

Abstract
Calcium handling in pancreatic β-cells is important for intracellular signaling, the control of electrical activity, and insulin secretion. The endoplasmic reticulum (ER) is a key organelle involved in the storage and release of intracellular Ca2+. Using mathematical modeling, we analyze the filtering properties of the ER and clarify the dual role that it plays as both a Ca2+ source and a Ca2+ sink. We demonstrate that recent time-dependent data on the free Ca2+ concentration in pancreatic islets and β-cell clusters can be explained with a model that uses a passive ER that takes up Ca2+ when the cell is depolarized and the cytosolic Ca2+ concentration is elevated, and releases Ca2+ when the cell is repolarized and the cytosolic Ca2+ is at a lower concentration. We find that Ca2+-induced Ca2+ release is not necessary to explain the data, and indeed the model is inconsistent with the data if Ca2+-induced Ca2+ release is a dominating factor. Finally, we show that a three-compartment model that includes a subspace compartment between the ER and the plasma membrane provides the best agreement with the experimental Ca2+ data.
INTRODUCTION
A key element of glucose homeostasis is the secretion of insulin by pancreatic β-cells in response to changes in the blood glucose level. The signal transduction from external glucose to insulin secretion is a multistep process, involving cell depolarization and a subsequent elevation of the intracellular Ca2+ concentration. Specifically, glucose is transported across the cell membrane and metabolized, resulting in an increase in the intracellular ATP/ADP ratio. This increased ratio closes ATP-sensitive K+ channels, depolarizing the cell and activating voltage-dependent Ca2+ channels. The resulting Ca2+ influx increases the intracellular Ca2+ concentration and evokes exocytosis of insulin granules (Ashcroft and Rorsman, 1989).
While Ca2+ influx is clearly important in the β-cell's response to depolarization, there is a large body of evidence that the endoplasmic reticulum (ER) also plays a role (Arredouani et al., 2002b; Bertram et al., 1995; Gilon et al., 1999; Lemmens et al., 2001; Miura et al., 1997; Tengholm et al., 2001). The contribution that the ER makes to Ca2+ handling in pancreatic islets was the focus, in particular, of a recent study in which the free cytosolic Ca2+ concentration was measured during an oscillatory potassium pulse protocol. Each K+ pulse depolarizes the islet, and at the end of the pulse the cell repolarizes. The oscillatory pattern of K+ pulses was chosen to resemble electrical bursting patterns typically observed in islets (Arredouani et al., 2002b). By controlling the electrical subsystem, the authors were able to focus on the Ca2+ dynamics. Calcium rose during each depolarization and decayed to a level above baseline during each repolarization. Experiments were repeated in the presence of the SERCA pump inhibitor thapsigargin (Tg), which blocks uptake of Ca2+ into the ER and ultimately leads to Ca2+ depletion in the organelle. In the presence of Tg, the amplitude of the Ca2+ response to depolarization was increased significantly, and the Ca2+ nadir between depolarizations was close to the baseline level. In a related study using longer depolarizations, Gilon et al. (1999) also demonstrated that Tg increases the amplitude of the Ca2+ response, and additionally increases the rate of rise of Ca2+ during the depolarization. Another study demonstrated that the Ca2+ dynamics in SERCA3 knockout mice are similar to those in control mice in the presence of Tg during short burst-like K+ pulses, suggesting that SERCA3 is the SERCA isoform primarily responsible for filling the ER when the cytosolic Ca2+ concentration is elevated by depolarization (Arredouani et al., 2002a).
In this study we use mathematical modeling to address several questions related to the contribution of the ER to Ca2+ handling in β-cells. First, we ask whether a simple model with two Ca2+ compartments, the ER and cytosol, can account for the recent Ca2+ data of Arredouani et al. (2002b) and Gilon et al. (1999). Second, whereas these authors focused on the effects of SERCA blockers, we ask what effect other modulators such as carbachol, Li+, and calmidazolium would have on the cytosolic Ca2+ dynamics. Carbachol promotes efflux of Ca2+ from the ER via the production of inositol 1,4,5-trisphosphate (IP3), whereas Li+ and calmidazolium inhibit Ca2+ exchangers and pumps in the plasma membrane, respectively (Nadal et al., 1994; Wolf et al., 1988). Third, we use the experimental data to constrain the speed at which the ER takes up and releases Ca2+, and find that ER Ca2+ must be much slower than cytosolic Ca2+, but not too slow.
Some have suggested that calcium-induced calcium release (CICR) from the ER is important in β-cells (Ämmälä et al., 1991; Gromada et al., 1996; Kang and Holz, 2003; Lemmens et al., 2001), and indeed there is little doubt that it is important when agonists such as acetylcholine or GLP-1 are present (Ämmälä et al., 1991; Gilon et al., 1995; Gromada et al., 1996; Kang and Holz, 2003; Lemmens et al., 2001). However, in the absence of these agonists the significance of CICR is unclear. A recent study (Beauvois et al., 2004) failed to find any CICR during brief depolarizations (<40 s) such as those that occur during a burst of action potentials. Also, mRNA for ryanodine receptors, the primary mechanism for CICR in the absence of cholinergic agonists, was only weakly expressed in β-cells. However, an atypical form of CICR was observed, which was not dependent on either IP3R or RyR, in response to supraphysiological KCl pulses, much longer and stronger than the ones in Arredouani et al. (2002b) that we simulate. This atypical CICR appeared only infrequently in response to glucose-induced calcium oscillations and was not required for those oscillations.
In the analysis presented here, we find that a passive two-compartment model (cytosol and ER only) is able to account for all of the recent time-dependent Ca2+ data. Thus, the Ca2+ filtering properties of an ER with a Ca2+-dependent SERCA and slow passive leak that depends on the gradient between the ER and cytosolic Ca2+ levels accounts for the reported effects of Tg on the cytosolic Ca2+ during trains of depolarizations. Although CICR is not necessary, a modest CICR contribution is compatible with the data. However, if CICR is dominant, such that the ER drains during depolarization and refills during repolarization, then the model does not reproduce the Ca2+ data. Thus, if CICR is present in β-cells during normal glucose stimulation, then it must be present at modest levels.
The simple two-compartment model predicts that during long sustained depolarizations the cytosolic Ca2+ concentration will approach the same value with or without SERCA pump blockers. This is true whether or not CICR is present. Thus, whereas the SERCA pumping rate has a profound effect on the dynamics of the cytosolic Ca2+, it has no impact on the equilibrium level of cytosolic Ca2+. A slightly more complex model, called the subspace model, on the other hand, predicts that the measured equilibrium Ca2+ concentration will be lower when the ER is depleted of Ca2+. The subspace model postulates that Ca2+ from the ER is released primarily into a compartment that lies between the ER and the plasma membrane and was motivated by the observation that a slow K+ current (IK(slow)) activated by burst-like trains of depolarizations (Goforth et al., 2002; Göpel et al., 1999; Kanno et al., 2002) is inhibited by SERCA pump blockade (Goforth et al., 2002). Our results suggest that the three-compartment subspace model is superior to a two-compartment model with or without CICR in reproducing the recent data on Ca2+ handling in β-cells.
METHODS
Basic two-compartment model
Whereas the experimental data that we simulate comes from intact islets, we use a single-cell model in our simulations. Although some phenomena, such as asynchronous Ca2+ releases, cannot be modeled with this approach, it suffices to explain most aspects of islet behavior because the cells are electrically coupled and their electrical activity and Ca2+ dynamics are synchronized. The basic two-compartment model is illustrated in Fig. 1 A. Calcium enters the cell through L-type Ca2+ channels in the plasma membrane, with flux (Jin) proportional to the Ca2+ current. Calcium is removed by Ca2+ pumps in the membrane, with flux (Jpmca) assumed to be proportional to the cytosolic free Ca2+ concentration (C). Thus,
 |
(1) |
where α converts current to concentration, and kpmca is the plasma membrane pump rate. The Ca2+ current is voltage (V)-dependent, and is given by
 |
(2) |
where gCa is the whole cell conductance, VCa is the Ca2+ reversal potential, and m is the channel activation variable. Since channel activation is known to be rapid, we assume for simplicity that it is instantaneous, so 
FIGURE 1.

Illustration of the Ca2+ fluxes incorporated in (A) the two-compartment model and (B) the three-compartment subspace model. The thin arrow in B represents a small flux from the ER to the bulk cytosol.
Calcium from the cytosol is transported into the ER by SERCA pumps in the ER membrane. Two SERCA genes are expressed in β-cells, SERCA2b and SERCA3. The SERCA2b ATPase has high Ca2+ affinity and is primarily responsible for setting the basal level of Ca2+ in the ER (Varadi and Rutter, 2002). The SERCA3 ATPase has lower Ca2+ affinity and is primarily responsible for transporting Ca2+ from the cytosol to the ER during Ca2+ elevations, such as those produced by bursts of action potentials (Arredouani et al., 2002a). We include terms for both SERCA2b and SERCA3 in our model. Flux through SERCA2b pumps is described by a small constant term (this pump is treated as saturated even at basal cytosolic Ca2+ levels), whereas flux through SERCA3 pumps is assumed to be proportional to C. Flux out of the ER (Jrelease) is assumed to be proportional to the gradient between the concentrations of free cytosolic Ca2+ and free ER Ca2+ (Cer). Thus,
 |
(3) |
where kserca2b and kserca3 are the SERCA2b and SERCA3 rate constants, and per is the rate of Ca2+ release from the ER. Release from the ER could be through inositol 1,4,5-trisphophate (IP3) or ryanodine channels in the ER membrane, or some other route of passive leak. Thapsigargin (Tg) is modeled by setting both kserca2b and kserca3 to 0, whereas SERCA3 knockout is modeled by setting only kserca3 to 0. The simple linear expressions used for ER fluxes demonstrate that no nonlinearities in the fluxes are needed to account for the data of Arredouani et al. (2002b). Similar linear ER fluxes were used by Gall and Susa (1999), but without the constant for SERCA2b.
The time-dependent concentrations of C and Cer are described using conservation equations. Thus,
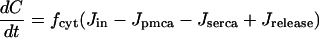 |
(4) |
 |
(5) |
where fcyt, fer are the fractions of free Ca2+ in the cytosolic and ER compartments, respectively, and νcyt and νer are the volumes of the cytosolic and ER compartments. All parameter values are listed in Table 1. The basic two-compartment model is used in Figs. 2–8.
TABLE 1.
Parameter values used in two-compartment model
| gCa | 1200 pS | VCa | 30 mV | α | 4.5 × 10−6μM fA−1 ms−1 |
| vm | −15 mV | sm | 8 mV | fcyt | 0.01 |
| fer | 0.01 | vcyt | 10 μm3 | ver | 0.4 μm3 |
| per | 10−4 ms−1 | pmin | 10−5 ms−1 | pmax | 3 × 10−4 ms−1 |
| Kcicr | 0.5 μM | kpmca | 0.08 ms−1 | kserca3 | 0.08 ms−1 |
| kserca2b | 0.02 μM ms−1 |
FIGURE 2.

(A) Simulated voltage protocol mimicking the biphasic response to an increase in glucose concentration. (B) Cytosolic Ca2+ response with SERCA pumps enabled (solid line) and disabled (dashed line). During each imposed oscillation, the nadir is defined as the lowest value of C during the repolarized phase. The amplitude is the difference between the peak and the nadir. (C) The ER Ca2+ concentration. No driving force is established between the ER and the cytosol when SERCA pumps are disabled (dashed line).
FIGURE 3.

(A) Doubling the rate of Ca2+ release from the ER, per, has a transient effect on the cytosolic Ca2+ response (dashed line) to the voltage protocol, but no long-term effect. (B) Cer adapts to the doubling in per (dashed line) by declining to a value one-half that of the control.
FIGURE 4.

(A) Reducing the rate of pumping across the plasma membrane by half (dashed line) slightly increases the amplitude of the Ca2+ response to depolarization and greatly increases the Ca2+ nadir. (B) The increase in the Ca2+ nadir is due to an increase in Cer.
FIGURE 5.

(A) An increase in the glucose concentration is mimicked (at arrow) by increasing the depolarized phase duration from 8 s to 12 s, while keeping the oscillation period fixed at 24 s. (B) In the control simulation (solid line), increasing the duration of depolarization results in an increase in the Ca2+ nadir, but not in the amplitude, whereas there is no effect when SERCA pumps are disabled (dashed line). (C) In the control simulation, an increase in the ER Ca2+ concentration is responsible for the increased cytosolic Ca2+ nadir.
FIGURE 6.

(A) When the Ca2+ channel conductance, gCa, is doubled (at arrow), both the Ca2+ amplitude and nadir are increased. This is true with SERCA pumps enabled (solid line) or disabled (dashed line), although the nadir increase in the latter case is quite small. (B) As in Fig. 5, the nadir increase is due primarily to an increase in the ER Ca2+ concentration, although increased Ca2+ entry also plays a role.
FIGURE 7.

(A) Voltage protocol for compound bursting. (B) In the control system the Ca2+ nadir rises during a compound burst and slowly falls between (solid line). This does not occur when the SERCA pumps are disabled (dashed line). (C) The rise and fall of the nadir reflects the dynamics of ER Ca2+.
FIGURE 8.

Demonstration that the slow rise and fall of the cytosolic Ca2+ nadir depends critically on the speed at which the ER takes up and releases Ca2+. (A) The speed of the ER is increased by setting fer = 0.1 (solid line), or decreased by setting fer = 0.0002 (dashed line). (B) There are no slow Cer dynamics when fer = 0.1, and there is only a small increase in Cer during a compound burst when fer = 0.0002.
Two-compartment model with CICR
In simulations performed with the basic two-compartment model the ER Ca2+ release rate, per, is constant. In simulations involving calcium-induced Ca2+ release (CICR), per is an increasing function of C,
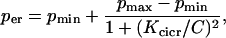 |
(6) |
which is constructed so that Ca2+ efflux from the ER is increased when cytosolic Ca2+ is elevated. The model with CICR is used in Figs. 9 and 10.
FIGURE 9.

Ca2+ response to a sequence of depolarizations when CICR dominates release from the ER. (A) Sequence of brief depolarizations. (B) Cytosolic Ca2+ rises during depolarization and falls during repolarization. (C) The ER releases Ca2+ during the depolarization due to CICR, and refills during the repolarization.
FIGURE 10.

(A) Biphasic voltage protocol applied to the model in which CICR dominates. (B) Even with active SERCA pumps the Ca2+ nadir is not elevated and the amplitude is large (solid line), so that blocking SERCA pumps has only a small effect (dashed line). (C) The cytosolic Ca2+ nadir is not elevated despite the fact that the ER is filled.
Three-compartment subspace model
The subspace model (Goforth et al., 2002) was motivated by experimental evidence for a Ca2+-activated K+ current (IK(slow)) that develops during burst-like voltage-clamp depolarizations (Goforth et al., 2002; Göpel et al., 1999; Kanno et al., 2002). In this model, Ca2+ released from the ER primarily enters a subspace compartment adjacent to the plasma membrane, and from here can enter the bulk cytosol (Fig. 1 B). The Ca2+ concentration in the subspace (Css) is elevated compared with the bulk cytosolic Ca2+ (C) due to this preferential influx from the ER. Here, we assume that there is also some release from the ER directly into the bulk cytosol. In Goforth et al. (2002) it was proposed that the Ca2+-activated K+ channels that comprise a large component of IK(slow) respond to this elevated Css rather than the lower bulk cytosolic Ca2+. We are interested here only in the Ca2+ handling, so ion channels other than the L-type channels are omitted from the model.
The Ca2+ flux terms in the subspace model are largely the same as in the two-compartment model, with the exception of the two release terms Jrelease,ss and Jrelease,cyt, and the new flux term between the subspace and the bulk cytosol (JX). Thus,
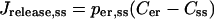 |
(7) |
 |
(8) |
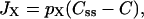 |
(9) |
where per,ss, per,cyt, and pX are constants. The differential equations describing the Ca2+ concentrations in the three compartments are
 |
(10) |
 |
(11) |
 |
(12) |
where fss is the fraction of free Ca2+ in the subspace and νss is the volume. The observable for this model is the average of the Ca2+ concentrations in the bulk cytosol and the subspace compartment weighted by the compartment volumes,
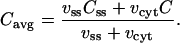 |
(13) |
All parameters for the subspace model are given in Table 2. This model is used in Fig. 11 only.
TABLE 2.
Parameter values used in the three-compartment subspace model
| gCa | 1450 pS | VCa | 30 mV | A | 4.5 × 10−6μM fA−1 ms−1 |
| kpmca | 0.12 ms−1 | kserca3 | 0.3 ms−1 | kserca2b | 0.02 μM ms−1 |
| fcyt | 0.01 | fer | 0.01 | fss | 0.01 |
| pX | 0.045 ms−1 | per,cyt | 5 × 10−5 ms−1 | per,ss | 1.5 × 10−6 ms−1 |
| νcyt | 3.2 μm3 | νer | 0.4 μm3 | νss | 0.8 μm3 |
| sm | 8 mV | vm | −13 mV |
FIGURE 11.

Ca2+ concentrations during a sustained depolarization from −70 mV to −30 mV with the three-compartment subspace model. (A) The weighted average of the bulk cytosolic Ca2+ and the subspace Ca2+ concentrations is lower when SERCA pumps are inhibited (dashed line) than in the control system (solid line). The ER Ca2+ concentration (B) and the subspace Ca2+ concentration (D) are lower when SERCA pumps are inhibited. (C) The bulk cytosolic concentration satisfies Eq. 16, so its steady state is independent of the Cer or ER parameters.
RESULTS
Ca2+ flux terms have differential effects on the Ca2+ profile
In the in vitro studies of pancreatic islets, when the bath glucose concentration is raised from a substimulatory to a suprastimulatory level the β-cell electrical response is typically biphasic. The cells first spike continuously for 1–2 min (phase 1), and then produce bursts of action potentials (phase 2) with periods ranging from 10 s to ∼1 min (Meissner and Atwater, 1976). This behavior was mimicked using K+ pulses in the experimental study by Arredouani et al. (2002b), and the average intracellular free Ca2+ concentration was measured. Each K+ pulse depolarizes the islet, and the islet repolarizes between pulses. Hence, the K+ pulse protocol is similar to a voltage-clamp protocol, with the advantage that it is applicable to intact islets. We simulated this voltage-clamp-like protocol with our mathematical models. As illustrated in Fig. 2 A, voltage is initially maintained at a resting potential of −70 mV. It is then elevated to −30 mV for 1 min, mimicking the first phase of electrical activity. After this, a voltage oscillation is imposed by periodically clamping V between −60 mV and −30 mV, mimicking the silent and active phases of bursting, respectively. The duration of the repolarized phase is 16 s, and that of the depolarized phase is 8 s.
The model cytosolic Ca2+ response to this protocol is shown in Fig. 2 B (solid curve), using the basic two-compartment model without CICR. (This model is used to generate Figs. 2–8.) Note that the Ca2+ nadir during the silent phases is elevated above the resting level. Also note the slow rise in Ca2+ during the initial depolarization, which reflects the time constant of ER filling. The model ER Ca2+ concentration is shown in Fig. 2 C. During the initial depolarization the ER fills, and during subsequent oscillatory activity Cer remains elevated. This maintained elevation of Cer provides the driving force for a net flux of Ca2+ from the ER into the cytosol during the repolarized phases of the oscillatory activity, elevating the cytosolic Ca2+ nadir.
Application of the SERCA pump blocker thapsigargin (Tg) is simulated by setting both kserca2b and kserca3 to 0 and allowing the system to equilibrate at the resting potential, so that the cytosolic and ER Ca2+ concentrations converge. When the voltage protocol is applied, the amplitude of the cytosolic Ca2+ response is much greater and the nadir is no longer elevated (Fig. 2 B, dashed curve). This twofold effect of blocking SERCA pumps reflects two important dynamical roles of the ER. First, the ER acts as a Ca2+ sink, slowing the rise of cytosolic Ca2+. When SERCA pumps are blocked, the filtering of cytosolic Ca2+ is reduced and C rises to a higher level. Second, the ER acts as a Ca2+ source when the cell is repolarized and there is little or no entry of Ca2+ into the cell. This latter role requires that a gradient be established between the ER and the cytosol, and this does not happen when SERCA pumps are blocked.
The simulation results in Fig. 2 reproduce the experimental data (Fig. 2, B and C, of Arredouani et al., 2002b, and Fig. 2 A of Gilon et al., 1999) very well, indicating that the simple two-compartment model with passive Ca2+ release from the ER is sufficient to describe the effects on cytosolic Ca2+ of disabling the SERCA pumps. Similar effects on cytosolic Ca2+ were observed in SERCA3 knockout mice (Arredouani et al., 2002b) and can be simulated with the model by setting kserca3 = 0 (not shown). However, the model indicates that the ER does not empty in that case, demonstrating that it is the rise and fall of ER Ca2+, not the level of ER Ca2+, that governs the filtering effect of the ER.
Another way to reduce Cer is to increase the release rate from the ER. We investigated whether increasing the Ca2+ release rate would increase the cytosolic Ca2+ amplitude and decrease the nadir during the voltage protocol, as did blocking the SERCA pumps. This was tested by doubling per. Fig. 3 shows the control response (solid line) and the response with per doubled (dashed line). Increasing the Ca2+ release rate had no effect on the cytosolic Ca2+ amplitude, and although the nadir is initially elevated, this effect is only transient. The lack of effect on the Ca2+ amplitude is explained by noting that the Ca2+ influx into the ER during a depolarization is mainly determined by kserca2b and kserca3, not per. The lack of a long-term effect on the Ca2+ nadir can be understood by noting that Jrelease = per (Cer−C) ≈ perCer. When per is doubled Cer adapts by falling to half its control value (Fig. 3 B), and Jrelease is ultimately the same as in the control simulation. Thus, the nadir is ultimately the same as in the control simulation. This model prediction can be readily tested experimentally by applying carbachol or some other muscarinic agonist to stimulate IP3 production and increase Ca2+ release from the ER through IP3 channels.
We next examined the effects of partially disabling the plasma membrane Ca2+ pumps by reducing kpmca by half. Although this maneuver did cause a small increase in the cytosolic Ca2+ amplitude in response to the pulse protocol, it raised the Ca2+ nadir by a much larger amount (Fig. 4 A). The former effect is due to the reduced pumping, whereas the latter effect is due to the increased Ca2+ release from the ER during the repolarized phase of the oscillation. Indeed, the main consequence of reducing pumping out of the cell is to increase C so that more Ca2+ gets pumped into the ER, setting up a larger gradient between the ER and cytosol compartments (Fig. 4 B). As a result, when the cell is repolarized the ER is a greater source of Ca2+, increasing the cytosolic Ca2+ nadir above its control value. This model prediction can be tested experimentally by replacing Na+ with Li+ to inhibit Na+/Ca2+ exchange (Nadal et al., 1994) or applying calmidazolium to inhibit calmodulin and thus the plasma membrane Ca2+ pumps (Wolf et al., 1988).
In summary, reducing the uptake of Ca2+ into the ER by disabling the SERCA pumps yields an increased cytosolic Ca2+ amplitude and reduced nadir in response to the voltage protocol, consistent with the effects of the SERCA pump blocker Tg or SERCA3 knockout during a similar K+ pulse protocol (Arredouani et al., 2002a,b). Increasing Ca2+ efflux from the ER has only a transient effect on the Ca2+ nadir; ultimately the Ca2+ nadir returns to its control level. Decreasing the Ca2+ efflux from the cell by partially inhibiting plasma membrane Ca2+ pumps yields a slightly increased Ca2+ amplitude and a significantly increased Ca2+ nadir in response to the voltage protocol. Thus, alterations in the level of Ca2+ release from the ER or in the plasma membrane pump rate have effects on the cytosolic Ca2+ that are quite different from the effect of disabling SERCA pumps. All of these effects can be understood in terms of the dual role of the ER as a Ca2+ sink and a Ca2+ source.
The effects of increased Ca2+ entry depend on how entry is increased
When the bath glucose concentration is increased from one suprastimulatory level to a higher level pancreatic islets typically respond by increasing the plateau fraction of bursting, i.e., the ratio of active phase duration to the total burst period. This effect on the membrane potential was mimicked by Arredouani et al. (2002b) by increasing the K+ pulse duration while keeping the oscillation period fixed. (The effects of increasing the glucose concentration on pumps were found to be negligible compared to the effect on the plateau fraction.) We simulate this by increasing the depolarized duration from 8 to 12 s while decreasing the repolarized duration from 16 to 12 seconds, so that the oscillation period remains fixed at 24 s (Fig. 5 A). In the control simulation (SERCA intact), the increase in plateau fraction causes an increase in the Ca2+ nadir, with little change in the Ca2+ amplitude (Fig. 5 B). This is consistent with data shown in Fig. 3 A of Arredouani et al. (2002b). The cause of the nadir increase is a greater influx of Ca2+ into the cell, and consequently a higher concentration of Ca2+ in the ER (Fig. 5 C). Thus, release of Ca2+ from the ER is greater during the repolarized phase, yielding an increased Ca2+ nadir. The Ca2+ amplitude is not increased by the longer depolarization because C equilibrates rapidly to the depolarized voltage; it has already nearly equilibrated by the end of an 8-s depolarization, so increasing the duration of depolarization will not significantly increase the cytosolic Ca2+ amplitude.
When the simulation is repeated with SERCA pumps disabled (Fig. 5 B, dashed line) there is again little change in the Ca2+ amplitude when the plateau fraction is increased. However, unlike the control simulation, the Ca2+ nadir is almost unchanged by the increased plateau fraction. This simulation is consistent with Fig. 3 B of Arredouani et al. (2002b).
Increasing the plateau fraction is one way to increase the total Ca2+ entry into the β-cell. Another way to do this is to increase influx through the Ca2+ channels. Arredouani et al. (2002b) achieved this by raising the external Ca2+ concentration. The approach we take in the simulations shown in Fig. 6 is to increase the Ca2+ channel conductance. In the control simulation, doubling gCa increases both the Ca2+ nadir and amplitude (Fig. 6 A). As when the plateau fraction was increased, the nadir is increased primarily because Cer is elevated (Fig. 6 B). An increase in the Ca2+ influx during the repolarized phase adds an additional (small) component to the nadir, which is evident as a small nadir increase when the SERCA pumps are disabled (dashed line). The increased Ca2+ amplitude is evident whether the SERCA pumps are enabled or disabled, and is due to an increase in the Ca2+ entry per unit time. Thus, in contrast to the effects of doubling the plateau fraction (Fig. 5), where the total Ca2+ entry is increased by increasing the duration of cell depolarization (and not the Ca2+ entry per unit time), increasing the conductance increases the influx (Jin) relative to efflux (Jpmca) during the depolarized phase, resulting in an increased Ca2+ amplitude. Results from this simulation, both in control and with SERCA pumps inhibited, are consistent with data in Fig. 4 of Arredouani et al. (2002b).
In summary, the two methods of increasing total Ca2+ entry reported in Arredouani et al. (2002b) and simulated here have different effects on the cytosolic Ca2+. These differential effects can be understood with the two-compartment model, and do not require additional features such as CICR.
A compound bursting pattern provides a good test protocol for Ca2+ handling
Mixed Ca2+ oscillations are sometimes observed in single β-cells or β-cell clusters (Jonkers et al., 1999; Krippeit-Drews et al., 2000) and in islets (Bergsten et al., 1994; Valdeolmillos et al., 1989; Zhang et al., 2003). These oscillations are most likely due to episodes of bursts (Barbosa et al., 1998; Cook 1983; Henquin et al., 1982), which we call compound bursts (Bertram et al., 2004). Compound bursts were mimicked by Arredouani et al. (2002b), and we simulate this protocol in Fig. 7. Here a series of five voltage oscillations is applied, with each oscillation consisting of a 16-s depolarization followed by an 8-s repolarization (Fig. 7 A). Two episodes of compound bursts were applied, separated in time by 2 min. In the control conditions, the cytosolic Ca2+ nadir rose during the first episode and slowly fell during the time between (Fig. 7 B). The slow rise and fall of the nadir reflects the dynamics of the ER Ca2+ concentration (Fig. 7 C). When the SERCA pumps were disabled (kserca2b = 0 and kserca3 = 0) the ER Ca2+ concentration remained flat, and consequently there was no rise in the nadir during the oscillations, and C immediately returned to its baseline value at the end of an episode of bursts (Fig. 7 B, dashed line). These results are consistent with data shown in Fig. 6, B and C, of Arredouani et al. (2002b).
The impact of the ER on the rise and fall of the Ca2+ nadir is further demonstrated in Fig. 8, where the rate of change of Cer has been modified by adjusting the fraction of free Ca2+ in the ER (fer). First, the speed of the ER was increased by a factor of 10 by changing fer from 0.01 to 0.1 (solid curves). In this case, the slow increase in Cer exhibited previously during a compound burst (Fig. 7 C) is replaced by oscillations in Cer around an elevated, but constant, mean. As a result, the cytosolic Ca2+ nadir rises quickly during a bursting episode, exhibiting no slow upward ramp (Fig. 8 A). Also, with the faster ER the cytosolic Ca2+ falls quickly to its baseline value at the end of a compound burst, reflecting the rapid decline in Cer (Fig. 8 B). To examine the other extreme, we decreased the speed of the ER by a factor of 50, setting fer = 0.0002 (dashed curves). In this case, the free ER Ca2+ concentration increases only slightly during a compound burst, and as a consequence the cytosolic Ca2+ nadir returns to baseline between each depolarization, so there is little elevation of the nadir. Thus, a very slow ER has effects on cytosolic Ca2+ that are in some ways similar to the effects of disabling the SERCA pumps. One key difference, however, is that the C amplitude is larger than that of the control when SERCA pumps are disabled (Fig. 7 B), whereas the amplitude is smaller than control when the ER dynamics are slowed by decreasing fer (Fig. 8 A). This is because the filtering performed by the ER is reduced in the former case and increased in the latter case.
In summary, the compound bursting simulations in conjunction with the experimental data of Arredouani et al. (2002b) show clearly the influence that the ER has on the cytosolic Ca2+ dynamics, both during an episode of bursts and between episodes. They also put constraints on the speed of the ER. If the ER fills and empties too rapidly or too slowly, the slow rise and fall of the Ca2+ nadir, which was shown clearly in Arredouani et al. (2002b), is not produced. Although decreasing the speed of the ER by increasing its filtering capacity greatly reduces the rise in the cytosolic Ca2+ nadir during a compound burst, it also decreases the Ca2+ amplitude, in contrast to the increase produced when SERCA pumps are inhibited. Indeed, blocking SERCA pumps is the only manipulation we have found that suppresses the rise in the Ca2+ nadir while increasing the Ca2+ amplitude.
The Ca2+ response is inconsistent with experimental data when CICR is a dominant factor
We next investigate the potential role of Ca2+-induced Ca2+ release (CICR) in β-cells, by making the rate of efflux from the ER, per, an increasing function of the cytosolic Ca2+ concentration (Eq. 6). The extent of the contribution of CICR to the total release from the ER depends on the choice of parameters. The parameter pmin sets the passive release, whereas the magnitude of CICR is determined by pmax−pmin. For example, if pmin = pmax then CICR will contribute nothing to Jrelease. Figures shown thus far have used the basic two-compartment model, where release is passive. All results can be reproduced by a model in which some degree of CICR is included (not shown). However, since CICR tends to reduce the ER Ca2+ concentration relative to that in the cytosol, the effects of blocking SERCA pumps are blunted when CICR is included.
We consider now an extreme case, in which Jrelease is dominated by CICR. We saw previously that when release is passive the ER fills during depolarization and drains during repolarization. On the contrary, when release is dominated by CICR the Ca2+ entering the cell during depolarization induces release from the ER, so the ER initially fills, but then releases once the cytosolic concentration becomes sufficiently large. During repolarization the ER fills since the cytosolic Ca2+ is lower, inducing less release. The response to a train of three depolarizations is demonstrated in Fig. 9.
This CICR model was used to simulate the Ca2+ response during the biphasic voltage protocol (Fig. 10 A). Except for an initial transient period during which the ER was filling, blocking SERCA pumps had little effect on either the Ca2+ nadir or the Ca2+ amplitude (Fig. 10 B). That is, C returned to baseline after each depolarization, whether SERCA pumps were activated or disabled, and the Ca2+ amplitude was slightly decreased when SERCA pumps were blocked. The nadir was not elevated because with this model the ER takes up Ca2+ during the repolarized phase, and it is release of Ca2+ that produces the elevated nadir. The amplitude was abnormally large because the ER does not take up Ca2+ when the cell is depolarized; instead it amplifies the cytosolic Ca2+. Thus, a model in which CICR dominates release from the ER does not reproduce the experimental Tg data (Arredouani et al., 2002b; Gilon et al., 1999).
Continuous depolarization data support the subspace model
The simulations presented thus far highlight dynamic or time-dependent properties of Ca2+ handling. A much simpler simulation, continuous depolarization, highlights steady-state properties and yields some surprising results. During continuous depolarization with elevated K+, Table 1 of Arredouani et al. (2002b) shows that the cytosolic Ca2+ concentration was lower in the presence of Tg. Although they did not report this difference as statistically significant, we note that reductions were observed in each of eight conditions. A statistically significant reduction in steady-state Ca2+ in the presence of Tg was reported in single β-cells by Lemmens et al. (2001).
Contrary to these data, the basic two-compartment model indicates that the steady-state values should be identical whether or not pumps are blocked. This is easily seen from the equations. At steady state, Eq. 5 yields
 |
(14) |
and combined with Eq. 4 we obtain
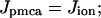 |
(15) |
so from Eq. 1,
 |
(16) |
where C∞ is the equilibrium or steady-state cytosolic Ca2+ concentration. Thus, the steady state depends only on the Ca2+ influx and efflux across the plasma membrane, and is independent of the ER Ca2+ concentration or any parameters relating to the ER. This is true whether release from the ER is passive, as in our basic model, or active through CICR. Thus, the two-compartment models fail to reproduce the continuous depolarization data (Arredouani et al., 2002b; Lemmens et al., 2001).
A three-compartment model has been developed (Goforth et al., 2002) to explain recent data on the buildup and decay of a Ca2+-activated K+ current during a burst of short action-potential-like depolarizations (Goforth et al., 2002; Göpel et al., 1999; Kanno et al., 2002). As described in Methods, this model contains an ER compartment, a bulk cytosolic compartment, and a Ca2+ subspace postulated to exist between the ER and the plasma membrane. In this model, most of the release from the ER enters the subspace compartment, so that the Ca2+ concentration in this compartment is elevated above that of the bulk cytosol (Fig. 1 B). Like the basic two-compartment model, the three-compartment subspace model reproduces all the experimental data related to the dynamics of Ca2+ handling (not shown). However, whereas the two-compartment model (with or without CICR) fails to reproduce the steady-state Ca2+ data, the subspace model correctly predicts a decrease in cytosolic Ca2+when SERCA pumps are blocked.
Fig. 11 shows the Ca2+ concentrations in the three compartments during a continuous depolarization from −70 to −30 mV. Also shown is Cavg, the weighted average of the bulk cytosolic Ca2+ (C) and the subspace Ca2+ (Css) concentrations (Eq. 13). This weighted average is what would be observed in experiments measuring the Ca2+ concentration in the cell. As with the two-compartment model, the bulk cytosolic Ca2+ attains the same steady-state level whether or not the SERCA pumps are inhibited (Fig. 11 C), since the steady-state concentration in this compartment satisfies Eq. 16. However, the subspace Ca2+ concentration has a lower steady-state value when SERCA pumps are blocked (dashed line, Fig. 11 D) since the Ca2+ influx to this compartment is less when the ER is depleted (Fig. 11 B). In fact, the steady-state subspace concentration equals the bulk cytosolic concentration when SERCA pumps are blocked, whereas it is higher when the subspace is intact. Since the observable Cavg is the weighted average of C and Css, it is lower when SERCA pumps are inhibited than in the control case (Fig. 11 A), consistent with the experimental data.
DISCUSSION
We have demonstrated that a simple two-compartment model with passive Ca2+ release from the ER can reproduce recent time-dependent Ca2+ data (Arredouani et al., 2002a,b; Gilon et al., 1999). Although a modest contribution of CICR is compatible with the data, a model in which CICR is the dominant efflux pathway from the ER is not compatible.
Although the two-compartment model, with or without modest CICR, reproduces the time-dependent data, it is not compatible with steady-state Ca2+ data showing that the cytosolic Ca2+ concentration is lower when SERCA pumps are inhibited by Tg (Arredouani et al., 2002b; Lemmens et al., 2001). Prolonged stimulation with high KCl can produce an atypical form of CICR that does not involve the IP3R or RyR in normal mouse β-cells (Beauvois et al., 2004). This could lead to a reduction in cytosolic Ca2+ when SERCA pumps are blocked. However, we note that the atypical CICR only gives a single, transient release in response to a maintained stimulus (Beauvois et al., 2004), which suggests that CICR is not the best explanation for the steady-state reduction. Here, we have shown a three-compartment model that includes a subspace compartment between the ER and the plasma membrane reproduces both the time-dependent and the steady-state data without recourse to CICR.
This subspace model was developed to account for data showing a transient rise and fall of a current tail that develops during a short train of action-potential-like depolarizations (Goforth et al., 2002), and that has been attributed largely to a voltage-independent, Ca2+-activated K+ current (Goforth et al., 2002; Göpel et al., 1999; Kanno et al., 2002). The subspace model accounts for this data, whereas the basic two-compartment model does not. If CICR is added to the two-compartment model and is the dominant factor in release, then the IK(slow) data is reproduced. That is, if a substantial part of the rise in cytosolic Ca2+ that activates IK(slow) is due to release from the ER, then Tg would reduce IK(slow). However, we have shown here that a model with dominant CICR is not compatible with time-dependent Ca2+ data. Thus, in our view, the subspace model most accurately reflects the handling of Ca2+ in pancreatic β-cells.
The ER serves as both a Ca2+ sink and source. When the cell is depolarized Ca2+ enters the cell through ion channels, elevating the cytosolic Ca2+ concentration. This causes the ER to take up Ca2+, therefore acting as a sink, which slows and blunts the rise in cytosolic Ca2+. When the cell is repolarized the influx through ion channels stops and the cytosolic Ca2+ concentration drops due to removal through membrane Ca2+ pumps. As a result Ca2+ is released from the ER, which now acts as a source and slows and antagonizes the fall in cytosolic Ca2+. These dual roles provide the framework for understanding the effects of agents known to modify Ca2+ pathways in β-cells. For example, when Tg is used to block SERCA pumps, the ER will not have any active uptake mechanism and will no longer serve as a Ca2+ sink. Also, since the ER does not fill with Tg present, it will not serve as a Ca2+ source when the cell is repolarized. These two effects of SERCA pump blockage can explain all the time-dependent Tg data.
Knockout experiments (Arredouani et al., 2002b) show that ER sourcing and sinking is also eliminated by removal of SERCA3 alone. Thus, this low affinity isoform is solely responsible for ER Ca2+ oscillations and their attendant effects on cytosolic Ca2+. The high affinity isoform, SERCA2b, is likely nearly saturated at basal Ca2+ and accounts for the ability of the ER to fill, albeit slowly, in low Ca2+, provided sufficient ATP is present (Tengholm et al., 2001).
In addition to accounting for the known data, we have made some new predictions. The model predicts that agents such as carbachol, that increase the release from the ER, will have no long-term effects on the cytosolic Ca2+ dynamics. This prediction can readily be tested. Other agents such as Li+ or calmidazolium reduce efflux across the plasma membrane (Nadal et al., 1994; Wolf et al., 1988) and are predicted to increase the Ca2+ nadir, but have no effect on the Ca2+ amplitude during the biphasic burst-like voltage protocol.
Finally, we have demonstrated that the cytosolic Ca2+ dynamics are affected greatly by the speed at which the ER takes up and releases Ca2+. If the ER is too fast then it will not contribute a slow component to the cytosolic Ca2+ dynamics. This slow component is evident in the slow fall of Ca2+ often observed after bursts of action potentials in free-running (not voltage-clamped) islets (Gilon et al., 1999; Nadal et al., 1994; Valdeolmillos et al., 1989; Zhang et al., 2003). If the ER is too slow then it will take up only a small amount of Ca2+ during burst-like depolarizations. This will have an effect on cytosolic Ca2+ that is somewhat similar to the effect of having no ER compartment at all (i.e., SERCA pumps blocked), and subsequently the ER will not contribute any observable slow component to the Ca2+ signal. Thus, the uptake and release of Ca2+ from the ER must be slow compared to the timescale of cytosolic Ca2+, but not too slow.
There is experimental evidence that a store-operated current (SOC) is activated in β-cells when the ER is drained either through activation of IP3 receptors or through inhibition of SERCA pumps (Mears and Zimliki, 2004; Miura et al., 1997; Worley et al., 1994b). This is a nonselective current, permeable to Ca2+ as well as other ions (Mears and Zimliki, 2004; Worley et al., 1994a). Although we have not included SOC in the simulations presented here, we have performed simulations to test its effects on the time-dependent Ca2+ dynamics (not shown). The extra Ca2+ that enters the model cell through SOC when the ER is depleted elevates both the Ca2+ peak and the Ca2+ nadir when the burst-like depolarization protocol is applied. However, if the Ca2+ component of SOC is relatively small, then the overall effect of draining the ER is to reduce the Ca2+ nadir. The Ca2+ amplitude is increased when the ER is drained regardless of the size of SOC. Also, the Ca2+ component of SOC increases the cytosolic Ca2+ concentration during long depolarizations when the ER is drained. Thus, the Tg-induced reduction in the Ca2+ concentration during long depolarizations reported by Arredouani et al. (2002b) and Lemmens et al. (2001) may be partially blunted by SOC; without SOC the reduction in Ca2+ concentration would be even greater. This provides additional support for the three-compartment subspace model, the only model that predicts a reduction in the steady-state cytosolic Ca2+ concentration when Tg is present. In this way, the effects of SOC make the case for a three-compartment subspace model more compelling.
Acknowledgments
This work was partially supported by National Science Foundation grant DMS-0311856 to R. Bertram.
References
- Ämmälä, C., P.-O. Larsson, P.-O. Berggren, K. Bokvist, L. Juntti-Berggren, H. Kindmark, and P. Rorsman. 1991. Inositol trisphosphate-dependent periodic activation of a Ca2+-activated K+ conductance in glucose-stimulated pancreatic β-cells. Nature. 353:849–852. [DOI] [PubMed] [Google Scholar]
- Arredouani, A., Y. Guiot, J.-C. Jonas, L. H. Liu, M. Nenquin, J. A. Pertusa, J. Rahier, J.-F. Rolland, G. E. Shull, M. Stevens, F. Wuytack, J. C. Henquin, and P. Gilon. 2002a. SERCA3 ablation does not impair insulin secretion but suggests distinct roles of different sarcoendoplasmic reticulum Ca2+ pumps for Ca2+ homeostasis in pancreatic β-cells. Diabetes. 51:3245–3253. [DOI] [PubMed] [Google Scholar]
- Arredouani, A., J.-C. Henquin, and P. Gilon. 2002b. Contribution of the endoplasmic reticulum to the glucose-induced [Ca2+]c response in mouse pancreatic islets. Am. J. Physiol. 282:E982–E991. [DOI] [PubMed] [Google Scholar]
- Ashcroft, F. M., and P. Rorsman. 1989. Electrophysiology of the pancreatic β-cell. Prog. Biophys. Mol. Biol. 54:87–143. [DOI] [PubMed] [Google Scholar]
- Barbosa, R. M., A. M. Silva, A. R. Tomé, J. A. Stamford, R. M. Santos, and L. M. Rosário. 1998. Control of pulsatile 5-HT/insulin secretion from single mouse pancreatic islets by intracellular calcium dynamics. J. Physiol. 510:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvois, M. C., A. Arredouani, J.-C. Jonas, J.-F. Rolland, F. Schuit, J. C. Henquin, and P. Gilon. 2004. Atypical Ca2+-induced Ca2+ release from a sarco-endoplasmic reticulum Ca2+-ATPase 3-dependent Ca2+ pool in mouse pancreatic β-cells. J. Physiol. 559:141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten, P., E. Grapengiesser, E. Gylfe, A. Tengholm, and B. Hellman. 1994. Synchronous oscillations of cytoplasmic Ca2+ and insulin release in glucose-stimulated pancreatic islets. J. Biol. Chem. 269:8749–8753. [PubMed] [Google Scholar]
- Bertram, R., L. Satin, M. Zhang, P. Smolen, and A. Sherman. 2004. Calcium and glycolysis mediate multiple bursting modes in pancreatic islets. Biophys. J. 87:3074–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram, R., P. Smolen, A. Sherman, D. Mears, I. Atwater, F. Martin, and B. Soria. 1995. A role for calcium release-activated current (CRAC) in cholinergic modulation of electrical activity in pancreatic β-cells. Biophys. J. 68:2323–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, D. L. 1983. Isolated islets of Langerhans have slow oscillations of electrical activity. Metabolism. 32:681–685. [DOI] [PubMed] [Google Scholar]
- Gall, D., and I. Susa. 1999. Effect of Na/Ca exchange on plateau fraction and [Ca]i in models for bursting in pancreatic β-cells. Biophys. J. 77:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilon, P., A. Arredouani, P. Gailly, J. Gromada, and J.-C. Henquin. 1999. Uptake and release of Ca2+ by the endoplasmic reticulum contribute to the oscillations of the cytosolic Ca2+ concentration triggered by Ca2+ influx in the electrically excitable pancreatic β-cell. J. Biol. Chem. 274:20197–20205. [DOI] [PubMed] [Google Scholar]
- Gilon, P., M. Nenquin, and J. C. Henquin. 1995. Muscarinic stimulation exerts both stimulatory and inhibitory effects on the concentration of cytoplasmic Ca2+ in the electrically excitable pancreatic β-cell. Biochem. J. 311:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth, P. B., R. Bertram, F. A. Khan, M. Zhang, A. Sherman, and L. S. Satin. 2002. Calcium-activated K+ channels of mouse β-cells are controlled by both store and cytoplasmic Ca2+: experimental and theoretical studies. J. Gen. Physiol. 114:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpel, S. O., T. Kanno, S. Barg, L. Eliasson, J. Galvanovskis, E. Renström, and P. Rorsman. 1999. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β-cells. J. Gen. Physiol. 114:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada, J., J. Frøkjær-Jensen, and S. Dissing. 1996. Glucose stimulates voltage- and calcium-dependent inositol trisphosphate production and intracellular calcium mobilization in insulin-secreting βTC3 cells. Biochem. J. 314:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin, J. C., H. P. Meissner, and W. Schmeer. 1982. Cyclic variations of glucose-induced electrical activity in pancreatic β-cells. Pflugers Arch. 393:322–327. [DOI] [PubMed] [Google Scholar]
- Jonkers, F. C., J.-C. Jonas, P. Gilon, and J. C. Henquin. 1999. Influence of cell number on the characteristics and synchrony of Ca2+ oscillations in clusters of mouse pancreatic islet cells. J. Physiol. 520:839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, G., and G. G. Holz. 2003. Amplification of exocytosis by Ca2+-induced Ca2+ release in INS-1 pancreatic β-cells. J. Physiol. 546:175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno, T., P. Rorsman, and S. O. Göpel. 2002. Glucose-dependent regulation of rhythmic action potential firing in pancreatic β-cells by KATP-channel modulation. J. Physiol. 545:501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krippeit-Drews, P., M. Dufer, and G. Drews. 2000. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic β-cells. Biochem. Biophys. Res. Commun. 267:179–183. [DOI] [PubMed] [Google Scholar]
- Lemmens, R., O. Larsson, P.-O. Berggren, and M. S. Islam. 2001. Ca2+-induced Ca2+ release from the endoplasmic reticulum amplifies the Ca2+ signal mediated by activation of voltage-gated L-type Ca2+ channels in pancreatic β-cells. J. Biol. Chem. 276:9971–9977. [DOI] [PubMed] [Google Scholar]
- Mears, D., and C. L. Zimliki. 2004. Muscarinic agonists activate Ca2+ store-operated and -independent ionic currents in insulin-secreting HIT-T15 cells and mouse pancreatic β-cells. J. Membr. Biol. 197:59–70. [DOI] [PubMed] [Google Scholar]
- Meissner, H. P., and I. Atwater. 1976. The kinetics of electrical activity of β-cells in response to “square wave” stimulation with glucose or glibenclamide. Horm. Metab. Res. 8:11–15. [DOI] [PubMed] [Google Scholar]
- Miura, Y., J. C. Henquin, and P. Gilon. 1997. Emptying of intracellular Ca2+ stores stimulates Ca2+ entry in mouse pancreatic β-cells by both direct and indirect mechanisms. J. Physiol. 503:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal, A., M. Valdeolmillos, and B. Soria. 1994. Metabolic regulation of intracellular calcium concentration in mouse pancreatic islets of Langerhans. Am. J. Physiol. 30:E769–E774. [DOI] [PubMed] [Google Scholar]
- Tengholm, A., B. Hellman, and E. Gylfe. 2001. The endoplasmic reticulum is a glucose-modulated high-affinity sink for Ca2+ in mouse pancreatic β-cells. J. Physiol. 530:533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdeolmillos, M., R. M. Santos, D. Contreras, B. Soria, and L. M. Rosario. 1989. Glucose-induced oscillations of intracellular Ca2+ concentration resembling electrical activity in single mouse islets of Langerhans. FEBS Lett. 259:19–23. [DOI] [PubMed] [Google Scholar]
- Varadi, A., and G. A. Rutter. 2002. Dynamic imaging of endoplasmic reticulum Ca2+ concentration in insulin-secreting MIN6 cells using recombinant targeted chameleons: roles of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)−2 and ryanodine receptors. Diabetes. 51:S190–S201. [DOI] [PubMed] [Google Scholar]
- Wolf, B. A., J. R. Colca, J. Turk, J. Florholmen, and M. L. McDaniel. 1988. Regulation of Ca2+ homeostasis by islet endoplasmic reticulum and its role in insulin secretion. Am. J. Physiol. 17:E121–E136. [DOI] [PubMed] [Google Scholar]
- Worley, J. F., M. S. McIntyre, B. Spencer, and I. D. Dukes. 1994a. Depletion of intracellular Ca2+ stores activates a maitotoxin-sensitive nonselective cationic current in β-cells. J. Biol. Chem. 269:32055–32058. [PubMed] [Google Scholar]
- Worley, J. F., M. S. McIntyre, B. Spencer, R. J. Mertz, M. W. Roe, and I. D. Dukes. 1994b. Endoplasmic reticulum calcium store regulates membrane potential in mouse islet β-cells. J. Biol. Chem. 269:14359–14362. [PubMed] [Google Scholar]
- Zhang, M., P. Goforth, A. Sherman, R. Bertram, and L. Satin. 2003. The Ca2+ dynamics of isolated mouse β-cells and islets: implications for mathematical models. Biophys. J. 84:2852–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]