

Abstract
Membrane proteins that belong to the major facilitator superfamily (MFS) are found in organisms across the evolutionary spectrum and mediate the transport of a variety of substrates ranging from small metabolites to neurotransmitters. The oxalate transporter (OxlT) is a representative MFS protein, and exchanges formate for oxalate across the cytoplasmic membrane of the organism Oxalobacter formigenes. Here, we present a structural model for the protein conformational changes that occur during oxalate transport by combining a three-dimensional map of the oxalate-bound, “closed” state of OxlT at 6.5 Å determined by cryo-electron microscopy with a model of the “open” state of OxlT based on the atomic structures of the related transporters, glycerol-3-phosphate transporter (GlpT) and lactose permease (LacY). We demonstrate that the principal structural change associated with substrate transport is a concerted rocking movement of the two structurally similar halves of the protein relative to each other. Our structural model places two positively charged residues, Arg-272 and Lys-355 in the central cavity, suggesting that electrostatic interactions between these residues and the oxalate anion is a key step in generating the conformational change between the open and closed states of the transporter.
INTRODUCTION
The major facilitator superfamily (MFS) (Henderson and Maiden, 1990; Paulsen et al., 2000) represents the largest evolutionarily related collection of secondary transporters that mediate the functions of uniport, anitiport, and symport in prokaryotic and eukaryotic cellular membranes. In contrast to primary transporters, such as those in the ABC superfamily that use the energy derived from making or breaking chemical bonds for solute transport, secondary transporters work by using only the electrochemical potentials of the transported substrates (Mitchell, 1967; Poolman and Konings, 1993). Proteins that function as uniporters generally mediate the uncoupled transport of a single substrate across the membrane. Those that function as antiporters have the ability to transport two substrates, i.e., one in each direction, whereas symporters mediate the coupled transport of two substrates in the same direction.
Huang et al. (2003) and Abramson et al. (2003) have recently reported atomic models for the structures of two bacterial MFS proteins: the glycerol-3-phosphate/inorganic phosphate antiporter (GlpT) (Huang et al., 2003) and the lactose/H+ symporter (LacY) (Abramson et al., 2003) in a “cytoplasmically open” conformation, in which the substrate binding site is preferentially exposed to the aqueous medium on the cytoplasmic side and closed to the aqueous medium on the periplasmic side. Insights into the architecture of MFS proteins have also come from determination of the structure of the oxalate-bound state of the oxalate/formate antiporter OxlT (Anantharam et al., 1989) determined at 6.5 Å using cryo-electron microscopy (Heymann et al., 2003, 2001; Hirai et al., 2002). The “open” conformation in which LacY and GlpT were crystallized is distinct from the conformation reported for the “closed” state of OxlT in which the central cavity tapers to a narrower opening at both periplasmic and cytoplasmic ends. Although OxlT and GlpT are antiporters, and LacY is a symporter, the overall architectures and helix arrangement of the three proteins are remarkably similar (Hirai et al., 2003). We have therefore combined knowledge of the atomic structures of the open states of GlpT and LacY with the map of the “closed” state of OxlT to derive a model for conformational changes that occur during oxalate transport.
METHODS
Crystallization and electron microscopy
The methods for crystallization and electron microscopic analysis have been described previously (Heymann et al., 2001; Hirai et al., 2002). The refinement described here was carried out using the data set collected from tubular OxlT crystals that led to the 6.5-Å map (Hirai et al., 2002).
Sequence alignment
The search for homology between OxlT and GlpT was carried out using PSI-BLAST (Altschul et al., 1997). Eight protein sequences from each of these protein families were selected to give a close and evenly distributed alignment. Initial alignment was done using CLUSTALW (Thompson et al., 1994). The sequence alignment was then checked and adjusted to give reasonable conservation pattern at each three-dimensional position. For example nonpolar residues that face the lipid bilayer appear periodically in the helical region as shown in Fig. 3 B, except in helices 3, 6, 9, and 12 where a high proportion of nonpolar residues appears throughout. Starting from whole-length alignment, alignment of two separate halves was also deduced based on the structural symmetry (Fig. 1). The regions of GlpT used for structural alignments and interpreted as equivalent regions are as follows, (H1, 33–53; H2, 67–90; H3, 93–110; H4, 122–142; H5, 155–178; H6, 187–204) and (H7, 257–277; H8, 293–316; H9, 322–339; H10, 349–369; H11, 383–406; H12, 414–431).
FIGURE 3.

(A) Sequence alignment between GlpT and OxlT protein families around helix 8 and 9. The sequences used are a selection from the most closely related transporter families such as UhpT. The sequence codes are from the Swiss-Prot or TREMBL database. Positions colored in red and green indicate negatively and positively charged residues, respectively. Positions colored in green are hydrophobic residues at sites where a hydrophobic residue is usually present. Positions colored in magenta represent nonhydrophobic, but conserved residues. Underlined letters indicate residues that are highly conserved in general. (B) Revised secondary structure model for OxlT showing the presence of two positively charged residues in the transmembrane region.
FIGURE 1.
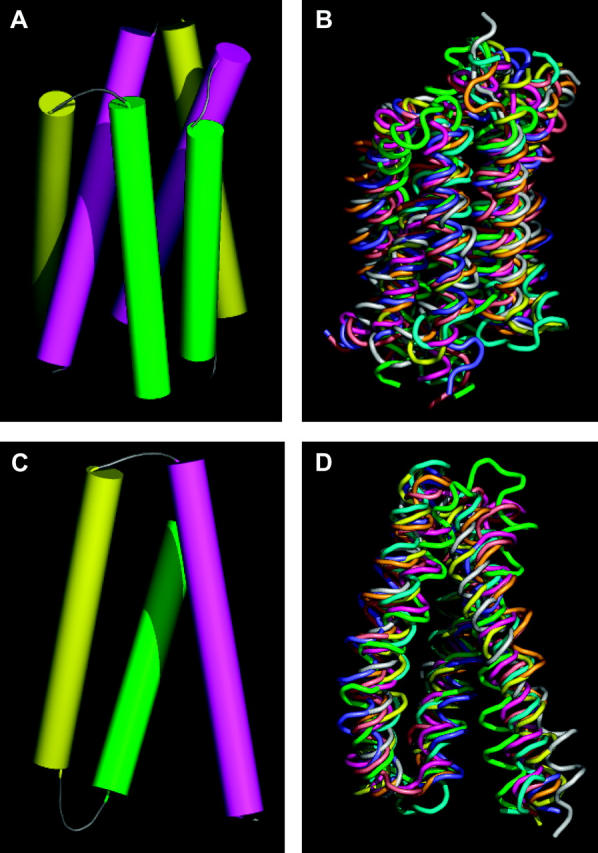
Six-helix and three-helix repeat unit in MFS proteins. (A) Helix packing derived from the density map of OxlT obtained by electron crystallography. The set of six cylinders shown corresponds to helices 7 (magenta, left), 8 (yellow, right), 9 (green, right), 10 (magenta, right), 11 (yellow, left), and 12 (green, left) of OxlT. (B) Superposition of the eight six-helix motifs from the structures of GlpT and LacY after alignment. Each six-helix unit was aligned using the C-terminal half of GlpT as a reference because it was closest to the average structure obtained from all four six-helix units as shown in Table 1. (C) Helix packing derived from the density map of OxlT obtained by electron crystallography. The set of three cylinders shown corresponds to helices 7 (magenta), 8 (yellow), and 9 (green) of OxlT. (D) Superposition of the eight three-helix motifs from the structures of GlpT and LacY after alignment. Each three-helix unit was aligned using helices 7–9 of GlpT as a reference, because it was closest to the average structure obtained from all eight three-helix units as shown in Table 2. The regions used for alignments are as follows: GlpT (H1, 33–53; H2, 67–90; H3, 93–110; H4, 122–142; H5, 155–178; H6, 187–204; H7, 257–277; H8, 293–316; H9, 322–339; H10, 349–369; H11, 383–406; H12, 414–431) and LacY (H1, 11–31; H2, 47–70; H3, 74–91; H4, 106–126; H5, 138–161; H6, 167–184; H7, 224–244; H8, 259–282; H9, 289–306; H10, 314–334; H11, 348–371; H12, 379–396).
Rigid-body refinement
Crystallography and NMR system (CNS) (Brunger et al., 1998) was used for refinement. At the resolution of the density map used here, differences in structure factors between x-ray and electron crystallographic data are not relevant (Subramaniam and Henderson, 2000). Either the N-terminal or C-terminal half of GlpT, LacY, or OxlT (residues 25–210 and 247–439 for GlpT, 1–194 and 214–406 for LacY, and 17–194 and 213–401 for OxlT) was treated as one rigid body. The starting positions of the N- and C-terminal halves were initially adjusted manually using O. Different starting positions were tested to ensure that the endpoint of refinement was independent of the position of the starting model as explained in the text. During the refinement, only atom types N, CA, C, O, and CB were included, and a maximum likelihood target using amplitudes and phase-probability distribution was selected. To meet the requirement of input format of CNS, phases derived by electron microscopy were converted to Hendrickson-Lattman coefficients. For the first phase of refinement, information to 7.6 Å data were used to locate the best fit of the model to the map. In subsequent refinement steps, data between 7.6 Å and 6.5 Å were included to arrive at the final model.
RESULTS AND DISCUSSION
To obtain a model for the structure of the substrate-bound, “closed” state of OxlT, we first carried out a comparison of the coordinates for GlpT and LacY with respect to each other and with the helix positions derived for OxlT. At first, N- and C-terminal halves of GlpT and LacY were compared to evaluate the similarity of each protein half and to assess the extent of variation between them. After alignment, root-mean-square deviation (RMSD) values were calculated between all possible N- or C-terminal-half symmetrical pairs. These calculations were performed using only the transmembrane regions of the structure. As shown in Table 1, the structure of the C-terminal half of GlpT is closest to the average structure, whereas the N-terminal half of GlpT is the furthest from the average. Fig. 1, A and B, presents a comparison of the six-helix unit proposed to constitute the underlying structural motif in OxlT (Hirai et al., 2002) (Fig. 1 A) with the corresponding units (Fig. 1 B) in GlpT (Huang et al., 2003) and LacY (Abramson et al., 2003). A similar comparison of the corresponding three-helix units is shown in Fig. 1, C and D. The eight three-helix motifs in Fig. 1 D thus represent a superposition of helices 1–3, 4–6, 7–9, and 10–12 from GlpT and LacY. The similarity of the three-helix unit architecture (see also Table 2) in all three proteins strongly supports the existence of a common theme in the structure in each case, and also is consistent with the notion that these 12-helix transporters may have developed from an ancestral precursor that contained three transmembrane segments as suggested previously (Hirai et al., 2003, 2002; Hvorup and Saier, 2002; Tamura et al., 2001). Despite these general similarities, there are also noticeable differences that could be relevant to the different functions of these proteins. Helix 1 of GlpT (green helix in Fig. 1 D), and helix 4 of LacY (gray helix in Fig. 1 D) show the most significant deviations in overall tilt relative to the membrane normal. Some of the loops such as those connecting helices 1 and 2, as well as helices 5 and 6 of GlpT also show subtle differences compared to the average structure. The close correspondence between the three-helix units in these proteins also explains why the respective six-helix units can have similar architectures when they are compared in the opposite orientation (i.e., upside down). The confirmation of conserved repeated units in the different proteins lends credence to the strategy of using the atomic structures of GlpT and/or LacY as useful starting points for the derivation of a plausible atomic model for the structure of OxlT. In other words, the variation in structure between OxlT and GlpT/LacY may therefore be described, to a first approximation in terms of the relative displacements of some or all of the respective repeated units.
TABLE 1.
Pairwise RMSD (Å) between each N- or C-half of GlpT and LacY
| GlpT N* | GlpT N′† | GlpT C | GlpT C′ | LacY N | LacY N′ | LacY C | LacY C′ | Average | |
|---|---|---|---|---|---|---|---|---|---|
| GlpT N | – | 4.79 | 2.44 | 4.18 | 3.31 | 4.86 | 3.34 | 4.33 | 3.89 |
| GlpT C | 2.44 | 4.18 | – | 3.49 | 2.46 | 3.83 | 2.83 | 3.44 | 3.16 |
| LacY N | 3.31 | 4.86 | 2.46 | 3.83 | – | 4.14 | 2.73 | 3.39 | 3.53 |
| LacY C | 3.34 | 4.33 | 2.83 | 3.44 | 2.73 | 3.39 | – | 3.14 | 3.31 |
The deviations were calculated using the program LSQMAN (Kleywegt and Jones, 1994).
The presence of a prime symbol (′) after N or C indicates that this six-helix unit was aligned in the opposite orientation.
TABLE 2.
Pairwise RMSD (Å) between each three-helix unit of GlpT and LacY: N1 (helices 1–3), N2 (helices 4–6), C1 (helices 7–9), and C2 (helices 10–12)
| GlpT N1 | GlpT N2 | GlpT C1 | GlpT C2 | LacY N1 | LacY N2 | LacY C1 | LacY C2 | Average | |
|---|---|---|---|---|---|---|---|---|---|
| GlpT N1 | – | 4.65 | 2.28 | 4.61 | 3.11 | 5.28 | 3.60 | 4.32 | 3.98 |
| GlpT N2 | – | 3.25 | 2.49 | 3.86 | 3.42 | 3.85 | 2.78 | 3.47 | |
| GlpT C1 | – | 3.21 | 2.25 | 3.75 | 2.86 | 3.14 | 2.96 | ||
| GlpT C2 | – | 3.26 | 2.57 | 3.24 | 2.49 | 3.12 | |||
| LacY N1 | – | 3.67 | 2.38 | 2.78 | 3.04 | ||||
| LacY N2 | – | 3.40 | 2.85 | 3.56 | |||||
| LacY C1 | – | 2.99 | 3.19 | ||||||
| LacY C2 | – | 3.05 |
To determine the nature of the conformational change, we started with simplest rigid-body refinement scenario, involving the use of two rigid bodies with six helices derived from the N- or C-terminal halves of GlpT or LacY, based on the idea of using the minimal number of independent parameters that are changed during refinement. A further reason for this choice was the fact that the interfaces between the three-helix units that make up each six-helix unit in GlpT/LacY and OxlT are much more interdigitated than the interfaces between the two six-helix halves of the proteins, suggesting that this latter interface was more likely to be involved in protein conformational changes.
Because the highly symmetric nature of the OxlT density map makes it difficult to guess the absolute orientation of the OxlT density map relative to the sequence simply by inspection, all possible locations of a given six-helix unit were tested without constraining their relative orientations. There are four possible choices of a model for each half of the density map, and each could be positioned facing upwards or downwards. For the whole map, this leads to a total of 2 × 4 × 2 × 4 = 64 possible unique starting positions for refinement. We determined the best fit of each of these 64 combinations to the OxlT density map by varying only the relative orientation and location of the six-helix units during refinement; i.e., a total of six translational and six rotational parameters. The best fit between the starting coordinates and the density map was obtained with one combination from the set of four that contained the C-terminal half of GlpT as a template for both N- and C-terminal halves of the OxlT density map. In this combination, the orientation of the six-helix halves was such that the C-terminal end of helix 6 and the N-terminal end of helix 7 were on the same face and the 6–7 loop region could therefore be connected and modeled using the x-ray structures of GlpT as a guide. Different starting positions were tested to ensure that the endpoint of refinement was independent of the position of the starting model as shown in Fig. 2.
FIGURE 2.

Test of convergence of refinement. (A) Three different starting positions of the GlpT model were intentionally shifted ∼4 Å to each other as shown in the superposition, and used as starting positions for refinement against the density map determined by electron crystallography for OxlT in the substrate-bound conformation. The same three N- and C-terminal pairs are shown in the panel B after rigid-body refinement, superposed with the OxlT density map. All three structures converged to very similar final positions, with RMSD values within 0.6 Å of each other. Although the absolute resolution of the density map is ∼6.5 Å in the plane and ∼12 Å in the vertical direction, there is clearly enough information for the refinement to converge because of the additional information derived from knowledge of the x-ray structure of GlpT. This figure was generated using the program PYMOL (http://pymol.sourceforge.net/).
Based on these results, we used the GlpT C-terminal half as a template to generate a homology model for OxlT. To obtain reliable alignment between OxlT and GlpT, we aligned a variety of sequences from both the OxlT and GlpT families (Fig. 3 A). Based on the repeated motif presented in Fig. 1, we could also align the sequence of OxlT to a tandem arrangement of the two C-terminal halves of GlpT. After alignment, a homology model for OxlT was constructed, with the primary purpose of generating a reliable Cα-trace for OxlT. The orientations of residues were selected from possible rotamers to match as much as possible with the corresponding conformations in GlpT, and to avoid steric conflicts between residues. We also altered the lengths of loop regions between helices by 1–4 residues to accommodate insertions and deletions when comparing the sequence of GlpT with OxlT. These loops were manually positioned to fit the density wherever possible, and subjected to energy minimization using CNS (Brunger et al., 1998) while keeping the helical regions fixed. The alignment of OxlT as described leads to a revised model for the secondary structure of OxlT as shown in Fig. 3 B. The major difference from previous secondary structural models such as that presented in Heymann et al. (2001) is that loop 2–3 and loop 8–9 are shorter and helices 2 and 8 are longer at the C-terminal side. A new finding from this analysis is the presence of two positively charged residues, K355 and R272, in the center of the lipid bilayer, with implications for interaction with the negatively charged primary substrates oxalate and formate.
The fit of the 12 helices to the density map after the two rigid-bodies refinement procedure starting with the coordinates of the cytoplasmically open conformation of OxlT is shown in Fig. 4, A–C. For comparison, the locations of the helices deduced independently (Hirai et al., 2002) using only the electron microscopic data are shown in Fig. 4, D–F. The excellent fit of the structure generated by the two rigid-bodies refinement procedure, and the close correspondence between the two sets of helix assignments (i.e., panels A–C vs. D–F) validates the most significant conclusion from the refinement exercise, i.e., that the structural changes associated with substrate transport primarily involve the relative motion of one set of six helices relative to the other set. In the course of refinement of this model against the density map, the contact position between helices 2 and 11, and between helix 5 and 8 were shifted by almost two turns. Thus, in the cytoplasmically open state, helices 2 and 11 are close to each other near residues Q56, T60, G364, A367 and I360. In the substrate-bound “closed” state, helices 2 and 11 are now close near residues V64, A67, A356, and I360. Likewise, the contact between helices 5 and 8 in the open state is near residues V154, P159, S262, and N265 but, in the substrate-bound “closed” state, they are replaced by residues A147, G151, G269, and L266.
FIGURE 4.

Selected horizontal sections from the 6.5-Å density map of OxlT in the oxalate-bound, “closed” state near the periplasmic end (A, D), center of membrane (B, E), and cytoplasmic end (C, F). The sections are 3-Å thick and are at intervals of 7.5 Å from the center. Helices are colored using the same color scheme used in Fig. 1 A. (A–C) Helix positions derived from the rigid-body refinement procedure described in the text starting from knowledge of the x-ray coordinates of GlpT. (D–F) Helix positions derived from a manual fit of helices into the density map as previously reported (Hirai et al., 2002).
The construction of a model for the substrate-bound, “closed” state using the OxlT density map provides an opportunity to dissect in detail the nature of the conformational changes that occur with oxalate binding. The overall nature of the switching between the two states is shown in Fig. 5, A and B. The main consequence of transition from cytoplasmically open to substrate-bound, “closed” state is to bring the cytoplasmic ends of the two halves of the protein together, and the conversion between the two states can be roughly described as involving a swiveling movement of one set of six helices relative to the other set of six helices. Each of two domain surfaces pointing toward the interior of the cavity is slightly convex. The rolling motion between these two slightly convex surfaces thus provides a plausible trajectory from a cytoplasmically open state to a “closed” state in which the central region is closed to both sides (see cartoon shapes in Fig. 6). The effect of this structural change on the internal cavities in the protein is shown in Fig. 5, C and D. In the cytoplasmically open state (Fig. 5 C), the cavity is very narrow on the periplasmic side as expected from the overall architecture of the protein. In the substrate-bound, “closed” state (Fig. 5 D), the cavity is of intermediate size on both sides, and is widest in the central region as already suggested by the helix arrangement in the central section of the protein (Fig. 4). No detectable density for the substrate is observed in this cavity in our density map, but this is likely to arise from a combination of the low resolution of the map and flexibility of the bound oxalate. A logical extension of these results is that the release of the substrate on the periplasmic side might involve a continuation of the same type of swiveling motion between the two halves of the protein, resulting in bringing the cytoplasmic parts of the protein closer together than the periplasmic halves. The global architecture of such a “periplasmically” open state is likely to be similar to that of the “cytoplasmically” open state because of the general equivalence of the structural environments of each of the four three-helix units in MFS proteins (Fig. 1). We therefore propose that a proper description of the cycle of substrate transport requires three distinct protein conformations, with the substrate-bound, “closed” state representing a key intermediate between cytoplasmically and periplasmically open conformations.
FIGURE 5.
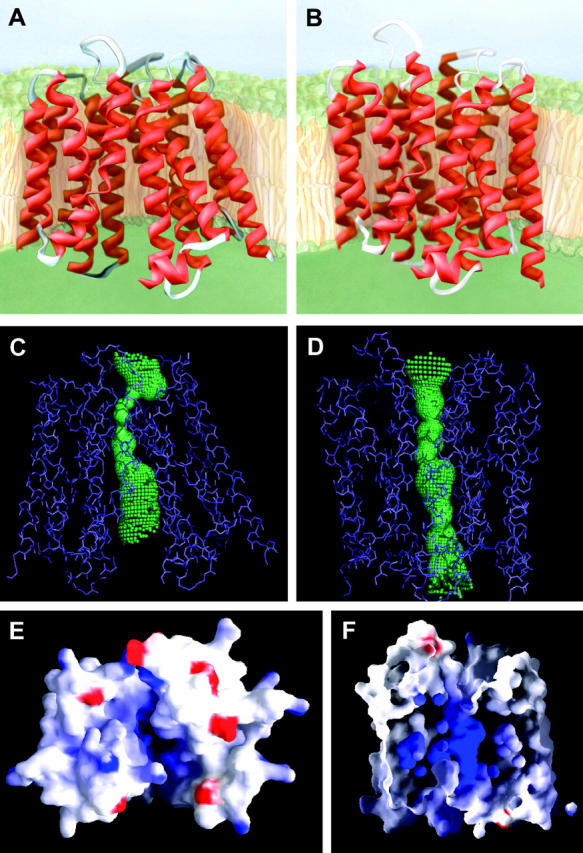
Comparison between structures of the cytoplasmically open and oxalate-bound, “closed” states of OxlT. (A and B) Ribbon presentations of models for the cytoplasmically open and oxalate-bound, “closed” states. The cytoplasmic side is at the top in these and in the subsequent panels C, D, and F. (C and D) Visualization of shape of internal cavity enclosed calculated using the program Hole (Smart et al., 1996) for the cytoplasmically open and oxalate-bound, “closed” states of OxlT. (E) Charge potential distribution in the cytoplasmically open state of OxlT as viewed from the cytoplasmic side, with blue and red colors denoting positive and negative potentials, respectively. Residues Lys-355 (helix 11) and Arg-272 (helix 8) make a major contribution to the positive potential in the cavity. Panels E and F were prepared using the program GRASP (Nicholls et al., 1991). For visual clarity, the cytoplasmic extended part of helices 1 (residues 1–16) and 12 (residues 403–418) and the majority of loop 6–7 (residues 195–207) are excluded in this view. (F) Sectional view of the charge potential distribution in the oxalate-bound, “closed” state as viewed from the front. The periplasmic side is at the top in this view. We propose that oxalate binds in the cavity by virtue of electrostatic interactions with Lys-355 and Arg-272.
FIGURE 6.

(A) Schematic drawing of the three proposed protein conformational states during a transport cycle by proteins that function as uniporters, antiporters, and symporters. (B) Specific description of sequence of events in uniport (top), antiport (middle), and symport (bottom). The states shown in green indicate “switchable” states, i.e., conformations in which the protein can transit across one or more of the states shown in panel A. The states shown in red indicate “unswitchable” states, referring to conditions that require a further event such as substrate binding before a “switchable” state is reached. Thus uniporters can be in “switchable” states with or without bound substrate. Antiporters are “switchable” with substrate but “unswitchable” without substrate. Symporters are “switchable” without substrate or with both substrates but “unswitchable” with only one substrate.
Examination of the structure of OxlT in more detail provides a clearer understanding of the chemistry underlying the transport of substrates such as oxalate and formate. Inspection of the electrostatic surface defining the entrance to the cavity in the open state reveals a concentration of positive charge that is expected to provide an attractive potential for the entry of negatively charged substrates such as oxalate into the cavity (Fig. 5 E). The cavity profile of the closed state (Fig. 5 D) suggests that the bound oxalate is likely to be located roughly in the center of the membrane where the cavity is widest. This is independently confirmed by inspection of the charge profile of the residues that are present at the center of the membrane, as demonstrated in Fig. 5 F. Two key residues in this central region are Lys-355 from helix 11 and Arg-272 from helix 8. Their presence in the cavity must be critical for substrate binding and transport. Replacement of Lys-355 by neutral residues is already known to abolish substrate transport (Fu et al., 2001); studies describing the effect of replacing Arg-272 are yet to be carried out. The locations of these two positively charged residues in the cavity provides a plausible mechanism for the conformational change enabled by charge neutralization by the bound substrates.
Our analysis implies that the sequence of events in transport by an antiporter involve progression through three distinct conformational states: cytoplasmically open form, substrate-bound, “closed” form, and periplasmically open form (Fig. 6 A). For a protein to function as an antiporter, the structural design must prevent transition between the two open states in the absence of bound substrate, and promote interconversion between the states in the presence of bound substrate. In a complete sequence of antiport, one can thus view the conformational changes in the context of the transition through “switchable” and “unswitchable” states, as shown schematically in the middle panel of Fig. 6 B.
In addition to the structural similarities between antiporters and symporter as evidenced by comparison of the structures of OxlT, GlpT, and LacY, it is also the case that uniporters, symporters, and antiporters are widely distributed in each subtree of the phylogenetic tree and do not make isolated large clusters separated from each other (Pao et al., 1998). We therefore propose that it is also very likely that the conformational changes that occur during transport involve a common in all three protein subfamilies, as summarized in Fig. 6 B. The central idea underlying our proposal for conformational changes is that the basic design as well as the type of conformational change is the same for all three types of transporter proteins, and that the differences in function originate from features that dictate whether the switch between the cytoplasmically and periplasmically open states can occur: i), in the absence or presence of bound substrates, as in the case of uniporters, ii), only in the presence of bound substrate, as in the case of antiporters, and iii), only in the absence or in the presence of both bound substrates, as in the case of symporters.
The similarity in the structures of the three different proteins (two antiporters and one symporter), as well as the derivation of the nature of conformational change in OxlT suggests that our findings will have general relevance to other members of the diverse family of MFS proteins. This will be especially valuable for development of plausible structural models for mammalian transporters that are likely to be less easily amenable to crystallographic methods for structure determination. Further, based on the trajectory of the conformational changes during transport, it should be possible to generate model structures for transporters in all three conformational states and generate testable hypotheses for the identities of key substrate binding sites in transporters whose structures have not been determined.
Acknowledgments
The 3D map of OxlT has been submitted in the EMBL-EBI (http://www.ebi.ac.uk/) Macromolecular Structure Database. Accession code assigned for the map entry is EMD-1098.
References
- Abramson, J., I. Smirnova, V. Kasho, G. Verner, H. R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam, V., M. J. Allison, and P. C. Maloney. 1989. Oxalate:formate exchange. The basis for energy coupling in Oxalobacter. J. Biol. Chem. 264:7244–7250. [PubMed] [Google Scholar]
- Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D. 54:905–921. [DOI] [PubMed] [Google Scholar]
- Fu, D., R. I. Sarker, K. Abe, E. Bolton, and P. C. Maloney. 2001. Structure/function relationships in OxlT, the oxalate-formate transporter of oxalobacter formigenes. Assignment of transmembrane helix 11 to the translocation pathway. J. Biol. Chem. 276:8753–8760. [DOI] [PubMed] [Google Scholar]
- Henderson, P. J., and M. C. Maiden. 1990. Homologous sugar transport proteins in Escherichia coli and their relatives in both prokaryotes and eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 326:391–410. [DOI] [PubMed] [Google Scholar]
- Heymann, J. A., T. Hirai, D. Shi, and S. Subramaniam. 2003. Projection structure of the bacterial oxalate transporter OxlT at 3.4 Å resolution. J. Struct. Biol. 144:320–326. [DOI] [PubMed] [Google Scholar]
- Heymann, J. A., R. Sarker, T. Hirai, D. Shi, J. L. Milne, P. C. Maloney, and S. Subramaniam. 2001. Projection structure and molecular architecture of OxlT, a bacterial membrane transporter. EMBO J. 20:4408–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, T., J. A. Heymann, P. C. Maloney, and S. Subramaniam. 2003. Structural model for 12-helix transporters belonging to the major facilitator superfamily. J. Bacteriol. 185:1712–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, T., J. A. Heymann, D. Shi, R. Sarker, P. C. Maloney, and S. Subramaniam. 2002. Three-dimensional structure of a bacterial oxalate transporter. Nat. Struct. Biol. 9:597–600. [DOI] [PubMed] [Google Scholar]
- Huang, Y., M. J. Lemieux, J. Song, M. Auer, and D. N. Wang. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 301:616–620. [DOI] [PubMed] [Google Scholar]
- Hvorup, R. N., and M. H. Saier, Jr. 2002. Sequence similarity between the channel-forming domains of voltage-gated ion channel proteins and the C-terminal domains of secondary carriers of the major facilitator superfamily. Microbiology. 148:3760–3762. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J., and T. A. Jones. 1994. A super position. CCP4/ESF-EACBM Newsletter on Protein Crystallography 31:9–14. [Google Scholar]
- Mitchell, P. 1967. Translocations through natural membranes. In Advances in Enzymology and Related Areas of Molecular Biology. Wiley Interscience, New York, NY. 33–87. [DOI] [PubMed]
- Nicholls, A., K. A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- Pao, S. S., I. T. Paulsen, and M. H. Saier, Jr. 1998. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen, I. T., L. Nguyen, M. K. Sliwinski, R. Rabus, and M. H. Saier, Jr. 2000. Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes. J. Mol. Biol. 301:75–100. [DOI] [PubMed] [Google Scholar]
- Poolman, B., and W. N. Konings. 1993. Secondary solute transport in bacteria. Biochim. Biophys. Acta. 1183:5–39. [DOI] [PubMed] [Google Scholar]
- Smart, O. S., J. G. Neduvelil, X. Wang, B. A. Wallace, and M. S. Sansom. 1996. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14:354–360, 376. [DOI] [PubMed] [Google Scholar]
- Subramaniam, S., and R. Henderson. 2000. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 406:653–657. [DOI] [PubMed] [Google Scholar]
- Tamura, N., S. Konishi, S. Iwaki, T. Kimura-Someya, S. Nada, and A. Yamaguchi. 2001. Complete cysteine-scanning mutagenesis and site-directed chemical modification of the Tn10-encoded metal-tetracycline/H+ antiporter. J. Biol. Chem. 276:20330–20339. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]