

Abstract
Grb14 belongs to the Grb7 family of adapters and was identified as a negative regulator of insulin signal transduction. Between the PH (pleckstrin homology) and SH2 (Src homology 2) domains is a new binding domain implicated in the interaction with receptor tyrosine kinases called PIR (phosphorylated insulin receptor interaction region). Both PIR and SH2 domains interact with the insulin receptor, but their relative role varies considering the member of the Grb7 family and the tyrosine kinase receptor. In the case of Grb14, PIR is the main binding domain and is sufficient to inhibit the insulin receptor kinase activity. We have proposed, on the basis of NMR measurements, that PIR lacks ordered structure and presents a high flexibility, although remaining fully active. To complement this first study, we have used small-angle x-ray scattering in solution together with a modeling approach representing the PIR domain as a chain of pseudo residues. Circular dichroism experiments were also performed in the presence of variable amounts of trifluoroethanol. These observations, together with an ensemble of sequence analyses and previous NMR results, all support the view of PIR as essentially unstructured but with a potentially structured short stretch encompassing residues 399–407. This stretch, which may be only structured transiently in the isolated molecule, could play a major role in Grb14 PIR binding to a biological partner by undergoing a structural transition.
INTRODUCTION
It has long been assumed that the function of a protein is closely linked to its three-dimensional structure. However, it is now recognized that numerous protein domains or even full length proteins are intrinsically disordered under physiological conditions with little or no ordered structure and high intramolecular flexibility, and furthermore that it is their normal and functional state (Dunker et al., 2001; Wright and Dyson, 1999). These proteins are referred to as “natively unfolded” (Uversky, 2002b), “intrinsically disordered” ((Dunker et al., 2002) and references therein), or “intrinsically unstructured” proteins (Tompa, 2002). Beyond minor differences, all these terms refer to proteins at least parts of which do not possess a unique three-dimensional structure in their native state. They are especially prevalent in eukaryotes (Dunker et al., 2000), mostly involved in transcription regulation, cell signaling, protein kinase activity, and binding to nucleic acids (Ward et al., 2004a,b).
In many cases, intrinsically disordered proteins or domains adopt folded structure upon binding to their biological targets, a process termed “induced folding”. These disordered-to-ordered transitions upon binding are becoming recognized as a common occurrence and appear to provide important functional advantages, including: i), the possibility of high specificity coupled with low affinity (Schulz, 1979), ii), the capacity of binding to numerous distinct biological partners by structural accommodations at the binding surfaces (Dyson and Wright, 2002; Kriwacki et al., 1996), and iii), the ability to overcome steric restrictions in the assembly of large complexes thanks to their potential for extensive intermolecular interface (Gunasekaran et al., 2003). Recently, the interacting domain PIR of the Grb14 molecular adaptor has been shown by NMR to be almost entirely disordered (Moncoq et al., 2003). Grb14 belongs to the Grb7 family of adapters comprising Grb7, Grb10, and Grb14. These proteins share a conserved multiple interacting domain structure including an N-terminal proline rich region, a Ras-associated-like domain, a pleckstrin homology (PH) domain, a C-terminal Src homology 2 (SH2) domain, and a more recently described domain known as PIR (phosphorylated insulin receptor interacting region) or BPS (between PH and SH2), located between the PH and SH2 domains.
All members of the Grb7 family are implicated in receptor tyrosine kinase signaling. Their biological role is mediated by the interaction with activated tyrosine kinase through their PIR and SH2 domains. Interestingly, the relative contribution of these domains is likely to be critical for the specificity of action of Grb7/10/14 proteins in receptor tyrosine kinase signaling.
It has been clearly established that PIR plays the predominant role in Grb14 binding to the insulin receptor (IR), whereas the interaction between Grb7 and IR mainly involves the SH2 domain and that between Grb10 and IR involves both SH2 and PIR domains. For all three proteins, binding to IR results in the inhibition of insulin action. Grb14 has been reported (Bereziat et al., 2002; Stein et al., 2001) to be the most potent and specific inhibitor of the Grb7 family toward the IR kinase activity. This observation suggests that PIR could be of main importance in the process. More recently, the loss of Grb14 function in mice was shown to result in improved glucose homeostasis and enhanced insulin signaling in liver and muscle (Cooney et al., 2004). This study points to the Grb14 protein, and especially its PIR domain, as new targets for therapies aimed to improve insulin action in vivo.
Although many reports have described the physiological function of the Grb7 family of proteins in receptor tyrosine kinase signaling, limited structural information is available concerning these proteins. Recently, the structures of the Grb10 and Grb7 SH2 domains have been solved by x-ray crystallography and NMR, respectively (Ivancic et al., 2003; Stein et al., 2003). However, these data are insufficient to fully explain the differences in binding specificity of Grb7/10/14 proteins, and further structural characterization is needed to clarify this point. In particular, understanding the specific inhibitory action of the Grb14 PIR domain toward IR catalytic activity requires structural study of the domain. To date, the PIR domain is structurally undercharacterized, and only one biophysical study has been reported (Moncoq et al., 2003). Indeed, PIR has been described as a largely unfolded and highly flexible polypeptide in solution using heteronuclear NMR spectroscopy, a powerful technique for the detection of unfolded or partially folded states of proteins, and for the characterization of internal molecular dynamics in the unstructured state.
Here, we report complementary information about the structure of PIR using small-angle x-ray scattering (SAXS) in solution experiments. SAXS studies are particularly well-adapted to study flexible and low compactness proteins, providing important structural parameters as the mean particle size (radius of gyration) and the maximal intramolecular distance (Dmax), as well as the shape of the scattering protein molecule. Analysis of SAXS data reveals that the PIR domain of Grb14 is a largely unfolded and extended protein with little or no residual structure in solution. A similar picture emerges from ab initio modeling of PIR by a chain of pseudo residues. In addition, the sequence of the PIR domains of the Grb7 family exhibits many features generally associated with disordered proteins. Finally, circular dichroism (CD) studies in the presence of trifluoroethanol (TFE) highlight the presence of a potential α-helical region that may be involved in the induced folding of PIR, and therefore in the interaction with biological partner(s).
MATERIALS AND METHODS
Cloning and expression of the PIR domain of Grb14
The cDNA fragment encoding the Grb14's PIR domain (aa 361–435) from rat was amplified by polymerase chain reaction using primers 5′-GCGCCTGCAGTTTCATGAGCGTATC-3′ and 5′-CATTGGTAAGCTTGGCAAATCACTGGGAC-3′. The polymerase chain reaction fragment was then digested with BspHI and HindIII and cloned into expression vector pET28b (Novagen, Darmstadt, Germany). After the DNA sequence was confirmed, Escherichia coli BL21-DE3 was transformed with plasmids, and cultures were grown in Luria Broth with 30 μg/ml kanamycine (Sigma, St. Louis, MO) at 37°C until an OD600nm of 1.0 was reached. Protein expression was induced by addition of 0.1 mM final isopropyl-β-d-thiogalactopyranoside (Sigma) for 4 h at 28°C. Cells were harvested by centrifugation at 8000 × g for 30 min and immediately frozen at −80°C.
Purification of recombinant PIR domain of Grb14
All purification procedures were carried out at 4°C. Cells (5 g) were suspended in 20 ml of 50 mM glycine buffer pH 9.5 containing 500 mM NaCl, 1% (v/v) Triton X100, 1 mM PMSF, 1 mM DTT, and were disrupted by sonication. CaCl2 was added to a final concentration of 0.6 mM to remove DNA before centrifugation at 8000 × g. The supernatant was dialyzed against a 50 mM glycine buffer pH 9.5 containing 500 mM NaCl, 0.1% (v/v) Triton X100, 5 mM EDTA to remove CaCl2, and then against a 50 mM Tris/HCl buffer pH 8.0 with 50 mM NaCl, 0.1% (v/v) Triton X100, 5 mM EDTA. The dialyzed supernatant was diluted threefold in 50 mM Tris/HCl buffer pH 8.0 (buffer A) and applied onto a cation exchange High Performance SP column (Pharmacia Biotech, Uppsala, Sweden). Proteins were eluted with a linear gradient (0 − 0.35 M) of NaCl in buffer A (total volume 140 ml). To perform SAXS studies in the same conditions as the previous Xenopus oocytes experiments establishing the biological activity of recombinant PIR, PIR fractions were further purified on a gel-filtration column (Sephacryl S-100, Pharmacia Biotech), equilibrated with 50 mM Tris/HCl pH 8.0, 100 mM NaCl, 1mM DTT, at a flow rate of 0.5 ml/min. Then, PIR was concentrated to 20 mg/ml in 50 mM Tris/HCl pH 8.0, 100 mM NaCl, 1 mM TCEP using Amicon Ultra 5000 (Millipore, Billerica, MA). For CD studies, the last step of the PIR purification was carried out on the same gel-filtration column equilibrated with 20 mM sodium phosphate pH 7.0, 1 mM DTT, at a flow rate of 0.5 ml/min as for the 2D NMR studies (Moncoq et al., 2003).
Every step of purification was verified by SDS-PAGE according to the Laemmli method (Laemmli, 1970). Protein concentrations were determined using the calculated absorption coefficient ɛ (0.73 mg cm−2) at 280 nm. Protein was characterized using N-terminal sequence determination.
SAXS measurements and data analysis
Scattering data were recorded on the small angle x-ray scattering instrument D24 at LURE (Laboratoire pour l'Utilisation du Rayonnement Electromagnétique, Orsay, France) using the radiation emitted by a bending magnet of the storage ring DCI. The wavelength λ was selected by a bent Ge(111) monochromator and adjusted to 1.488 Å, calibrated by the nickel absorption edge. X-ray patterns were recorded using a linear position-sensitive detector with delay-line readout. The sample-to-detector distance was 1374 mm, corresponding to the scattering vector range: 0.015 Å−1 < Q < 0.34 Å−1, where Q = 4 πsinθ/λ; 2 θ is the scattering angle). The sample was placed in a quartz capillary temperature-controlled (T = 20°C) via water circulation. Air and window scattering was virtually eliminated by inserting the cell in an evacuated beam path (Dubuisson et al., 1997). Several successive frames (usually eight) of 200 s each were recorded for both the sample and the corresponding buffer. Each frame was carefully inspected to check for any protein damage induced by x rays (none was found) before calculating the average intensity and the associated experimental error. Each scattering spectrum was corrected for the detector response and scaled to the transmitted intensity, using the scattering intensity from a reference carbon-black sample integrated over a given angular range. The scattering from the buffer was measured and subtracted from the corresponding protein sample pattern.
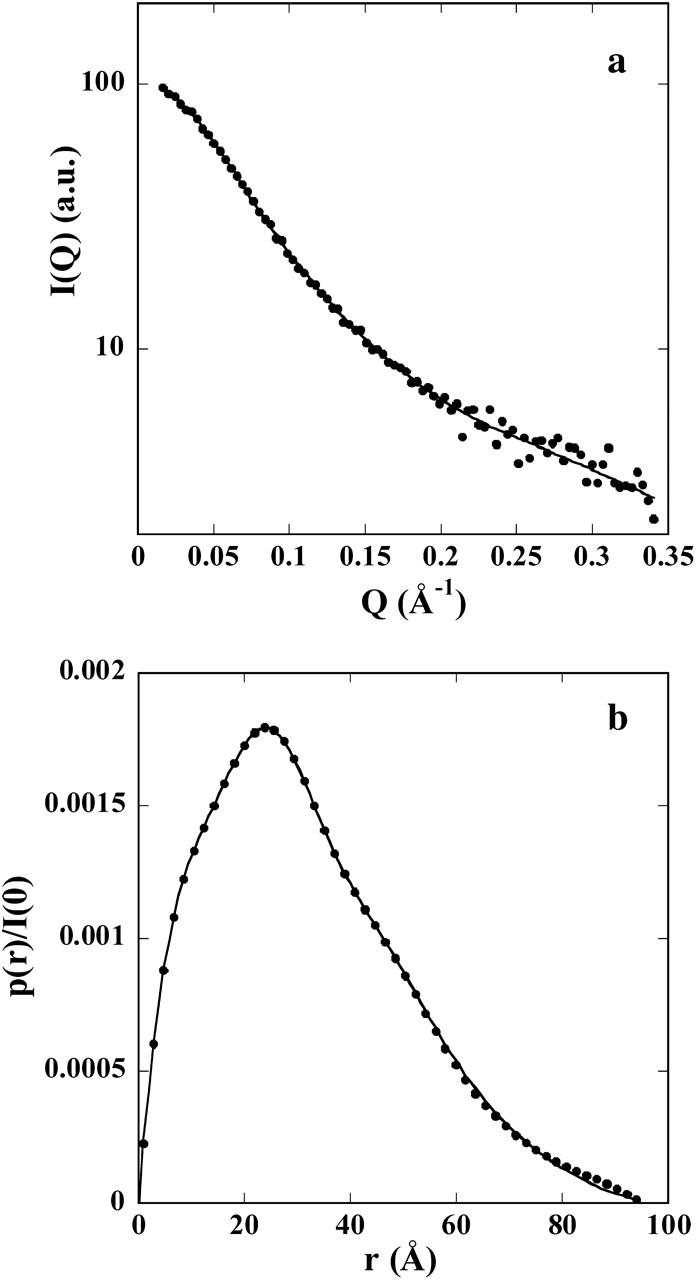
Scattering patterns were recorded at three different protein concentrations from 6 to 20 mg/ml. After scaling for concentration, the smallest angle data exhibited concentration dependence, which was indicative of moderate attractive interactions between molecules. Data were extrapolated to zero concentration following standard procedures using the second virial coefficient (Mangenot et al., 2002). The scattering from a solution of lysozyme in 50 mM acetate buffer, pH 4.5, 100 mM NaCl was recorded and used as a calibration sample to derive the molecular mass from the intensity at the origin. The radius of gyration Rg was first evaluated using the Guinier approximation (Guinier and Fournet, 1955). It is worth noting that in the case of an unstructured protein, the Guinier approximation holds true on a very restricted angular range corresponding to RgQ < 0.8. Such a very narrow range contains a small number of experimental points, thereby limiting the accuracy of the Rg determination. It has been shown that the scattering function is well described over a more extended domain Q ≤ 1.4/Rg by the Debye equation
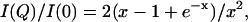 |
where x = (QRg)2, yielding a more accurate determination of the radius of gyration (Pérez et al., 2001). Finally, Rg is also evaluated using the indirect transform package GNOM (Svergun, 1992), which provides the distance distribution function p(r) of the proteins that represents the histogram of distances within the particle. Its value is equal to zero when r exceeds Dmax, the maximum dimension of the protein.
The conformation in solution of PIR was determined using two ab initio approaches. The program DAMMIN represents the protein as an assembly of closely packed small spheres (dummy atoms) of radius r0 ≪ Dmax inside a sphere of diameter Dmax (Svergun, 1999). The DAM structure is defined by a configuration vector X with N ≈ (Dmax/r0)3 components; X(i) = 1 if the ith dummy atom belongs to the protein and X(i) = 0 otherwise. Using simulated annealing, the program searches for a configuration that fits the experimental data while a looseness penalty ensures the compactness and connectivity of the solution. No particular condition of particle shape was imposed as constraint in these calculations.
The program GASBOR uses a protein representation as a chain of dummy residues (DR) centered at the Cα positions. Starting from a gaslike distribution of DRs inside the same search volume as DAMMIN, the program condenses this distribution so as to fit the experimental data under constraints that ensure the chainlike character of the DRs spatial distribution. Thus, each elementary step moves a DR to a new location 0.38 nm from another randomly chosen DR. Furthermore, the penalty function imposes a proteinlike distribution of nearest neighbors, minimizing the number of discontinuities along the chain while maintaining the center of mass close to the center of the search volume (Svergun et al., 2001). The program was run in default mode using standard values of the parameters. It was also used with a reduced weight of the penalty term, ensuring that the nearest neighbor distribution would resemble that of a compact protein.
Sequence analysis
The SwissProt sequence accession numbers for the Grb7 proteins family are, respectively, human-Grb7, Q14451; mouse-Grb7, Q03160; human-Grb10, Q13322; mouse-Grb10, Q60760; human-Grb14, Q14449; mouse-Grb14, Q9JLM9; and rat-Grb14, O88900.
A consensus secondary structure prediction was performed for the PIR domain of rat-Grb14 with the JPred program (http://www.compbio.dundee.ac.uk/∼www-jpred/) and the Network Protein Sequence @nalysis (Institut de Biologie et Chimie des Protéines, Lyon, France, http://npsa-pbil.ibcp.fr/). The prediction of the helical behavior of PIR was carried out using the AGADIR program (http://www.embl-heidelberg.de/Services/serrano/agadir/agadir-start.html) (Muñoz and Serrano, 1997).
Sequence of PIR was submitted to the PONDR (Predictors of Natural Disordered Regions) server (http://www.pondr.com/) using the integrated predictor VL-XT (Li et al., 1999; Romero et al., 1997, 2001). Access to PONDR® was provided by Molecular Kinetics (IUETC, 351 West 10th Street, Suite 318, Indianapolis, IN 46202; 317-638-0244; E-mail: main@molecularkinetics.com). PONDR® is copyright ©1999 by the WSU Research Foundation, all rights reserved.
Circular dichroism studies
The CD spectra were recorded on a Jasco (Nantes, France) J-715 dichrograph using 1-mm-thick quartz cells. Grb14 PIR samples (17 μM) were equilibrated in 10 mM sodium phosphate buffer, pH 7.0 at 20°C. Secondary structure variations were monitored as a function of changes in the initial CD spectrum upon addition of increasing concentrations of TFE (Fluka, Buchs, Switzerland). CD spectra were recorded between 185 nm and 250 nm using a wavelength width of 2.0 nm and a scanning rate of 20 nm/min. For all spectra, an average of five scans was obtained and the background spectrum of the buffer was removed. The resulting curves were averaged with a bin width of 1 nm before display and analysis performed using the CDSSTR program available at the DICHROWEB web site (http://www.cryst.bbk.ac.uk/cdweb/html/) (Lobley and Wallace, 2001; Lobley et al., 2002).
RESULTS
SAXS studies
The scattering pattern after correction for interparticle effects is shown in Fig. 1 a (circles), whereas the corresponding distance distribution function p(r) is shown in Fig. 1 b (circles). The value of the maximal diameter of the molecule was found to be 95 Å ± 5 Å. The values of the radius of gyration derived from Guinier's law, Debye's law, and the p(r) function are respectively 26 Å ± 1 Å, 27 Å ± 0.5 Å, and 26.1 Å ± 0.2 Å. Using lysozyme as a reference sample, the molecular mass for PIR derived from the intensity at the origin was found equal to 8.0 kDa, in close agreement with the known molecular mass derived from the primary sequence (7881 Da), thereby ruling out any detectable aggregation.
FIGURE 1.
(a) X-ray scattering pattern of PIR extrapolated to zero concentration (•). The line represents the average of the five calculated scattering patterns of the DR models shown in Fig. 3. (b) Distance distribution function of PIR. The solid line represents the distance distribution function corresponding to the calculated scattering curve in a.
Both Dmax and Rg values are very large for a protein of 7.9 kDa molecular mass, suggestive of an extremely extended conformation. The Rg value can be compared with the value of ∼12 Å expected for a compact globular protein with the molecular mass of PIR, an estimate obtained by using small compact proteins of known radius of gyration. In contrast, the experimental value can be compared with that expected for a polymer chain with persistence length given by the following equation:
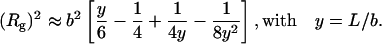 |
L, the chain contour length, is given by L = n × a × f, where n = 74 is the number of PIR residues, a = 3.78 Å is the characteristic dimension of one residue, and f = 0.95 takes into account the constraints of the polypeptide chain (Pérez et al., 2001). This yields L = 266 Å. The b value, that is twice the persistence length, expresses the rigidity of the polypeptide chain. Assuming b = 20 Å, a reasonable value judging from the literature (Rowe and Pineiro, 1990) that corresponds to slightly less than 3 residues within the persistence length, one derives a value of 28 Å for the radius of gyration, only slightly higher than the experimental determinations.
The so-called Kratky plot (Q2I(Q) versus Q) is particularly useful to emphasize the compactness of a molecule. The plot of the PIR pattern, shown in Fig. 2, displays no trace of the bell shape associated with compact, globular particles. Indeed, the curve is very similar to the pattern observed with a solution of a small (113 residues) protein, neocarzinostatin (NCS) (Pérez et al., 2001), denatured at high temperature, and very different from the sharp maximum observed on the native compact pattern of the same protein. Finally, very similar observations are made when comparing the PIR distance distribution function p(r) with those of the native and the heat-denatured NCS after scaling to the intensity at the origin I(0). Indeed, the p(r) of the unfolded NCS and PIR are practically identical (compare our Fig. 1 b and Fig. 5 from Pérez et al., 2001).
FIGURE 2.

Kratky plots. Thick solid line, PIR; thin continuous line with circles, NCS in its native conformation (T = 22.8°C); and dashed line, denatured NCS (T = 76.8°C).
FIGURE 5.
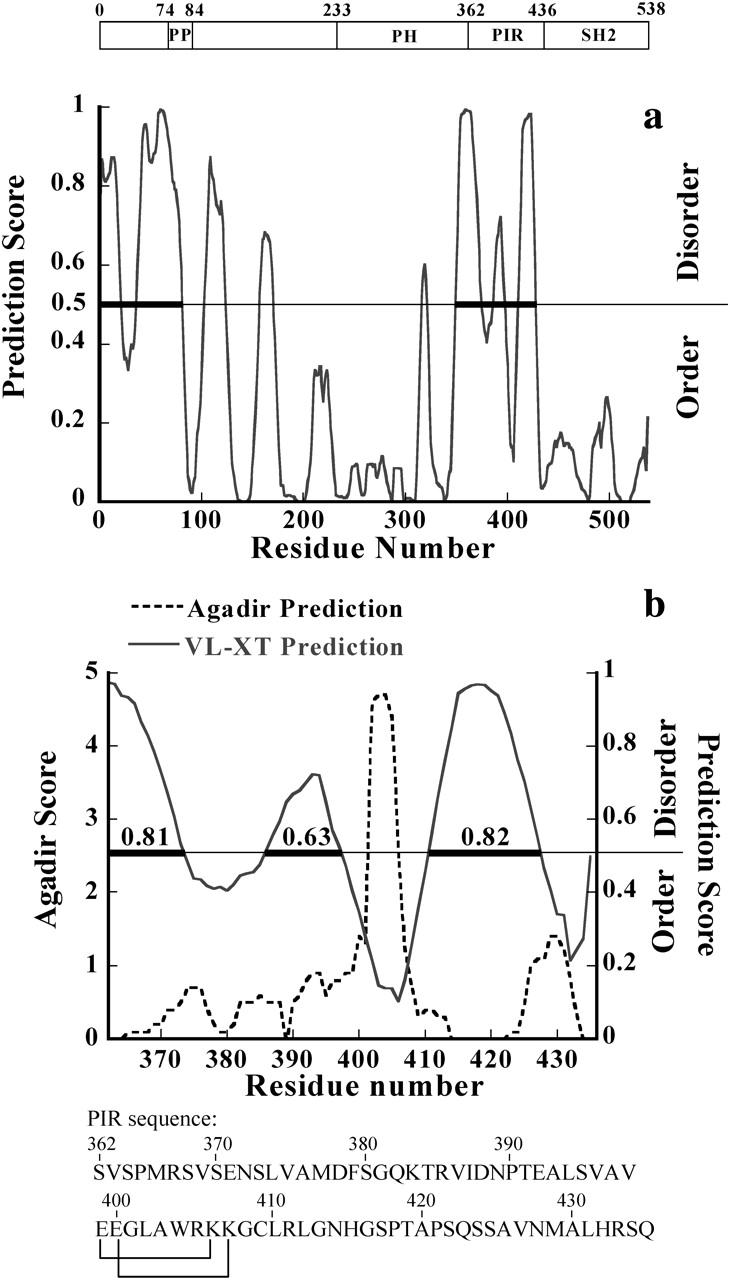
PONDR prediction of unstructured regions. (a) PONDR prediction of unstructured regions in Grb14, using VL-XT predictor (Li et al., 1999; Romero et al., 1997, 2001). The prediction score is plotted against residue number. Residues with a score above a threshold of 0.5 are considered disordered. The disordered regions are marked by thick bars. (b) Same representation for the PIR domain on which the AGADIR prediction is superimposed. The PIR sequence is also indicated. The residues connected are in close proximity when the sequence is represented on a helix wheel.
All these observations and simple calculations support the view of PIR as an unfolded molecule with little or no residual structure in solution.
Ab initio modeling
PIR appears to explore a large conformational space similar to a denatured protein. Despite the absence of a unique, stable, three-dimensional structure, we tried to model its average conformation using two ab initio approaches implemented in the programs DAMMIN and GASBOR. Both approaches yielded qualitatively similar results, and we therefore restrict our presentation to the results obtained with GASBOR. The most attractive feature of this program is its representation of the protein as a chain of dummy residues, a description still valid in the case of a statistical polymer, whereas the notion of a protein shape seems better suited to compact, folded proteins. The program was run using various sets of parameters (see Materials and Methods). Resulting models were all very similar, showing a disordered chain of dummy residues, although those obtained in default mode exhibited a shorter maximal diameter Dmax (smaller than its estimate from the p(r)) together with a poorer fit to the data at small angles, due to the constraint imposing a proteinlike nearest neighbor distribution. A set of models is displayed in Fig. 3. Although the conformations are different, they all appear to be very extended, with a small but distinct accumulation of dummy residues in the central part of the chain. This last feature should be considered with some caution, since it might, at least in part, result from the necessary constraints to preserve the connectivity of the chain. The calculated curve and the experimental pattern are shown in Fig. 1 a, whereas the p(r) distribution of the calculated curve is shown in Fig. 1 b superimposed on the experimental p(r). The agreement is excellent in both cases, supporting the relevance of the resulting models in representing the average conformation of PIR.
FIGURE 3.
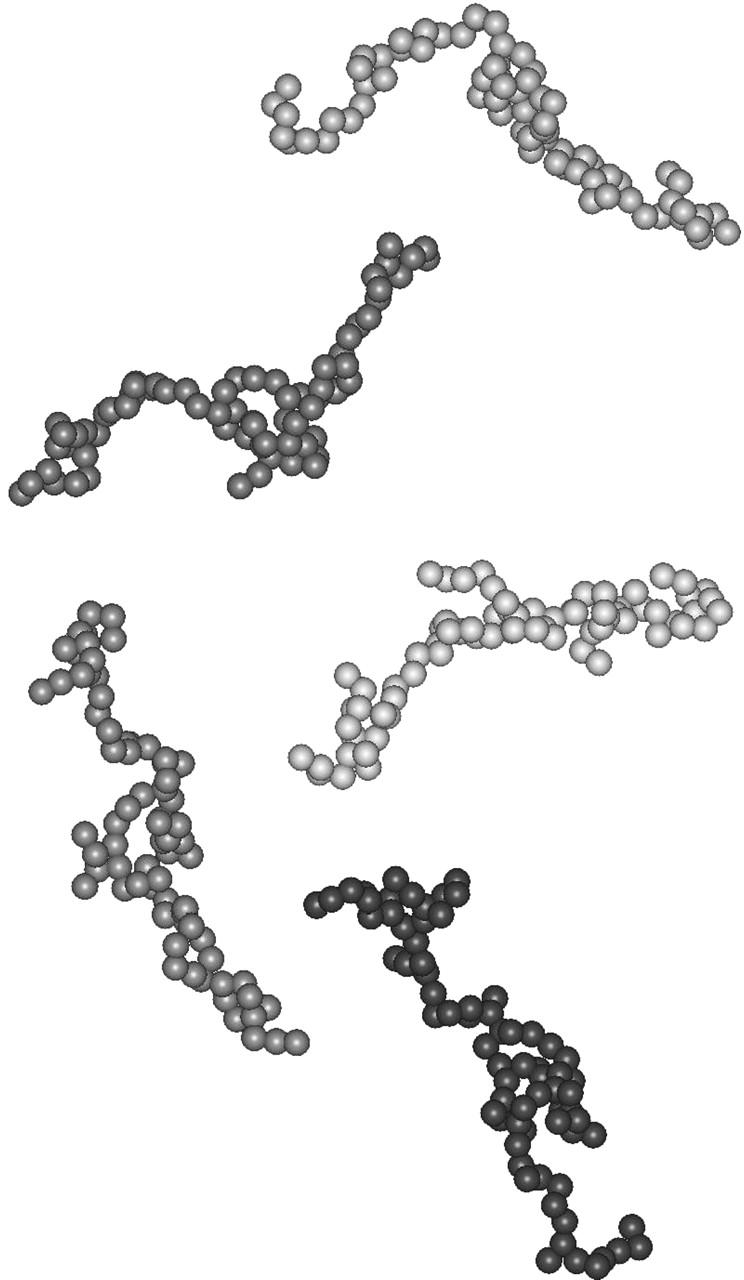
Five models of PIR obtained by GASBOR (see text for details).
CD studies and folding of Grb14 PIR domain induced by TFE
Circular dichroism can provide insights into the secondary structure of nonglobular proteins. Far ultraviolet-CD spectra of polypeptides exhibit two characteristic minima near 208 and 222 nm for extensive α-helical structure, one minimum at 215 nm for β-sheet structure, and one negative peak in the vicinity of 200 nm for random coil structure. The CD spectrum of Grb14 PIR exhibits characteristics typical of a random coil, like the large negative ellipticity at 200 nm and the low ellipticity at 185 nm. This is confirmed by the estimate of the helical structure content (∼10%) derived from the spectrum using the program CDSSTR (Lobley and Wallace, 2001; Lobley et al., 2002).
It has been reported that most intrinsically disordered proteins adopt folded structures upon binding to their biological targets (Dyson and Wright, 2002). TFE is widely used as a probe to discover regions that have a propensity to undergo an induced folding, since TFE has been reported to mimic the hydrophobic environment experienced by proteins in protein-protein interaction (Hua et al., 1998). Therefore the question whether PIR has a potential for such structural transition was addressed by recording CD spectra in the presence of increasing TFE concentrations ranging from 0 to 30% (v/v). The addition of TFE up to 30% causes significant modifications of the CD pattern (Fig. 4 a), suggesting the formation of increasing percentage of α-helix structures. Observation of an isodichroic point at 203 nm is a strong indication that TFE induces a shift in the equilibrium between two conformational states. The fraction of helix structure estimated using CDSSTR is shown in Fig. 4 b as a function of TFE concentration. In conclusion, the behavior of PIR in the presence of moderate concentrations of TFE reveals an α-helix forming propensity.
FIGURE 4.
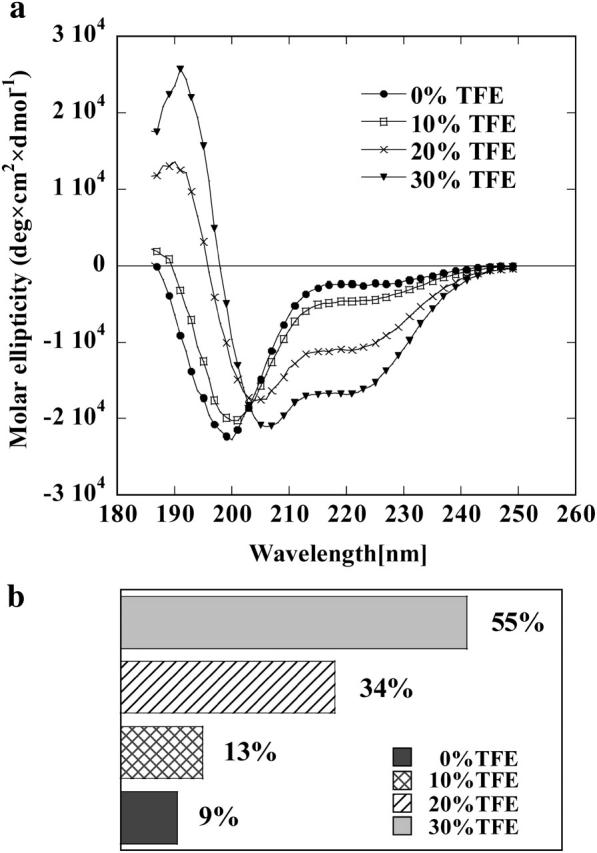
(a) Far ultraviolet-CD spectra of Grb14 PIR in the presence of increasing concentration of TFE recorded at 20°C. Mean residue ellipticity is given in deg × cm2 × dmol−1. (b) Percentage of α-helical conformation of Grb14 PIR in the presence of increasing concentrations of TFE as deduced from ultraviolet-CD spectra using CDSSTR (Lobley and Wallace, 2001; Lobley et al., 2002).
The potential for a structural transition can also be theoretically estimated using the AGADIR program, which estimates the probability of a coil-to-helix transition on the basis of the analysis of short-range interactions (Muñoz and Serrano, 1997). The results shown in Fig. 5 b point to a small region (aa 402–406 in Grb14 sequence numbering) with a very high score together with a much lower score stretch (aa 426–431) close to the C-terminal end of PIR.
Disorder and secondary structure prediction
Neural Network Predictors of Naturally Disordered Regions have been developed to discriminate ordered from disordered regions in proteins (Dunker et al., 2001; Li et al., 1999; Romero et al., 1997, 2001). These predictors, which make use of several amino-acid attributes such as coordination number, hydropathy, and flexibility, are self-trained on experimentally reported disordered proteins or regions of proteins. The predictor VL-XT of the PONDR program was applied to both the entire Grb14 protein and the PIR domain. As shown in Fig. 5 a, Grb14 is predicted to have two main disordered regions located in the N-terminal region and in the PIR domain. In fact, more than two-thirds of the PIR domain is predicted to be disordered with a score higher than the 0.5 threshold or is barely below the threshold (Fig. 5 b). The region encompassing residues 398–410 is strongly predicted not to be disordered, whereas the C-terminal stretch (428–434) is distinctly, though less strongly, below the threshold. Noticeably, the first region corresponds to the helical region predicted by the AGADIR program (residues 402–406).
Finally, the program JPred for secondary structure prediction (Cuff and Barton, 1999) predicts two regions of helical structure (residues 391–407 and 427–430). The agreement between the predictions by three different programs is good enough to be noticed.
Sequences of a set of disordered proteins have been compared to those of a set of compact, globular proteins (Dunker et al., 2002, 2001). This comparative analysis allowed the characterization of specific sequence features shared by natively unfolded proteins. Indeed, disordered protein sequences have been found to be substantially depleted in “order-promoting” residues (Trp, Cys, Phe, Ile, Tyr, Val, Leu, Asn) and enriched in “disorder-promoting” residues (Ala, Arg, Gly, Gln, Ser, Pro, Glu, Lys) (Dunker et al., 2001). As shown in Table 1, 28% of Grb14 PIR sequence is composed of order-promoting residues, compared to an average of 36% for globular proteins. Conversely, almost 60% of the Grb14 PIR is composed of disorder-promoting residues, compared to an average of 47% for globular proteins. This is not a specific property of Grb14 PIR, as it extends to all PIR domains of the Grb7 family, which exhibit very similar compositions down to the level of individual residue, as shown in the profiles of relative amino-acid enrichment (Fig. 6). These profiles represent the difference between the composition of studied sequences and the average composition of ordered globular proteins (studied sequences - ordered/ordered). This distribution is very similar to those shown by Dunker (compare our Fig. 6 with Fig. 10 from Dunker et al., 2001) for disordered proteins, with the notable exception of Lys, in which PIR sequences are depleted, in contrast to disordered sequences.
TABLE 1.
Frequency of order-promoting and disorder-promoting residues
| Amino acids | Grb14 PIR | PIRs | Globular |
|---|---|---|---|
| Order | 28.0 | 26.5 | 36.2 |
| Disorder | 58.7 | 58.3 | 47.4 |
Frequencies in percent of order-promoting residues (Trp, Cys, Phe, Ile, Tyr, Val, Leu, Asn) and disorder-promoting residues (Ala, Arg, Gly, Gln, Ser, Pro, Glu, Lys) are given for globular proteins (Tompa, 2002), Grb14 PIR and for the average sequence of all PIR domains of the Grb7 family (rGrb14, mGrb14, hGrb14, hGrb7, mGrb7, mGrb10, and hGrb10-IRSV1).
FIGURE 6.

Analysis of the amino-acid composition of Grb14 PIR and of the average of all PIR domains of Grb7 family. The amino-acid relative enrichment for Grb14 PIR (light-shaded bars) and for all Grb7 family PIR domains (dark-shaded bars) is plotted relative to their differences from the composition of the globular data set (as described in Materials and Methods). A negative peak indicates that the given data set is depleted in that amino acid as compared with the globular data set, whereas a positive peak indicates enrichment.
Following Uversky et al. (2000), we calculated the mean net charge R and the mean hydrophobicity H of Grb14 PIR and found values of 0.027 and 0.46, respectively. According to this author, a protein is predicted as disordered when H − (R + 1.151)/2.785 is negative. The value found for PIR is 0.037, which places the point for PIR in the (R, H) plane in the region for ordered proteins, though not very far from the border, together with a few other disordered proteins (see Fig. 1 in Uversky, 2002b).
DISCUSSION
Although members of Grb7 family have been studied for almost a decade, and despite growing evidence for an inhibitory role of Grb14 in insulin signaling, few structural studies have been performed on these proteins, and especially on the PIR domain. In this work, we provide further information on the conformational state of Grb14 PIR using SAXS, a powerful method for the analysis of conformation, shape, and dimensions of biopolymers in solution (Koch et al., 2003). Analysis of x-ray scattering data yields structural parameters such as the radius of gyration and the maximum diameter whose large values show that PIR adopts an extended conformation in solution. These values are, in fact, too large to be compatible with a globular, albeit very elongated, shape for such a small (74 residue long) protein, and strongly suggest that PIR is essentially unstructured. This is illustrated by the shape of the Kratky plot, which exhibits a plateau instead of the bell shape associated with globular objects. Indeed, it is very similar to that of a small heat-denatured protein. Finally, ab initio modeling of PIR conformation by a chain of pseudo residues yields models that, although different, are all predominantly disordered and much extended. In conclusion, SAXS analysis of PIR supports the view of a protein that, although functional, is essentially unstructured. This is in excellent agreement with the lack of significant ordered structure of Grb14 PIR detected by CD experiments and with recent heteronuclear NMR experiments carried out on 15N-labeled PIR showing that PIR is highly flexible and largely unstructured in solution (Moncoq et al., 2003). In this previous work, the biological activity of recombinant PIR had been established in vivo using Xenopus laevis oocytes.
We further proceeded to the sequence analysis of the Grb14 PIR domain and all PIR domains of the Grb7 family. Compared with globular proteins, Grb14 PIR is significantly depleted in “order-promoting” residues and are enriched in “disorder-promoting” residues, as already described for natively unfolded proteins (Dunker et al., 2001). The underrepresentation of order-promoting residues comprising hydrophobic and aromatic amino acids decreases one of the basic contributions to the thermodynamic stabilization of the hydrophobic core. As a result, such proteins have large dimensions in solution as shown here for Grb14 PIR using SAXS experiments. In addition, the peculiar amino-acid composition of Grb14 PIR is found in all PIR domains of the Grb7 family of adaptor proteins. The comparison between the sequence properties and the experimental observations deserves two additional remarks. First, the results of PONDR sequence analysis, although pointing toward a mainly disordered protein, predicts three disordered regions with three intervening sections below the threshold of 0.5. Therefore. the program would not detect a continuous disordered region longer than 40 residues, the criterion proposed by Dunker et al. (2001) for a native disordered protein. Second, the values of the mean net charge and of the mean hydrophobicity of PIR (Uversky 2002a,b, 2000), would not predict PIR as a natively unstructured protein, following Uversky's criterion. Despite these predictions, PIR adopts an almost fully disordered conformation in solution, as seen by different experimental approaches, as CD, NMR, and SAXS.
Accumulating studies support the view that most intrinsically unstructured proteins or protein domains undergo some degree of folding in the presence of a physiological partner (Dyson and Wright, 2002). We used CD spectroscopy to study the possible structural transitions of PIR upon addition of TFE. CD is a useful method to monitor such structural variation, since secondary structure (preferentially helical) changes involving a small fraction (on the order of 10–20%) of the sequence can be detected. It is generally accepted that TFE-induced helix formation may reveal hidden structural propensities of the protein sequence, similar to those revealed by the interaction with a partner (Buck, 1998; Dahlman-Wright and McEwan, 1996; Tell et al., 1998). This approach was recently applied in the case of other disordered proteins, the nucleoprotein C-terminal domain of measles virus (Bourhis et al., 2004) and most notably thymosin β4, a 43 amino-acid polypeptide that binds to monomeric actin (Domanski et al., 2004). The isolated polypeptide in solution is mostly unstructured with the exception of an N-terminal helix in equilibrium with an extended conformation and of the 31–37 stretch, which exhibits a weak tendency to fold into an α-helix (Domanski et al., 2004). This portion of the molecule has also been shown to fold into an α-helix in TFE (Zarbock et al., 1990). NMR studies of thymosin bound to G-actin show that it folds upon binding with the same C-terminal stretch forming an α-helix (Domanski et al., 2004), supporting in this case the physiological relevance of observations made in the presence of TFE. The evolution of the CD spectrum as a function of TFE concentration clearly shows that Grb14 PIR undergoes helical structure formation in the presence of TFE. This observation is corroborated by other results from both computational analysis and experiments on the protein in aqueous buffer: the AGADIR prediction of helix formation points toward a stretch of strong (score ≥2.5) helix nucleation (402–406), which represents 6.7% of the protein. Using a weaker score of 1.0 as a cutoff, this can be extended to 399–407, and an additional helical region (426–431) can be observed at the C-terminal end. The results of PONDR analysis suggest a mainly unstructured sequence with the exception of the stretch (398–410) and a less convincing one at the C-terminal extremity (428–434). JPred analysis predicts two potentially helical sections around residues (391–407) and (427–430). Experimentally, heteronuclear 15N-1H NOE effects measured on 15N labeled-PIR protein have shown that 5–7% of the PIR domain is less mobile than the rest of the protein, suggesting a potential nucleation point for PIR folding (Moncoq et al., 2003). This estimated percentage of lower mobility residues is consistent with the AGADIR prediction with a high cutoff value of 2.5. The intersection of the three program predictions (399–407, 428–430) and the larger limits (391–410, 426–434) represent 16% and 39% of PIR, respectively, consistent with the CD experiments in the presence of 10% and 20% TFE. Finally, the experimental value of the radius of gyration is slightly smaller than that derived for a polymer chain with persistence length, suggesting a small amount of residual structure. Taken together, these observations suggest that the PIR region encompassing residues 399–407 may represent a potential secondary structure element. This sequence does not exhibit any distinctive feature in comparison with the rest of the PIR domain. The only noticeable feature is the presence of two pairs of charge residues of opposite sign located at each extremity of the sequence and which are in close proximity on a helical wheel representation, a situation compatible with the helix prediction.
This secondary structure element, perhaps only present transiently in the isolated molecule, can be stabilized in the complex with a target molecule. To date, only two proteins were identified as binding partners of the PIR domain of Grb14: the tyrosine kinase domain of the insulin receptor (Bereziat et al., 2002; Kasus-Jacobi, 1998) and ZIP, the PKC-ζ interacting protein (Cariou et al., 2002). Concerning the insulin receptor, accumulating studies show that PIR recognizes specifically the phosphorylated tyrosine kinase domain of IR and functions as an noncompetitive inhibitor of IR catalytic activity (Bereziat et al., 2002; Kasus-Jacobi, 1998). A model of PIR-IR binding has been recently reported (Moncoq et al., 2003), although the structural basis for the interaction of PIR with IR is not yet clearly elucidated. This model was derived from crystallographic studies of IR performed by S. R. Hubbard (Hubbard, 1997), where it was observed that the rearrangement of the insulin receptor kinase loop upon self-phosphorylation reorients the C- and N-terminal lobes of the kinase. This movement exposes residues of α-helix C to the solvent, making them available for protein-protein interactions. Due to its extended form, PIR could insert in the cleft between the N- and C-terminal lobes of the kinase and interact with the solvent-exposed residues of α-helix C. The present SAXS data, showing that PIR adopts an extremely extended conformation, provide a strong support for this model.
It remains to be seen whether the presence of one or two of PIR partners may induce its folding. In any event, the presence of an unfolded protein or domain in the cell and its escaping recognition by the proteasome constitutes a pending question.
Acknowledgments
We thank Dr. A. F. Burnol for the cDNA fragment encoding Grb14.
This work was supported by Action Concertée Incitative of the French Ministry of Research (MENRT), and by a grant from the “Association pour la Recherche sur le Cancer” (grant No. 5217 to A.F.B.). K.M. is the recipient of a fellowship from MENRT and from the “Ligue Nationale contre le Cancer”.
References
- Bereziat, V., A. Kasus-Jacobi, D. Perdereau, B. Cariou, J. Girard, and A. F. Burnol. 2002. Inhibition of insulin receptor catalytic activity by the molecular adapter grb14. J. Biol. Chem. 277:4845–4852. [DOI] [PubMed] [Google Scholar]
- Bourhis, J. M., K. Johansson, V. Receveur-Bréchot, C. J. Oldfield, K. A. Dunker, B. Canard, and S. Longhi. 2004. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 99:157–167. [DOI] [PubMed] [Google Scholar]
- Buck, M. 1998. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 31:297–355. [DOI] [PubMed] [Google Scholar]
- Cariou, B., D. Perdereau, K. Cailliau, E. Browaeys-Poly, V. Bereziat, M. Vasseur-Cognet, J. Girard, and A. F. Burnol. 2002. The adapter protein ZIP binds Grb14 and regulates its inhibitory action on insulin signaling by recruiting protein kinase Czeta. Mol. Cell. Biol. 22:6959–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney, G. J., R. J. Lyons, A. J. Crew, T. E. Jensen, J. C. Molero, C. J. Mitchell, T. J. Biden, C. J. Ormandy, D. E. James, and R. J. Daly. 2004. Improved glucose homeostasis and enhanced insulin signaling in Grb14-deficient mice. EMBO J. 23:582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff, J. A., and G. J. Barton. 1999. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins. 34:508–519. [DOI] [PubMed] [Google Scholar]
- Dahlman-Wright, K., and I. J. McEwan. 1996. Structural studies of mutant glucocorticoid receptor transactivation domains establish a link between transactivation activity in vivo and alpha-helix-forming potential in vitro. Biochemistry. 35:1323–1327. [DOI] [PubMed] [Google Scholar]
- Domanski, M., M. Hertzog, J. Coutant, I. Gutsche-Perelroizen, F. Bontems, M. F. Carlier, E. Guittet, and C. Van Heijenoort. 2004. Coupling of folding and binding of thymosin β-4 upon interaction with monomeric actin monitored by nuclear magnetic resonance. J. Biol. Chem. 279:23637–23645. [DOI] [PubMed] [Google Scholar]
- Dubuisson, J. M., T. Decamps, and P. Vachette. 1997. Improved signal-to-background ratio in small-angle X-ray scattering experiments with synchrotron radiation using an evacuated cell for solutions. J. Appl. Crystallogr. 30:49–54. [Google Scholar]
- Dunker, A. K., C. J. Brown, J. D. Lawson, L. M. Iakoucheva, and Z. Obradovic. 2002. Intrinsic disorder and protein function. Biochemistry. 41:6573–6582. [DOI] [PubMed] [Google Scholar]
- Dunker, A. K., J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner, and Z. Obradovic. 2001. Intrinsically disordered protein. J. Mol. Graph. Model. 19:26–59. [DOI] [PubMed] [Google Scholar]
- Dunker, A. K., Z. Obradovic, P. Romero, E. C. Garner, and C. J. Brown. 2000. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 11:161–171. [PubMed] [Google Scholar]
- Dyson, H. J., and P. E. Wright. 2002. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv. Protein Chem. 62:311–340. [DOI] [PubMed] [Google Scholar]
- Guinier, A., and G. Fournet. 1955. Small Angle Scattering of X-Rays. Wiley, New York.
- Gunasekaran, K., C. J. Tsai, S. Kumar, D. Zanuy, and R. Nussinov. 2003. Extended disordered proteins: targeting function with less scaffold. Trends Biochem. Sci. 28:81–85. [DOI] [PubMed] [Google Scholar]
- Hua, Q. X., W. H. Jia, B. P. Bullock, J. F. Habener, and M. A. Weiss. 1998. Transcriptional activator-coactivator recognition: nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry. 37:5858–5866. [DOI] [PubMed] [Google Scholar]
- Hubbard, S. R. 1997. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16:5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivancic, M., R. J. Daly, and B. A. Lyons. 2003. Solution structure of the human Grb7–SH2 domain/erbB2 peptide complex and structural basis for Grb7 binding to ErbB2. J. Biomol. NMR. 27:205–219. [DOI] [PubMed] [Google Scholar]
- Kasus-Jacobi, A. 1998. Study of the first steps of insulin signaling pathway. Cloning of a new insulin effector: the Grb14 protein. PhD thesis, Université Paris-XI.
- Koch, M. H., P. Vachette, and D. I. Svergun. 2003. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Q. Rev. Biophys. 36:147–227. [DOI] [PubMed] [Google Scholar]
- Kriwacki, R. W., L. Hengst, L. Tennant, S. I. Reed, and P. E. Wright. 1996. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA. 93:11504–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Li, X., P. Romero, M. Rani, A. K. Dunker, and Z. Obradovic. 1999. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 10:30–40. [PubMed] [Google Scholar]
- Lobley, A., and B. A. Wallace. 2001. DICHROWEB: a web site for the analysis of protein secondary structure from circular dichroism spectra. Biophys. J. 80:373d. (Abstr.) [DOI] [PubMed] [Google Scholar]
- Lobley, A., L. Whitmore, and B. A. Wallace. 2002. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 18:211–212. [DOI] [PubMed] [Google Scholar]
- Mangenot, S., A. Leforestier, P. Vachette, D. Durand, and F. Livolant. 2002. Salt-induced conformation and interaction changes of nucleosome core particles. Biophys. J. 82:345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncoq, K., I. Broutin, V. Larue, D. Perdereau, K. Cailliau, E. Browaeys-Poly, A. F. Burnol, and A. Ducruix. 2003. The PIR domain of Grb14 is an intrinsically unstructured protein: implication in insulin signaling. FEBS Lett. 554:240–246. [DOI] [PubMed] [Google Scholar]
- Muñoz, V., and L. Serrano. 1997. Development of the multiple sequence approximation within the AGADIR model of a-helix formation. Comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers. 41:495–509. [DOI] [PubMed] [Google Scholar]
- Pérez, J., P. Vachette, D. Russo, M. Desmadril, and D. Durand. 2001. Heat-induced unfolding of neocarzinostatin, a small all-beta protein investigated by small-angle X-ray scattering. J. Mol. Biol. 308:721–743. [DOI] [PubMed] [Google Scholar]
- Romero, P., Z. Obradovic, and A. K. Dunker. 1997. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inform. Ser. Workshop Genome Inform. 8:110–124. [PubMed] [Google Scholar]
- Romero, P., Z. Obradovic, X. Li, E. Garner, C. Brown, and A. K. Dunker. 2001. Sequence complexity of disordered protein. Proteins. 42:38–48. [DOI] [PubMed] [Google Scholar]
- Rowe, G., and A. Lopez Pineiro. 1990. Influence of the solvent on the conformational-dependent properties of random-coil polypeptides. I. The mean-square of the end-to-end distance and of the dipole moment. Biophys. Chem. 36:57–64. [DOI] [PubMed] [Google Scholar]
- Schulz, G. E. 1979. Nucleotide binding proteins. In Molecular Mechanism of Biological Recognition. M. Balaban, editor. Elsevier/North-Holland Biomedical Press, New York. 79–94.
- Stein, E. G., R. Ghirlando, and S. R. Hubbard. 2003. Structural basis for dimerization of the Grb10 Src homology 2 domain. Implications for ligand specificity. J. Biol. Chem. 278:13257–13264. [DOI] [PubMed] [Google Scholar]
- Stein, E. G., T. A. Gustafson, and S. R. Hubbard. 2001. The BPS domain of Grb10 inhibits the catalytic activity of the insulin and IGF1 receptors. FEBS Lett. 493:106–111. [DOI] [PubMed] [Google Scholar]
- Svergun, D. I. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503. [Google Scholar]
- Svergun, D. I. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun, D. I., M. V. Petoukhov, and M. H. Koch. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80:2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tell, G., L. Perrone, D. Fabbro, L. Pellizzari, C. Pucillo, M. De Felice, R. Acquaviva, S. Formisano, and G. Damante. 1998. Structural and functional properties of the N transcriptional activation domain of thyroid transcription factor-1: similarities with the acidic activation domains. Biochem. J. 329:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa, P. 2002. Intrinsically unstructured proteins. Trends Biochem. Sci. 10:527–533. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N. 2002a. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V. N. 2002b. What does it mean to be natively unfolded? Eur. J. Biochem. 269:2–12. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., J. R. Gillespie, and A. L. Fink. 2000. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 41:415–427. [DOI] [PubMed] [Google Scholar]
- Ward, J. J., L. J. McGuffin, K. Bryson, B. F. Buxton, and D. T. Jones. 2004a. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 20:2138–2139 [DOI] [PubMed] [Google Scholar]
- Ward, J. J., J. S. Sodhi, L. J. McGuffin, B. F. Buxton, and D. T. Jones. 2004b. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337:635–645. [DOI] [PubMed] [Google Scholar]
- Wright, P. E., and H. J. Dyson. 1999. Intrinsically unstructured proteins: re-assessing the protein structure- function paradigm. J. Mol. Biol. 293:321–331. [DOI] [PubMed] [Google Scholar]
- Zarbock, J., H. Oschkinat, E. Hannapel, H. Kalbacher, W. Voelter, and T. A. Holak. 1990. Solution conformation of thymosin b4: a nuclear magnetic resonance and simulated annealing study. Biochemistry. 29:7814–7821. [DOI] [PubMed] [Google Scholar]