluster diameter increased to 0.6–0.9 μm when cells were activated with epidermal growth factor. Similarly, NSOM imaging of dye-labeled lipids and proteins in plasma membranes of fixed fibroblasts (Hwang et al., 1998) revealed the existence of small patches and membrane domains on a scale of tens to hundreds of nanometers. More recently, NSOM has demonstrated that the segregation of β4 integrin in the membrane of human keratinocytes increases upon exposure to a magnetic field (Rieti et al., 2004). These examples, and related work (Kirsch et al., 1998; Lei et al., 2001; Rothery et al., 2003), clearly indicate that cell surfaces are amenable to near-field imaging. However, few groups have been able to take advantage of NSOM to resolve features that cannot be visualized by diffraction-limited optical microscopy.
Several technical challenges must be solved before NSOM can be routinely used for the nanoscale interrogation of cellular membranes (Edidin 2001; de Lange et al., 2001). One of the main problems is the difficulty of imaging soft biological samples in a physiological environment using shear-force techniques. Furthermore, issues related to the interpretation of NSOM images must be clarified to employ NSOM for imaging thick samples with complex morphology, such as cells. These include accounting for topography-induced artifacts in the optical image (Novotny et al., 1998, Hecht et al., 1997) and understanding the contributions from chromophores that are significantly below the cell surface (Doyle et al., 2001). Our recent work has shown that bent double-etched probes provide a solution to some of the problems associated with NSOM imaging in an aqueous environment (Burgos et al., 2003a,b; Ianoul et al., 2003). We have now extended applications of these probes to the challenges of imaging cell surfaces, initially for dry, fixed cells.
We have carried out a detailed study of the distribution of voltage-gated L-type Ca2+ channels on the surface of H9C2 cells. This cell type is a clonal cardiac cell line derived from embryonic rat hearts (Kimes and Brandt, 1976) that was developed as an in vitro model of the cardiac muscle phenotype and has been frequently used to study cardiac ion channel function (Hescheler et al., 1991; Menard et al., 1999). The contraction of cardiac myocytes is initiated by a calcium gradient that is mediated by voltage-gated L-type Ca2+ channels (Davare et al., 2001). The channels consist of a central pore-forming α1c subunit and two additional subunits (α2δ and β) and are regulated by messengers that activate signal transduction networks through G proteins, protein kinases, and phosphatases (Cong et al., 2001; Catterall, 1995, 2000). Influx of calcium through the L-channels regulates the release of calcium from the closely coupled ryanodine channels, leading to contraction. Although much functional data is available for these calcium channels, understanding the mechanisms that regulate calcium channel function requires knowledge of the distribution of various components of the signaling network. Near-field microscopy provides one approach to this problem.
The NSOM results show that the L-type Ca2+ channels are organized in clusters (60–250 nm in diameter) with an average size of ∼100 nm. We have considered background optical signal that could arise from a variety of sources, including topography-induced artifacts, areas with low levels of membrane ion channels (rather than clusters), or labeled fluorophores significantly below the membrane surface. Imaging unstained cells indicates that the topography-induced signal is typically <10% of the NSOM fluorescence intensity. We have determined the distance dependence of the NSOM signal intensity for 40-nm dye-labeled spheres to assess the importance of contributions from fluorophores below the sample surface in a well-defined model system. The data are used to define an effective fluorescence interaction volume and an associated NSOM probe depth of field that demonstrates that contributions from fluorophores >100–200 nm below the surface are likely to be negligible for typical imaging conditions. Although NSOM theory predicts both evanescent and radiative components for light emerging from an NSOM probe aperture, our measurements are consistent with only a radiative component. In fact, we show that in many illumination configuration fluorescence experiments of biological samples it is technically very difficult to measure the evanescent field-fluorophore interaction.
EXPERIMENTAL
Cell culture
H9C2 cells (ATCC) were grown in culture flasks in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, glutamine (2 mM), penicillin (100 IU/ml), and streptomycin (100 mg/ml) under standard culture conditions (37°C, 5% CO2). The H9C2 cells were seeded using the following procedure. The medium was aspirated from cells growing in a T75 flask (∼70% confluent). The monolayer of cells was washed with 10 ml phosphate-buffered saline (PBS). One ml of trypsin was added and the sample was incubated at 37°C until cells had detached. The cells were suspended in 10 ml of H9C2 medium, transferred to a 15-ml tube, and centrifuged at 1000 rpm for 5 min at room temperature. The medium was aspirated and the cell pellet was suspended in 3 ml of fresh medium. The number of viable cells was determined by trypan blue exclusion. The H9C2 cells were then seeded onto 18-mm round glass coverslips in a 12-well plate at a density of 4 × 104 cells per well in a total volume of 2 ml medium. These samples were incubated at 37°C and 5% CO2 until the cells had attached and reached 50–70% confluency. The cells were fixed by aspirating the medium from the wells, washing twice with 1 ml PBS, and then adding 2 ml of 3.7% formaldehyde at 4°C. After 20 min the formaldehyde was removed and the samples were washed three times (3 min per wash) with 1 ml of PBS.
Immunofluorescence
H9C2 cells fixed on coverslips were aspirated to remove the PBS and one well was covered with 1 ml of PBS, which served as the no-primary-antibody control. The remaining monolayers were covered with a 1-ml solution of rat (rabbit) primary antibody against voltage-gated L-type Ca2+ channel, α1c-subunit (Calbiochem, San Diego, CA) at 1:250 dilution in PBS, sealed, and left at 4°C overnight. The primary-antibody solution was removed by aspiration and the samples were washed three times for 3 min with 1 ml PBS. All monolayers were then covered with 1 ml of Cy 3-labeled secondary-antibody solution (4–6 dye molecules/antibody, Jackson ImmunoResearch Laboratories, West Grove, PA) and stained at room temperature for 2 h. The secondary-antibody solution was aspirated and the cells were washed three times for 3 min with 1 ml PBS. For fluorescence microscopy the monolayers were covered with 1 ml of 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes, Eugene, OR) solution (300 nM in PBS) and stained for 1 min at room temperature. The monolayers were washed three times for 3 min with 1 ml PBS and 1 ml of 50% glycerol was added to keep the cells hydrated until use.
Fluorescence images were collected using a Zeiss Axiovert 200M confocal inverted microscope with Axiovision 3.1 software (Zeiss, Oberkochen, Germany). The cells were imaged in 50% glycerol in PBS and were photographed with an AxioCam connected to the inverted microscope.
Polymer film samples
FluoSpheres (dye-labeled polymer spheres, nominally 40 nm in diameter) were purchased from Molecular Probes. A 1-wt % aqueous solution of polyvinyl alcohol (PVA, 70,000–100,000 mol wt, Sigma, St. Louis, MO) containing 0.0001–0.0005 wt % of the spheres was used to prepare 40 nm thick films by depositing 5 μl on an 18-mm diameter glass slide and spin-coating at 4000 rpm for 30 s.
Near-field scanning optical microscopy
Bent NSOM probes were prepared from high GeO2-doped fibers with a core diameter of 3 μm using a two-step chemical etching method followed by aluminum deposition and focused ion beam milling to produce a flat circular aperture (Burgos et al., 2003a). Probes with aperture diameters of ∼60 and ∼150 nm were used in this work (estimated from scanning electron microscopy images). The estimated spring constant for these probes is ∼100 N/m (Burgos et al., 2003a).
NSOM experiments were carried out on a combined atomic force microscopy (AFM)/NSOM microscope based on a Digital Instruments bioscope (Santa Barbara, CA) mounted on an inverted fluorescence microscope (Zeiss Axiovert 100), as described previously (Yuan and Johnston, 2002). An x-y piezo scanner (Polytec PI, Auburn, MA; 50-μm lateral scan range) was used for sample scanning. A continuous-wave mixed-gas ion laser (Innova 70 Spectrum, Coherent, Santa Clara, CA) was used for excitation (568.5 nm, 20 mW, linear polarization). Fluorescence for the sphere samples was collected at ≈590 nm with 100× oil immersion (1.3 NA) or 40× (0.65 NA) objectives, with appropriate bandpass (Omega Optics, Brattleboro, VT) and notch filters (Kaiser Optical Systems, Ann Arbor, MI) to remove residual excitation and red-alignment laser light, and detected using an avalanche photodiode detector (SPCM-AQR-15, Perkin Elmer Optoelectronics, Vaudreuil, Canada). Images were recorded in tapping mode at a scan rate of 0.25 Hz and a resolution of 512 × 512 for large-scale images and 256 × 256 for 5 × 5 μm2 images. Using the interleave LiftMode feature of the bioscope we were able to acquire two optical signals and topography in a single experiment. In the first line scan one obtains the topography and the fluorescence signal with the probe tip on the sample surface. A second interleaved line scan yields the fluorescence with the probe tip lifted a known height (h). The topographic data obtained on the first line scan was used to keep the tip a fixed height above the surface. The amplitude of the NSOM tip oscillation in the interleave mode was reduced to match the oscillation amplitude of the engaged tip (∼10 nm).
Images were processed using Nanoscope and Image J (National Institutes of Health) software. The fluorescence color-coded surface plots of the cells were obtained with a WS×M 2.1 scanning probe microscopy program (Nanotec Electrónica, Madrid, Spain). The 3-D images display a combination of the topographic data (height) and fluorescence data (color-coded). Cluster-size analysis was performed using original nonprocessed NSOM images with custom-made software that determines the number of clusters and their location in the image, as well as their height (intensity) and halfwidth. The halfwidth distributions of the clusters are presented as histograms with the size increments equal to the pixel size in the NSOM images (∼80 nm for a 40 × 40 μm2 image, ∼30 nm for a 15 × 15 μm2 image, and ∼20 nm for a 5 × 5 μm2 image).
RESULTS
NSOM imaging of the L-type Ca2+ channel
Rat cardiac myocytes (H9C2 cells) were fixed with formaldehyde and stained for the L-type Ca2+ channel as described in the Experimental section. Fluorescence microscopy images of the cells are shown in Fig. 1. The nucleus has been counterstained with DAPI (shown in blue). The cells show a diffuse fluorescence due to the Cy3 antibody-labeled calcium channels (shown in red). A similar set of cells was used for NSOM imaging. Fig. 2 presents topography (A and D) and the corresponding NSOM fluorescence (B and E) images for an individual cell. The nucleus is clearly visible as a raised (brighter) area in the large-scale topography image (Fig. 2 A). The H9C2 cells are relatively flat, reaching 600–800 nm at the highest point, with a lateral size of 20–40 μm (Fig. 1). Their relative flatness prevents large jumps in the position of the NSOM tip, reducing the risk of tip damage and also minimizing the contribution from topography-induced artifacts (see next section).
FIGURE 1.

Fluorescence microscopy image of H9C2 cells stained for the L-type Ca2+ channel.
FIGURE 2.
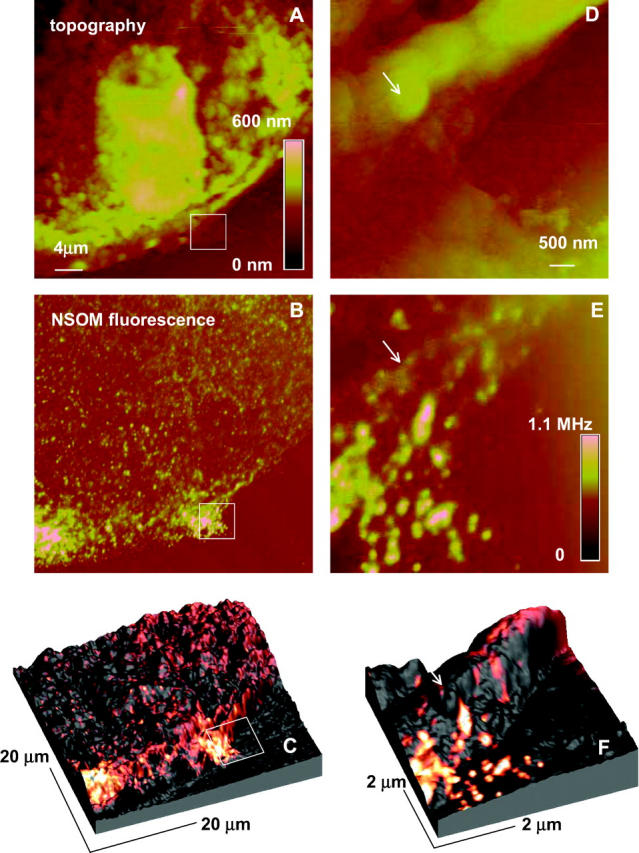
Topography (A and D), NSOM (B and E), and composite 3-D (C and F) images of H9C2 cells stained for the L-type Ca2+ channel, excitation wavelength 568.5 nm, probe aperture size ∼60 nm. The 5 × 5 μm2 images (D and E) were obtained by zooming the indicated area of the cell. The 3-D images display a combination of the topographic data (height) and fluorescence data (color-coded).
The large-scale NSOM image (Fig. 2 B, 40 × 40 μm2) shows a number of small, well-separated (≥200 nm) domains that are randomly distributed across the cell surface in addition to several large patches of more intense fluorescence near the edge of the cell. Similar patterns were observed for several different cells. Fig. 2 E shows a small-scale image recorded by rescanning the area in the box in Fig. 2 B. The smaller-scale image shows many small individual clusters that cannot be resolved in the larger-scale image; the smallest, weakest feature is ∼70 nm in diameter (measured at the full width at half maximum, FWHM, Fig. 3), which is comparable to the diameter of the NSOM probe aperture (∼60 nm). Note that in addition to the many small bright areas of fluorescence there are also patches with much weaker fluorescence (see arrows in Fig. 2, E and F), some of which appear to be correlated with features in the corresponding topography scan (Fig. 2 D). This is clearly visible in Fig. 2, C and F, where the topography and NSOM fluorescence images are superimposed to generate 3-D surface plots of the cell surface color-coded corresponding to the fluorescence intensity. The bright features that correspond to the Ca2+ channel clusters are frequently (but not always) located at the peaks and ridges of the topography plots, whereas weaker features tend to be located in lower areas.
FIGURE 3.

Cross section through one of the smallest features observed in the NSOM fluorescence images of H9C2 cells stained for the L-type Ca2+ channel.
Fig. 4 illustrates the size distribution for the clusters observed for three NSOM images of the same cell: 40 × 40 μm2 (Fig. 2 B), 15 × 15 μm2 (image not shown), and 5 × 5 μm2 (Fig. 2 E). The data are presented as histograms of the percentage of clusters of a particular size, with the bar width equal to the pixel size for the image. Since our data acquisition is limited to 512 × 512 data points per image, the resolution of the large 40 × 40 μm2 image is limited primarily by the pixel size of ∼80 nm, which is larger than the diameter of the probe aperture (∼60 nm). Therefore, the average cluster diameter of ∼250 nm for the large-scale image significantly overestimates the actual cluster size. For the medium- and small-scale images the pixel sizes are ∼30 and 20 nm, respectively, making the aperture size the limiting factor. For both of these images the Ca2+ channel clusters ranged in size from 60 nm in diameter (limited by the probe aperture) to 250 nm, with an average size of ∼100 nm. Similar size distributions were observed for images of several other cells (not shown).
FIGURE 4.
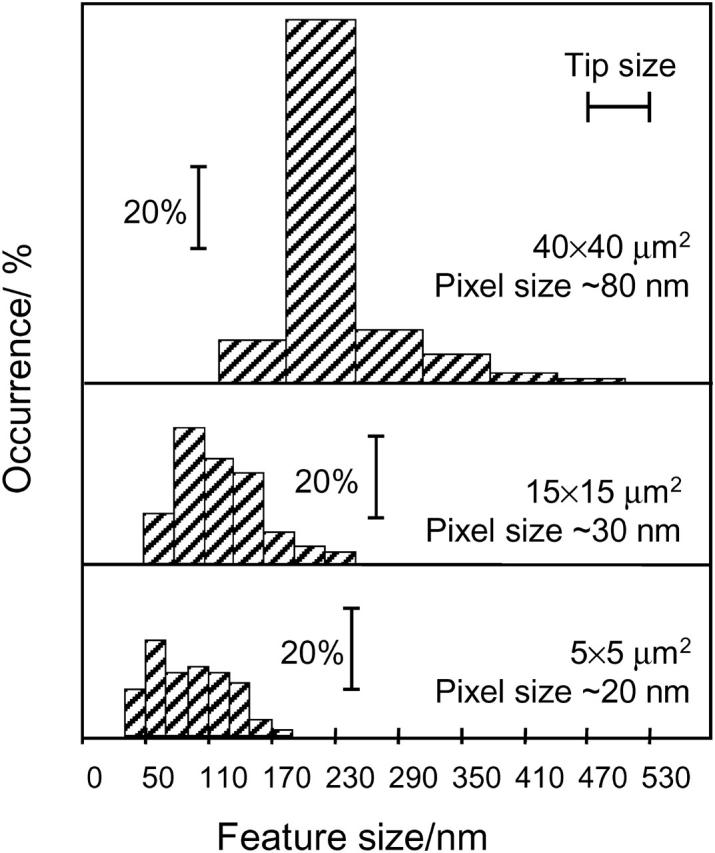
Histograms of cluster size for three NSOM images of H9C2 cells stained for the L-type Ca2+ channel.
The total number of detected clusters was ∼2500, ∼400, and ∼80 for the large-, medium-, and small-scale images, respectively, in Fig. 4, corresponding to a cluster density of 1–3 clusters/μm2. Similar values were obtained for several other images. The range of values is at least partly due to the fact that some areas of the membrane have more closely packed clusters. The intensity of the clusters varies by more than an order of magnitude. Based on NSOM imaging of samples of the secondary antibody at various dilutions on a glass slide, we estimate that the weakest signals that we detect (e.g., Fig. 3) are due to a single antibody (with 4–6 dyes/antibody). The integrated intensities for small (60-nm) and large (∼200-nm) clusters indicate that the number of channels/cluster varies between 1 and 20, assuming uniform labeling of one secondary antibody/channel.
Topography-induced artifacts in NSOM images of cells
The level of nonspecific binding of the secondary antibody and the contributions from topography-induced artifacts were determined by NSOM imaging of cells that were treated similarly to those shown in Figs. 1 and 2, but without addition of the primary Ca2+ channel antibody. A comparison of topographic and optical images for part of one cell is shown in Fig. 5. As can be clearly seen from the section analysis, there is a good correlation between the features in the topography and optical images. Since our NSOM images are recorded in constant gain mode, correlation between the topography and optical features suggests that the apparent optical signal is a topography-induced artifact (Hecht et al, 1997) related to changes in the transmission of the excitation light. However, the level (intensity) of the topography-induced signal is much lower than the fluorescence intensity observed in immunostained cells. The maximum artifact signal in Fig. 5 is only ∼10% of the average intensity of the fluorescence signal for the images in Fig. 2. Since the background and the fluorescence are spectrally separated, in principle it is possible to independently measure contributions from the topography-induced artifacts and real fluorescence signal by using appropriate combinations of bandpass filters. However, it is practically quite difficult to completely remove residual excitation, especially at the laser intensities required for fluorescence imaging.
FIGURE 5.
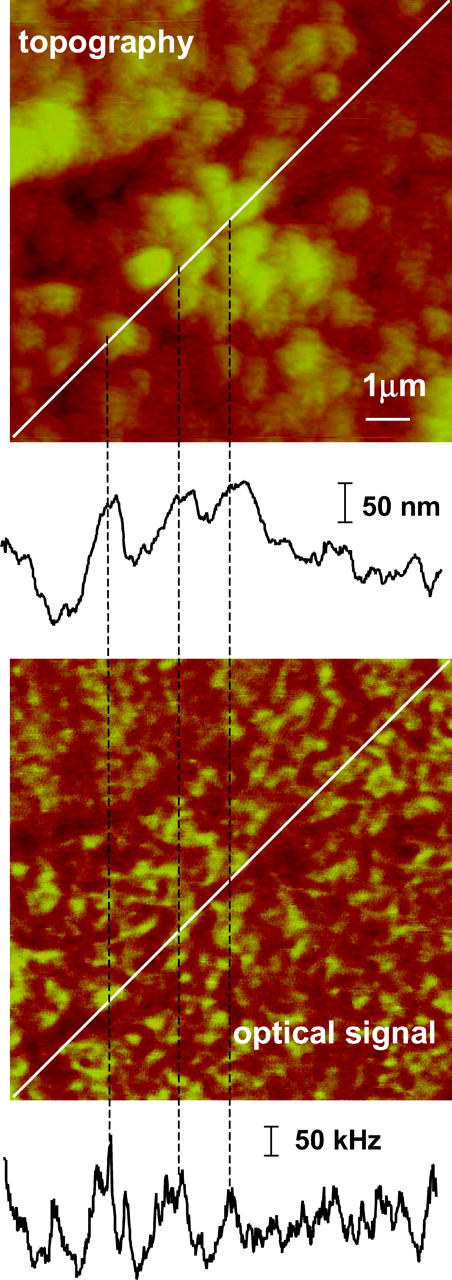
Topography and optical images of H9C2 cells stained with secondary antibody only, excitation wavelength 568.5 nm, probe aperture size ∼60 nm. The contrast in the optical image correlates with topography changes, as shown by the coincidence of the features in the cross sections shown below each image.
Interleave LiftMode imaging of spheres embedded in a polymer film
To measure the dependence of the NSOM fluorescence signal on the fluorophore-to-tip separation we used the LiftMode feature of the bioscope (Fig. 6) to scan samples of dye-labeled 40-nm polymer spheres embedded in a thin (∼40-nm) polymer layer. The dye loading is ∼350 molecules/sphere, thus minimizing any effects due to photobleaching and the polarization dependence of NSOM fluorescence. The size of the spheres is smaller than the probe aperture (d) to adequately probe the field distribution around the tip. The LiftMode measurement allows the simultaneous acquisition of two optical images, one with the tip engaged at the surface and the second with the probe tip lifted off the surface to a desired height. Thus, one can measure the intensity changes as the tip-to-sample distance (h) changes, with the top of the sample containing the fluorophores always in the focal plane of the collecting optics (Fig. 6). This has the advantage of ensuring that changes in fluorescence intensity arise from variations in the excitation intensity and not from changes in the collection efficiency. This approach also provides a reference image at zero height for each sample, thus avoiding errors due to variations in fluorescence intensity caused by the distribution of sphere sizes and dye loading. Imaging was performed at a laser power low enough to provide a linear dependence of the fluorescence signal on the excitation intensity.
FIGURE 6.

Schematic representation of the interleave mode experiment: the sample (PVA film containing 40-nm diameter fluorescent spheres) is kept in the focal plane of the microscope objective, while the aperture (diameter d) is moved off the surface to a desired distance (h).
The series of 5 × 5 μm2 LiftMode images presented in Fig. 7 was obtained using the ∼60-nm diameter aperture probe with a 100× oil immersion objective (NA = 1.3). Similar images (not shown) were obtained for the 150-nm diameter probe aperture and with a 60-nm aperture probe but using a 40×, NA = 0.65 light collection lens rather than the NA = 1.3 lens. The images shown in the left column of Fig. 7 were all obtained with the tip engaged at the surface (separation is zero), and the corresponding images in the right column were measured in the interleave mode with the tip lifted by the height indicated. The separation between spheres is generally >500 nm to ensure that we collect light from one sphere at a time even at the highest tip heights. Comparison of the images clearly demonstrates that as the tip-to-sample distance increases, the intensity of the fluorescence signal decreases, with a concomitant increase in the size of the spheres.
FIGURE 7.
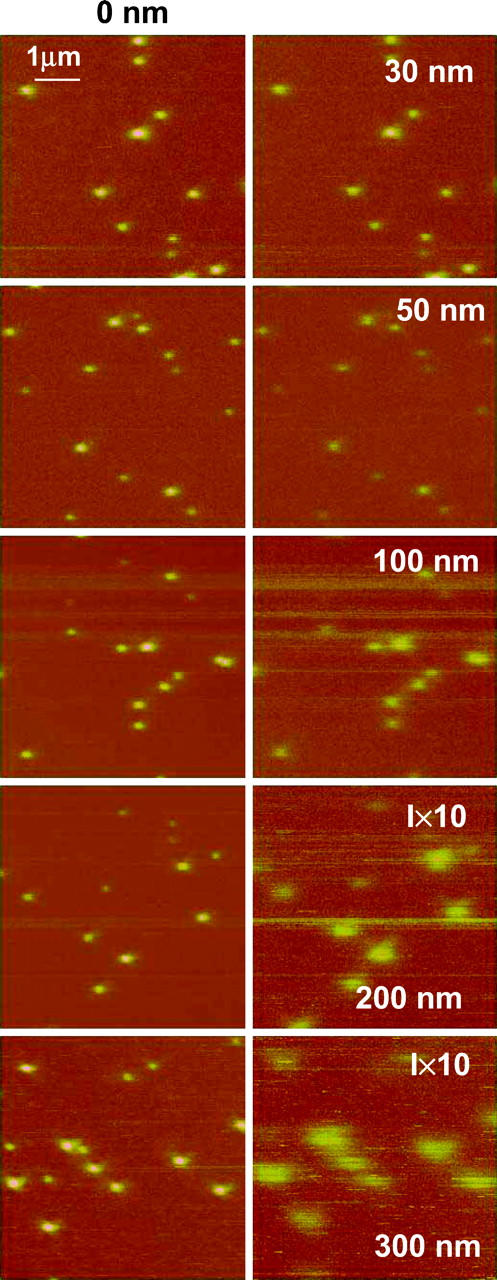
NSOM fluorescence images of 40-nm fluorescent spheres embedded in a 40-nm layer of PVA for zero separation (left column) and different interleave heights (right column): 30, 50, 100, 200, and 300 nm. The excitation wavelength is 568.5 nm, objective 100× oil immersion, 1.3 NA, and probe aperture size ∼60 nm.
The dependence of the peak fluorescence intensity on the probe-to-sample separation is presented in Fig. 8 A for experiments with both the 60- and 150-nm tips using the NA 1.3 lens. Each data point represents an average of many individual comparable-sized fluorescent spheres. The decrease of the fluorescence signal intensity shows a slower than exponential decrease and can be fit using a simple normalized function (d/d + h)2 (solid lines, Fig. 8 A), where d is the aperture diameter (60 and 150 nm for the two curves) and h is the tip-to-sample separation. Fig. 8 A also shows a good fit to the same function for the 0.65 numerical aperture lens using the 60-nm aperture probe. The quadratic decrease of the fluorescence intensity with separation is very well demonstrated by the linear dependence of (I0/Ilift)1/2 on separation (Fig. 8 A, inset).
FIGURE 8.
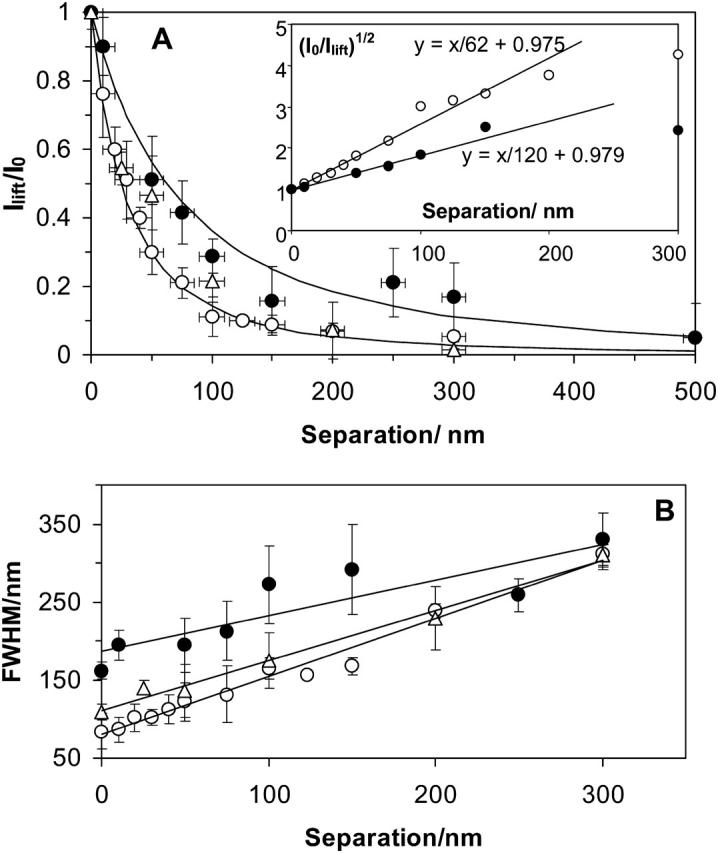
(A) Dependence of the LiftMode relative intensity of the maximum fluorescence signal across a sphere (Ilift/I0, averaged over many spheres) on the tip-to-sample separation for 60- (○) and 150-nm (•) probes. The solid lines show the decay predicted by a (d/(d + h))2 function, where d is the NSOM probe aperture diameter and h is the tip-to-sample separation. Inset: the dependence of (I0/Ilift)1/2 on separation for ∼60 nm and ∼150 nm probe apertures. (B) Dependence of the measured sphere size (FWHM) on the separation between the tip and the sample for aperture diameters of 60 nm (○ and ▵) and 150 nm (•). 100× NA = 1.3 (○) and 40× NA = 0.65 (• and ▵) objectives were used.
The dependence of the average sphere size observed in the images (measured as FWHM) on the separation between the tip and the polymer film is presented in Fig. 8 B. As the separation increases, the size of the spheres increases approximately linearly from an initial value limited by the diameter of the probe aperture to a final size of 250–350 nm that is determined by the diffraction-limited resolution of the light collection optics.
DISCUSSION
Ca2+ channel clustering
Our high-resolution NSOM fluorescence images of the membrane of cardiac myocytes stained for L-type Ca2+ channels indicate that the ion channel is localized in small clusters that vary in size from <60 to 250 nm, with an average size of ∼100 nm. Most of the clusters are randomly distributed across the cell membrane, although there are occasional larger patches near the edges of some cells. The resolution achievable with NSOM allows one to detect a range of clusters of differing sizes and intensity, many of which are too close together to be resolved by conventional fluorescence microscopy. The smallest and weakest features that we observe most likely correspond to a single antibody-labeled channel. These are relatively rare, and clusters of 5–10 channels are much more typical. The channel clustering is in good agreement with several previous electron microscopy studies of the organization of the L-type Ca2+ channel in cardiac myocyte membranes (Block et al., 1988; Gathercole et al., 2000). In the most recent study, label fracture electron microscopy showed that the channels in guinea pig cardiomyocytes were clustered rather than randomly distributed (Gathercole et al., 2000). Two distributions of cluster sizes were observed. The smaller, with nearest-neighbor distances of ∼20 nm, was explained by multiple binding of secondary gold-labeled antibodies to a single primary antibody. Note that NSOM will detect these clusters as weaker features with size determined by the probe aperture, but may underestimate their number if they are within 50 nm of each other. The larger clusters had an average diameter of 300 nm, in qualitatively good agreement with the NSOM results. However, the results from this study provide more convincing evidence for the clustering than do those of the previous study, which required mathematical analysis to distinguish between clusters and a random distribution. Furthermore, NSOM is compatible with imaging intact cells (fixed and live), a significant advantage over the sample preparation techniques required for electron microscopy.
Several electron cryomicroscopy experiments have provided high-resolution (2- to 3-nm) structures of individual L-type Ca2+ channels from both skeletal muscle (Serysheva et al., 2002; Wolf et al., 2003; Wang et al., 2002) and cardiac cells (Wang et al., 2004a). As summarized in a recent review (Wang et al., 2004b), dimeric and several monomeric structures have been reported for the skeletal muscle channel. These variations probably reflect differences in solubilization and purification procedures as well as data acquisition and analysis methods. Furthermore, it remains to be established whether any of the structures are representative of the functional channel within a cellular membrane. The L-type Ca2+ channel is believed to be physically coupled to ryanodine channels in skeletal muscle and a tetrameric arrangement of four calcium channels over every other ryanodine tetramer has been proposed (Serysheva et al., 2002). The tetrads are ∼30 nm across (Wolf et al, 2003) and have been shown by electron microscopy to form large membrane clusters of variable size and shape in differentiated BC3H1 cells. Individual clusters showed an orthogonal arrangement of tetrads with center-to-center spacing of 41 nm and diagonal spacing of 58 nm (Protasi et al., 1997).
The only high-resolution study for the cardiac L-type Ca2+ channel indicates a dimeric structure with a width of 14.5 nm and a height of 19 nm (Wang et al., 2004a). This is actually slightly smaller than the dimer structure obtained by the same group for the skeletal muscle channel (Wang et al., 2002), consistent with the fact that the cardiac channel lacks the γ-membrane subunit. Although cardiac channels have been shown by immunofluorescence to colocalize with ryanodine channels, there does not appear to be direct mechanical coupling between the two, nor is there any evidence for the tetrad arrangement suggested for skeletal muscle channels (Woo et al., 2003; Gathercole et al., 2000).
The NSOM results do not allow us to distinguish between the various monomeric, dimeric, and tetrameric structures suggested by high-resolution electron microscopy and modeling experiments for Ca2+ channels. Although we observe occasional features that are likely to correspond to a channel with a single antibody, this does not preclude either dimeric or higher oligomeric structures as the close packing of such dimers and oligomers could at least in some cases prevent complete antibody labeling of each α1C subunit. Note that if the channels were in a close-packed array, the surface area defined by the cluster diameter could accommodate significantly more channels than we observe based on intensity measurements. This observation, plus the variable size and intensity of the clusters, argues for a more random clustering than is observed for arrays of skeletal muscle tetrads, and is consistent with previous studies on cardiac channels (Gathercole et al., 2000). It is also interesting to note that clustering of L-type Ca2+ channels occurs even in the absence of the ryanodine receptors and other elements of the excitation-contraction machinery of muscle cells (Harms et al., 2001). A recent single-molecule fluorescence study of cardiac eYFP-labeled L-type Ca2+ channels overexpressed in HEK 293 cells showed clusters in the membrane of live cells with an average of three molecules per cluster, even at the low expression levels required for single-molecule studies.
Two previous high-resolution NSOM fluorescence studies have examined membrane protein clusters in fixed cells. Our results for the Ca2+ channel are qualitatively similar to those for erbB2, a member of the epidermal growth factor receptor tyrosine kinase family, for which a distribution of cluster sizes, intensities, and separations was observed (Nagy et al., 1999). However, the average size of the calcium channel clusters is ∼5 times smaller than the value of 0.5 μm for the ErbB2 receptor and each cluster contains many fewer proteins. Similarly, NSOM studies of the distribution of HLA class 1 molecules in the membrane of fixed fibroblasts showed patches with distributions centered at ∼70 nm and ∼600 nm, with an estimated 25–125 antibodies per patch (Hwang et al., 1998). These examples and the work reported here illustrate the potential of NSOM for obtaining detailed information on protein clustering on a length scale that is intermediate between the use of FRET to study protein complexes and dimers and the large-scale clustering that can be probed by conventional fluorescence microscopy. Colocalization studies with the 50-nm resolution of NSOM will facilitate the investigation of larger signaling complexes that cannot be studied by either FRET or conventional fluorescence microscopy.
Practical issues for NSOM imaging of cells
In this work we have focused on resolving some of the practical issues for near-field imaging of cell surfaces. In particular, we have examined the contributions of topography-induced artifacts to the fluorescence images. We find that the topography-induced signal is <10% of the average fluorescence intensity for the labeled ion channels in the relatively flat cardiac myocytes. Although scanning unlabeled cells is a useful approach for assessing the level of interference from the sample topography, it does not allow for subtraction of the artifact since the artifact and fluorescence signal are measured for two different cells. Alternately, the same sample can be scanned sequentially with matched intensities at two different excitation wavelengths, only one of which is absorbed by the fluorophore. The fluorescence image can then be corrected by subtracting the optical artifact. Similar results could also be obtained by scanning the sample at a sufficiently high excitation intensity to completely bleach the fluorophore and then rescanning the same area to obtain the signal due to topography. It should be noted that although cardiac myocytes are relatively flat compared to other cell types, topographic features are still too large for application of topography artifact-free contact height mode of NSOM (Hecht et al., 1997).
Our results indicate that the smallest clusters observed for the Ca2+ channel are very similar in size to the probe aperture, even though the ion channel is labeled on its intercellular side, using a primary and secondary antibody combination. The fluorophore attached to the secondary antibody will therefore be separated from the NSOM probe by the membrane thickness plus the dimension of the primary antibody and the height of the channel that protrudes from the membrane. This distance is likely to be in the range of 10–20 nm depending on the orientation of the two antibodies. This, in addition to the possibility of having chromophores below the membrane surface, makes it important to understand the depth dependence of the NSOM signal intensity.
NSOM theory predicts that the light emerging from an NSOM probe aperture will consist of an evanescent component and a radiative or propagating component, with the former usually assumed to predominate for small-aperture diameters (Novotny et al., 1998; Obermüller et al., 1995). According to Novotny et al. (1998), the evanescent decay length will be a small fraction (10%) of the aperture diameter. However, the distance dependence that we have measured for the NSOM signal intensity (Figs. 7 and 8) does not show the rapid exponential decay characteristic of an evanescent field component which would lead to extremely small fluorescence excitation volumes, on the order of 10 zeptoliters (1 zl = 10−21 liters). Despite the potential for such small interaction volumes to allow single molecule detection at high analyte concentrations (Levene et al., 2003), they are rarely realized in practice due to a variety of reasons. These include the fact that the probe must be kept very close to the sample surface to remain in the evanescent zone. This may be prevented by the probe characteristics (large size, metal protrusions), by difficulties associated with imaging samples with complex morphology or in a liquid environment, or by failure to mount the probe so that the axis is orthogonal to the surface. Operation of the probe in tapping mode, although advantageous for biological samples, will lead to signal averaging unless the oscillation amplitude is small compared to the evanescent decay length. Finally, a complicated light-collection apparatus is required to observe light originating from the perturbed evanescent field, which is emitted at large angles sometimes beyond the angle for total internal reflection in the sample and referred to as forbidden light (Hecht et al., 1998).
The fact that we do not observe a rapid exponential decay in our distance dependence experiments is not surprising given the many difficulties associated with measuring the evanescent component. We believe that the flat end of our nanomachined probes and our ability to track the sample surface will minimize the probe-sample separation. However, the difficulty in ensuring that the probe is normal to the surface and the fact that there will be some averaging over the evanescent zone with tapping-mode operation, even with small (≈10 nm) amplitudes, may contribute to the absence of an evanescent component. The efficiency of light collection from the evanescent zone and the large size of the fluorescent spheres relative to the probe aperture will also limit our ability to detect an evanescent component. We conclude that the data in Figs. 7 and 8 unambiguously show the decay of only the radiative component. Given the extreme precision and near ideal probes required to measure the evanescent component it seems likely that most NSOM experiments of biological samples will also be unable to measure the interaction of the evanescent component with the sample. A number of researchers have performed controlled probe-to-sample distance experiments with NSOM probes and observed a nonlinear increase in the fluorescence signal as the aperture comes into near contact with surface fluorophores (Doyle et al., 2001; Moerner et al., 1994). Although this nonlinear increase has been attributed to local excitation of the sample by the evanescent field-fluorophore interaction, we believe that it is the nonlinear increase in the radiative signal that is measured as the tip approaches the sample. Despite the fact that one can devise (Hecht et al., 1998) an experimental apparatus to probe the evanescent component (Obermüller et al., 1995) it is rarely undertaken by experimentalists working on complex samples, largely due to the technical problems outlined above.
The radiative component emerging from an NSOM probe decays less rapidly than the evanescent component and is therefore considerably more amenable to measurement. For apertures larger and distances greater than the excitation wavelength (λexc) the light intensity should decay like a point source, i.e., as 1/h2, where h is the distance from the probe aperture. However, it is not clear from various theoretical treatments how fast the radiative component falls off for subwavelength distances h < λexc and subwavelength apertures. Our data clearly show that a similar d2/(d + h)2 dependence is followed for separations h < λexc with two different apertures (60 nm and 150 nm) and two different lens numerical apertures (1.3 and 0.65). This allows us to define an effective depth of field for an NSOM probe as the distance where the excitation intensity is nearly one-half its peak value and the spatial resolution is nearly constant. A reasonable value for the depth of field is h = d/2, i.e., equal to the radius of the probe aperture since over this separation the spatial resolution of the 60-nm probe decreased only slightly from 60 nm to 77 nm, whereas the excitation intensity dropped to 0.44 of the peak value.
For d = 60 nm and assuming that the excitation volume can be approximated as a hemisphere of radius equal to the radius of the probe aperture we obtain a fluorescence excitation volume of only ≈50 zl. However, a consequence of the d2/(d + h)2 dependence is that significant excitation can penetrate several hundred nanometers into thick samples such as cells. Thus, one expects contributions from fluorophores within 100–200 nm of the membrane, although the intensity will be much less than that for an equivalent chromophore at the cell surface. From a practical point of view, this could be problematic in cases where membrane receptors undergo internalization in response to ligand activation, leading to background signals from proteins in endocytic vesicles beneath the plasma membrane. Since the NSOM resolution will depend on the depth of the fluorophore, it may be difficult to assess whether diffuse fluorescence signals arise from a low level of randomly distributed fluorophore in the membrane or from fluorophores significantly below the cell surface. Nevertheless, the dependence of the width of the fluorescence signal on distance from the probe aperture may be used to determine the depth of a chromophore from the surface under certain controlled situations. For example, if a thick sample is loaded with very small strong fluorescers such as quantum dots and if aggregation can be avoided, it should be possible to determine the depth distribution of the dots from the correlation between the fluorescence width and depth.
CONCLUSIONS
A combination of near-field scanning optical microscopy and immunofluorescence have been used to study the localization of voltage-gated L-type Ca2+ channels on the surface of fixed cardiac myocytes. The channels are organized in small clusters that vary in size from <60 to 250 nm, with an average diameter of 100 nm. Although we do observe some signal that is correlated with sample topography, the artifact is <10% of the intensity of the average NSOM fluorescence. We have also investigated the dependence of the NSOM resolution and the near-field fluorescence intensity on the fluorophore-to-NSOM tip separation. Using a small 60-nm aperture probe it is possible to selectively excite fluorescence from a tiny 50-zl volume, thus providing excellent 3-D resolution to study membrane localization of ion channels and other membrane proteins. The distance dependence experiments allow us to define a depth of field of approximately half the probe diameter and indicate that fluorophores up to 200 nm below the cell surface may contribute to the observed fluorescence. This may make it difficult to distinguish between weak fluorescence signals arising from the cell surface and fluorophores below the surface.
In this article, we have presented a practical approach to understanding some of the potential problems encountered with NSOM imaging of cells. This work, together with our successful development of NSOM imaging in liquid (Ianoul et al., 2003), will allow us to attempt NSOM imaging of cells in liquid, and eventually of live cells, and to pursue more detailed studies of the organization of signaling complexes in cell membranes. Here we have provided quantitative data on the distribution of ion channel clusters on the surface of cardiac myocytes. It is now possible to examine changes in the distribution of ion channels and associated membrane proteins in response to stimulus of cardiac function and at various developmental stages.
Acknowledgments
We thank Zhengfang Lu for NSOM probe fabrication and Doug Moffatt for help with image analysis.
M.S. acknowledges support from the Reactive Intermediate Student Exchange program.
Anatoli Ianoul's present address is Dept. of Chemistry, Carleton University, Ottawa, ON K1S 5B6 Canada.
References
- Block, B. A., T. Imagawa, K. P. Campbell, and C. Franzini-Armstrong. 1988. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107:2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos, P., Z. Lu, A. Ianoul, C. Hnatovsky, M. L. Viriot, L. J. Johnston, and R. S. Taylor. 2003a. Near-field scanning optical microscopy probes: a comparison of pulled and double-etched bent NSOM probes for fluorescence imaging of biological samples. J. Microsc. 211:37–47. [DOI] [PubMed] [Google Scholar]
- Burgos, P., C. Yuan, M.-L. Viriot, and L. J. Johnston. 2003b. Two-color near-field fluorescence microscopy studies of microdomains (‘Rafts’) in model membranes. Langmuir. 19:8002–8009. [Google Scholar]
- Catterall, W. A. 1995. Structure and function of voltage-gated channels. Annu. Rev. Biochem. 64:493–531. [DOI] [PubMed] [Google Scholar]
- Catterall, W. A. 2000. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16:521–555. [DOI] [PubMed] [Google Scholar]
- Cong, M., S. J. Perry, F. T. Lin, I. D. Fraser, L. Y. A. Hu, W. Chen, J. A. Pitcher, J. D. Scott, and R. J. Lefkowitz. 2001. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J. Biol. Chem. 276:15192–15199. [DOI] [PubMed] [Google Scholar]
- De Lange, F., A. Cambi, R. Huijbens, B. de Bakker, W. Rensen, M. Garcia-Parajo, N. van Hulst, and C. G. Figdor. 2001. Cell biology beyond the diffraction limit: near-field scanning optical microscopy. J. Cell Sci. 114:4153–4160. [DOI] [PubMed] [Google Scholar]
- Davare, M. A., V. Avdonin, D. D. Hall, E. M. Peden, A. Burette, R. J. Weinberg, M. C. Horne, T. Hoshi, and J. W. Hell. 2001. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 293:98–101. [DOI] [PubMed] [Google Scholar]
- Doyle, R. T., M. J. Szulzcewski, and P. G. Haydon. 2001. Extraction of near-field fluorescence from composite signals to provide high resolution images of glial cells. Biophys. J. 80:2477–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, R. C. 1999. Near-field scanning optical microscopy. Chem. Rev. 99:2891–2927. [DOI] [PubMed] [Google Scholar]
- Edidin, M. 2001. Near-field scanning optical microscopy, a siren call to biology. Traffic. 2:797–803. [DOI] [PubMed] [Google Scholar]
- Enderle, T., T. Ha, D. F. Ogletree, D. S. Chemla, C. Magowan, and S. Weiss. 1997. Membrane specific mapping and colocalization of malarial and host skeletal proteins in the Plasmodium falciparum infected erythrocyte by dual-color near-field scanning optical microscopy. Proc. Natl. Acad. Sci. USA. 94:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathercole, D. V., D. J. Colling, J. N. Skepper, Y. Takagishi, A. J. Levi, and N. J. Severs. 2000. Immunogold-labeled L-type calcium channels are clustered in the surface plasma membrane overlaying junctional sarcoplasmic reticulum in guinea-pig myocytes—implications for excitation-contraction coupling in cardiac muscle. J. Mol. Cell Cardiol. 32:1981–1994. [DOI] [PubMed] [Google Scholar]
- Harms, G. S., L. Cognet, P. H. M. Lommerse, G. A. Blab, H. Kahr, R. Gamsjäger, H. P. Spaink, N. M. Soldatov, C. Romanin, and T. Schmidt. 2001. Single-molecule imaging of L-type Ca2+ channels in live cells. Biophys. J. 81:2639–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht, B., H. Bielfeldt, Y. Inouye, D. W. Pohl, and L. Novotny. 1997. Facts and artifacts in near-field optical microscopy. J. Appl. Phys. 81:2492–2498. [Google Scholar]
- Hecht, B., H. Bielfeldt, D. W. Pohl, L. Novotny, and H. Heinzelmann. 1998. Influence of detection conditions on near-field optical imaging. J. Appl. Phys. 84:5873–5882. [Google Scholar]
- Hell, S. W. 2003. Toward fluorescence nanoscopy. Nat. Biotechnol. 21:1347–1355. [DOI] [PubMed] [Google Scholar]
- Hescheler, J., R. Meyer, S. Plant, D. Krautwurst, W. Rosenthal, and G. Schultz. 1991. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 69:1476–1486. [DOI] [PubMed] [Google Scholar]
- Hwang, J., L. A. Gheber, L. Margolis, and M. Edidin. 1998. Domains in cell plasma membranes investigated by near-field scanning optical microscopy. Biophys. J. 74:2184–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianoul, A., P. Burgos, Z. Lu, R. S. Taylor, and L. J. Johnston. 2003. Phase separation in complex supported phospholipid bilayers visualized by near-field scanning optical microscopy in liquid. Langmuir. 19:9246–9254. [Google Scholar]
- Kimes, B. W., and B. L. Brandt. 1976. Properties of a clonal muscle-cell line from rat heart. Exp. Cell Res. 98:367–381. [DOI] [PubMed] [Google Scholar]
- Kirsch, A. K., V. Subramanian, G. Striker, C. Schnetter, D. J. Arndt-Jovin, and T. M. Jovin. 1998. Continuous wave two-photon scanning near-field optical microscopy. Biophys. J. 75:1513–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, F. H., G. Y. Shang, M. Troyon, M. Spajer, H. Morjani, J. F. Angiboust, and M. Manfait. 2001. Nanospectrofluorometry inside single living cell by scanning near-field optical microscopy. Appl. Phys. Lett. 79:2489–2491. [Google Scholar]
- Levene, M. J., J. Korlach, S. W. Turner, M. Foquet, H. G. Craighead, and W. W. Webb. 2003. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 299:682–686. [DOI] [PubMed] [Google Scholar]
- Lewis, A., M. Isaacson, and A. Lewis. 1984. Development of a 500 angstrom spatial resolution light microscope. Ultramicroscopy. 13:227–232. [Google Scholar]
- Lewis, A., H. Taha, A. Strinkovski, A. Manevitch, A. Khatchatouriants, R. Dekheter, and E. Ammann. 2003. Near-field optics: from subwavelength illumination to nanometric shadowing. Nat. Biotechnol. 21:1378–1386. [DOI] [PubMed] [Google Scholar]
- Menard, C., S. Pupier, D. Mornet, M. Kitzmann, J. Nargeot, and P. Lory. 1999. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J. Biol. Chem. 274:29063–29070. [DOI] [PubMed] [Google Scholar]
- Moerner, W. E., T. Plakhotnik, T. Irngartinger, U. P. Wild, D. W. Pohl, and B. Hecht. 1994. Near-field optical spectroscopy of individual molecules in solids. Phys. Rev. Lett. 73:2764–2767. [DOI] [PubMed] [Google Scholar]
- Nagy, P., A. Jenei, A. K. Kirsch, J. Szollosi, S. Damjanovich, and T. M. Jovin. 1999. Activation-dependent clustering of the erbB2 receptor tyrosine kinase detected by scanning near-field optical microscopy. J. Cell Sci. 112:1733–1741. [DOI] [PubMed] [Google Scholar]
- Novotny, L., B. Hecht, and D. W. Pohl. 1998. Implication of high resolution to near-field optical microscopy. Ultramicroscopy. 71:341–344. [Google Scholar]
- Obermüller, C., K. Karrai, G. Kolb, and G. Abstreiter. 1995. Transmitted radiation through a subwavelength-sized tapered optical fiber tip. Ultramicroscopy. 61:171–177. [Google Scholar]
- Pohl, D. W., W. Denk, and M. Lanz. 1984. Optical stethoscopy: image recording with a resolution of λ/20. Appl. Phys. Lett. 44:651–653. [Google Scholar]
- Protasi, F., C. Franzini-Armstrong, and B. E. Flutcher. 1997. Coordinated incorporation of skeletal muscle dihydropyridine receptors and ryanodine receptors in peripheral couplings of BC3H1 cells. J. Cell Biol. 137:859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieti, S., V. Manni, A. Lisi, L. Giuliani, D. Sacco, E. D'Emilia, A. Cricenti, R. Generosi, M. Luce, and S. Grimaldi. 2004. SNOM and AFM microscopy techniques to study the effect of non-ionizing radiation on the morphological and biochemical properties of human keratinocytes cell line (HaCaT). J. Microsc. 213:20–28. [DOI] [PubMed] [Google Scholar]
- Rothery, A. M., J. Gorelik, A. Bruckbauer, W. Yu, Y. E. Korchev, and D. Klenerman. 2003. A novel light source for SICM-SNOM of living cells. J. Microsc. 209:94–101. [DOI] [PubMed] [Google Scholar]
- Serysheva, I. I., S. J. Ludtke, M. R. Baker, W. Chiu, and S. L. Hamilton. 2002. Structure of the voltage-gated L-type Ca2+ channel by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA. 99:10370–10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiya, E., S. Iwabuchi, N. Nagatani, Y. Murakami, T. Sakaguchi, K. Yokoyama, N. Chiba, and H. Muramatsu. 1997. Simultaneous topographic and fluorescence imaging of recombinant bacterial cells containing a green fluorescent protein gene detected by a scanning near-field optical/atomic force microscope. Anal. Chem. 69:3697–3701. [DOI] [PubMed] [Google Scholar]
- Tsien, R. Y. 2003. Imagining imaging's future. Nat. Rev. Mol. Cell Biol. 4:SS16–SS21. [PubMed] [Google Scholar]
- Wang, M.-C., R. F. Collins, R. C. Ford, N. S. Berrow, A. C. Dolphin, and A. Kitmitto. 2004a. The three-dimensional structure of the cardiac L-type voltage-gated calcium channel. J. Biol. Chem. 279:7159–7168. [DOI] [PubMed] [Google Scholar]
- Wang, M.-C., A. Dolphin, and A. Kitmitto. 2004b. L-type voltage-gated calcium channels: understanding function through structure. FEBS Lett. 564:245–250. [DOI] [PubMed] [Google Scholar]
- Wang, M.-C., G. Velarde, R. C. Ford, N. S. Berrow, A. C. Dolphin, and A. Kitmitto. 2002. 3D structure of the skeletal muscle dihydropyridine receptor. J. Mol. Biol. 323:85–89. [DOI] [PubMed] [Google Scholar]
- Wolf, M., A. Eberhart, H. Glossman, J. Striessnig, and N. Grigorieff. 2003. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. J. Mol. Biol. 332:171–182. [DOI] [PubMed] [Google Scholar]
- Woo, S.-H., N. M. Soldatov, and M. Morad. 2003. Modulation of Ca2+ signalling in rat atrial myocytes: possible role of the α1C carboxyl terminal. J. Physiol. 552:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, C., and L. J. Johnston. 2002. Phase evolution in DPPC/cholesterol monolayers: AFM and NSOM studies. J. Microsc. 205:136–146. [DOI] [PubMed] [Google Scholar]
- Zenobi, R., and V. Deckert. 2000. Scanning near-field optical microscopy and spectroscopy as a tool for chemical analysis. Angew. Chem. Int. Ed. 39:1746–1756. [DOI] [PubMed] [Google Scholar]