

Abstract
The interactions between membrane proteins and their lipid bilayer environment play important roles in the stability and function of such proteins. Extended (15–20 ns) molecular dynamics simulations have been used to explore the interactions of two membrane proteins with phosphatidylcholine bilayers. One protein (KcsA) is an α-helix bundle and embedded in a palmitoyl oleoyl phosphatidylcholine bilayer; the other (OmpA) is a β-barrel outer-membrane protein and is in a dimyristoyl phosphatidylcholine bilayer. The simulations enable analysis in detail of a number of aspects of lipid-protein interactions. In particular, the interactions of aromatic amphipathic side chains (i.e., Trp, Tyr) with lipid headgroups, and “snorkeling” interactions of basic side chains (i.e., Lys, Arg) with phosphate groups are explored. Analysis of the number of contacts and of H-bonds reveal fluctuations on an ∼1- to 5-ns timescale. There are two clear bands of interacting residues on the surface of KcsA, whereas there are three such bands on OmpA. A large number of Arg-phosphate interactions are seen for KcsA; for OmpA, the number of basic-phosphate interactions is smaller and shows more marked fluctuations with respect to time. Both classes of interaction occur in clearly defined interfacial regions of width ∼1 nm. Analysis of lateral diffusion of lipid molecules reveals that “boundary” lipid molecules diffuse at about half the rate of bulk lipid. Overall, these simulations present a dynamic picture of lipid-protein interactions: there are a number of more specific interactions but even these fluctuate on an ∼1- to 5-ns timescale.
INTRODUCTION
Membrane proteins play key roles in a wide range of processes in cells, including transport and signaling. It has been estimated (Wallin and von Heijne, 1998; Jones, 1998) that ∼25% of genes code for membrane proteins, reflecting their biological significance. From a biomedical perspective one may note that ∼50% of drug targets correspond to membrane proteins (Terstappen and Reggiani, 2001). Despite their functional significance, ongoing difficulties in expression and crystallization mean that only a few (∼50, see http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html) membrane protein structures have been determined at high resolution. It is therefore of crucial importance that we extract the maximum information possible from those structures that have been determined. Membrane proteins fall into two broad families. The α-helix bundle family is by far the larger of the two and includes nearly all of the membrane proteins of higher organisms, and the membrane proteins of the inner membrane of (Gram negative) prokaryotes. The outer membranes of Gram negative bacteria host a second class of membrane protein, namely the β-barrel outer membrane proteins (OMPs).
It is important to recall that membrane proteins exist in a more complex environment than the approximately isotropic environment provided by the cytoplasm for water-soluble proteins. Thus, membrane proteins generally span a lipid bilayer, and so must contain regions on their surfaces that interact with water (on either side of the membrane), with polar lipid headgroups, and with the hydrophobic core of the lipid bilayer (see Wiener and White, 1992; and White, 1994, for a discussion of the environment presented by a phospholipid bilayer). There have been a number of studies of the importance of protein-lipid interactions in the context of structure and stability of membrane proteins. For example, Killian and colleagues have used simple model peptides and biophysical methods to probe in some detail the nature of such interactions (Killian and von Heijne, 2000; Killian, 2003; Strandberg and Killian, 2003). Analysis of those crystal structures of membrane proteins that contain lipids provides a detailed structural perspective on lipid-protein interactions (Fyfe et al., 2001; Lee, 2003). However, it should be remembered that structural biology provides a biased sample of lipid-protein interactions, focusing in on those interactions that are sufficiently strong to be retained after cooling of the protein crystal to a temperature of ∼100 K (Halle, 2004). These studies have revealed, e.g., the importance of amphipathic aromatic residues (Trp and Tyr) at membrane/water interfaces (Yau et al., 1998a,b). A number of experimental studies have also revealed the importance of bound lipid molecules for the stability and function of some membrane proteins (O'Keeffe et al., 2000; Lange et al., 2001; Fernandez et al., 2002; daCosta et al., 2002; de Planque and Killian, 2003; Costa-Filho et al., 2003; Bulieris et al., 2003). For example, in the case of the K channel KcsA, acidic phospholipids appear to bind to specific (nonannular) sites at which they play a role in refolding and possibly in function (Valiyaveetil et al., 2002; Demmers et al., 2003; Alvis et al., 2003). Lipid bound to this nonannular site can be observed within the crystal. Similarly, some crystal forms of the outer membrane transport protein FhuA have revealed the presence of a tightly bound lipid A molecule (Ferguson et al., 1998, 2000), indicating a specific interaction between an outer membrane protein and an outer membrane lipid. Interestingly, bound lipid A has been suggested to be required for activation of the outer membrane protease OmpT (Kramer et al., 2002).
Molecular dynamics (MD) simulations (Karplus and McCammon, 2002) provide an opportunity to study the conformational dynamics and interactions of membrane proteins under approximately physiological conditions. Building on earlier simulation studies of pure lipid bilayers (reviewed in, e.g., Tieleman et al. 1997; Tobias et al., 1997), MD simulations have been extended to increasingly complex membrane proteins (Roux and Woolf, 1996; Woolf and Roux, 1996; Belohorcova et al., 1997; Tieleman and Berendsen, 1998; also see reviews by, e.g., Forrest and Sansom, 2000; Domene et al., 2003a). These simulations are able to provide insights into the nature of the interactions between membrane proteins and their lipid environment (Woolf and Roux, 1996; Woolf, 1997, 1998; Tieleman et al., 1999; Petrache et al., 2000, 2002). Furthermore, MD simulations can enable us to compare the behavior of a membrane protein in a lipid bilayer and a detergent micelle environment (Bond and Sansom, 2003). Current MD simulations of membrane proteins are able to address timescales of >10 ns. This provides improved sampling of details of protein-lipid interactions (Saiz and Klein, 2002; Tang and Xu, 2002; Feller et al., 2003; Crozier et al., 2003; Allen et al., 2003; Huber et al., 2004; Saiz et al., 2004) on a timescale comparable to that observed in NMR studies (Fernandez et al., 2002; Tamm et al., 2003). Recent simulations of lipid bilayers (Mukhopadhyay et al., 2004) and of peptide/bilayer systems (Jensen et al., 2004) suggest that even longer timescales are becoming addressable via simulation.
Recent preliminary studies (Domene et al., 2003b) suggest that ∼10-ns duration simulations can reveal details of the interactions of membrane lipids with inner and outer membrane proteins Here, we present the results of a detailed comparative analysis of the lipid-protein interactions of an α-helical membrane protein (KcsA) versus an OMP (OmpA). The results suggest that such simulations can indeed provide molecular details of lipid-protein interactions and dynamics. We attempt to relate these simulation results to experimental studies of how membrane proteins interact with their lipid bilayer environment.
METHODS
Two simulations are analyzed and compared in this study (Table 1): 1), an ∼15-ns simulation of the potassium channel KcsA (pdb code 1K4C; Zhou et al., 2001) in a palmitoyl oleoyl phosphatidylcholine (POPC) bilayer; and 2), an ∼20-ns simulation of OmpA (pdb code 1BXW; Pautsch and Schulz, 1998) in a dimyristoyl phosphatidylcholine (DMPC) bilayer. In each case the protein was inserted in a preformed cavity in an equilibrated lipid bilayer, using the methods described in detail in Faraldo-Gómez et al. (2002). The proteins were oriented interactively, such that the long axis of the protein was parallel to the bilayer normal and the bands of aromatic/amphipathic (i.e., Trp and Tyr) side chains were located in the lipid headgroup regions (Fig. 1). Note that these are continuations of simulations described, from a functional perspective, in other articles (Domene and Sansom, 2003; Bond and Sansom, 2003). Here the focus is upon what these simulations reveal concerning lipid-protein interactions.
TABLE 1.
Summary of simulations
| Simulation | Duration (ns) | Lipids | Waters and ions | Atoms | Cα RMSD* (nm) | |
|---|---|---|---|---|---|---|
| OmpA | ∼20 | 111 DMPC | 5055 waters and 9 Na+ + 9 Cl− | 22013 | All residues | 0.20 |
| Core TM† | 0.10 | |||||
| KcsA | ∼15 | 243 POPC | 7938 waters and 3 K+ + 15 Cl− | 40376 | All residues | 0.23 |
| Core TM‡ | 0.17 | |||||
Cα RMSDs are versus the starting structure for the simulation, averaged over the final 5 ns of each simulation.
For OmpA, the core TM residues were defined as those in the β-barrel.
For KcsA, the core TM residues were defined as those in the two TM helices plus the selectivity filter.
FIGURE 1.
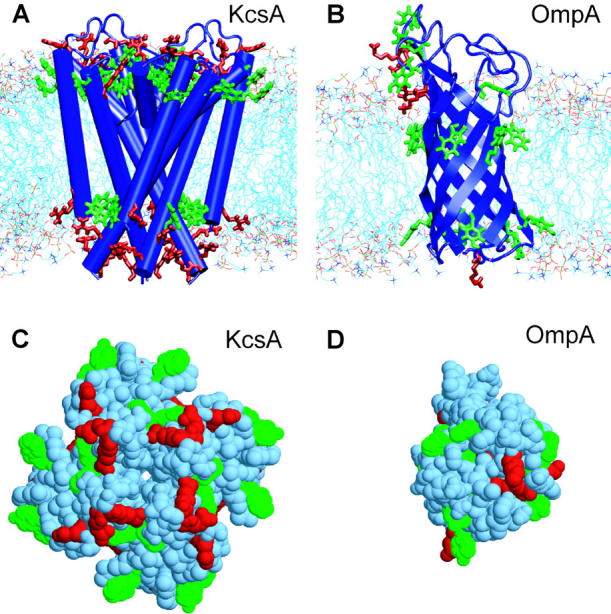
In A and B, the simulation systems are shown, namely (A) KcsA/POPC, which consisted of the KcsA transmembrane tetramer embedded in a bilayer of 243 POPC molecules, solvated with 7938 waters; and (B) OmpA/DMPC, which consisted of the OmpA N-terminal domain embedded in a bilayer of 111 DMPC molecules, solvated with 5055 waters. In C and D, surface representations of (C) KcsA and (D) OmpA are shown, both seen from “below” (i.e., from the cytoplasmic surface for KcsA, from the periplasmic surface for OmpA). The color code for amino acids is green for Trp and Tyr, and red for Arg and Lys.
Simulation details: OmpA
Simulations were performed as described in Bond and Sansom (2003). Briefly, the 2.5-Å OmpA structure (1BXW) was used as a starting model, and pKA calculations, performed using the University of Houston Brownian Dynamics program (Davis et al., 1991), were used to aid assignment of side-chain ionization states. The resulting model was neutral overall. Thirty-nine crystal waters were localized in the asymmetric unit. Of the 39 waters in the crystallographic asymmetric unit, 27 were retained in the starting model because they were approximately within the bounds of the protein surface. OmpA and the 27 crystal waters were embedded in a preequilibrated DMPC bilayer. Further details of the protein set-up can be found in Bond et al. (2002).
The simulation was conducted using the GROMACS v2.0 (Berendsen et al., 1995) MD simulation package (www.gromacs.org). An extended united atom version of the GROMOS96 force field was used (Hermans et al., 1984). The protein-lipid system was energy-minimized before MD, using ∼100 steps of the steepest descent method, to relax any steric conflicts generated during setup. The system was solvated with SPC (simple point charge) waters (Berendsen et al., 1981) and system-neutralizing sodium and chloride ions (corresponding to ∼1 M NaCl) were added. During restrained runs, the protein was harmonically restrained with a force constant of 1000 kJ mol−1 nm−2. Electrostatic interactions were calculated using particle mesh Ewald (PME; Darden et al., 1993) with a 0.9-nm cutoff for the real-space calculation. A cutoff of 1.0 nm was used for van der Waals interactions. The simulation was performed at constant temperature, pressure, and number of particles. The temperatures of the protein, DMPC, and solvent (including both crystal and bulk water molecules, along with ions) were each coupled separately, using the Berendsen thermostat (Berendsen et al., 1984), at 310 K for the DMPC simulation, with coupling constant τT = 0.1 ps. The pressure was coupled using the Berendsen algorithm at 1 bar with coupling constant τP = 1 ps. The compressibility was set to 4.5 × 105 bar−1 in all box dimensions. The time step for integration was 2 fs, and coordinates and velocities were saved every 5 ps. The LINCS algorithm was used to restrain bond lengths (Hess et al., 1997). Simulations were performed on a Linux workstation, an eight-node Beowulf cluster, or an SGI Origin 2000 (Mountain View, CA) using either four or eight parallel 195-MHz R10000 processors.
Simulation details: KcsA
The KcsA simulation was performed as described previously (Domene and Sansom, 2003). Briefly, the simulation system consisted of the 2-Å resolution high [K+] structure (1K4C) of KcsA, embedded in a POPC bilayer consisting of 116 lipid molecules of POPC in the periplasmic leaflet, and 127 in the intracellular leaflet. An acetyl group was attached to the N-terminus of KcsA (residue 22) and the C-terminal carboxylate was protonated. The side chain of Glu71 was protonated, so as to form a diacid hydrogen bond with the carboxylate group of Asp80, in agreement with the earlier simulation studies (Ranatunga et al., 2001; Bernèche and Roux, 2002) and with structural data (Zhou et al., 2001). The rest of the ionizable residues were in their default ionization state.
The simulation used GROMACS v3 (Lindahl et al., 2001). An initial energy minimization was followed by a 0.2-ns equilibration period during which the protein and the cation positions were restrained. After this, unrestrained molecular dynamics simulation was performed of duration 15 ns in the NPT ensemble. Long-range electrostatic interactions were calculated using PME. The time step was 2 fs with the LINCS algorithm to constrain bond lengths. A constant pressure of 1 bar independently in all three directions was used with a coupling constant of τP = 1.0 ps. Water, protein, and lipid were coupled separately to a temperature bath at 300 K using a coupling constant τT = 1.0 ps. Coordinate sets were saved every 0.1 ps for analysis. Lipid parameters were based on those used previously (Berger et al., 1997; Marrink et al., 1998). Structural diagrams were prepared using VMD (Humphrey et al., 1996) and RasMol (Sayle and Milner-White, 1995).
RESULTS
KcsA versus OmpA
The two proteins selected for this analysis represent the two major classes of membrane protein: KcsA is an α-helix bundle, a fold found in the great majority of membrane proteins; OmpA is a β-barrel, and is a simple representative of the outer membrane proteins of Gram negative bacteria. In both cases the simulations employed only the transmembrane (TM) domains of the proteins (Fig. 1), corresponding to the available crystal structures. Thus, the KcsA TM domain is missing an N-terminal helix (residues 1–22) and a 37-residue C-terminal domain, whereas OmpA is missing an ∼150-residue C-terminal domain (the structure of which is homologous to that of RmpM; Grizot and Buchanan, 2004).
For both KcsA and OmpA, the TM structure represents the majority of those residues interacting with the membrane. It is useful to compare the cross-sectional areas of OmpA and of KcsA for the interfacial regions of the inner and outer leaflets. For OmpA the cross-sectional area at both aromatic belts is ∼4.5 nm2. For KcsA the cross-sectional at the upper (i.e., periplasmic) aromatic belt is ∼15.2 nm2, whereas at the lower (i.e., cytoplasmic) belt it is ∼4.8 nm2. Thus, the OmpA structure may be approximated as a regular cylinder, whereas the KcsA structure is more akin to a truncated cone.
In terms of potential interaction sites with lipid headgroups (see below) we can consider the surface distributions of the side chains of two classes of amino acid, namely amphipathic aromatic (Trp and Tyr), and basic residues (Arg and Lys). Considering first the aromatics, in OmpA the region of the protein surface corresponding to the extracellular interface has five (Trp + Tyr) residues, whereas the inner (periplasmic) interface has six (Trp + Tyr) residues. There are also four (Trp + Tyr) residues on the surface of the region defined by the extracellular loops. Turning to the KcsA tetramer, the outer (periplasmic) interface contains 12 (Trp + Tyr) residues, whereas the inner (cytoplasmic) interface has eight (Trp + Tyr) residues. Thus the larger protein, KcsA, presents twice as many amphipathic aromatics on its surface. If one considers the basic side chains, for OmpA there are three (Arg + Lys) at the outer interface, with an additional two Lys in the extracellular loops, whereas the inner interface has just one Lys residue. For KcsA, the outer interface contains 12 Arg residues, and the inner interface 16 Arg residues. Thus KcsA has nearly five times as many surface-exposed basic side chains as does OmpA.
In both cases it was necessary to simplify the in vivo membrane environment by embedding the protein in a simple phosphatidylcholine membrane. DMPC was used for OmpA as it has a thinner transmembrane zone (∼2.1 nm in OmpA versus ∼2.7 nm in KcsA, as indicated by the spacing between the aromatic belts). For DMPC the average distance between the two lipid/water interfacial regions (measured as the glycerol to glycerol distance) is ∼2.7 nm whereas for POPC it is ∼3.3 nm.
The conformational stability of the proteins during these extended simulations may be assessed, albeit crudely, by measurement of the conformational drift from the initial crystal structures as given by the Cα atom root mean-square deviation (RMSD; see Table 1). In both cases there is an initial rise in RMSD over the first 5 ns. For the last 5 ns of each simulation the RMSD for all Cα atoms (i.e., both the transmembrane core and the extramembraneous loops) is ∼0.2 nm. This provides evidence of relatively small overall conformational drift, i.e., a “stable” simulation, for both proteins, giving us confidence in the use of these simulations to further analyze the interactions of the proteins with their bilayer environment. Indeed, for the Cα atoms of the core TM residues, the RMSDs are <0.2 nm (0.17 nm for KcsA and 0.10 for OmpA). Of course, even on a >10-ns timescale we realize that sampling of protein motions is incomplete. The degree of convergence of the simulations can be estimated from block analysis of the Cα mean-square fluctuations (as described in more detail in, e.g., Bond and Sansom, 2003; and Faraldo-Gómez et al., 2004). For OmpA this analysis suggests better sampling of the motions of the core TM domain residues than for the extracellular loops. In particular, the extracellular loops are 2.5–5 times more mobile (on a 0.5- to 20-ns timescale, respectively) than the TM β-barrel. For KcsA the difference in loop and core TM mobility is less pronounced.
Contacts and interactions
Having established the stability of the proteins within the simulations, we may examine their interactions with the surrounding phospholipid molecules. A simple measure of such interactions can be obtained by estimating the number of interactions as a function of time (Fig. 2). We have defined an interaction as occurring when a lipid-protein interatomic distance is ≤0.35 nm. We have divided interactions into those of the protein with the hydrophobic alkyl tails and those with the polar lipid headgroups (which include the glycerol and the acyl oxygens) as we anticipate that these may behave differently. It is also informative to look at both the number of lipid molecules making these two classes of contact (Fig. 2, A and B) and the number of interatomic contacts falling into the two classes (Fig. 2, C and D).
FIGURE 2.

Overall numbers of lipid-protein interactions (cutoff 0.35 nm, sampled every 0.1 ns). (A and B) Numbers of lipid molecules making contact with protein. (C and D) Numbers of lipid atoms making contact with protein. Black lines are for lipid headgroups; shaded lines are for lipid tails.
If we look first at the number of lipid molecules forming interactions with the two proteins, the first thing we notice is that, after the first ∼2 ns, the number of interacting lipid molecules is about constant for both simulations, implying successful equilibration of the lipid-protein interactions in the simulation. The number of interacting molecules for KcsA is ∼30 (for headgroup interactions) and ∼40 for acyl tail interactions (Fig. 2 A). For OmpA the corresponding figures are somewhat lower (∼20 and ∼25, respectively) reflecting the smaller cross-sectional size of OmpA relative to KcsA (see Fig. 1, C and D).
If we focus on the number of interacting atoms (Fig. 2, C and D) then some complexities emerge. For KcsA, the total number of atomic interactions gradually increases over the course of the simulation. More detailed analysis reveals that most of this increase is due to a steady increase in protein-headgroup interatomic interactions over the first ∼5 ns of the simulation, from a total of ∼350–600 within ∼5 ns. For OmpA the number of interatomic contacts increases significantly over the first ∼12 ns of the simulations. In contrast to KcsA, for OmpA this increase is due to both headgroup and tail atoms. Furthermore, whereas for KcsA the increase is approximately monotonic, for OmpA there are significant fluctuations in the number of interatomic contacts as a function of time, on a timescale of ∼5 ns. Interestingly, these fluctuations originate both from residues of the TM β-barrel and of the extracellular loops. This may represent breathing-like fluctuations of the lipid annulus of this simple membrane protein. Correlated lipid motions within the bilayer on such timescales have been described (Lindahl and Edholm, 2000); our results may represent related fluctuations of protein-lipid interactions.
Hydrogen bonds
A more detailed picture of the interactions of lipid headgroups with the proteins can be obtained by analysis of H-bonds. A priori one would expect substantial H-bonding between the lipid headgroups (which contain several possible H-bond acceptors but no potential donors) and protein side-chain and backbone donors. Analysis of the total numbers of H-bonds versus time (with the cutoffs used to define H-bonds being 0.25 nm for the hydrogen-acceptor distance, and 60° for the donor-hydrogen-acceptor angle) for both simulations shows a rise during the simulations over the first ∼10 ns to an approximate plateau (Fig. 3) of ∼45 H-bonds for KcsA and ∼15 for OmpA. If we break the H-bonds down into those formed by different regions of the headgroup (data not shown) we find for KcsA that H-bonds formed by the acyl carbonyl groups (∼15 in total toward the end of the simulation) and those formed by the phosphate oxygens (∼20 in total) are the largest classes. In contrast, for OmpA, on average there are ∼8 H-bonds formed by the acyl group carbonyls, compared with only ∼3.5 H-bonds with phosphate oxygens, and ∼2 H-bonds with the glycerol backbone. The smaller number of apparent “anchoring” interactions for OmpA may reflect the absence, in the current simulations, of interactions with the more complex lipid A headgroup present in the in vivo outer membrane.
FIGURE 3.

Number of H-bonds between protein and lipid. The cutoffs used to define H-bonds are 0.25 nm for the hydrogen-acceptor distance, and 60° for the donor-hydrogen-acceptor angle.
Analysis of the lifetimes of each individual H-bond made during each simulation (data not shown) reveals that there are two main kinds of interaction. At any time, most H-bonds made between protein and lipid are very stable ones that have lifetimes on the order of ∼2–5 ns or longer. Additionally, more transient H-bonds (with lifetimes of ∼0.1 ns or less) occur between different donors and acceptors, presumably due to the thermal “breathing motions” of lipids and polar side chains. Exponential fits to average autocorrelation functions derived from H-bond existence functions yielded relaxation times of ∼10 ns in both simulations, reflecting the longevity of most lipid-protein H-bond partners. Of course, it should be remembered that two H-bond populations, short- and long-timescale, are contributing to the estimated relaxation behavior. Nevertheless, the long relaxation times found indicate that protein and lipid in its immediate vicinity are tightly bound, consistent with the stability of the annular shell revealed by our analysis of lipid lateral diffusion (see below).
Interactions versus position and time
The location of the headgroup interactions with the protein, and the changes in these interactions with respect to time, can be seen by generating a contour plot of the number of interactions as a function of position along the bilayer normal and of time. The contour plot for KcsA (Fig. 4 A) shows two broad (∼1 nm—cf. Wiener and White, 1992; and White, 1994) bands corresponding to the two interfaces. For both interfaces there is an increase in the density of interactions over the first ∼2 ns, consistent with the observations above. For OmpA (data not shown) the interactions with lipid headgroups also show two bands. That for the extracellular surface is broader, reflecting interactions with the long extracellular loops of the protein (see below).
FIGURE 4.

Protein side-chain interactions with lipid headgroups. In each case the number of interactions (≤0.35 nm) are shown as a function of position along the bilayer normal (z) and time. Sampling is every 0.1 nm and 0.1 ns. (A) Interactions of side chains of KcsA with lipid headgroups; (B) interactions of aromatic belt side chains of KcsA with lipid headgroups; (C) interactions of side chains of OmpA with lipid headgroups; and (D) interactions of aromatic belt side chains of OmpA with lipid headgroups.
Comparable contour plots of the interactions with the lipid tails reveal a broader range of lower-number interactions than with the headgroups. Toward the center of the bilayer, the number of interactions drops, reflecting the fluidity gradient of the lipid tails. Comparing KcsA and OmpA, the latter shows a greater number of lipid tail contacts along the entire length of the protein, in contrast with a smaller number of localized interactions with KcsA. This reflects the differences in shape between the two proteins—an approximately regular cylinder (OmpA) versus a truncated cone (KcsA). The more symmetric shape of the β-barrel domain may enable establishment of a more tightly bound lipid annulus without requiring changes in global lipid conformation or bilayer packing.
Aromatic side chains
Amphipathic aromatic amino acids (i.e., Trp and Tyr) have been shown to form bands at either end of the transmembrane domains of membrane proteins, corresponding to the location of the lipid/water interface (Schiffer et al., 1992; Ulmschneider and Sansom, 2001). It has been suggested that Trp and Tyr residues “lock” the protein into its correct orientation within the membrane by forming interactions with the lipid headgroups and water molecules in the interfacial region (Yau et al., 1998b; de Planque et al., 2003). The current simulations provide an opportunity to extend previous analysis of such aromatic-interface interactions (Tieleman et al., 1999; Grossfield and Woolf, 2002).
We first analyzed the orientations of the aromatic side chains at the interfaces. In particular, we have calculated the time-dependent angles with respect to bilayer normal of the aromatic belt residue rings and their normals. For both OmpA and KcsA, the aromatic ring planes are aligned roughly parallel to the bilayer normal, oscillating on a picosecond timescale around their mean values. Occasional (once every few nanoseconds) aromatic ring flips of 180° occur, the actual transition from one orientation to another being relatively rapid, i.e., over in a few tens of picoseconds. Similar behavior has been observed in, e.g., simulations of single TM helices in a membrane (Ulmschneider et al., 2004). In all cases, the aromatic rings are oriented so that the polar moieties of the Trp (i.e., ring nitrogen) and Tyr (i.e., hydroxyl group) side chains are nearest to the interfacial region (see, e.g., Fig. 9 A). This is consistent with the proposed roles of these aromatic belt residues as membrane protein “anchors,” with their hydrophobic regions interacting with the lipid acyl chain region of the bilayer and their polar regions interacting with lipid headgroup and solvent. Interestingly, there are Tyr residues that lie in the extracellular loop regions of OmpA, above the upper aromatic belt and lying over the membrane surface. Consistent with the aqueous location, their behavior is far less predictable than for the Tyr residues in the aromatic belts, with the angles of both the ring plane and orientation with respect to the bilayer normal varying between 0° and 180° over the simulation. The variation is characterized by approximately constant angles being maintained on the nanosecond timescale, followed by sudden flips in orientation to a new angle and/or direction, which again lasts for another few nanoseconds. This is consistent with transient interactions between these residues and polar headgroups on the bilayer surface, but indicates no one set of interactions is more favorable than the other.
FIGURE 9.
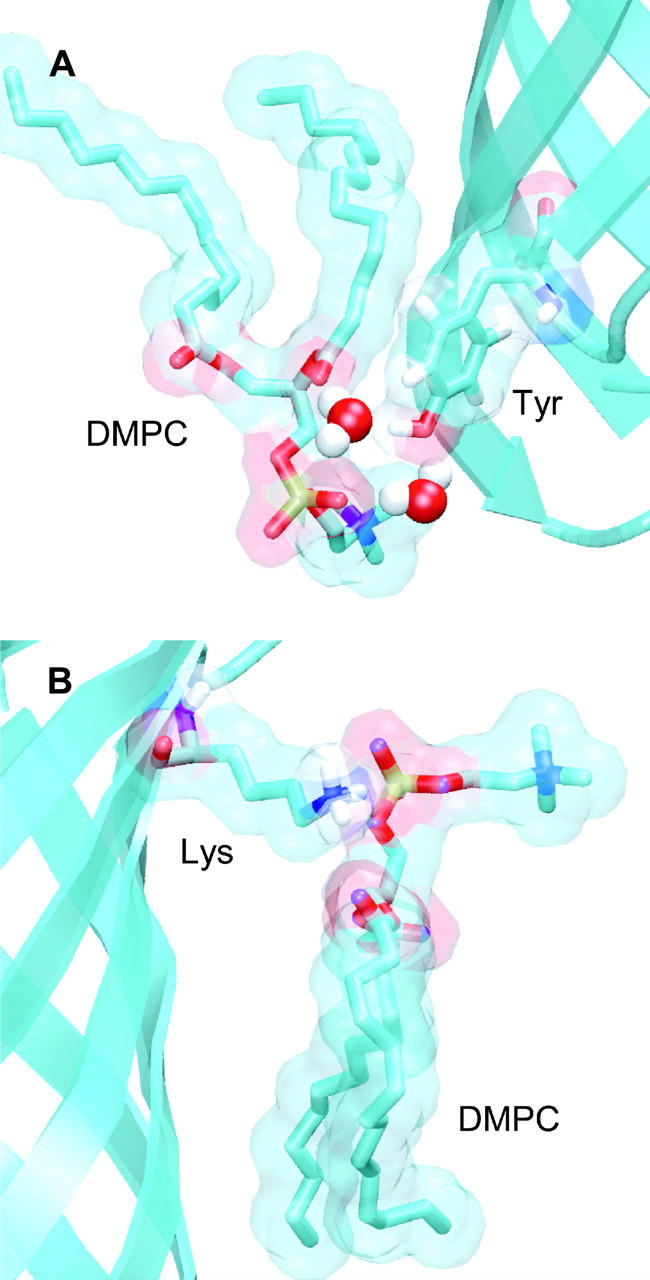
Illustrative snapshots of protein-lipid interactions, taken from the OmpA simulations. In both diagrams the protein backbone is shown in “ribbons” format (in cyan). Selected side chains and lipids are shown in “bonds” format, with nitrogen and oxygen atoms colored blue and red respectively. The lipid and side-chain molecular surfaces (probe radius of 0.1 nm) are also shown. (A) Tyrosine (Y48) residue in the lower aromatic belt of OmpA interacting with a lipid molecule. The tyrosine hydroxyl group is H-bonded with the DMPC fatty acyl carbonyl and glycerol groups via a bridging water molecule, whereas the aromatic ring forms van der Waal's contacts with the acyl chain. (B) The amine group of a lysine side chain (K73) at the extracellular membrane surface forms an electrostatic interaction with a DMPC phosphate group.
We have used contour plot analysis of the frequency of headgroup interactions of aromatics versus (z,t) similar to that described above to explore the spatial and temporal variation in such interactions. For KcsA, as anticipated, the interactions between the two aromatic belts and lipid headgroups result in two clear zones of interaction (data not shown), each of ∼1-nm width. The outer (periplasmic) belt is involved in more interactions than the inner (cytoplasmic) one, reflecting the greater number of solvent-exposed Trp and Tyr residues in the outer belt (see above).
For OmpA (Fig. 4 B) the patterns of interaction between aromatic side chains and lipid headgroups are more complex. In particular, there is an additional aromatic belt formed by the extracellular loops of OmpA. Thus there are three bands of interaction between the aromatic side chains and the headgroups. The lower band, centered at ∼−1.5 nm, is similar to the equivalent band in the KcsA simulation, although the higher solvent accessibility of the OmpA aromatic belt means that more long-lasting, tighter interactions can be established during the course of the simulation, occasionally forming ∼50 contacts during the latter half of the simulation. At the outer surface, two zones of contact (i.e., the outer-barrel aromatic belt and the extracellular loop belt) are seen. Although the number of interactions in these zones is less than for the inner belt, it is clear that the location of the aromatic belts in OmpA result in a broad range of contacts with headgroups. The broad upper zone of interactions due to the aromatic side chains of the extracellular loops may be biologically significant, in that it may help to relieve a bilayer/protein mismatch, thereby enabling the narrow hydrophobic transmembrane region of OmpA to stably exist in a lipid bilayer. Additionally, extensive fluctuations in contacts between the extracellular aromatic belt and polar headgroups are apparent over the course of the simulation, leading to a reduction in the width of this zone but a concomitant increase in interaction number. This flexibility is afforded by the highly mobile extracellular loops, resulting in a narrower, more uniform aromatic belt, matching the bilayer interface. This may be important for adjusting to the heterogeneous lipopolysaccharide (LPS) environment.
Basic side chains
The other side chains that are thought to play a key role in interactions with lipid headgroups are the basic side chains, especially Lys, which are proposed to “snorkel” to the membrane surface where they can interact with the phosphate groups of phospholipid headgroups (Mishra et al., 1994; Strandberg and Killian, 2003). Such interactions have been observed in, e.g., simulations of models of membrane proteins made up of parallel α-helix bundles (Saiz et al., 2004).
As noted above, KcsA has nearly five times as many surface exposed basic side chains as does OmpA. Furthermore, whereas in OmpA both Lys and Arg residues are present, in KcsA only Arg residues are present. These differences are reflected in the frequency of basic side chain-headgroup interactions for the two proteins (Fig. 5).
FIGURE 5.

Total number of atomic contacts (≤0.35 nm) between lipid headgroups and snorkeling lysine and arginine side chains (sampled every 0.1 ns), for (A) KcsA (Arg only), and (B) OmpA (black line, Arg; shaded line, Lys).
For KcsA, there is a steady rise in the number of Arg-headgroup interactions over the first half of the simulation. As was noted above, H-bonds to the oxygens of the phosphates are the major class of H-bonds for this protein in a POPC bilayer. More detailed examination suggests that the increase in such interactions is largely due to those Arg residues located at the lower (intracellular) surface of the KcsA molecule. This increase seems to be due to relatively small changes in Arg side-chain conformation so as to maximize the number of H-bonds to lipid headgroups.
In contrast, OmpA forms fewer interactions of basic side chains (both Lys and Arg) with headgroups, and these interactions fluctuate on a timescale of ∼0.5 ns. These fluctuations may reflect the location of the basic side chains predominantly in the mobile extracellular loops rather than in the TM barrel domain, thus leading to more transient interactions. It is interesting that basic side chains on the surfaces of OMPs have been implicated in specific binding of LPS via its constituent phosphates, both from crystallographic analysis of lipid A bound to FhuA (Ferguson et al., 2000) and from simulation studies of the outer membrane protease OmpT (Baaden and Sansom, 2004).
The location of the Lys and Arg side chains along the bilayer normal may be compared with the location of the phosphate groups (Fig. 6). For both proteins it can be seen that the basic side chains are approximately coincident with the interfaces defined by the phosphate groups. Thus, these simulations extend the earlier studies of model TM helices (Saiz et al., 2004) to reveal the importance of basic-phosphate interactions for both of the major classes (α-helix bundle and β-barrel) of integral membrane protein.
FIGURE 6.
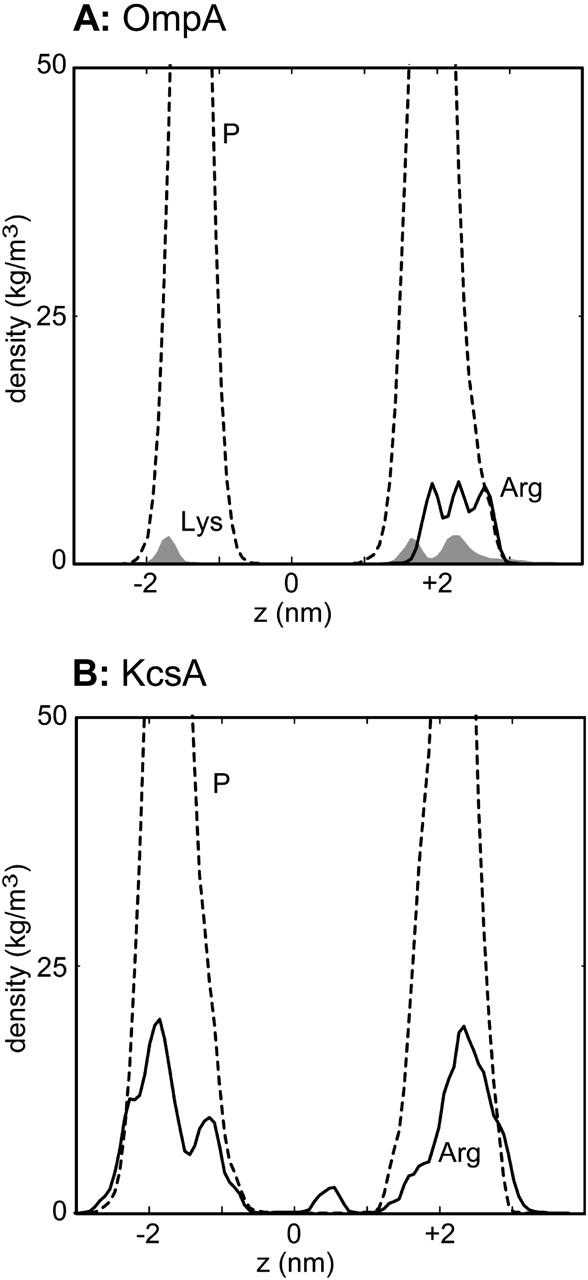
Simulation-averaged atomic densities versus bilayer normal axis for (A) OmpA and (B) KcsA. In A, densities are shown for the Lys side-chain amine group (shaded, solid), for the guanidinium group of Arg (black solid line), and for lipid headgroup phosphorus atoms (black broken line). In B, densities are shown for the Arg guanidinium (black solid line) and for lipid headgroup phosphorus atoms (black broken line).
Analyzing in more detail the interactions between Lys and Arg side chains and the lipids reveals that such interactions predominantly (90% for KcsA; from 70% to 90% over time for OmpA) involved the charged groups at the end of the side chains. If one examines the orientations of the lipid-interacting basic side chains, a complex picture emerges. For OmpA most of these side chains are located in the long, mobile extracellular loops region of the protein, above the membrane surface. Of these, one Arg and one Lys lie with their side chains approximately perpendicular to the bilayer normal during the simulation (see Fig. 9 B). The Arg side chain lies very close to the headgroup region, at the bottom of a loop, whereas the Lys side chain is in the middle of a very mobile loop, which is therefore able to move toward the membrane surface to allow tight interactions with headgroups. Additionally, a Lys and Arg residue located at the tops of the loops both make interactions with headgroups by pointing downward toward the membrane surface. Finally, one Lys side chain is located in the short periplasmic turn region, and points downward/outward so as to interact with phosphate groups of lipid headgroups. This, therefore, is the only side chain that shows classical “snorkeling” characteristics in OmpA. A similar situation is seen in simulations of OmpT (Baaden and Sansom, 2004). Thus, snorkeling may be less important for outer membrane proteins where, instead, anchoring interactions between charged and aromatic groups of the mobile loops and lipopolysaccharide may occur.
For KcsA, the orientations of lipid-interacting Arg residues may be grouped according to the location of the Arg residues. First, for those Arg residues that lie in the “turret” loops at the extracellular surface of the protein, the side chains are approximately perpendicular to the bilayer normal, pointing downward/inward so that their charged groups interact with lipid headgroups. Second, transmembrane helix M2 extends beyond the membrane on the intracellular side. This results in a range of orientations of Arg residues at the end of M2, which change on a nanosecond timescale and allow interaction with lipid headgroups in a similar manner to the Arg and Lys residues in the extracellular loops of OmpA. Third, Arg residues at the intracellular end of helix M1, on the intracellular side, are embedded in the membrane region and exhibit classical snorkeling, with their side chains pointing downward/outward so that their charged groups can interact with lipid headgroups. There is a final class of Arg residues that are more deeply buried, but still able to interact with lipid headgroups. These residues are located at the extracellular end of the protein. The orientations of these Arg side chains are quite constant during the simulation, all lying at ∼20–30° with respect to the bilayer normal. Further simulations, in the presence of anionic lipids (Deol and Sansom, unpublished data) suggest that these Arg residues may be responsible for the role of anionic lipids in stabilizing the structure of KcsA (Valiyaveetil et al., 2002; Demmers et al., 2003; Alvis et al., 2003).
Lateral motions of lipids
Having demonstrated interactions between both integral membrane proteins and the surrounding lipids, it is of interest to determine to what extent the mobility of the lipids is perturbed by the inserted proteins. For example, Saiz et al. have shown that the presence of an inserted TM helix bundle increases the orientational order of the adjacent lipids (Saiz et al., 2004). The extended nature of our simulations enables us to address a somewhat different question, namely whether the presence of an integral membrane protein reduces the lateral mobility of those lipids “bound” to the surface of the protein.
At a qualitative level, we can distinguish between the lateral mobility of two extreme classes of bound and free lipid molecules, where bound lipids are defined as those which have atoms within 0.35 nm of the protein in every 5-ns snapshot from a simulation, and free lipids are defined as those which are never within 0.35 nm of protein in any 5-ns trajectory snapshot. We can then plot sample trajectories for the two classes of lipid in the bilayer plane relative to the membrane protein (Fig. 7). For OmpA a clear difference in lateral mobility can be seen between the examples of the bound and free classes of lipid. For KcsA, the distinction is similar, if a little less clear cut. Thus, the bound lipids seem to have a restricted mobility.
FIGURE 7.
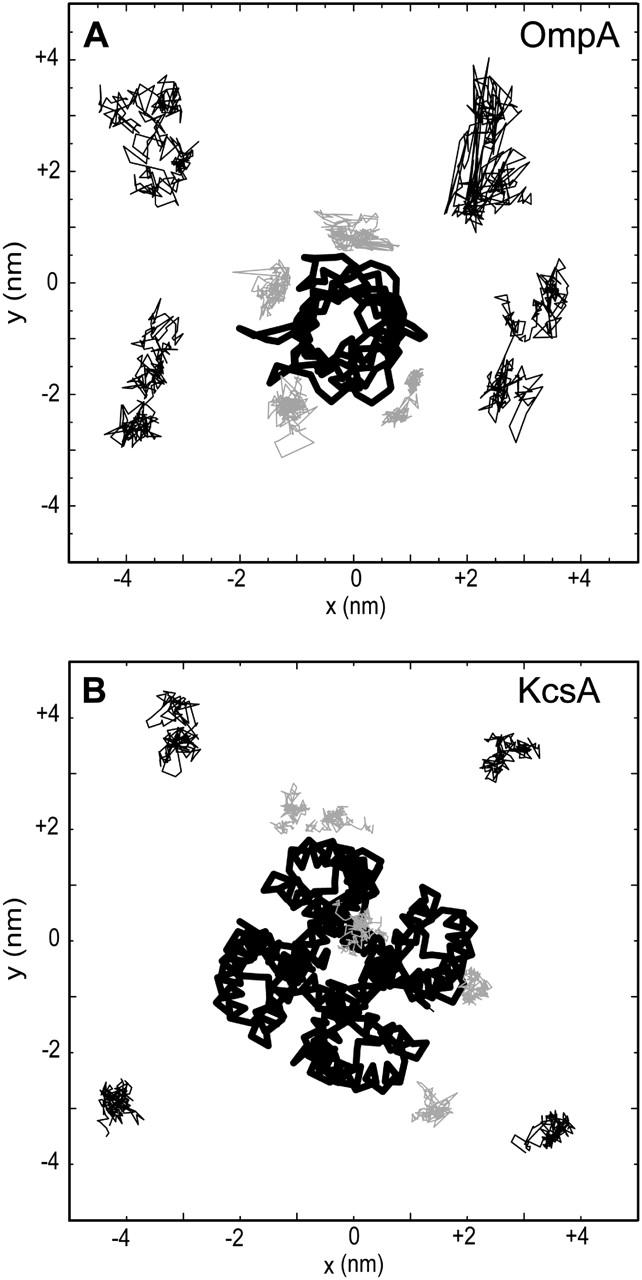
Trajectories in the bilayer (xy) plane for (A) OmpA and (B) KcsA of four selected bound (shaded lines) and four free (thin black lines) lipid headgroups. The lines join positions of the lipid headgroups (saved every 0.1 ns). In each case, the protein Cα trace is shown as a thick black line.
At a more quantitative level, we may examine mean-square displacements (MSDs) in the bilayer plane for the examples of the two classes of lipid (Fig. 8) and so calculate lateral diffusion coefficients (see Table 2). For the plots of MSDs versus time we have divided each simulation into 5-ns sections. For the OmpA simulation (Fig. 8 A) for all four sections the MSDs are higher for the free than the bound lipids. For OmpA the situation is a little more complex, in that the first 5-ns section shows a raised MSD for both the bound and free lipids. This may reflect the changes in lipid-protein interactions seen during the first part of the OmpA simulation (see above). However, for all three 5-ns sections, the MSDs are higher for the free than for the bound lipids, as was the case for OmpA as well. From these MSD curves it is possible to derive lateral diffusion coefficients and to compare these for control simulations where a pure lipid bilayer has been simulated (Table 2). For OmpA the bound lipids have a diffusion coefficient about half that of the free lipids, which in turn are the same as for the pure lipid controls. For KcsA the situation is a little less clear cut, but the overall trend is the same.
FIGURE 8.
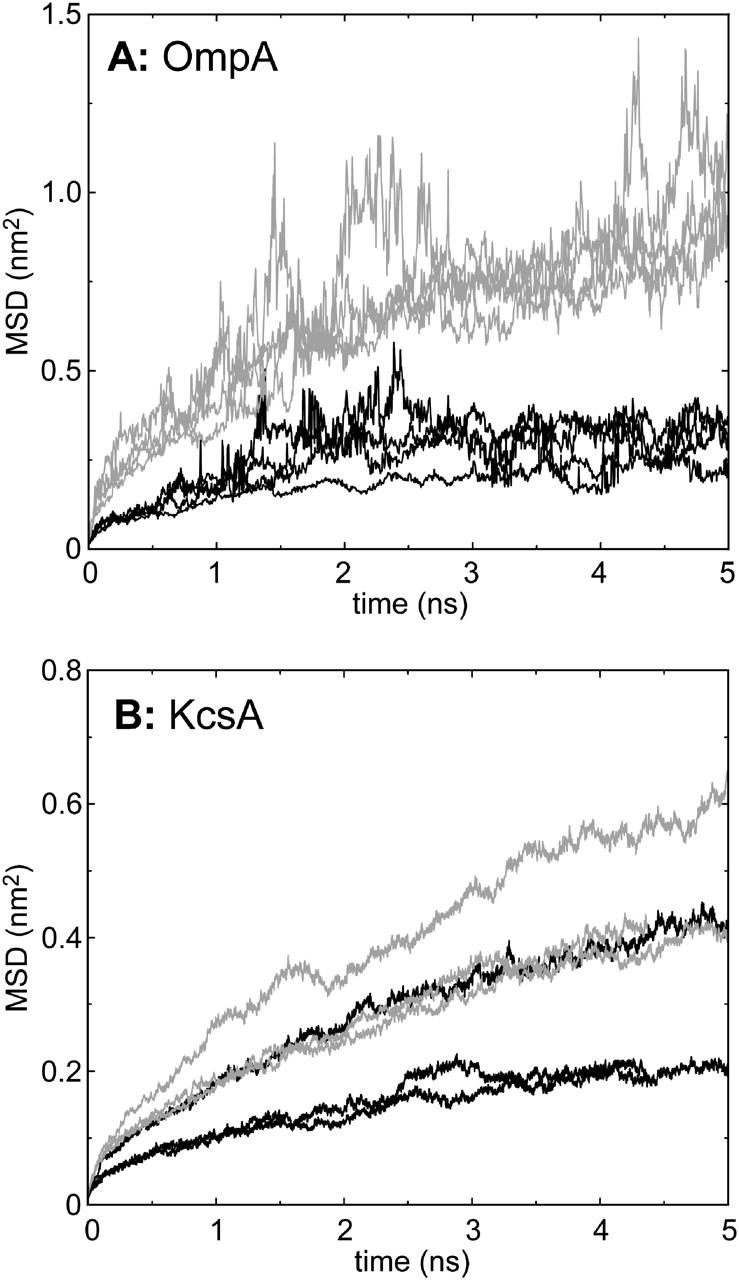
Mean-square deviations (MSDs) of two categories of lipid: black lines, bound lipids; shaded lines, free lipids. Bound lipid were defined as those whose phosphate atoms were within 0.35 nm of protein in every 5-ns trajectory snapshot, whereas free lipids were those that were never within 0.35 nm of protein in any 5-ns trajectory snapshot. Separate lines are shown for each 5-ns period within a trajectory.
TABLE 2.
Lateral diffusion coefficients of lipids
| Lateral diffusion coefficient (10−5 cm2 s−1)
|
|||
|---|---|---|---|
| OmpA/DMPC at 310 K | Bound | Free | Pure lipid control* |
| 0–5 ns | 0.019 (±0.029) | 0.055 (±0.093) | 0.053 (±0.023) |
| 5–10 ns | 0.028 (±0.007) | 0.056 (±0.045) | |
| 10–15 ns | 0.038 (±0.035) | 0.081 (±0.141) | |
| 15–20 ns | 0.036 (±0.043) | 0.053 (±0.080) | |
| KcsA/POPC at 300 K | Bound | Free | Pure lipid control |
| 0–5 ns | 0.026 (±0.005) | 0.032 (±0.035) | 0.031 (±0.004) |
| 5–10 ns | 0.009 (±0.010) | 0.020 (±0.015) | |
| 10–15 ns | 0.012 (±0.004) | 0.023 (±0.006) | |
Each control simulation consisted of a bilayer of 128 lipid molecules, run for 5 ns at either 310 K (for DMPC) or 300 K (for POPC).
In the OmpA simulation there are ∼14 bound DMPC molecules. This correlates well with recent studies of the association of spin-labeled lipids with β-barrel proteins (Ramakrishnan et al., 2004) which suggested there were 11 motionally restricted dimyristoyl phosphatidylglycerols per OmpA molecule. Thus, the simulation results correlate with the available experimental data on bound lipids.
DISCUSSION
The results of these simulations provide molecular level details of the nature of interaction of two integral membrane proteins, representing the two main classes of such protein, with their lipid bilayer environment. These results facilitate the interpretation of recent experimental data. For example, the simulations reveal that both OmpA and KcsA form significant interactions between their belts of aromatic side chains and the lipid headgroups (Fig. 9 A). This is in agreement with recent experimental literature on the interactions of Trp-containing TM peptides with lipid bilayers (de Planque et al., 1999; Killian and von Heijne, 2000; de Planque et al., 2003; Killian, 2003), on the basis of which it was concluded that “the tryptophan indole ring was consistently found to be positioned near the lipid carbonyl moieties” (de Planque et al., 2003). Our studies extend previous simulation studies of tryptophan-bilayer interactions that focused on model systems (Grossfield and Woolf, 2002) or idealized TM helix peptides (Petrache et al., 2002). In KcsA and other ion channels (e.g., MscL, MscS, and KvAP), the aromatic side chains must “lock” the protein in the membrane while at the same time enabling the dynamic changes in ipid-protein interactions that will occur during channel-gating conformational transitions. For OmpA, and other outer membrane proteins, aromatic interactions with lipid bilayers may be anticipated to be somewhat more complex, as the native LPS membrane presents an asymmetric transmembrane environmental profile.
Our studies have also revealed the importance of snorkeling interactions of basic side chains with the phosphate groups of lipids (Fig. 9 B). Again, this has been studied in some detail experimentally (Mishra et al., 1994; Liu et al., 2002; Strandberg and Killian, 2003) and has been observed in simulations of model TM peptide systems (Saiz et al., 2004). Our results indicate that this mechanism is likely to be important for a range of membrane proteins and can involve both lysine and arginine side chains.
Because of the extended nature of our simulations we are able to distinguish between bound and free lipid molecules in terms of their lateral mobility. These two categories have been the subject of recent experimental (spectroscopic) studies (Costa-Filho et al., 2003). The simulation data on the number of lipid molecules bound to OmpA correlates well with experimental estimates of the number of motionally restricted lipids (Ramakrishnan et al., 2004), and so this aspect of the simulation data may merit further investigations for other membrane proteins. Recent crystallographic and biochemical data have suggested a role for tightly bound lipids in the stability of some membrane proteins (Fyfe et al., 2004; Valiyaveetil et al., 2002; Demmers et al., 2003; Alvis et al., 2003). Furthermore, extended simulation studies of rhodopsin indicate that some selectivity may be present in terms of lipid-fatty acyl chain interactions (Feller et al., 2003).
The simulation approaches we have used are standard for current membrane protein simulations, and the proteins appeared to be stable. However, it is important to be aware of the possible limitations of such methods. Long-range electrostatics have been treated using PME (Darden et al., 1993; Essmann et al., 1995; Sagui and Darden, 1999). There is something of a consensus that this is the best available method for membrane simulations (Tobias et al., 1997; Tobias, 2001). However, one should be aware that it is not without potential artifacts, both for peptides (Hunenberger and McCammon, 1999; Weber et al., 2000; Kastenholz and Hünenberger, 2004) and for membrane systems (Bostick and Berkowitz, 2003). Further studies are needed to explore the sensitivity of lipid-protein interactions to the exact simulation condition employed.
A second limitation is that simulations of the order of ∼20 ns are still relatively short, and provide incomplete sampling of protein motions (Faraldo-Gómez et al., 2004). However, our simulations do suggest that lipid-protein interactions are able to relax to a stable state on this timescale. As substantially longer simulations (e.g., >100 ns) become available it will be of interest to see whether/how our picture of lipid-protein interactions changes.
From a biological perspective, a limitation in the current study is the use of simple (phosphatidylcholine) lipid bilayers. Although this may provide an adequate representation of lipid bilayers used in vitro it is a considerable simplification to the in vivo membrane environment. Recent development of more complex lipid models (Lins and Straatsma, 2001) lends hope that future simulation studies will be able to address this limitation. There are therefore a number of challenges for future simulation studies of membrane-protein interactions to address. In particular, it will be important to extend such studies to a wider range of membrane proteins. Such studies will benefit from more automated methodologies (Wu et al., 2003) for comparative analysis of multiple simulations.
Acknowledgments
Our thanks to all of our colleagues for their continued interest and discussions concerning these studies. We thank the Oxford Supercomputing Centre for access to resources.
We thank The Wellcome Trust, the Biotechnology and Biological Sciences Research Council, and the Engineering and Physical Sciences Research Council for their financial support of research in M.S.P.S.'s laboratory. P.B. is a Wellcome Trust research student; S.D. is an Engineering and Physical Sciences Research Council research student; and C.D. is a Royal Society University research fellow.
Sundeep S. Deol, Peter J. Bond, and Carmen Domene contributed equally to this work.
References
- Allen, T. W., O. S. Andersen, and B. Roux. 2003. Structure of gramicidin A in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR data. J. Am. Chem. Soc. 125:9868–9877. [DOI] [PubMed] [Google Scholar]
- Alvis, S. J., I. M. Williamson, J. M. East, and A. G. Lee. 2003. Interactions of anionic phospholipids and phosphatidylethanolamine with the potassium channel KcsA. Biophys. J. 85:3828–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baaden, M., and M. S. P. Sansom. 2004. OmpT: molecular dynamics simulations of an outer membrane enzyme. Biophys. J. 87:2942–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belohorcova, K., J. H. Davis, T. B. Woolf, and B. Roux. 1997. Structure and dynamics of an amphiphilic peptide in a lipid bilayer: a molecular dynamics study. Biophys. J. 73:3039–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Intermolecular Forces. Reidel, Dordrecht, The Netherlands.
- Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 95:43–56. [Google Scholar]
- Berger, O., O. Edholm, and F. Jahnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2002. The ionization state and the conformation of Glu-71 in the KcsA K+ channel. Biophys. J. 82:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond, P. J., J. D. Faraldo-Gómez, and M. S. P. Sansom. 2002. OmpA: A pore or not a pore? Simulation and modelling studies. Biophys. J. 83:763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond, P. J., and M. S. P. Sansom. 2003. Membrane protein dynamics vs. environment: simulations of OmpA in a micelle and in a bilayer. J. Mol. Biol. 329:1035–1053. [DOI] [PubMed] [Google Scholar]
- Bostick, D. L., and M. L. Berkowitz. 2003. The implementation of slab geometry for membrane-channel molecular dynamics simulations. Biophys. J. 85:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulieris, P. V., S. Behrens, O. Holst, and J. H. Kleinschmidt. 2003. Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J. Biol. Chem. 278:9092–9099. [DOI] [PubMed] [Google Scholar]
- Costa-Filho, A. J., R. H. Crepeau, P. P. Borbat, M. Ge, and J. H. Freed. 2003. Lipid-gramicidin interactions: dynamic structure of the boundary lipid by 2D-ELDOR. Biophys. J. 84:3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier, P. S., M. J. Stevens, L. R. Forrest, and T. B. Woolf. 2003. Molecular dynamics simulation of dark-adapted rhodopsin in an explicit membrane bilayer: Coupling between local retinal and larger scale conformational change. J. Mol. Biol. 333:493–514. [DOI] [PubMed] [Google Scholar]
- daCosta, C. J. B., A. A. Ogrel, E. A. McCardy, M. P. Blanton, and J. E. Baenziger. 2002. Lipid-protein interactions at the nicotinic acetylcholine receptor: a functional coupling between nicotinic receptors and phosphatidic acid-containing lipid bilayers. J. Biol. Chem. 277:201–208. [DOI] [PubMed] [Google Scholar]
- Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- Davis, M. E., J. D. Madura, B. A. Luty, and J. A. McCammon. 1991. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian dynamics program. Comput. Phys. Commun. 62:187–197. [Google Scholar]
- de Planque, M. R., B. B. Bonev, J. A. Demmers, D. V. Greathouse, R. E. Koeppe, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- Demmers, J. A. A., A. van Dalen, B. de Kruijiff, A. J. R. Heck, and J. A. Killian. 2003. Interaction of K channel KcsA with membrane phospholipids as studied by ESI mass spectrometry. FEBS Lett. 541:28–32. [DOI] [PubMed] [Google Scholar]
- Domene, C., P. J. Bond, S. S. Deol, and M. S. Sansom. 2003b. Lipid-protein interactions and the membrane/water interfacial region. J. Am. Chem. Soc. 125:14966–14967. [DOI] [PubMed] [Google Scholar]
- Domene, C., P. J. Bond, and M. S. Sansom. 2003a. Membrane protein simulation: ion channels and bacterial outer membrane proteins. Adv. Protein Chem. 66:159–193. [DOI] [PubMed] [Google Scholar]
- Domene, C., and M. S. P. Sansom. 2003. A potassium channel, ions and water: simulation studies based on the high resolution x-ray structure of KcsA. Biophys. J. 85:2787–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Faraldo-Gómez, J. D., L. R. Forrest, M. Baaden, P. J. Bond, C. Domene, G. Patargias, J. Cuthbertson, and M. S. P. Sansom. 2004. Conformational sampling and dynamics of membrane proteins from 10-nanosecond computer simulations. Proteins. In press. [DOI] [PubMed]
- Faraldo-Gómez, J. D., G. R. Smith, and M. S. P. Sansom. 2002. Setup and optimisation of membrane protein simulations. Eur. Biophys. J. 31:217–227. [DOI] [PubMed] [Google Scholar]
- Feller, S. E., K. Gawrisch, and T. B. Woolf. 2003. Rhodopsin exhibits a preference for solvation by polyunsaturated docosohexaenoic acid. J. Am. Chem. Soc. 125:4434–4435. [DOI] [PubMed] [Google Scholar]
- Ferguson, A. D., E. Hofmann, J. W. Coulton, K. Diederichs, and W. Welte. 1998. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science. 282:2215–2220. [DOI] [PubMed] [Google Scholar]
- Ferguson, A. D., W. Welte, E. Hofmann, B. Lindner, O. Holst, J. W. Coulton, and K. Diederichs. 2000. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Struct. Fold. Des. 8:585–592. [DOI] [PubMed] [Google Scholar]
- Fernandez, C., C. Hilty, G. Wider, and K. Wuthrich. 2002. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 99:13533–13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest, L. R., and M. S. P. Sansom. 2000. Membrane simulations: bigger and better? Curr. Opin. Struct. Biol. 10:174–181. [DOI] [PubMed] [Google Scholar]
- Fyfe, P. K., N. W. Isaacs, R. J. Cogdell, and M. R. Jones. 2004. Disruption of a specific molecular interaction with a bound lipid affects the thermal stability of the purple bacterial reaction centre. Biochim. Biophys. Acta. 1608:11–22. [DOI] [PubMed] [Google Scholar]
- Fyfe, P. K., K. E. McAuley, A. W. Roszak, N. W. Isaacs, R. J. Codgell, and M. R. Jones. 2001. Probing the interface between membrane proteins and membrane lipids by X-ray crystallography. Trends Biochem. Sci. 26:106–112. [DOI] [PubMed] [Google Scholar]
- Grizot, S., and S. K. Buchanan. 2004. Structure of the OmpA-like domain of RmpM from Neisseria meningitidis. Mol. Microbiol. 51:1027–1037. [DOI] [PubMed] [Google Scholar]
- Grossfield, A., and T. B. Woolf. 2002. Interaction of tryptophan analogs with POPC lipid bilayers investigated by molecular dynamics calculations. Langmuir. 18:198–210. [Google Scholar]
- Halle, B. 2004. Biomolecular cryocrystallography: Structural changes during flash-cooling. Proc. Natl. Acad. Sci. USA. 101:4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans, J., H. J. C. Berendsen, W. F. van Gunsteren, and J. P. M. Postma. 1984. A consistent empirical potential for water-protein interactions. Biopolymers. 23:1513–1518. [Google Scholar]
- Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- Huber, T., A. V. Botelho, K. Beyer, and M. F. Brown. 2004. Membrane model for the G-protein-coupled receptor rhodopsin: hydrophobic interface and dynamical structure. Biophys. J. 86:2078–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Hunenberger, P. H., and J. A. McCammon. 1999. Effect of artificial periodicity in simulations of biomolecules under Ewald boundary conditions: a continuum electrostatics study. Biophys. Chem. 78:69–88. [DOI] [PubMed] [Google Scholar]
- Jensen, M. Ø., O. G. Mouritsen, and G. H. Peters. 2004. Simulations of a membrane-anchored peptide: structure, dynamics, and influence on bilayer properties. Biophys. J. 86:3556–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D. T. 1998. Do transmembrane protein superfolds exist? FEBS Lett. 423:281–285. [DOI] [PubMed] [Google Scholar]
- Karplus, M. J., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- Kastenholz, M. A., and P. H. Hünenberger. 2004. Influence of artificial periodicity and ionic strength in molecular dynamics simulations of charged biomolecules employing lattice-sum methods. J. Phys. Chem. B. 108:774–788. [Google Scholar]
- Killian, J. A. 2003. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 555:134–138. [DOI] [PubMed] [Google Scholar]
- Killian, J. A., and G. von Heijne. 2000. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25:429–434. [DOI] [PubMed] [Google Scholar]
- Kramer, R. A., K. Brandenburg, L. Vandeputte-Rutten, M. Werkhoven, P. Gros, N. Dekker, and M. R. Egmond. 2002. Lipopolysaccharide regions involved in the activation of Escherichia coli outer membrane protease OmpT. Eur. J. Biochem. 269:1746–1752. [DOI] [PubMed] [Google Scholar]
- Lange, C., J. H. Nett, B. L. Trumpower, and C. Hunte. 2001. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc(1) complex structure. EMBO J. 20:6591–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, A. G. 2003. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 1612:1–40. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., and O. Edholm. 2000. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 79:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- Lins, R. D., and T. P. Straatsma. 2001. Computer simulation of the rough lipopolysaccharide membrane of Pseudomonas aeruginosa. Biophys. J. 81:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F., R. N. Lewis, R. S. Hodges, and R. N. McElhaney. 2002. Effect of variations in the structure of a polyleucine-based alpha-helical transmembrane peptide on its interaction with phosphatidylcholine bilayers. Biochemistry. 41:9197–9207. [DOI] [PubMed] [Google Scholar]
- Marrink, S. J., O. Berger, D. P. Tieleman, and F. Jahnig. 1998. Adhesion forces of lipids in a phospholipid membrane studied by molecular dynamics simulations. Biophys. J. 74:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, V., M. Palgunachari, J. Segrest, and G. Anantharamaiah. 1994. Interactions of synthetic peptide analogs of the class A amphipathic helix with lipids: evidence for the snorkel hypothesis. J. Biol. Chem. 269:7185–7191. [PubMed] [Google Scholar]
- Mukhopadhyay, P., L. Monticelli, and D. P. Tieleman. 2004. Molecular dynamics simulation of a palmitoyl-oleoyl phosphatidylserine bilayer with Na+ counterions and NaCl. Biophys. J. 86:1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keeffe, A., J. M. East, and A. G. Lee. 2000. Protein-lipid interactions of beta-barrel membrane proteins. FASEB J. 14:898. [Google Scholar]
- Pautsch, A., and G. E. Schulz. 1998. Structure of the outer membrane protein A transmembrane domain. Nat. Struct. Biol. 5:1013–1017. [DOI] [PubMed] [Google Scholar]
- Petrache, H. I., A. Grossfield, K. R. MacKenzie, D. M. Engelman, and T. B. Woolf. 2000. Modulation of glycophorin A transmembrane helix interactions by lipid bilayers: molecular dynamics calculations. J. Mol. Biol. 302:727–746. [DOI] [PubMed] [Google Scholar]
- Petrache, H. I., D. M. Zuckerman, J. N. Sachs, J. A. Killian, R. E. Koeppe, and T. B. Woolf. 2002. Hydrophobic matching mechanism investigated by molecular dynamics simulations. Langmuir. 18:1340–1351. [Google Scholar]
- Ramakrishnan, M., C. L. Pocanschi, J. H. Kleinschmidt, and D. Marsh. 2004. Association of spin-labeled lipids with β-barrel proteins from the outer membrane of Escherichia coli. Biochemistry. 43:11630–11636. [DOI] [PubMed] [Google Scholar]
- Ranatunga, K. M., I. H. Shrivastava, G. R. Smith, and M. S. P. Sansom. 2001. Side-chain ionization states in a potassium channel. Biophys. J. 80:1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B., and T. B. Woolf. 1996. Molecular dynamics of Pf1 coat protein in a phospholipid bilayer. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz, editor. Birkhäuser, Boston. 555–587.
- Sagui, C., and T. A. Darden. 1999. Molecular dynamics simulations of biomolecules: Long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 28:155–179. [DOI] [PubMed] [Google Scholar]
- Saiz, L., S. Bandyopadhyay, and M. L. Klein. 2004. Effect of the pore region of a transmembrane ion channel on the physical properties of a simple membrane. J. Phys. Chem. B. 108:2608–2613. [Google Scholar]
- Saiz, L., and M. L. Klein. 2002. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- Sayle, R. A., and E. J. Milner-White. 1995. RasMol: Biomolecular graphics for all. Trends Biochem. Sci. 20:374–376. [DOI] [PubMed] [Google Scholar]
- Schiffer, M., C. H. Chang, and F. J. Stevens. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5:213–214. [DOI] [PubMed] [Google Scholar]
- Strandberg, E., and J. A. Killian. 2003. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 544:69–73. [DOI] [PubMed] [Google Scholar]
- Tamm, L. K., F. Abildgaard, A. Arora, H. Blad, and J. H. Bushweller. 2003. Structure, dynamics and function of the outer membrane protein A (OmpA) and influenza hemagglutinin fusion domain in detergent micelles by solution NMR. FEBS Lett. 555:139–143. [DOI] [PubMed] [Google Scholar]
- Tang, P., and Y. Xu. 2002. Large-scale molecular dynamics simulations of general anesthetic effects on the ion channel in the fully hydrated membrane: The implication of molecular mechanisms of general anesthesia. Proc. Natl. Acad. Sci. USA. 99:16035–16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terstappen, G. C., and A. Reggiani. 2001. In silico research in drug discovery. Trends Pharmacol. Sci. 22:23–26. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., and H. J. C. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 74:2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman, D. P., L. R. Forrest, H. J. C. Berendsen, and M. S. P. Sansom. 1999. Lipid properties and the orientation of aromatic residues in OmpF, influenza M2 and alamethicin systems: molecular dynamics simulations. Biochemistry. 37:17554–17561. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., S. J. Marrink, and H. J. C. Berendsen. 1997. A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta. 1331:235–270. [DOI] [PubMed] [Google Scholar]
- Tobias, D. J. 2001. Electrostatics calculations: recent methodological advances and applications to membranes. Curr. Opin. Struct. Biol. 11:253–261. [DOI] [PubMed] [Google Scholar]
- Tobias, D. J., K. C. Tu, and M. L. Klein. 1997. Atomic-scale molecular dynamics simulations of lipid membranes. Curr. Opin. Colloid Interface Sci. 2:15–26. [Google Scholar]
- Ulmschneider, M. B., and M. S. P. Sansom. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta. 1512:1–14. [DOI] [PubMed] [Google Scholar]
- Ulmschneider, M. B., D. P. Tieleman, and M. S. P. Sansom. 2004. Interactions of a transmembrane helix and a membrane: comparative simulations of bacteriorhodopsin helix A. J. Phys. Chem. B. 108:10149–10159. [Google Scholar]
- Valiyaveetil, F. I., Y. Zhou, and R. MacKinnon. 2002. Lipids in the structure, folding and function of the KcsA channel. Biochemistry. 41:10771–10777. [DOI] [PubMed] [Google Scholar]
- Wallin, E., and G. von Heijne. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archean, and eukaryotic organisms. Protein Sci. 7:1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, W., P. H. Hunenberger, and J. A. McCammon. 2000. Molecular dynamics simulations of a polyalanine octapeptide under Ewald boundary conditions: influence of artificial periodicity on peptide conformation. J. Phys. Chem. B. 104:3668–3675. [Google Scholar]
- White, S. N. 1994. Hydropathy plots and the prediction of membrane protein topology. In Membrane Protein Structure: Experimental Approaches. S. N. White, editor. Oxford University Press, New York.
- Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, T. B. 1997. Molecular dynamics of individual α-helices of bacteriorhodopsin in dimyristoyl phosphatidylcholine. I. Structure and dynamics. Biophys. J. 73:2376–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, T. B. 1998. Molecular dynamics of individual α-helices of bacteriorhodopsin in dimyristoyl phosphatidylcholine. II. Interaction energy analysis. Biophys. J. 74:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1996. Structure, energetics, and dynamics of lipid-protein interactions: a molecular-dynamics study of the gramicidin-A channel in a DMPC bilayer. Proteins. 24:92–114. [DOI] [PubMed] [Google Scholar]
- Wu, B., K. Tai, S. Murdock, M. G. Ng, S. Johnston, H. Fangohr, P. Jeffreys, J. Cox, J. Essex, and M. S. P. Sansom. 2003. BioSimGRID: a distributed database for biomolecular simulations. In Proc. UK e-Science All Hands Meeting 2003, Nottingham, UK. 412–419.
- Yau, W. M., P. J. Steinbach, W. C. Wimley, S. H. White, and K. Gawrisch. 1998a. Indole and N-methyl indole orientation in lipid bilayers. Biophys. J. 74:A303. [Google Scholar]
- Yau, W. M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998b. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]