

Abstract
The formation of amyloid fibrils is often encountered in Alzheimer's disease, type II diabetes, and transmissible spongiform encephalopathies. In the last few years, however, mounting evidence has suggested that the soluble oligomers of amyloid-forming peptides are also cytotoxic agents. Understanding the early pathway steps of amyloid self-assembly at atomic detail might therefore be crucial for the development of specific inhibitors to prevent amyloidosis in humans. Using the activation-relaxation technique and a generic energy model, we study in detail the aggregation of a hexamer of KFFE peptide. Our simulations show that a monomer remains disordered, but that six monomers placed randomly in an open box self-associate to adopt, with various orientations, three possible distant low-energy structures. Two of these structures show a double-layer β-sheet organization, in agreement with the structure of amyloid fibrils as observed by x-ray diffraction, whereas the third one consists of a barrel-like curved single-layer hexamer. Based on these results, we propose a bidirectional growth mode of amyloid fibril, involving alternate lateral and longitudinal growths.
INTRODUCTION
Amyloid fibrils are often encountered in the late steps of Alzheimer's disease, type II diabetes, and transmissible spongiform encephalopathies (Hardy and Selkoe, 2002; Taylor et al., 2002). This phenomenon is not limited to a few sequences, however, and recent data show that the ability to form these amyloid structures is a general property of any polypeptide chain under destabilizing conditions (Stefani and Dobson, 2003); even peptides with as few as four to six residues can form well-defined fibrils showing all the characteristics of amyloid fibrils built from large proteins of 100 residues or more (Lopez De La Paz et al., 2002; Reches et al., 2002; Tjernberg et al., 2002). Although a relation of causality between the amyloid fibrils and diseases still remains to be fully established, mounting evidence suggests that both insoluble fibrils and soluble oligomers are toxic in cell cultures (Kirkitadze et al., 2002). The structural characterization of these aggregation intermediates raises considerable challenges, however, as oligomers typically occur with a very low concentration and tend to be rather short-lived, making it very difficult to obtain precise structural information experimentally. For example, solid-state NMR has only been used to study the structural properties of stable fibrils (Antzutkin, 2004), and, despite many efforts, the structure of oligomers remains mostly unknown (Kayed et al., 2003).
In view of these limitations, computer simulations have provided a very convenient complement to experimental approaches and the aggregation of peptides/proteins has been studied by using a range of protein models. Although investigations based on lattice models have provided numerous insights into the thermodynamic and kinetic properties of protein aggregation (Bratko and Blanch, 2001; Harrison et al., 2001; Dima and Thirumalai, 2002), simplified off-lattice studies with a single-bead residue model and a Gō potential (Jang et al., 2003; Friedel and Shea, 2004) have helped clarify the kinetics and assembly processes of the β-sheet complex. Due to the simplicity of these models, however, it is not possible to extract detailed information on the structure and growth of oligomeric intermediates and more realistic representations must be used. Proceeding along these lines, various groups have studied the aggregation of dimers and trimers of amyloid-forming peptides using different atomic peptide models (Gsponer et al., 2003; Klimov and Thirumalai, 2003; Santini et al., 2003, 2004). These simplified trajectories with only two or three strands help understand the first steps of aggregation, but they fall well below the size of the critical nucleus from which growth is rapid (O'Nuallain et al., 2004). Taking a different approach, Ma and Nussinov have studied the stabilities of various oligomers (Ma and Nussinov, 2002; Zanuy et al., 2003) using molecular dynamics (MD) in explicit solvent, identifying stable structures of hexamers and octamers.
Complementing these previous studies, we examine here the self-assembly pathways of oligomers using an off-lattice peptide model (Derreumaux, 1999, 2000), which goes beyond the Cα representation. This peptide model, where each amino acid is represented by its N, H, Cα, C, O, and one bead for its side chain, allows us to study the explicit formation of hydrogen bonds (H-bonds), but also to simulate aggregation in systems with more than three peptides, the current system size. More precisely, we study the aggregation process of a hexamer of the KFFE peptide. This tetrapeptide was shown to form amyloid fibrils (Tjernberg et al., 2002) and yet is small enough for simulations at an atomic level. The choice of a hexamer is based on results of a recent mathematical model of the nucleated polymerization mechanism which identifies this as the critical size for fibril formation (Masel et al., 1999).
Using the activation-relaxation technique (ART) (Barkema and Mousseau, 1996; Malek and Mousseau, 2000) combined with an optimized-potential for efficient peptide structure prediction (OPEP) (Derreumaux, 1999, 2000), we identify various self-assembly pathways leading to various hexameric metastable structures, including a two β-sheet structure displaying the structural characteristics of amyloid fibril. Our results demonstrate that small oligomers are in equilibrium between many different aggregation structures in addition to the fibril-like conformation, in agreement with recent structural-recognition measurements using antibody agents (Chromy et al., 2003), and in situ atomic force microscopy (Zhu et al., 2004).
METHODS
We have simulated the self-assembly of the KFFE hexamer using ART-OPEP. The N- and C-termini were neutralized using acetyl and amine groups, respectively. Twenty simulations were performed with open boundary conditions; each run generates 9000 events, starting from four different initial states and using different random number seeds. These four initial states include six monomers placed with random orientations in a sphere of diameter 40 Å (IN1, 10 runs), an antiparallel dimer and four monomers with random orientations (IN2, four runs), a double-layer three-stranded β-sheet structure with almost perpendicular orientations of the inter β-sheets (IN3, four runs), and a tetramer-dimer structure (IN4, two runs). In this study, all the structures were produced using the MolMol software (Koradi et al., 1996) except for the structure in Fig. 5 d produced using Rasmol (Bernstein, 1999), and the N-terminal end of each chain is located by a sphere.
FIGURE 5.

Three lowest-energy structures generated in our simulations: (a) the double-layer three-stranded β-sheet; (b) the double-layer tetramer-dimer structure; and (c) the β-barrel hexamer with the main-chain atoms shown. (d) The β-barrel hexamer in a ball-and stick-representation and the side chains of Phe residue labeled by F. The thick arrows indicate pathways generated in our simulations.
Simulation method—ART
ART is a generic method for exploring the energy landscape of complex systems. An ART event consists of four basic steps: starting from a local minimum, a randomly chosen chain of the system is pushed and moves along a random direction until the lowest eigenvalue of the Hessian matrix becomes negative, indicating the vicinity of a saddle point; the system is then pushed along the eigenvector associated with the lowest eigenvalue until the total force approaches zero, and the system converges onto a first-order saddle point; the system is pushed slightly over the saddle point and is relaxed to a new local minimum, using standard minimization technique; the new local minimum is accepted or rejected using the Metropolis criterion.
Because ART neglects the entropic contribution associated with thermal fluctuations, the Metropolis temperature used in the simulation does not correspond to a precise experimental temperature and the ART-generated trajectories do not sample a well-defined statistical mechanical ensemble. We therefore adjust the simulation temperature to ensure that it is high enough to sample efficiently the configurational space while converging toward well-ordered low-energy structures. All simulations presented here are conducted at Metropolis temperature 310 K.
Despite this limitation, ART trajectories are well-controlled. Since each event is constructed by going over a first-order saddle point, in accordance with transition-state theory, each aggregation trajectory represents a physically possible path. Moreover, we have shown previously that the folding trajectories generated this way are qualitatively identical to those obtained by all-atom folding MD simulations for a 14-residue α-helix (Wei et al., 2002) and by several approaches for the second β-hairpin of protein G (Wei et al., 2004). In the case of the latter system, for example, all-atom folding MD, dynamics ensemble, replica exchange MC, and MD simulations predicted two contradicting folding mechanisms. With ART-OPEP, we could produce the first complete picture of the folding process for this peptide, showing that three (and not two) folding mechanisms were possible; which one is preferred depends, in simulations, on the energy model and the simulation protocol and, in experiments, on the amino-acid composition and the solvent conditions (Wei et al., 2004). This third mechanism was observed by another group on a related peptide using multicanonical MD simulations with explicit solvent (Ikeda and Higo, 2003).
A detailed description of the algorithm and its implementation can be found in Barkema and Mousseau (1996), Malek and Mousseau (2000), and Wei et al. (2002, 2004).
Energy model—OPEP
Our protein model is a coarse-grained off-lattice model where the main chain of each amino acid is represented explicitly and all side chains are represented by beads with appropriate van der Waals radii and geometrical parameters with respect to the main chain (Derreumaux, 1999, 2000). The OPEP energy function, which includes solvent effects implicitly and was trained on the structures of monomeric peptides with various secondary structures, is expressed as a function of four types of interactions: harmonic potentials for maintaining the geometry of peptides (bond lengths and bond-angles for all particles, and improper dihedral angles of side chains with respect to the backbone); backbone two-body and four-body (cooperative) hydrogen-bonding interactions (Derreumaux, 1999; Wei et al., 2003); pairwise potential between side chains, considering all 20 amino-acid types, represented by 12-6 potential if the interactions are hydrophobic in character and by a 6-potential, otherwise; and excluded-volume potentials between all particles.
Because of the coarse-grained nature of the atomic representation used, OPEP is an approximation to the real energy surface; OPEP does not take into account H-bonds between the solvent and solute, for example, and it cannot capture the full complexity of hydrophobic and electrostatic interactions between all-atom side chains. Nonetheless, OPEP with ART identified folding mechanisms of helix and hairpin models similar to those found using all-atom models and explicit solvent conditions.
Moreover, OPEP is not biased toward the formation of specific topologies. This potential has predicted correctly the native state of proteins models with β-hairpin, three-helix bundle, and α/β conformations (Derreumaux, 1999, 2000; Wei et al., 2004). OPEP can also discriminate between sequences: although ART-OPEP simulations on the tetramer of the wild-type sequence KFFE lead to antiparallel β-sheets, similar simulations on the mutant sequence KPGE find only disordered structures because the β-sheet structure is destabilized by the β-sheet breaker property of the proline residue (Melquiond et al., 2004), in agreement with drug-design experiments (Adessi et al., 2004). More details on the impact of the force field on the ART trajectories and the energy barriers are discussed in Wei et al. (2004).
RESULTS
Structure characterization of monomer
We first characterize the structure of the monomeric peptide. We run four simulations for a total of 8000 accepted events. The secondary structure, computed using the φ and ψ angles of the two inner residues following the criteria of Srinivasan and Rose (1995), shows that ∼72% of all monomeric structures adopt a random coil conformation and 28% a β-strand structure; the population of α-helical conformations is negligible (0.12%). These results are consistent with CD analysis in solution at pH 6.0, with β-strands and random coil structures coexisting (no data shown) (Tjernberg et al., 2002). The KFFE peptide in solution is therefore well described by OPEP.
Self-assembly pathways for the hexamers
All simulations lead to organized conformations with a total energy between −55 and −60 kcal/mol. Although each simulation generates a unique trajectory, it is possible to categorize them as belonging to one of three generic interconnected self-assembly pathways as discussed separately below. The first identified pathway leads to the formation of a double-layer three-stranded β-sheet with various in-plane orientations; this structure displays the characteristics of amyloid fibrils. The second pathway leads to a double-layer tetramer-dimer structure, and the third one generates a monolayer β-sheet in a barrel-like shape. These three pathways are not independent: for example, we find that the double-layer three-stranded β-sheet can transform into a tetramer-dimer, and that both structures are possible intermediates toward the monolayer β-sheet.
Double-layer three-stranded parallel-antiparallel β-sheets
Of the 10 runs starting from the random state IN1, two aggregate into a double-layer three-stranded parallel-antiparallel β-sheet. Although the detailed molecular structure of amyloid fibrils has not yet been determined experimentally—their noncrystalline and insoluble nature makes their characterization by x-ray diffraction or liquid-state NMR very difficult—x-ray fiber diffraction shows that amyloid fibrils contain a cross-β structural motif. This motif is characterized by extended β-sheets in which the β-strands run perpendicular to, and the intermolecular H-bonds run parallel to, the fibril axis (Sunde et al., 1997).
These structural characteristics are present in both of our generated double-layer three-stranded parallel-antiparallel-β-sheet hexamers. Fig. 1, a and b, show two different views of one hexamer (T1) which displays a perfect parallel match between layers even though each layer has a mixture of parallel and antiparallel β-strand. The other hexamer (T2), with one antiparallel pair across the plane (figure not shown), is less stable due to the misalignment of this pair. We describe below the assembly process for the parallel hexamer T1; results are qualitatively similar for T2. As shown in Fig. 1, a and b, the distance between antiparallel β-strands is 4.5 Å, and the distance between two nearest nitrogen atoms located in the two parallel β-strands belonging to the two different layers is ∼10–12 Å . These two distances are in agreement with the x-ray diffraction measured distances of 4.7 Å between β-strands and 10–11 Å between two β-sheets in fibrils (Sunde et al., 1997; Balbach et al., 2000).
FIGURE 1.

Two different views of one double-layer three-stranded β-sheet hexamer (T1). The two layers are perfectly parallel. (a) View perpendicular to the fibril axis and the β-strands; (b) view perpendicular to the fibril axis and parallel to the β-strands. Dotted line represents H-bonds formed between the carbonyl oxygen and the amide hydrogen. The N-terminal end of each chain is located by a sphere.
As can be seen from Fig. 2, there is considerable energy relaxation in the first 1000 events (Fig. 2 a), with most peptides (1, 3, 4, 5, and 6) becoming extended, with an end-to-end distance close to 12 Å the fully extended value (Fig. 2 b); however, no peptide shows a stable β-strand conformation (Fig. 2 c). Although the end-to-end distance continues to fluctuate over the next 500 events, at event 1500, peptides 1, 4, 5, and 6 have moved in a β-strand conformation, with the remaining two adopting a random coil state. Fig. 2 c shows that the peptides only oscillate between these two states, with very little α-helical component. During these first 1500 events, there is considerably reorientation of the various peptides, as the first trimer, composed of 3, 5, and 6, stabilized into a parallel-antiparallel conformation. For the following 1500 events, this trimer adopts a more stable, fully extended state, whereas the second trimer (1, 2, 4), stabilizes, also in a parallel-antiparallel conformation, before locking into place, parallel to the first trimer. As is shown in Fig. 2 d, after these two trimers have settled into a two β-sheet structure, they rotate about each other by a full 360°, adopting in passing a fully perpendicular orientation around event 2600 and antiparallel orientation around event 2900. During the whole self-assembly process, the configurational energy decreases steadily and converges to a plateau around −55 kcal/mol, avoiding being trapped in high-energy structures.
FIGURE 2.

Characterization of the self-assembly pathway as a function of accepted event number leading to the formation of fibril-like hexamer. Evolution of (a) the conformational energy; (b) end-to-end Cα distance for the six monomers; (c) secondary structure of each residue in the six monomers—the secondary structure is assigned according to the values of dihedral angles φ and ψ in Srinivasan and Rose (1995), black for random coil, white for α-helix, and shaded for β-strand, Fji is the jth (j = 2, 3, the two inner residues in each peptide) residue Phe in peptide i (i = 1–6); and (d) the intersheet orientation for the three final pairs. Orientation is calculated using the scalar product between the end-to-end unit vectors of each chain.
We summarize the assembly pathway for the fibril-like hexamer in Fig. 3: starting from a random conformation (Fig. 3 a), an antiparallel dimer forms (Fig. 3 b) and attracts a third monomer in a parallel orientation (Fig. 3 c). At the same time, the other monomers come together and form an amorphous aggregate state (Fig. 3 c). After interpeptide interactions, a second trimer almost forms (Fig. 3 d). The first formed antiparallel-parallel trimer helps stabilize the formation of the second trimer (Fig. 3 e) which, after some rotation, forms a β-sheet perpendicular to the first trimer (Fig. 3 e). This structure is not very stable and the two trimers rotate about each other and rearrange themselves into a parallel β-sheet structure (Fig. 3 f).
FIGURE 3.

Assembly pathway (a → f) leading to a fibril-like hexamer with a parallel-antiparallel β-sheet/parallel layer structure: (a) six monomers with random orientations; (b) a dimer with four monomers; (c) a trimer with three monomers; (d) a trimer with another trimer, which is not well-aligned; (e) two trimers with perpendicular interstrand arrangement; and (f) two trimers with parallel interstrand arrangement (view parallel to fibril axis and perpendicular to β-strands). Dotted line represents H-bonds formed between the carbonyl oxygen and amide hydrogen.
Double-layer tetramer-dimer and monolayer hexamer β-sheets
Four out of the 10 runs starting from the random state IN1 lead to a tetramer-plus-dimer structure; two out of four runs starting from IN2 and one out of four starting from IN3 also aggregate into a similar structure. Fig. 4 shows the details of one representative trajectory from IN1. The conformational energy (Fig. 4 a) drops rapidly and the number of H-bonds increases to 16 as the tetramer forms. After 900 accepted events, four out of the six monomers become extended (the end-to-end distance is ∼12 Å) (Fig. 4 b), and the tetramer is already almost in its most stable state, in a well-aligned antiparallel β-sheet (Fig. 4 d). The two remaining peptides continue to move with respect to each other until event 2000, when they lock into an antiparallel dimer rotating into a parallel orientation with respect to the tetramer (Fig. 4 c). The distance between two nearest nitrogen atoms between the two layers, is again 10–12 Å. The width of the β-sheets (defined as the length of β-strand) is ∼12 Å which is consistent with the width 12–16 Å of 10 measured fibrils experimentally (Tjernberg et al., 2002).
FIGURE 4.

Characterization of the self-assembly pathway as a function of accepted event number leading to the formation of a tetramer plus a dimer. Evolution of (a) the conformational energy and number of H-bonds; (b) end-to-end distance for the six monomers; (c) intrasheet orientation for selected pairs in the top (5–6) and the bottom layer (2–4, 4–1, 1–3), intersheet orientation between a monomer in the tetramer and one in the dimer (5–4). (d) A close-up view of the tetramer, in a well-aligned four-stranded antiparallel configuration. From left to right, the four monomers are 2, 4, 1, and 3, respectively. Dotted line represents H-bonds formed between the carbonyl oxygen and amide hydrogen.
We can summarize qualitatively the assembly process of this structure by a sequence of structural steps. As in the case of the fibril-like hexamer, an antiparallel structure forms first. Two more peptides aggregate to this dimer, in sequence, with an antiparallel orientation, leading to a fully antiparallel tetramer. The two remaining peptides then assemble into a separate antiparallel dimer. During the whole process, the two peptides at the edge of the tetramer show considerably more flexibility than those in the center and will often break H-bonds.
The antiparallel four-stranded β-sheet (Fig. 4 d) generated by ART during the folding of the KFFE hexamers is consistent with the proposed model of Tjernberg et al. (2002). Moreover, its assembly pathway is also in agreement with that proposed by Jang et al. (2003) for a tetrameric β-sheet complex using MD simulations and a Go-energy model: four monomers → dimer and two monomers → trimer and monomers → tetramer.
Starting from IN4, a tetramer-dimer structure (Fig. 5 b), both simulations, run over 9000 events, lead to a third low-energy structure: a β-barrel structure (Fig. 5, c and d). It is a curved monolayer hexamer, mostly antiparallel, of energy similar to the tetramer-dimer structure (−60 kcal/mol) and stabilized by H-bond interactions. Looking at the all-atom ball and stick representation of this structure (Fig. 5 d), we found six out of 12 Phe side chains (labeled by F) inside the barrel.
DISCUSSION
Interpeptide organization
Although we identify three low-energy metastable structures in our simulations, there is a considerable fluctuation in the details of the interpeptide organization, both in terms of orientation and H-bond network. The existence of out-of-register alignments is to be expected for such short peptides. This observation has been reported during the aggregation of the trimeric heptapeptide GNNQQNN from yeast prion Sup35 by MD with implicit solvent (Gsponer et al., 2003), and the assembly of the dimeric heptapeptide KLVFFAE from Aβ Alzheimer's peptide by ART-OPEP (Santini et al., 2004).
These fluctuations are also found in other amyloid-forming systems. Experimental and modeling studies suggest, for example, that the β-sheet of the human islet polypeptide could be arranged in a parallel or as a mixture of both parallel and antiparallel conformation (Jaikaran and Clark, 2001; Sumner Makin and Serpell, 2004). Similarly, an off-register staggered arrangement in Aβ34–42-fibrils was proposed recently (Antzutkin, 2004). It was also shown by NMR solid-state analyses that the molecular conformations of fibrils depend strongly on the pH conditions. In particular, the human calcitonin fibril structure may be composed mainly of antiparallel β-sheets at pH 7.5 and 4.1, and a mixture of antiparallel and parallel β-sheets at pH 3.3 (Naito et al., 2004). Petkova et al. showed that Aβ11–25 fibrils adopt distinct antiparallel β-sheet registries as a function of pH (Petkova et al., 2004). Although the most favored arrangement might depend on the specificity of the environment, these results imply that all alignments are thermodynamically reachable.
Role of side chain-side chain and backbone-backbone interactions
Following the aggregation pathway, it is possible to assess the role of side chain-side chain and backbone-backbone interactions in the self-assembly process of fibril-like oligomers. Fig. 6 shows the percentage of native side chain-side chain contacts, the percentage of native alpha carbon (CA)-alpha carbon (CA) contacts and the number of total side chain-side chain contacts as a function of event number for the two runs leading to a fibril-like trimer-trimer β-sheet. We define a CA-CA contact between two CAs separated by at least two residues if their distance lies within 6.0 Å; two side chains interact if their respective distance is <6.5 Å. Native contacts are those present in the lowest-energy structure for each run. Identical results are obtained with other cutoffs.
FIGURE 6.
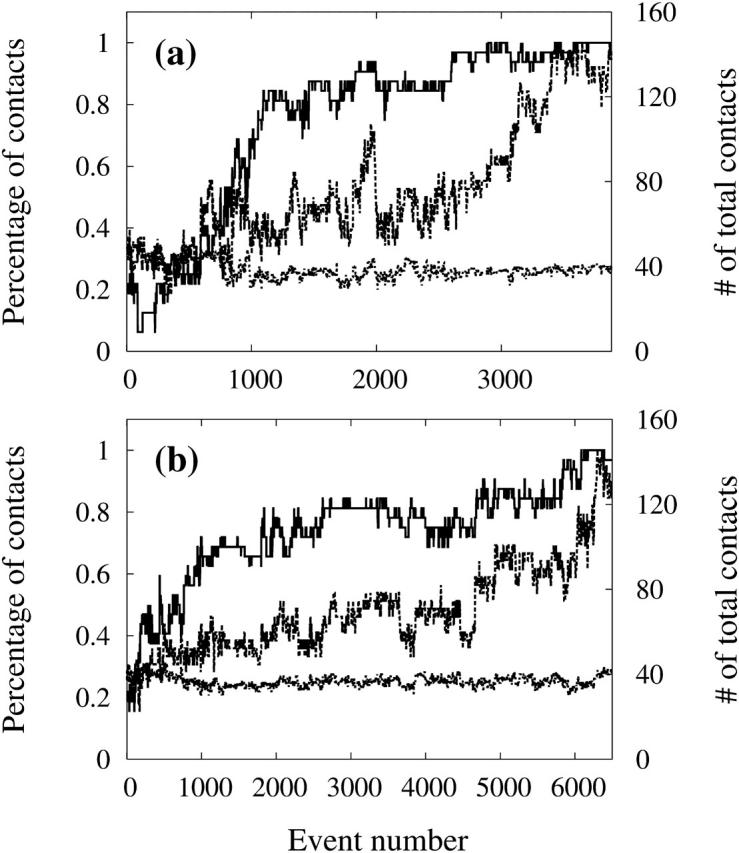
Percentage of native side chain-side chain contacts (dashed line), the percentage of native CA-CA contacts (solid line), and the number of total side chain-side chain contacts (dotted line) as a function of event number for the two runs which generated a fibril-like trimer-trimer hexamer.
Fig. 6, a and b, show a very similar results. In both cases, the total number of side chain-side chain contacts remains almost constant throughout the aggregation process. Starting from a random assembly of peptides, the side-chain interactions pull the molecules together. Slowly, each peptide rearranges, but remains in tight contact with the others at all times. Nonnative side chain-side chain interactions seem therefore to play an important role in the early stage of hexamer formation.
The percentage of native CA-CA contacts (or backbone-backbone interactions) increases rapidly in the first 1000 to 1200 events. As backbone-backbone contacts are closely associated with the formation of β-sheets, this evolution indicates that the sheets form first, stabilizing the structural elements required in the assembly of the fibril-like hexamer. At ∼1200 events, more than 70% of the native backbone-backbone interactions are formed, whereas only 40% native side chain-side chain bonds are in place. For the rest of the simulation, the β-sheets will stabilize themselves and more around until the two trimers adopt a parallel orientation, forming a fibril-like structure. We clearly see the role of native side chain-side chain contacts in the final organization of the fibril.
The self-assembly process of the hexamer seems therefore to take place in steps. Nonnative side chain-side chain interactions bring the peptides together, allowing the native backbone-backbone interactions to start the assembly of β-sheets. Once these are formed, the overall three-dimensional organization appears to be driven by native side chain-side chain interactions.
Oligomer growth process
The early steps of the oligomeric assembly consist in the association of monomers, resulting in the formation of different arrangement of hexamers. In our simulations, starting from randomly chosen conformations, the peptides undergo a dramatic increase in β-strand content, consistent with the hypothesis proposed by Serpell and Smith (2000), leading to two distinct double-layers with trimer-trimer or tetramer-dimer β-sheets. Some simulations show that the trimer-trimer structure can transform into a tetramer-dimer and, from there, into a curved monolayer six-stranded β-sheet. The connection between the three different lowest-energy hexamers is shown in Fig. 5. Interestingly, all these conformations are structurally relevant intermediates in the aggregation of fibrils, in the sense that they represent building blocks that are fully compatible with the structural properties of fibrils.
The three different oligomeric structures generated in our simulations are consistent with a large body of data. The double-layer three-stranded β-sheets and the tetramer-dimer intermediate have the structural characteristics of fibril. In a recent work of Zanuy et al. (2003), the stabilities of all-atom double-layer and triple-layer β-sheets were studied by MD simulations in explicit solvent. The monolayer hexamer was not considered in this theoretical study, but it has been observed by light scattering and atomic force microscopy for a related peptide (Fung et al., 2003). Fung et al. (2003) have investigated the concentration effects on the aggregation of the EAK16-II peptide. Three types of nanostructures were observed: the filaments, the globular aggregates, and the fibrils. They also found that the filaments are monolayers of β-sheets, but the globular aggregates are made of at least two layers of β-sheets.
Although all our simulations require first the formation of an antiparallel dimer intermediate, they suggest that there does not exist a single route to amyloid fibrils: these can form by the addition of single monomer or small oligomers to preformed oligomers of various size, in contradiction with the unique addition-reaction mechanism proposed in Gsponer et al. (2003) and Ferrone (1999). Even for a hexamer, dimers and trimers are seen to attach to existing structures in a single step.
Based on our simulations, we can identify an assembly mechanism. Two different models for the assembly of human amylin (hA) fibril have been proposed based on morphological studies using time-lapse atomic force microscopy (Goldsbury et al., 1999; Green et al., 2004). The first model is that the hA fibril forms by lateral growth of oligomers followed by longitudinal growth into mature fibrils (Green et al., 2004). The second scenario postulates that profibrils elongate (longitudinal growth) and subsequently associate laterally to form thicker fibrils (Goldsbury et al., 1999). Also using atomic force microscopy, Khurana et al. (2003) studied the fibril assembly of three proteins: α-synuclein, insulin, and the B1 domain of protein G. They observed that the fibrils formed by the lateral association of preformed protofilaments. From our simulations of the self-assembly of KFFE hexamers, we find that the growth of the fibril-like hexamer is by a bidirectional growth mode, with an alternate longitudinal growth and lateral growth. We propose that the growth of fibrils made of short peptides follows a bidirectional growth mode as shown by a cartoon in Fig. 7, which complements the two previously proposed fibril growth modes.
FIGURE 7.

Schematic diagram showing a bidirectional growth mode of fibril. The growth can take place by adding monomer, dimers, or larger oligomers. It also alternates between lateral and longitudinal growth. A newly generated β-strand is shaded, an old one is in black.
CONCLUSION
Understanding the process of aggregation is the first step for the rational design of therapeutics preventing neurodegenerative diseases. In this study, we have simulated the assembly of six monomers of the KFFE peptide into ordered oligomers using ART-OPEP. To our knowledge, this is the first aggregation study that takes into account more than three monomers using a more realistic chain representation than one- or two-beads and without using a Gō-energy model. Because of the simplicity of the side chains and solvent effects, the assembly process generated here might not reproduce the exact aggregation pathway as by experiments. Nevertheless, we can extract some qualitative features about its self-assembly pathway and the structures of oligomers. We find that the self-assembly involves nonnative and native backbone-backbone and side chain-side chain interactions in a process where each of these interactions dominate in turn, bringing the peptides together, organizing the β-sheets and assembling the fibril-like structures.
We identify three different low-energy hexameric structures with both parallel and antiparallel β-strand arrangements which should coexist during the aggregation process in experimental conditions. Two of these oligomers are double-layer β-sheets with structural characteristics of amyloid fibrils observed by x-ray diffraction, and one is a monolayer β-sheet. These results indicate that oligomers can adopt many distinct aggregation intermediates, in agreement with recent experimental observations using different techniques, whose populations will be determined by the solvent condition and the crowded environment. The assembly of ordered hexamers follows three different processes: 1), six monomers → dimer and four monomers → trimer and three monomers → two trimers → bilayer hexamer; 2), six monomers → dimer and four monomers → trimer and three monomers → tetramer and two monomers → bilayer hexamer; and 3), bilayer trimer trimer or bilayer tetramer dimer → single-layer hexamer. Finally, from our simulations, we propose a bidirectional growth mode of amyloid fibril, alternate lateral growth, and longitudinal growth.
Acknowledgments
G.W. and N.M. are supported in part by the Fonds québécois de la recherche sur la nature et les technologies and the Natural Sciences and Engineering Research Council of Canada. N.M. also acknowledges funding from the Canada Research Chair Program. Most of the calculations were done on the computers of the Réseau québécois de calcul de haute performance (RQCHP). N.M. is a Cottrell Scholar of the Research Corporation.
References
- Adessi, C., M. Frossard, C. Boissard, S. Fraga, S. Bieler, T. Ruckle, F. Vilbois, S. M. Robinson, M. Mutter, W. A. Banks, and C. Soto. 2004. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer's disease. J. Biol. Chem. 278:13905–13911. [DOI] [PubMed] [Google Scholar]
- Antzutkin, O. N. 2004. Amyloidosis of Alzheimer's aβ peptides: solid-state nuclear magnetic resonance, electron paramagnetic resonance, transmission electron microscopy, scanning transmission electron microscopy and atomic force microscopy studies. Magn. Reson. Chem. 42:231–246. [DOI] [PubMed] [Google Scholar]
- Balbach, J. J., Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed, and R. Tycko. 2000. Amyloid fibril formation by aβ16–22, a seven-residue fragment of the Alzheimer's β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 39:13748–13759. [DOI] [PubMed] [Google Scholar]
- Barkema, G. T., and N. Mousseau. 1996. Event-based relaxation of continuous disordered systems. Phys. Rev. Lett. 77:4358–4361. [DOI] [PubMed] [Google Scholar]
- Bernstein, H. J. 1999. Rasmol Version 2.7.1 Mods Copyright Herbert J. Bernstein, 1998–2001. http://www.bernstein-plus-sons.com/software/RasMol/.
- Bratko, D., and H. W. Blanch. 2001. Competition between protein folding and aggregation: a three-dimensional lattice-model simulation. J. Chem. Phys. 114:561–569. [Google Scholar]
- Chromy, B. A., R. J. Nowak, M. P. Lambert, K. L. Viola, L. Chang, P. T. Velasco, B. W. Jones, S. J. Fernandez, P. N. Lacor, P. Horowitz, C. E. Finch, G. A. Krafft, and W. L. Klein. 2003. Self assembly of aβ1–42 into globular neurotoxins. Biochemistry. 42:12749–12760. [DOI] [PubMed] [Google Scholar]
- Derreumaux, P. 1999. From polypeptide sequences to structures using Monte Carlo simulations and an optimized potential. J. Chem. Phys. 11:2301–2310. [Google Scholar]
- Derreumaux, P. 2000. Generating ensemble averages for small proteins from extended conformations by Monte Carlo simulations. Phys. Rev. Lett. 85:206–209. [DOI] [PubMed] [Google Scholar]
- Dima, R. I., and D. Thirumalai. 2002. Exploring protein aggregation and self-propagation using lattice models: phase diagram and kinetics. Protein Sci. 11:1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrone, F. 1999. Analysis of protein aggregation kinetics. Methods Enzymol. 309:256–274. [DOI] [PubMed] [Google Scholar]
- Friedel, M., and J. E. Shea. 2004. Self-assembly of peptides into β-barrel motif. J. Chem. Phys. 120:5809–5823. [DOI] [PubMed] [Google Scholar]
- Fung, S. Y., C. Keyes, J. Duhamel, and P. Chen. 2003. Concentration effect on the aggregation of a self-assembling oligopeptide. Biophys. J. 85:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsbury, C., J. Kistler, U. Aebi, T. Arvinte, and G. J. Cooper. 1999. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J. Mol. Biol. 285:33–39. [DOI] [PubMed] [Google Scholar]
- Green, J. D., C. Goldsbury, J. Kistler, G. S. Cooper, and U. Aebi. 2004. Human amylin oligomer growth and fibril elongation define two distinct phases in amyloid formation. J. Biol. Chem. 279:12206–12212. [DOI] [PubMed] [Google Scholar]
- Gsponer, J., U. Haberthúr, and A. Caflisch. 2003. The role of side-chain interactions in the early steps of aggregation: molecular dynamics simulations of an amyloid-forming peptide from the yeast prion sup35. Proc. Natl. Acad. Sci. USA. 100:5154–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, J., and D. J. Selkoe. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 297:353–356. [DOI] [PubMed] [Google Scholar]
- Harrison, P. M., H. S. Chan, S. B. Prusiner, and F. E. Cohen. 2001. Conformational propagation with prion-like characteristics in a simple model of protein folding. Protein Sci. 10:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, K., and J. Higo. 2003. Free-energy landscape of a chameleon sequence in explicit water and its inherent α/β bifacial property. Protein Sci. 12:2542–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaikaran, E., and A. Clark. 2001. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta. 1537:179–203. [DOI] [PubMed] [Google Scholar]
- Jang, H., C. H. Hall, and Y. Zhou. 2003. Assembly and kinetic folding pathways of a tetrameric β-sheet complex: molecular dynamics simulations on simplified off-lattice protein models. Biophys. J. 86:31–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed, R., E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman, and C. G. Glabe. 2003. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 300:486–489. [DOI] [PubMed] [Google Scholar]
- Khurana, R., C. Ionescu-Zanetti, M. Pope, J. Li, L. Nielson, M. Ramirez-Alvarado, L. Regan, A. L. Fink, and S. A. Carter. 2003. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys. J. 85:1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze, M. D., G. Bitan, and D. B. Teplow. 2002. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 69:567–577. [DOI] [PubMed] [Google Scholar]
- Klimov, D., and D. Thirumalai. 2003. Dissecting the assembly of aβ16–22 amyloid peptide into antiparallel β sheets. Structure. 11:295–307. [DOI] [PubMed] [Google Scholar]
- Koradi, R., M. Billeter, and K. Wuthrich. 1996. Molmol: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55. [DOI] [PubMed] [Google Scholar]
- Lopez De La Paz, M., K. Goldie, J. Zurdo, E. Lacroix, C. M. Dobson, A. Hoenger, and L. Serrano. 2002. De novo designed peptide-based amyloid fibrils. Proc. Natl. Acad. Sci. USA. 99:16052–16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, B., and R. Nussinov. 2002. Stabilities and conformations of Alzheimer's β-amyloid peptide oligomers aβ16–22, aβ16–35, aβ10–35. Proc. Natl. Acad. Sci. USA. 99:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek, R., and N. Mousseau. 2000. Dynamics of Lennard-Jones clusters: A characterization of the activation-relaxation technique. Phys. Rev. E. Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 62:7723–7728. [DOI] [PubMed] [Google Scholar]
- Masel, J., V. A. A. Jansen, and M. A. Nowark. 1999. Quantifying the kinetic parameters of prion replication. Biophys. Chem. 77:139–152. [DOI] [PubMed] [Google Scholar]
- Melquiond, A., G. Boucher, N. Mousseau, and P. Derreumaux. 2004. (in preparation).
- Naito, A., M. Kamihira, R. Inoue, and H. Saito. 2004. Structural diversity of amyloid fibril formed in human calcitonin as revealed by site-directed 13c solid-state NMR spectroscopy. Magn. Reson. Chem. 42:247–257. [DOI] [PubMed] [Google Scholar]
- O'Nuallain, B., A. D. Williams, P. Westermark, and R. Wetzel. 2004. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 279:17490–17499. [DOI] [PubMed] [Google Scholar]
- Petkova, A. T., G. Buntkowsky, F. Dyda, R. D. Leapman, W. M. Yau, and R. Tycko. 2004. Solid state nmr reveals a ph-dependent antiparallel β-sheet registry in fibrils formed by a β-amyloid peptide. J. Mol. Biol. 335:247–260. [DOI] [PubMed] [Google Scholar]
- Reches, M., Y. Porat, and E. Gazit. 2002. Amyloid fibril formation by pentapeptide and tetrapeptide fragment of human calcitonin. J. Biol. Chem. 277:35475–35480. [DOI] [PubMed] [Google Scholar]
- Santini, S., G. Wei, N. Mousseau, and P. Derreumaux. 2003. Exploring the folding pathways of proteins through energy landscape sampling: application to Alzheimer's β-amyloid peptide. Internet Electron. J. Mol. Des. 2:564–577. [Google Scholar]
- Santini, S., G. Wei, N. Mousseau, and P. Derreumaux. 2004. Pathway complexity of Alzheimer's β-amyloid aβ16–22 peptide assembly. Structure. 12:1245–1255. [DOI] [PubMed] [Google Scholar]
- Serpell, L., and J. Smith. 2000. Direct visualization of the β-sheet structure of synthetic Alzheimer's amyloid. J. Mol. Biol. 299:225–231. [DOI] [PubMed] [Google Scholar]
- Srinivasan, R., and G. D. Rose. 1995. Linus: a hierarchic procedure to predict the fold of a protein. Proteins. 22:81–99. [DOI] [PubMed] [Google Scholar]
- Sumner Makin, O., and L. C. Serpell. 2004. Structural characterization of islet amyloid polypeptide fibrils. J. Mol. Biol. 335:1279–1288. [DOI] [PubMed] [Google Scholar]
- Stefani, M., and C. M. Dobson. 2003. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81:678–699. [DOI] [PubMed] [Google Scholar]
- Sunde, M., L. Serpell, M. Bartlam, P. Fraser, M. Pepys, and C. Blake. 1997. Common core structure of amyloid fibrils by synchrotron x-ray diffraction. J. Mol. Biol. 273:729–739. [DOI] [PubMed] [Google Scholar]
- Taylor, J. P., J. Hardy, and K. H. Fischbeck. 2002. Toxic proteins in neurodegenerative disease. Science. 296:1991–1995. [DOI] [PubMed] [Google Scholar]
- Tjernberg, L., W. Hosia, N. Bark, J. Thyberg, and J. Johansson. 2002. Charge attraction and β propensity are necessary for amyloid fibril formation from tetrapeptides. J. Biol. Chem. 277:43243–43246. [DOI] [PubMed] [Google Scholar]
- Wei, G., P. Derreumaux, and N. Mousseau. 2003. Sampling the complex energy landscape of a simple β-hairpin. J. Chem. Phys. 119:6403–6406. [Google Scholar]
- Wei, G., N. Mousseau, and P. Derreumaux. 2002. Exploring the energy landscape of proteins: a characterization of the activation-relaxation technique. J. Chem. Phys. 117:11379–11387. [Google Scholar]
- Wei, G., N. Mousseau, and P. Derreumaux. 2004. Complex folding pathways in a simple β-hairpin. Proteins. 56:464–474. [DOI] [PubMed] [Google Scholar]
- Zanuy, D., B. Ma, and R. Nussinov. 2003. Short peptide amyloid organization: stabilities and conformations of the islet amyloid peptide nfgail. Biophys. J. 84:1884–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, M., S. Han, F. Zhou, S. A. Carter, and A. L. Fink. 2004. Annular oligomeric amyloid intermediates observed by in-situ AFM. J. Biol. Chem. 279:24452–24459. [DOI] [PubMed] [Google Scholar]