

Abstract
Single-molecule manipulation techniques have enabled the characterization of the unfolding and refolding process of individual protein molecules, using mechanical forces to initiate the unfolding transition. Experimental and computational results following this approach have shed new light on the mechanisms of the mechanical functions of proteins involved in several cellular processes, as well as revealed new information on the protein folding/unfolding free-energy landscapes. To investigate how protein molecules of different folds respond to a stretching force, and to elucidate the effects of solution conditions on the mechanical stability of a protein, we synthesized polymers of the protein ubiquitin and characterized the force-induced unfolding and refolding of individual ubiquitin molecules using an atomic-force-microscope-based single-molecule manipulation technique. The ubiquitin molecule was highly resistant to a stretching force, and the mechanical unfolding process was reversible. A model calculation based on the hydrogen-bonding pattern in the native structure was performed to explain the origin of this high mechanical stability. Furthermore, pH effects were studied and it was found that the forces required to unfold the protein remained constant within a pH range around the neutral value, and forces decreased as the solution pH was lowered to more acidic values.
INTRODUCTION
Experimental evidence suggests that force-induced protein unfolding is an essential step in several important cellular processes, such as protein degradation by ATP-dependent proteases and protein translocation across certain membranes (Matouschek, 2003). The mechanisms of some of these mechanical functions have been investigated by direct measurements of the response of protein molecules to externally applied forces using single-molecule manipulation techniques such as atomic force microscopy (AFM) and laser tweezers. The single-molecule approach also offers a novel means to investigate the protein-folding problem through the direct observation of the unfolding events of individual protein molecules induced by an externally applied stretching force. This method can provide complementary information on the unfolding-refolding pathways of a protein to that obtainable from bulk biochemical and biophysical measurements. The giant protein titin from muscle was the first protein to be investigated using this approach (Kellermayer et al., 1997; Rief et al., 1997; Tskhovrebova et al., 1997). The globular domains of titin have since been used in several studies, yielding new information on the folding/unfolding free-energy landscape as well as the function mechanisms for this protein (Williams et al., 2003; Marszalek et al., 1999; Oberhauser et al., 1999). In addition to titin domains, a few other proteins have also been studied using the mechanical unfolding technique, including the all-α cytoskeletal protein spectrin (Rief et al., 1999; Law et al., 2003), the intracellular matrix protein tenascin (Oberhauser et al., 1998), the lysozyme from bacteriophage T4 (Yang et al., 2000), and the enzyme barnase (Best et al., 2001). It was found from these studies that proteins with certain folds seem to require a higher force to unfold. Molecular dynamics simulations have been performed on several of these systems to provide an atomistic view of the force-induced unfolding and refolding of protein molecules (Gao et al., 2002; Lu et al., 1998), albeit on a different timescale. However, it is still not clear what factors determine the mechanical stability of a protein molecule, and more importantly, what information on the unfolding and refolding pathways can be extracted from the mechanical unfolding/refolding data. To take full advantage of this technique, it is essential to investigate a wider range of protein molecules with different structural and thermodynamic properties, and under various solution conditions.
In the mechanical unfolding experiments, the macromolecules are tethered between two surfaces. Ideally, only one protein molecule should be tethered and studied. It has been reported that the force-induced unfolding of a single protein molecule was studied with AFM (Hertadi and Ikai, 2002; Hertadi et al., 2003). However, because radii of curvature of the tethering surfaces (a bead in the laser tweezers or an AFM tip) are all much larger than the dimensions of a typical globular protein molecule, the nonspecific interactions between the surfaces often conceal the force exerted on the protein molecule and thus make it difficult to unequivocally interpret the experimental data. To clearly observe the unfolding events the two surfaces need to be kept far apart. For this reason, the first mechanical protein-unfolding studies used proteins that naturally occur as tandem arrays of globular domains (Rief et al., 1997), and most of the mechanical unfolding studies since have also used proteins in the polymeric form. The naturally occurring polymeric proteins are not ideal to investigate protein folding because the heterogeneity in the domains complicates the data interpretation, thus limiting the information contents of the experiments. To circumvent this problem, polymers of identical globular protein molecules have been synthesized by cloning multiple copies of the gene using a protein-engineering technique (Carrion-Vazquez et al., 2000) or by linking the domains via disulfide bonds using the solid-state synthesis method (Yang et al., 2000). When a polymer of globular protein molecules is subject to a stretching force, it is still possible to observe the unfolding of individual molecules due to the stochastic nature of protein-folding events and the experimental scheme. When pulled from its ends, the tension in the polymer is the same throughout its length and the extension of the polymer is the sum of the extensions of all the molecules in the polymer. When using a relative stiff force sensor, such as an AFM cantilever, unfolding of a protein molecule in the polymers leads to a sudden lengthening of the chain and thus an abrupt drop in the tension. Such a process makes it unlikely that two or more molecules unfold simultaneously or closely following each other in time. The next unfolding event is mostly likely to occur only after the tension rises again to a certain level in the ensuing pulling of the chain. Thus, these pulling experiments readily yield the unfolding behaviors of individual molecules. Up to now, there are only a limited number of protein systems that have been successfully polymerized and studied in mechanical unfolding experiments. The difficulties may arise from several sources. For example, significant changes in the protein's structure and stability might be induced from polymerization; the linked multiple copies of the gene might not be expressed; the mechanical stability of the protein could be too low to generate detectable signals; and the polymerized protein may not generate single-molecule unfolding data due to aggregation and/or weak attachment to the surfaces.
We have synthesized polymers of the protein ubiquitin and characterized the unfolding behaviors of this protein when subjected to mechanical forces. Ubiquitin is a protein that has been extensively studied with various methods. Several features make ubiquitin an excellent model protein for protein-folding investigations: 1), it is a small protein, consisting of 76 amino acid residues (molecular weight of 8433), without disulfide bonds or other structural complications; 2), its high-resolution three-dimensional structure is known from x-ray crystallography and NMR studies; 3), thermal unfolding of ubiquitin is reversible and conforms closely to the two-state equilibrium model in most experimental measurements; 4), ubiquitin is very stable at neutral pH, with denaturation temperature >100°C (Makhatadze et al., 1998); and 5), a library of mutants has been developed and characterized. Recently, several articles were published by the Fernandez group on single-molecule studies of ubiquitin. Carrion-Vazquez et al. (2003) reported their results on the mechanical unfolding of ubiquitin. Using both N-C-linked and K48-C-linked polymers, they found that the forces required to unfold ubiquitin are strongly dependent on the direction along which the force is applied, which may indicate a general mechanism of macromolecular mechanical function in biological systems. Fernandez and Li (2004) used the force-clamp atomic force microscopy to characterize the folding pathways of ubiquitin. These experiments provided the first direct observations of the folding trajectory of single-protein molecules. They found that ubiquitin folding occurs through a series of continuous stages instead of well-defined states. Schlierf et al. (2004) studied the kinetics of unfolding of ubiquitin and found that, at the single-protein level, ubiquitin unfolding is well described by a simple two-state kinetic model. However, rare events not following the simple two-state kinetics did occur, revealing the diversity of pathways available to a protein undergoing forced unfolding.
In this work, we have studied the mechanical unfolding of ubiquitin molecules, in N-C-linked polymers, in more detail; we especially characterized the refolding as well as the unfolding behaviors of ubiquitin and made measurements in solutions with different pH values. In addition, we calculated the unfolding forces based on the strength of the hydrogen bonds between the two β-strands at the N- and C-termini.
MATERIALS AND METHODS
Cloning of polymeric ubiquitin gene
The clone containing the ubiquitin (UBI) gene, pTOBUBI, was purchased from American Type Culture Collection (Manassas, VA). The polymerase chain reaction (PCR) technique was used to amplify the monomeric UBI gene using the pTOBUBI as a template. Two primers used for the PCR reaction were 5′-cgggatccatgcagatattcgtgaaa accc-3′ and 5′-cgtctgagaggtggtagatcttgctgctgatgacggccg-3′. The former, which was used as the 5′-end primer, contained a BamHI restriction enzyme cleavage site. The latter, the 3′-end primer, contained a BglII restriction enzyme cleavage site, two Cys codons, a stop codon, and an XhoI restriction enzyme cleavage site sequentially. The PCR-amplified monomeric UBI gene was purified and ligated to a pGEM-T vector (Promega, Madison, WI). The constructs were screened by the α-complementation and restriction enzyme digestion map. The selective clones were verified by automatic DNA sequencing of the entire coding region and the restriction enzyme cleavage sites. The verified construct was designated as pGEMTUBI_1. The dimeric and tetrameric UBI genes were constructed by iterative cloning based on a previously published protocol (Carrion-Vazquez et al., 1999) with modifications. The BamHI/XhoI doubly digested monomeric UBI gene was ligated to the BglII/XhoI doubly digested pGEMTUBI_1 plasmid. The constructs containing dimeric UBI genes were screened by the restriction enzyme digestion map and verified by automatic DNA sequencing of the entire coding region. The verified construct was designated as pGEMTUBI_2. The procedures to construct the tetrameric and octameric UBI genes, pGEMTUBI_4 and pGEMTUBI_8, were the same as those for pGEMTUBI_2. The structure and restriction enzyme digestion analysis of the expression plasmids pETUBI_1, pETUBI_2, pETUBI_4, and pETUBI_8 are shown in Fig. 1.
FIGURE 1.

(A) Structure of expression vectors, pETUBI_N containing N tandem repeats of human ubiquitin cDNA. The insert region containing ubiquitin gene and the cloning sites are shown in the black arrow. The cDNA inserted between the T7 promoter and T7 terminator is under control of strong bacteriophage T7 transcription. Kan and ori represent the kanamycin resistance gene and the origin of replication, respectively. (B) Restriction enzyme digestion and agarose gel analysis of pETUBI_N. The uncut plasmids, pETUBI_1, pETUBI_2, pETUBI_4, pETUBI_8 are shown in lanes 2, 4, 6, and 8. The BamHI/XhoI doubly digested plasmids are shown in lanes 3, 5, 7, and 9. The upper bands show the pET30a vehicle, and lower bands show the ubiquitin monomer gene (lane 3), dimer gene (lane 5), tetramer gene (lane 7), and octamer gene (lane 9), respectively.
Overexpression and purification of recombinant monomeric, dimeric, tetrameric, and octameric ubiquitin
The BamHI/XhoI doubly digested monomeric, dimeric, tetrameric, and octameric UBI genes were subcloned into a pET30a expression vector separately (Studier et al., 1990). The verified plasmids, designated as pETUBI_1, _2, _4, and _8, were transformed into Escherichia coli strain BL21(DE3). The transformed cells were grown at 37°C to 1 OD600nm in Luria Broth, then 0.5 mM isopropyl-β-d-thiogalactopyranoside was added to induce gene expression. After 3 h of induction, cells were harvested by centrifugation. The harvested cells were resuspended in 50 mM Tris-HCl buffer (pH 8.0), containing 1 mM EDTA, 1 mM β-mercaptoethanol, and 0.1 mM phenylmethanesulfonyl fluoride, and lysed by repeat “freeze-and-thaw.” After centrifugation, the supernatant was collected and applied to a Ni-NTA agarose column. The column was washed with 5 mM imidazol and eluted with 40–60 mM imidazol. Enterokinase (6U) was then added to the eluting sample and incubated overnight. The sample was reapplied to the Ni column to remove the fragment containing His-tag. The final construct contains AMADIGS residues on the N-terminus, single or multiple repeats of ubiquitin, Arg-Ser residues in between each repeat of ubiquitin, and two cysteine residues on the C-terminus. Each of the purified monomeric, dimeric, and tetrameric UBI proteins ran as a single band in SDS-PAGE as shown in Fig. 2. The octameric UBI protein was only partially purified, with a purity of ∼60–80%, as shown in Fig. 2, but the octameric samples were readily usable in the experiments of mechanical unfolding of single molecules.
FIGURE 2.

Overproduction and purification of human monomeric and polymeric ubiquitins as analyzed by SDS-PAGE. SDS-PAGE analysis of monomeric (lanes 1–5), dimeric (lane 6), tetrameric (lane 7), and octameric (lane 8) ubiquitins was shown. The descriptions of individual lanes are as follows. Molecular weight marker proteins (M. W.), total cell lysate of E. coli. BL21(DE3) carry pETUBI_1 after induction (lane 1), the fusion proteins bound to Ni-NTA agarose column (lane 2), the fusion proteins eluted from Ni-NTA agarose column by use of elution buffer containing 40 mM imidazole (lane 3), the eluted fusion proteins after enterokinase cleavage (lane 4), the purified recombinant ubiquitin after removing the His-containing tag region by Ni-NTA affinity chromatography (lane 5), purified dimeric (lane 6), tetrameric (lane 7), and octameric (lane 8) ubiquitins. Recombinant human ubiquitin polymers were purified from E. coli lysate as described under Materials and Methods. Lanes 6, 7, and 8 were cut from separate gels with the positions of the bands adjusted to the markers on the left.
Circular dichroism study and secondary structure analysis
CD spectra were recorded on a Jasco J-715 circular dichroism spectrometer. CD spectra were collected using a cylindrical quartz cuvette with a 1-mm pathlength. The step resolution was 0.2 nm with 1.0-nm bandwidth at a scan speed of 50 nm/min. Each CD spectrum was averaged over 16 measurements and corrected for the appropriate buffer baseline. All spectra are presented as the molar CD absorption coefficient (ΔɛM). Concentrations of monomeric, dimeric, and tetrameric ubiquitins were determined by ultraviolet (UV) absorption using the extinction coefficient, ɛ276nm, 1450 M−1 cm−1 per single ubiquitin monomer. Sample concentrations were 10 μM. The contents of secondary structures were calculated from the neural network program CDNN (Bohm et al., 1992).
Thermodynamic stability measurements
The equilibrium constant for the unfolding of ubiquitin in the presence of GdnHCl can be described as follows using a two-state model.
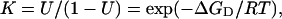 |
where U is the fraction of unfolded state, and ΔGD represents the free energy of unfolding of proteins in the presence of GdnHCl. It has been found experimentally that the free energy of unfolding of proteins in the presence of GdnHCl is linearly related to the concentration of GdnHCl (Pace, 1986):
 |
where 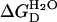 is the apparent free energy of unfolding in the absence of denaturant, and m is a measure of the dependence of free energy on GdnHCl concentration. By combining these two equations, the fraction of the unfolded state (U) can be written as the following equation:
is the apparent free energy of unfolding in the absence of denaturant, and m is a measure of the dependence of free energy on GdnHCl concentration. By combining these two equations, the fraction of the unfolded state (U) can be written as the following equation:
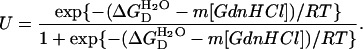 |
Experimental data can be fitted according to this equation using a Boltzmann regression analysis algorithm in the program Origin 6.0 (Microcal Software, Northampton, MA).
Mechanical unfolding measurements
The mechanical unfolding experiments were performed using a modified commercial Nanoscope III scanning probe microscope (Digital Instruments/Veeco, Santa Barbara, CA). A desktop PC, running programs written in LabView (National Instruments, Austin, TX), was employed to control the movements of the AFM tip relative to the sample surface. The cantilevers used in the experiments were triangular, Si3N4 cantilevers (purchased from ThermoMicroscopes/Veeco, Sunnyvale, CA), with a nominal spring constant of 50 pN/nm. The value of the spring constant of each cantilever was calibrated individually using the method of thermal energy equipartition (Hutter and Bechhofer, 1993). The ubiquitin polymer was dissolved in PBS buffer (126 mM NaCl, 7.2 mM Na2HPO4, 3 mM NaH2PO4, pH 7.0) with a protein concentration of 50 μg/ml. The specimen for the mechanical unfolding experiments was prepared by depositing 20 μl of the protein solution on a fresh gold surface and allowing the molecules to adsorb for 10 min. After washing off the unbound molecules with PBS, the sample was placed in the liquid chamber of the AFM. The experiments were carried out with both the sample and the tip immerged in the same buffer. The tip was first pushed onto the sample surface with a force of a few nanonewtons for 5 s to allow the molecules to interact with and attach to the tip. The tip was then retracted from the surface at a specified speed and the force was measured as a function of the tip-sample separation. For unfolding ubiquitin molecules in solutions with different pH values, the samples were prepared in two different ways. In the first method, the sample was prepared in PBS buffer of pH 7. After the sample was mounted in liquid chamber of the AFM, the pH 7 buffer was washed out with a buffer of the desired pH value, followed by the mechanical unfolding measurement. In the second method, the ubiquitin polymer was directly dissolved in a buffer of the desired pH value (adjusting using HCl or NaOH), and this same buffer was used throughout the sample preparation and the subsequent measurement. The results obtained after these two procedures were not distinguishable.
For the refolding experiments, the tethered polymer chain was relaxed after several unfolding events had been observed in the force curve, by bringing the AFM tip to a specified position near the sample without touching the surface. After holding the tip at that position for a specified period of time, the protein polymer was stretched again. This process was repeated until the polymer detached from the surfaces.
Data analysis
The raw data were first screened for curves showing multiple unfolding events, with the characteristic “saw-tooth” pattern. The selected data curves were further processed by eliminating any artifacts from the thermal drifts, and converting the scales into force and distance from the experimental parameters. To evaluate the structural changes upon the unfolding of a globular protein domain in the polymer, the force-versus-extension relationship was fitted to the wormlike-chain (WLC) model (Bustamante et al., 1994):
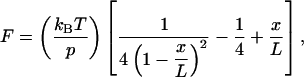 |
where L is the contour length, x is the end-to-end distance of the chain, p is the persistence length, and kB and T are the Boltzmann constant and the absolute temperature, respectively. This formula describes the relationship between the tension (F) and the relative extension (x/L) of an ideal entropic chain, which is used to fit the data to obtain the contour length of the protein polymer.
Monte Carlo simulation
Monte Carlo simulation was performed to elucidate the unfolding rate of the protein. In the simulation, force-versus-extension curves are generated by assuming the polymer to be a WLC chain, and the cantilever to be a linear spring. To determine if a still-folded protein molecule will unfold, a probability is calculated according to the theory developed by Bell (1978) and elaborated by Evans and Ritchie (1999) for two-state unfolding:
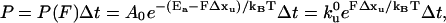 |
where Ea is the activation energy barrier for unfolding, F is the pulling force experienced by the protein, Δxu is the distance between the folded state and the transition state along the pulling direction, Ao is an attempt frequency,  is the unfolding rate when no external force is present, and Δt is the time interval over which force F is acting on the protein. At each force level, each folded molecule in the polymer is checked for unfolding by comparing the unfolding probability with a randomly generated number before the chain is pulled further. One-hundred force curves on pulling an octameric chain were generated, which yielded 800 points, for each set of parameters. The values of
is the unfolding rate when no external force is present, and Δt is the time interval over which force F is acting on the protein. At each force level, each folded molecule in the polymer is checked for unfolding by comparing the unfolding probability with a randomly generated number before the chain is pulled further. One-hundred force curves on pulling an octameric chain were generated, which yielded 800 points, for each set of parameters. The values of 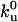 and Δxu for the protein are obtained as the adjusting parameters in fitting the Monte Carlo simulation to the experimental data. The simulation provides distribution of the unfolding force at a particular pulling speed, as well as the dependence of the unfolding forces on the pulling speed. Both sets of data are fitted to the experimental results in obtaining the parameters
and Δxu for the protein are obtained as the adjusting parameters in fitting the Monte Carlo simulation to the experimental data. The simulation provides distribution of the unfolding force at a particular pulling speed, as well as the dependence of the unfolding forces on the pulling speed. Both sets of data are fitted to the experimental results in obtaining the parameters 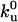 and Δxu.
and Δxu.
Calculation of the unfolding forces
The native structure of ubiquitin (1UBQ) contains a five-stranded β-sheet, a 3.5-turn α-helix, and a 310-helix. The five β-strands are arranged in the order of β4-β3-β5-β1-β2, with β1 and β5 parallel and other strands packed in an antiparallel arrangement (see, e.g., Fig. 3 in Cordier and Grzesiek, 2002). The β1-β5 strands are connected by five backbone hydrogen bonds: Q2 → E64, S65 → F4, F4 → L67, L67 → K6, K6 → L69. Here the notation is such that the arrows point in the direction from the carbonyl oxygen acceptor toward the amide proton donor, and the numbers indicate the sequence positions of the amino acids. In the native structure, the β1 and β5 strands are approximately parallel to each other in their spatial arrangements. This conformation is similar to the relative position between A′ and G strands of the cardiac titin I27 immunoglobulin domain in its native structure (Lu et al., 1998).
FIGURE 3.

(A) Far-UV CD spectra of recombinant monomeric and polymeric ubiquitin. Monomeric ubiquitin is represented in solid line, dimeric ubiquitin in dashed line, and tetrameric ubiquitin in dotted line. (B) The equilibrium unfolding of tetrameric ubiquitin monitored by CD at the wavelength of 215 nm (•) and 222 nm(○) was compared with the equilibrium unfolding of monomeric ubiquitin by fluorescence with the excitation at the wavelength of 274 nm and the emission of 305 nm (▴).
In our simplified model, it is assumed that the first and last amino acid residues of a folded-protein molecule in the polymer are pulled away from each other. The direction from R72 to M1 in the native structure is chosen as the direction of pulling. This choice is based on the native structure of ubiquitin and the pulling geometry in the experiment. Nevertheless, the choice does involve a certain degree of arbitrariness. Let 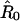 be a unit vector in the direction of pulling, it is assumed that the β1 strand moves along
be a unit vector in the direction of pulling, it is assumed that the β1 strand moves along 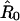 and β5 strand remains stationary during the pulling process. A reaction coordinate can thus be defined by the movement
and β5 strand remains stationary during the pulling process. A reaction coordinate can thus be defined by the movement 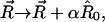 where
where 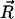 is the location of any atom on the β1 strand and the reaction coordinate, α, goes from 0 to a positive value much larger than unity.
is the location of any atom on the β1 strand and the reaction coordinate, α, goes from 0 to a positive value much larger than unity.
The force field of hydrogen bonds used to calculate the potential change along this reaction coordinate is that of Hagler et al. (1974). The potential energy of each of the backbone hydrogen bonds is given by
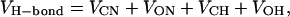 |
where C, O, N, H are the atoms involved in H-bonding and VAB is given by
 |
The values of the Lennard-Jones parameters (r* and ɛ) and the point charge (q) used in our calculation are from Hagler et al. (1974), where the Lennard-Jones parameters for the H atoms are zero. Because the positions of hydrogen atoms are not given in Protein Data Bank (PDB) data, they are hereto determined by assuming that the geometry of the hydrogen bond, O–H-N, is linear and NH bond length is 0.99 Å. Furthermore, it is assumed that the stretching of the protein molecules is carried out quasistatically and the unfolding force obtained in our model will be for those cases where the unfolding force is solely determined by the five backbone hydrogen bonds, without contributions from side-chain interactions. For real protein molecules, the unfolding force is determined by various interactions, including hydrogen bonding and side-chain interactions. For certain protein molecules, hydrogen bonds form a “clamp” between two β-strands, such as that between strands A′ and G in titin domain I27 and that between strands β1 and β5 in ubiquitin. When the two β-strands are pulled along the parallel direction, the breaking of these hydrogen bonds forms the barrier to the mechanical unfolding and dominates the maximum unfolding force as indicated by experimental evidence (Carrion-Vazquez et al., 1999) and the molecular dynamics simulations (Lu et al., 1998; Lu and Schulten, 2000). Side-chain interactions can have significant effects on the mechanical unfolding results because proteins with very similar arrangement of secondary structures were found to unfold at different forces (Li et al., 2000). The calculation here is to show that the breaking of the five hydrogen bonds between strands β1 and β5 dominates the forces required to unfolding a ubiquitin molecule. All the calculations reported in the Results and Discussion section were done using MAPLE (Maplesoft, Waterloo, Ontario, Canada).
RESULTS AND DISCUSSION
Secondary structure determination
To assess the effects of the polymerization on the native structure of ubiquitin, CD was used to characterize the secondary structure contents of monomeric, dimeric, and tetrameric ubiquitin in the far-UV region, as shown in Fig. 3 A. The spectra in the far-UV region were used to estimate the percentage of secondary structure. The estimated α-helix and β-sheet content are 16% and 33%, respectively, for the commercially obtained ubiquitin, 14% and 34% for the engineered monomeric, 14% and 33% for the engineered dimeric, and 13% and 36% for the engineered tetrameric ubiquitin. As shown in Table 1, the estimated α-helical and β-sheet contents of the recombinant ubiquitin monomer, dimer, and tetramer are very similar and also very close to the secondary structure content determined from the x-ray structure (1UBQ) and NMR structure (1D3Z).
TABLE 1.
Summary of the estimated percentage of secondary structure of commercial and recombinant ubiquitins
| X-ray | Commercial | Monomer | Dimer | Tetramer | |
|---|---|---|---|---|---|
| α-Helix* | 16† | 16 | 14 | 14 | 13 |
| β-Sheet | 32 | 33 | 34 | 33 | 36 |
| Turn and random coil | 52 | 51 | 52 | 53 | 51 |
Percentages of the secondary structure of recombinant ubiquitins were estimated from the far-UV CD spectra (195–260 nm) using the neural network program, CDNN. Thirty-three basis sets were used in the calculations.
Percentage was calculated from the x-ray structure (pdb ID, 1ubq).
Bulk thermodynamic stability measurements
We first compared the stabilities of monomeric and tetrameric ubiquitin by analyzing their unfolding by denaturant, as monitored by a decrease in the intrinsic fluorescence or the secondary structure content of the proteins. The fraction of unfolded state was plotted as a function of GdnHCl concentration (Fig. 3 B). The unfolding curves of both the monomeric and tetrameric ubiquitin coincide and show cooperative characteristics. The transition curve was quantified by the methods described in Materials and Methods, and the corresponding values of 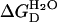 for monomeric and tetrameric ubiquitin are similar, 6.7, and 7.2 kcal/mol, respectively. These observations indicate that the polymerization has no significant effect on the free energy of unfolding of ubiquitin.
for monomeric and tetrameric ubiquitin are similar, 6.7, and 7.2 kcal/mol, respectively. These observations indicate that the polymerization has no significant effect on the free energy of unfolding of ubiquitin.
Mechanical unfolding of ubiquitin molecules
Fig. 4 A shows several force-versus-extension curves obtained when individual polymers of ubiquitin were stretched in the AFM. Each peak corresponds to the sequential unfolding of an individual protein molecule. The mechanical unfolding was observed to be an “all-or-none” or a two-state process, without any detectable unfolding intermediate states. As predicted by the Bell model (Bell, 1978; Evans and Ritchie, 1999), the force required to unfold the protein is linearly dependent on the logarithm of the force-loading rate, which is equal to the product of the pulling speed and the effective spring constant of the polymer-cantilever system. Fig. 4 B shows the dependence of the unfolding forces as a function of the pulling speed.
FIGURE 4.

(A) Force curves from pulling octameric ubiquitin molecules. The top curve was generated from Monte Carlo simulation, whereas the other three curves were from experiments. In the experiments, the ubiquitin octamer could be tethered between the surface and the tip at any two points on the chain; thus the number of unfolding events observed was different for each pulling. The level portion of each curve corresponds to zero force where the tethered polymer chain detached from the tip or the sample. The pulling speed was 1000 nm/s. (B) The pulling speed dependence of the average unfolding forces. The solid circles are experimental data and the squares are simulation data. The parameters used for the Monte Carlo simulation were Δxu = 0.225 nm and, 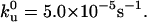
When a protein molecule in the tethered polymer unfolds, the contour length of the polymer increases by an amount of ΔL, which is equal to the distance between the two terminal residues in a fully extended unfolded protein minus the distance between the two terminal residues in a native protein molecule. Because of this length increment, there is a sudden drop in the tension of the polymer chain for each unfolding event, resulting in the saw-tooth pattern. Because the polymers chain is never fully extended during the pulling process, the distance between two adjacent peaks in a force curve is not equal to ΔL. To extract the values of ΔL, we fit the stretching part of each peak to the WLC model because it has been shown that WLC is an adequate model to describe the elastic behavior of a polypeptide chain (Carrion-Vazquez et al., 2000). Fig. 5 A shows a force curve with each peak fitted to the WLC. The distribution of ΔL values obtained in this way is plotted in Fig. 5 B. The average value determined from the force curves, ΔL = 24.5 ± 1.7 nm, is in good agreement with the expected value of 24.4 nm, which is obtained from the crystal structure (1UBQ) of ubiquitin and polypeptide conformation as described below. In the native structure, the first and last amino acids are separated by a distance of 3.7 nm. In the unfolded polypeptide chain, the distance between two adjacent α-carbon atoms is 0.38 nm (Voet and Voet, 1995). However, the tetrahedral geometry reduced the maximal extension of a polypeptide chain to ∼0.37 nm per residue, as in a fully extended β-sheet conformation (Voet and Voet, 1995). For an unfolded ubiquitin molecule, the fully extended length (contour length) is thus 76 × 0.37 nm = 28.1 nm. Therefore, each unfolding event will increase the length by ΔL = 28.1 − 3.7 nm = 24.4 nm. We have also checked the distances between α-carbon atoms along the axes of β-sheet strands in the crystal structures of ubiquitin and titin domain I27 using their native structure data, and found that the maximum value of the inter-α-carbon distances was ∼0.35 nm. Thus, it appears that polypeptide chains do not assume the fully extended conformation possible in a native protein structure.
FIGURE 5.

(A) A force curve from mechanical unfolding of octameric ubiquitin with the stretching parts of each peak fitted with the WLC model. The persistence length used in the model fitting is p = 0.4 nm. (B) The distribution of the contour length increments of the polymer chain upon each unfolding event. The values were obtained from WLC fitting of experimental curves as shown in panel A.
From force-extension curves similar to those of Fig. 4 A, the forces required to unfold the ubiquitin molecules were determined and the results are plotted in Fig. 6. The average unfolding force is 230 ± 34 pN (mean ± SD, number of points n = 169) when stretched at a speed of 1000 nm/s. This value is in good agreement with the results reported by Carrion-Vazquez et al. (2003) (203 ± 35 pN at a pulling speed range of 250–410 nm/s). It should be noted that the distribution of the measured unfolding forces was mainly due to the stochastic nature of the individual unfolding events, with a minor contribution from the instruments, as shown by the Monte Carlo simulation results also shown in Fig. 6. By comparing the distribution and the pulling speed dependence of the unfolding forces from experimental measurements with that from Monte Carlo simulations, it was found that an optimal agreement was obtained if the parameters were Δxu = 0.225 nm and, 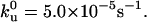 These results are consistent with previously measured results in mechanical unfolding experiments and in bulk kinetics measurements (Sivaraman et al., 2001). The average unfolding force for ubiquitin is slightly larger than that measured for titin I27 (−200 pN) under similar conditions (Carrion-Vazquez et al., 2000), and much higher than the unfolding forces for spectrin (∼30 pN) (Rief et al., 1999) and for T4 lysozyme (Yang et al., 2000). These values of unfolding forces reflect the structural properties that determine the mechanical stability of the protein molecules. Titin Ig domains possess a β-barrel motif, spectrin repeats have a triple-helical coiled-coils structure, T4 lysozyme is mainly α-helical, whereas ubiquitin has a mixed α-β structure. A steered molecular dynamics simulation (SMD) of the mechanical unfolding of I27 domain of titin revealed that the force peaks in the force-extension curves observed in atomic force spectroscopy experiments were mainly due to the initial disruption of the backbone hydrogen bonds between antiparallel β-strands A and B and between the parallel β-strands A′ and G. (Fowler et al., 2002). The existence of a so-called “mechanical clamp” involving the A′ and G strands in titin I27 seems to be responsible for the domain's resistance to force. When an ubiquitin molecule is pulled from the termini, a similar force “mechanical clamp” exists between strands β1 and β5, which may be responsible for the high unfolding force (Li and Makarov, 2004). As detailed later, a theoretical model was utilized to calculate the force required to rupture the hydrogen bonds between the two strands.
These results are consistent with previously measured results in mechanical unfolding experiments and in bulk kinetics measurements (Sivaraman et al., 2001). The average unfolding force for ubiquitin is slightly larger than that measured for titin I27 (−200 pN) under similar conditions (Carrion-Vazquez et al., 2000), and much higher than the unfolding forces for spectrin (∼30 pN) (Rief et al., 1999) and for T4 lysozyme (Yang et al., 2000). These values of unfolding forces reflect the structural properties that determine the mechanical stability of the protein molecules. Titin Ig domains possess a β-barrel motif, spectrin repeats have a triple-helical coiled-coils structure, T4 lysozyme is mainly α-helical, whereas ubiquitin has a mixed α-β structure. A steered molecular dynamics simulation (SMD) of the mechanical unfolding of I27 domain of titin revealed that the force peaks in the force-extension curves observed in atomic force spectroscopy experiments were mainly due to the initial disruption of the backbone hydrogen bonds between antiparallel β-strands A and B and between the parallel β-strands A′ and G. (Fowler et al., 2002). The existence of a so-called “mechanical clamp” involving the A′ and G strands in titin I27 seems to be responsible for the domain's resistance to force. When an ubiquitin molecule is pulled from the termini, a similar force “mechanical clamp” exists between strands β1 and β5, which may be responsible for the high unfolding force (Li and Makarov, 2004). As detailed later, a theoretical model was utilized to calculate the force required to rupture the hydrogen bonds between the two strands.
FIGURE 6.

The distribution of the unfolding forces of ubiquitin molecules. The dashed line shows the results from Monte Carlo simulation. The pulling speed was 1000 nm/s, and the parameters used in the Monte Carlo simulation were Δxu = 0.225 nm and 
Reversibility of mechanical unfolding of ubiquitin
Mechanical unfolding experiments show that the force-induced unfolding of ubiquitin molecules is reversible. In such an experiment, the protein polymers are first stretched and then relaxed after unfolding events have been observed. Fig. 7 shows the unfolding-refolding of the ubiquitin molecules in a polymer. For this set of data, each cycle of unfolding/refolding took ∼1 s, while the protein chain remained in the relaxed state for ∼0.3 s. When the polymer was stretched for the first time, six molecules were observed to have been unfolded. In the subsequent stretching of the same polymer, unfolding events were again observed, indicating that certain unfolded protein molecules properly refolded (as judged from the unfolding forces) during the time of chain relaxation. However, there were fewer than six peaks observed in the ensuing force curves, i.e., not all of the unfolded protein molecules were refolded. This is most likely due to the fact that the polymer was not completely relaxed (the tip remained 40 nm above the sample surface) in the experiment to avoid nonspecific interactions between the tip and the sample. Any tension in the polymer will reduce the refolding rate of the protein (Carrion-Vazquez et al., 1999; Rief et al., 1998). Even with the less-than-perfect refolding efficiency, the data still demonstrate that the folding process of ubiquitin is robust; a large fraction of the protein molecules can still refold with the degrees of freedom of the peptide chain severely reduced by the polymerization and the residual tension in the chain.
FIGURE 7.

The unfolding and refolding of the ubiquitin molecules in a polymer. When the polymer was stretched for the first time for a distance of 160 nm, six unfolding events were observed (the extra peak in the fifth event was due to a nonspecific interaction). The polymer was then relaxed to 40-nm extension before being stretched again. The procedure was repeated until the chain became detached. The incomplete relaxation yields a low refolding efficiency of the ubiquitin molecules in the polymer.
Theoretical calculation of the unfolding force
As described in Materials and Methods, the maximal force for unfolding can be calculated according to a simple model using the force field and parameters developed for hydrogen bonds by Hagler et al. (1974). Fig. 8 A shows a schematic of strands β1 and β5 at several positions as they are pulled away from each other. In Fig. 8 B, the calculated force and the potential energy along the pulling direction are shown as a function of the stretching distance. The maximal force calculated is 308 pN at a stretching distance of 2.5 Å between the strands β1 and β5 (see Fig. 8 B). Both values are in reasonable agreement with experimental data, if it is assumed, as in the titin I27 domain, that the rupture of the backbone hydrogen bonds between β-strands 1 and 5 is the dominating factor responsible for the peak values in the experimentally observed force-extension curves of ubiquitin. The smaller force peak of 67 pN at an extension Δr = 0.4 Å arises from the fact that, when the β1 strand is pulled away from the β5 strand, the O–H distances of three hydrogen bonds, Q2-E64, S65-F4, and L67-K6, become shorter than the equilibrium values (PDB) at small extensions. Further pulling of the β1 strand increases these O–H distances to larger values, after passing the equilibrium lengths. This process produces a minimum in the force-extension curves and a plateau in the potential energy. The native structure of ubiquitin shows that these three hydrogen bonds are tilted in the opposite direction to that of the pulling, and the tilt angles are similar (Cordier and Grzesiek, 2002). The other two hydrogen bonds, F4-L67 and K6-L69, are tilted in the direction of pulling, thus, they are stretched beyond the equilibrium lengths from the beginning. This low force peak was not observed experimentally, due to either its low values or the oversimplification of the model.
FIGURE 8.

(A) Schematic showing the relative positions of strands β1 and β5 at a stretching distance of Δr = 0, 1.0 Å, 2.5 Å, and 4.0 Å. The drawing is based on the native structure of ubiquitin and Fig. 3 in the article by Cordier and Grzesiek (2002). The hydrogen bonds are represented by dashed lines between the amide proton donor (•) and the oxygen acceptor (○). At Δr = 4.0 Å, the hydrogen bonds are much weakened or broken. It should be noted that the relative lengths of the hydrogen bonds shown are not proportional to the calculated values due to projection of a three-dimensional structure onto a two-dimensional plane. (B) The calculated force and potential energy as a function of the distance moved by β1-strand relative to β5-strand in a molecule of ubiquitin. The small peak in the force curve is due to the geometry of the hydrogen bonds between the two strands as discussed in the text.
To assess the appropriateness of using the Hagler force field or that of AMBER in this calculation, we have also carried out a cruder calculation, in which it is simply assumed that all five H-bonds connecting the two β-strands are ruptured at the same time. In such a calculation the results based on the Hagler's force field are: maximum force = 620 pN and Δr = 0.4 Å; those based on AMBER are: maximum force = 1328 pN and Δr = 0.4 Å. It should be noted that the net charges on the carbonyl group (C=O) and the amide (NH) group of the hydrogen bonds involved are nonzero in the AMBER force field, thus, we have to take the average so that no net charges exist in these functional groups. The Hagler force field fares better than the AMBER in this crude calculation and, therefore is used in the refiner calculation presented above. This refiner model is, of course, an oversimplified depiction of the mechanical unfolding process of proteins, as compared with the SMD calculations; however, it does include the essential features of the process in estimating the unfolding force based solely on backbone hydrogen bonds and provides further evidence that the rupture of the hydrogen bonds between β1 and β5 is the dominant factor. In this calculation, we consider the static limit of the pulling experiments, thus, the unfolding force obtained should be the lower bound to the observed value. The fact that the theoretical predicted value is actually higher than the observed value can be attributed to the defects of the model employed, the oversimplifications of our model, as described in Materials and Methods. The unfolding forces calculated from SMD are ∼1 order of magnitude larger than the observed values, because the pulling rates used in the SMD are ∼6–7 orders of magnitude larger than the experimental pulling rates.
Dependence of the unfolding forces on pH
It has been demonstrated in various experiments that the pH value has dramatic effects on the thermodynamic behaviors of ubiquitin and other proteins in vitro (Ibarra-Molero et al., 1999; Sundd et al., 2002; Itzhaki and Evans, 1996). To elucidate the effects of pH on the mechanical stability of ubiquitin, we have carried out mechanical unfolding experiments in solutions of different pH values. Fig. 9 presents the unfolding forces and the unfolding rates of ubiquitin as a function of the pH value. The unfolding force became lower as the pH of the solution was decreased from the neutral value. However, within the range around the neutral value between pH 6 and pH 10, the unfolding force did not change significantly (single-molecule unfolding events were not observed above pH 10, probably due to unfolding and aggregation of the polymers). The zero-force unfolding rate constants of ubiquitin, obtained via Monte Carlo simulation, do not change substantially within the pH range around the neutral value. Using stopped flow and magnetization transfer in native ubiquitin (Sivaraman et al., 2001) showed that the stability (K = ku/kf) of ubiquitin did not appreciably change between pH 6 and pH 9.5. They expected that the values of ku and kf were probably constant over this pH range, which is in agreement with our results, although the absolute values of ku from our measurements (∼10−4) are different from that reported by Sivaraman et al. (2001) (∼10−3); this is partially due to the fact that their measurements were made in the presence of GdnDCl. Below pH 6, the pH-dependence of the unfolding rate is due to the uptake of protons upon reaching the transition state from the native state, thus reflecting the extent of electrostatic interactions in the transition state relative to the native state. The change in free energy as a function of pH values can be expressed in terms of the number of bound protons (Tanford, 1968, 1970; Tan et al., 1996):
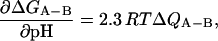 |
where ΔGA−B is the free-energy change as the protein goes from state A to state B, due to the pH changes, and ΔQA−B is the change in the number of mol of protons bound to the protein. According to the transition-state theory, the unfolding rate constant is
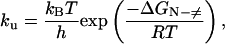 |
where ΔGN-≠ is the activation energy on going from the native state to the transition state. Consequently, the change of unfolding rate constant with pH can be expressed as
 |
where ΔQN-≠ is the change in bound protons on going from the native state to the transition state. As shown in Fig. 9, the average unfolding force increases with pH values in the acidic range, and the intrinsic unfolding rate (at zero force) of ubiquitin decreases as the pH increases in this range. According to the expression above, ΔQN-≠ = Q≠ − QN is positive, indicating that the native state is less protonated than the transition state below pH 6. It can be estimated from Fig. 9 that the transition state possesses ∼0.4 extra charge between pH 2 and pH 4, and ∼0.2 extra charge between pH 4 and pH 6, respectively, as compared with the native state. The higher degree of protonation of the transition state in the acidic pH range indicates that it requires more energy to protonate the native state than the transition state, with the extra free energy equal to the integration of ΔQN-≠ over a pH range. Around neutral pH, the protonation level of the native state and the transition state is similar, thus the electrostatic interactions make comparable contributions to both states.
FIGURE 9.

The unfolding force and the unfolding rate at zero force as a function of the solution pH value. The each point in the force curve is the average of the experimentally obtained forces from the force-versus-extension curves. The zero force unfolding rate data were obtained from Monte Carlo simulation, by fitting the simulated data to the experimental data at a specific pH value, with the unfolding rate as the fitting parameter at the optimal fitting.
The effects of pH on thermodynamic and kinetic properties of proteins arise from the pH-dependence of charge-charge interactions. Experimental determination of the pH effects can be complicated if the electrostatic interaction is altered by the measurements. Ibarra-Molero et al. (1999) characterized the folding and unfolding behaviors of ubiquitin using both denaturant-induced unfolding and thermal unfolding methods at different pH values within an acidic pH range. The results using denaturant showed that the stability of the protein (ΔG) was almost independent on pH. However, in the thermal unfolding experiments using differential scanning calorimetry, the stability and the unfolding temperature showed a strong dependence on pH, i.e., both the unfolding free energy ΔG and the denaturation temperature increased significantly as the pH value changed from 2 to 4. This discrepancy was attributed to the fact that the charge-charge interactions were screened by the high concentration of denaturant molecules (guanidine) used in the experiments. In the mechanical unfolding experiments, force is used as the agent to induce the unfolding-refolding transition while the solution is not altered, thus the measured pH-dependent properties are not influenced by solution condition changes. An accurate determination of the pH-dependence of the thermodynamic and kinetic parameters of proteins is important to understand the effects of the charge-charge interactions on the stability, folding kinetics, and functions of proteins. It has been suggested that some enzymes might stabilize the transition-state structures of reacting macromolecules primarily via electrostatic interactions (Borman, 2004). For ubiquitin, the electrostatic interactions are important for its stability and folding/unfolding kinetics because both the unfolding free energy and the unfolding rates have a strong pH-dependence. In native ubiquitin, the charged atoms of ionizable groups are mostly exposed to the solvent (Rashin and Honig, 1984), the free-energy cost of desolvation on folding is not significant, and the charge-charge interaction in the native state is stabilizing at neutral pH and destabilizing at acidic pH (Ibarra-Molero et al., 1999). This contribution is believed to be a major factor responsible for the high stability of ubiquitin. The results in Fig. 8 show that the effect of pH on the mechanical stability of the protein is significant only in the acidic range. Around neutral pH the mechanical stability remains practically constant. This may indicate that the function of ubiquitin is not weakened or inactivated if the cellular pH value changes slightly around the neutral value, as it has been suggested that mechanical forces are utilized in proteasomal degradation of targeted proteins and ubiquitin molecules are subject to a pulling force during the process (Carrion-Vazquez et al., 2003).
CONCLUSION
To fully characterize the folding and unfolding pathways of a protein molecule, it will be necessary to determine the properties of the native state, the denatured states, the transition states, and any intermediate states. Although there are many techniques to study the structural and thermodynamic properties of the various conformational species during protein folding and unfolding, the mechanical unfolding approach adds some unique capabilities. The force-induced unfolding experiments monitor the reaction along a well-defined reaction coordinate, i.e., the end-to-end distance of a tethered protein polymer, and the unfolding-refolding reactions can be observed in physiologically relevant solution environment. As the spatial and temporal resolutions are further improved, the heterogeneity of the unfolding and refolding pathways could be determined.
In this work we have synthesized ubiquitin polymers and studied the reversible mechanical unfolding behaviors of individual ubiquitin molecules. The force required to unfold a ubiquitin molecule was found to be close to that for titin domain I27, although the two molecules have different secondary structure contents. The results suggest that the unfolding forces might be determined mostly by the hydrogen-bonding pattern between the two strands being pulled directly. Our model calculation is consistent with this assumption. The pH-dependent measurements show that the unfolding forces do not change appreciably within a pH range from 6–10, indicating that the protein could function in various cellular environments. Furthermore, the characterization of the mechanical properties of proteins with various folds and under various environmental conditions may have important implications on designing protein-based artificial materials for various applications (Carrion-Vazquez et al., 2003; Hochstrasser and Wang, 2001; Lee et al., 2001).
Acknowledgments
The authors thank Dr. Feng-Yin Li for insightful suggestions and helpful discussions during the course of the work. We also thank Ensheng Liu, Jian-Wen Huang, and Sneha Rajan for their technical assistance.
This work was supported in part by the Nanotechnology Institute of Southeast Pennsylvania (G.Y.) and by Drexel University's Antelo Devereus Award for Young Faculty (to G.Y). The work was also supported in part by the National Sciences Council of the Republic of China (NSC91-2113-M-259-007 to C.-L.C.).
References
- Bell, G. I. 1978. Models of the specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- Best, R. B., B. Li, A. Steward, V. Daggett, and J. Clarke. 2001. Can non-mechanical proteins withstand force? Stretching barnase by atomic force microscopy and molecular dynamics simulation. Biophys. J. 81:2344–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm, G., R. Muhr, and R. Jaenicke. 1992. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5:191–195. [DOI] [PubMed] [Google Scholar]
- Borman, S. 2004. Much ado about enzyme mechanisms. Chem. Eng. News. 82:35–39. [Google Scholar]
- Bustamante, C., J. F. Marko, E. D. Siggia, and S. Smith. 1994. Entropic elasticity of lambda-phage DNA. Science 265:1599–1601. [DOI] [PubMed] [Google Scholar]
- Carrion-Vazquez, M., H. Li, H. Lu, P. E. Marszalek, A. F. Oberhauser, and J. M. Fernandez. 2003. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol. 10:738–743. [DOI] [PubMed] [Google Scholar]
- Carrion-Vazquez, M., A. F. Oberhauser, T. E. Fisher, P. E. Marszalek, H. Li, and J. M. Fernandez. 2000. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Bio. 74:63–91. [DOI] [PubMed] [Google Scholar]
- Carrion-Vazquez, M., A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke, and J. M. Fernandez. 1999. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 96:3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordier, F., and S. Grzesiek. 2002. Temperature-dependence of protein hydrogen bond properties as studied by high-resolution NMR. J. Mol. Biol. 715:739–752. [DOI] [PubMed] [Google Scholar]
- Evans, E., and K. Ritchie. 1999. Strength of a weak bond connecting flexible polymer chains. Biophys. J. 76:2439–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, J. M., and H. Li. 2004. Force-clamp spectroscopy monitors the folding trajectory of a single protein. Science. 303:1674–1678. [DOI] [PubMed] [Google Scholar]
- Fowler, S. B., R. Best, J. L. Toca-Herrera, T. J. Rutherford, A. Steward, E. Paci, M. Karplus, and J. Clarke. 2002. Mechanical unfolding of a titin Ig domain: structure of unfolding intermediate revealed by combining AFM, molecular dynamic simulations, NMR and protein engineering. J. Mol. Biol. 322:841–849. [DOI] [PubMed] [Google Scholar]
- Gao, M., W. Matthias, and K. Schulten. 2002. Steered molecular dynamics studies of titin i1 domain unfolding. Biophys. J. 83:3435–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagler, A. T., E. Huler, and S. Lifson. 1974. Energy functions for peptides and proteins. I. Derivation of a consistent force field including the hydrogen bond from the amide crystals. J. Am. Chem. Soc. 96:5319–5327. [DOI] [PubMed] [Google Scholar]
- Hertadi, R., F. Gruswitz, L. Silver, A. Koide, S. Koide, H. Arakawa, and A. Ikai. 2003. Unfolding mechanics of multiple OspA substructures investigated with single molecule force spectroscopy. J. Mol. Biol. 333:993–1002. [DOI] [PubMed] [Google Scholar]
- Hertadi, R., and A. Ikai. 2002. Unfolding mechanics of holo- and apocalmodulin studied by the atomic force microscope. Protein Sci. 11:1532–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser, M., and J. Wang. 2001. Unraveling the means to the end in ATP-dependent proteases. Nat. Struct. Biol. 8:294–296. [DOI] [PubMed] [Google Scholar]
- Hutter, J. L., and J. Bechhofer. 1993. Calibration of atomic force microscope tips. Rev. Sci. Instrum. 64:1868–1873. [Google Scholar]
- Ibarra-Molero, B., V. V. Lolazde, G. I. Makhatadze, and J. M. Sanchez-Ruiz. 1999. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 38:8138–8149. [DOI] [PubMed] [Google Scholar]
- Itzhaki, L. S., and P. A. Evans. 1996. Solvent isotope effects on the refolding kinetics of hen egg-white lysozyme. Protein Sci. 5:140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermayer, M. S., S. B. Smith, H. L. Granzier, and C. Bustamante. 1997. Folding-unfolding transition in single titin modules characterized with laser tweezers. Science. 276:1112–1116. [DOI] [PubMed] [Google Scholar]
- Law, R., G. Liao, S. Harper, G. Yang, D. Speicher, and D. Discher. 2003. Pathway shifts and thermal softening in temperature-coupled forced unfolding of spectrin domains. Biophys. J. 86:3286–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C., M. P. Schwartz, S. Prakash, M. Iwakura, and A. Matouschek. 2001. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell. 7:627–637. [DOI] [PubMed] [Google Scholar]
- Li, P.-C., and D. E. Makarov. 2004. Ubiquitin-like protein domains show high resistance to mechanical unfolding similar to that of the i27 domain in titin: evidence from simulations. J. Phys. Chem. B. 108:745–749. [Google Scholar]
- Li, H., A. F. Oberhauser, S. B. Fowler, J. Clarke, and J. M. Fernandez. 2000. Atomic force microscopy reveals the mechanical design of a modular protein. Proc. Natl. Acad. Sci. USA. 97:6527–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H., B. Isralewitz, A. Krammer, V. Vogel, and K. Schulten. 1998. Unfolding of titin immunoglobulin domains by steered molecular dynamics. Biophys. J. 75:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H., and K. Schulten. 2000. The key event in force-induced unfolding of titin's immunoglobulin domains. Biophys. J. 79:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhatadze, G. I., M. M. Lopez, J. M. Richardson, and S. T. Thomas. 1998. Anion binding to the ubiquitin molecule. Protein Sci. 7:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek, P., H. Lu, H. Li, M. Carrion-Vazquez, A. Oberhauser, K. Schulten, and J. Fernandez. 1999. Mechanical unfolding intermediates in titin modules. Nature. 402:100–103. [DOI] [PubMed] [Google Scholar]
- Matouschek, A. 2003. Protein unfolding: an important process in vivo? Curr. Opin. Struct. Biol. 13:98–109. [DOI] [PubMed] [Google Scholar]
- Oberhauser, A. F., P. E. Marszalek, M. Carrion-Vazquez, and J. M. Fernandez. 1999. Single protein misfolding events captured by atomic force microscopy. Nat. Struct. Biol. 6:1025–1028. [DOI] [PubMed] [Google Scholar]
- Oberhauser, A. F., P. E. Marszalek, H. P. Erickson, and J. M. Fernandez. 1998. The molecular elasticity of the extracellular matrix protein tenascin. Nature. 393:181–185. [DOI] [PubMed] [Google Scholar]
- Pace, C. N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131:266–280. [DOI] [PubMed] [Google Scholar]
- Rashin, A. A., and B. Honig. 1984. On the environment of ionizable groups in globular proteins. J. Mol. Biol. 173:515–521. [DOI] [PubMed] [Google Scholar]
- Rief, M., J. Fernandez, and H. E. Gaub. 1998. Elastically coupled two-level systems as a model for biopolymer extensibility. Phys. Rev. Lett. 81:4764–4766. [Google Scholar]
- Rief, M., M. Gautel, F. Oesterheld, J. Fernandez, and H. Gaub. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 276:1109–1112. [DOI] [PubMed] [Google Scholar]
- Rief, M., J. Pascual, M. Saraste, and H. E. Gaub. 1999. Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles. J. Mol. Biol. 286:553–561. [DOI] [PubMed] [Google Scholar]
- Schlierf, M., H. Li, and J. M. Fernandez. 2004. The unfolding kinetics of ubiquitin captured with single-molecule force-clamp techniques. Proc. Natl. Acad. Sci. USA. 101:7299–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman, T., C. B. Arrington, and A. D. Robertson. 2001. Kinetics of unfolding and folding from amide hydrogen exchange in native ubiquitin. Nat. Struct. Biol. 8:331–333. [DOI] [PubMed] [Google Scholar]
- Studier, F. W., A. Rosenberg, J. J. Dunn, and J. W. Dubendoff. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60–89. [DOI] [PubMed] [Google Scholar]
- Sundd, M., N. Iverson, B. Ibarra-Molero, J. M. Sanchez-Ruiz, and A. D. Robertson. 2002. Electrostatic interactions in ubiquitin: stabilization of carboxylates by lysine amino groups. Biochemistry. 41:7586–7596. [DOI] [PubMed] [Google Scholar]
- Tan, Y., M. Oliveberg, and A. R. Fersht. 1996. Titration properties and thermodynamics of the transition state for folding: comparison of two-state and multi-state folding pathways. J. Mol. Biol. 264:377–389. [DOI] [PubMed] [Google Scholar]
- Tanford, C. 1968. Protein denaturation. Part A and B. Adv. Protein Chem. 23:121–282. [DOI] [PubMed] [Google Scholar]
- Tanford, C. 1970. Protein denaturation. Part C. Adv. Protein Chem. 24:1–95. [PubMed] [Google Scholar]
- Tskhovrebova, L., J. Trinick, J. Sleep, and R. Simmons. 1997. Elasticity and unfolding of single molecules of the giant protein titin. Nature. 387:308–312. [DOI] [PubMed] [Google Scholar]
- Williams, P. M., S. B. Fowler, R. B. Best, J. L. Toca-Herrera, K. A. Scott, A. Steward, and J. Clarke. 2003. Hidden complexity in the mechanical properties of titin. Nature. 422:446–449. [DOI] [PubMed] [Google Scholar]
- Voet, D., and J. Voet. 1995. Biochemistry, 2nd Ed. John Wiley and Sons, New York.
- Yang, G., C. Cecconi, W. A. Baase, I. R. Vetter, W. A. Breyer, J. A. Haack, B. W. Matthews, F. W. Dahlquist, and C. Bustamante. 2000. Solid-state synthesis and mechanical unfolding of polymers of T4 lysozyme. Proc. Natl. Acad. Sci. USA. 97:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]