

Abstract
Proteasome-dependent proteolysis is essential for a number of key cellular processes and requires a sophisticated biogenesis pathway to function. Here, we have arrested the assembly process in its dynamic progression at the short-lived 16S state. Structural analysis of the 16S proteasome precursor intermediates by electron microscopy, and single particle analysis reveals major conformational changes in the structure of the β-ring in comparison with one-half of the 20S proteasome. The individual β-subunits in the 16S precursor complex rotate with respect to their positions in the x-ray crystallographic structure of the fully assembled 20S. This rearrangement results in a movement of the catalytic residue threonine-1 from the protected location in 16S precursor complexes to a more exposed position in the 20S structure. Thereby, our findings provide a molecular explanation for the structural rearrangements necessary for the dimerization of two 16S precursor complexes and the subsequent final maturation to active 20S proteasomes.
INTRODUCTION
The proteasomal system is central to the proteolysis machinery in eukaryotic cells, and the 26S proteasome complex is responsible for most cytosolic and nuclear protein degradation. The 26S proteasomes are formed by the association of 20S subcomplex representing the proteolytically active core with one or two 19S regulator complexes. The regulator components confer substrate recognition, unfolding, and accessibility to the proteolytic active sites of the proteasome and connect this machinery to the ubiquitin system (Glickman and Ciechanover, 2002). Thus, an important prerequisite for the selective degradation of proteins resides in the structure and the specific catalytic features of the 20S complex whose architecture is conserved from Archaea to humans. The 20S complex comprises four staggered rings each of seven subunits (α or β) that form a barrel-shaped structure. The α- and β-subunits reside in the two outer and inner rings, respectively. The outer rings function to gate substrate entrance and product release whereas the inner rings have proteolytic activity. The α- and β-rings form two so-called antechambers through which substrates have to pass to reach the active site centers that are oriented toward the proteasomal lumen (Groll et al., 1997; Lowe et al., 1995).
Pro- and eukaryotic proteasomes differ in complexity. Whereas prokaryotic 20S complexes mostly contain only one α- and one β-type subunit, eukaryotic proteasomes are built up by seven different α- and seven different β-type subunits. A catalytically important active residue is a threonine located at the N-termini of the β-subunits that characterizes the proteasome as a member of the family of N-terminal nucleophile (Ntn) hydrolases. In prokaryotes, the 14 β-subunits exhibit proteolytic activity, whereas in eukaryotes only three of the seven different β-subunits, namely β1, β2, and β5, have the N-terminal threonine, yielding a total of six active sites.
As in many proteinases, the active sites of the β-subunits are preceded by prosequences that have to be removed to generate the active-site threonine (Ditzel et al., 1998; Groll et al., 1999). This activation is an autocatalytic event that takes place during the final steps of proteasome assembly, protecting the cell against an uncontrolled activation of the enzyme complex (Schmidtke et al., 1996; Seemuller et al., 1996).
Proteasome-dependent proteolysis is in part regulated at the level of subunit incorporation. Thus, proteasome biogenesis is a precisely ordered multistep event involving the biosynthesis of all subunits, their assembly, and maturation processes. Current models of early assembly events are mainly based on the knowledge of simple proteasome structures such as that of the archaebacterium Thermoplasma acidophilum and Archeoglobus fulgidus (Groll et al., 2003; Zuhl et al., 1997; Zwickl et al., 1994). Initially, the cooperative formation of the heptameric α-ring takes place serving as matrix for the subsequent docking of the correct β-subunits. In an alternative model, a given α-subunit binds to a defined β-subunit, which in turn determines the binding to the next αβ-heterodimer (Gerards et al., 1997; Mayr et al., 1998). In Rhodococcus erythropolis, the β-propeptide appears to act as an assembly promoting factor by linking its own β-subunit to two adjacent α-subunits and thus explains the structural basis for the α-subunits not forming an α-ring when expressed alone (Kwon et al., 2004). In eukaryotes, where the assembly process is slower, more complex, and requires helper proteins, the following assembly steps are determined by two distinct precursor intermediates. These intermediates represent partially assembled complexes of all seven α-subunits and some β-subunits with sedimentation coefficients of ∼13S and 16S. Three of the seven β-subunits, i.e., β2, β3, and β4 could be identified as part of the 13S precursor complex, whereas β1, β5, β6, and β7 are incorporated later, forming the 16S precursor complex. The dimerization of two 16S precursor complexes into the preholoproteasome is coupled with the final processing of the N-terminal propeptides (Frentzel et al., 1994; Nandi et al., 1997; Schmidtke et al., 1997). These steps are assisted by the maturation proteins Ump1p in yeast or POMP in mammalia, which are associated with precursor complexes and are degraded upon completion of proteasome maturation (Krüger et al., 2003; Ramos et al., 1998; Witt et al., 2000).
An important regulatory role in the assembly process is played by the prosequences of the different β-subunits that appear to be species specific with respect to sequence and length. For example, the N-terminal propeptides of β-subunits were dispensable for Thermoplasma proteasome assembly in vitro (Seemuller et al., 1996). In eukaryotes, the N-terminal propeptides influence the efficiency and timeliness of subunit incorporation and maturation to different degrees. The absence or exchange of the β5 prosequence exerts the strongest effect that reflects a hierarchy of active site functions as follows: β5 ≫ β2 > β1 (Chen and Hochstrasser, 1996; Heinemeyer et al., 1997; Jager et al., 1999; Kingsbury et al., 2000; Schmidtke et al., 1997). The β5 active site is archetypical and has the greatest effect on proteasome maturation and proteolysis. During assembly of the eukaryotic 20S proteasome, these prosequences are removed by a two-step mechanism in cis- and trans-autocatalysis (Ditzel et al., 1998; Jager et al., 1999; Schmidtke et al., 1996). In the first step, neighboring active sites cleave within the prosequences whereas the second step is autocatalytic, generating the active-site threonine and a functional 20S complex (Heinemeyer et al., 1997; Nandi et al., 1997; Schmidt and Kloetzel, 1997).
Structural rearrangements have been assumed to allow dimerization of two 16S precursor complexes and final maturation (Groll et al., 2003). This would imply that 16S precursor complexes differ in their structural features in comparison to the half 20S complexes. However, direct evidence for this hypothesis is missing. To explore the structural organization of 16S proteasome precursor intermediates we constructed a chimeric β-subunit of the simple archaebacterial A. fulgidus proteasome carrying the prosequence of the human β5 subunit. Substitution of the archaebacterial prosequence resulted in the formation of proteolytically inactive complexes in the size of 16S precursor intermediates unable to dimerize.
Analysis of the 16S precursor intermediates by electron microscopy (EM) and single particle analysis revealed conformational changes in the individual subunits during proteasomal maturation.
MATERIALS AND METHODS
Preparation of 16S precursor complexes
The compatible plasmids encoding the A. fulgidus α- and β-subunits were kindly provided (Groll and Huber, 2003). The chimeric β-subunit was constructed by PCR amplifying the coding sequence of the A. fulgidus proteasomal β-subunit and the fragment encoding the propeptide of the human β5 subunit. Both fragments were cloned into pRSET A (Invitrogen, Carlsbad, CA). For cloning, a 5′ NdeI and a 3′ KpnI site (encoding GT residues) were introduced in the 177-bp propeptide sequence (full-length) whereas a 5′ KpnI site and a 3′ BamHI site were generated for the 509-bp β-subunit sequence. Truncations of the propeptides were performed by mismatches in the forward primers and the chimeric full-length construct as template. The correct plasmids were verified by sequencing.
The two subunits and their derivatives were overproduced by coexpression of the α-and the respective β-subunit in Escherichia coli BL21 (DE3) using T7 polymerase and purified as described (Groll and Huber, 2003). Proteins were stored in a buffer containing 20 mM Tris-HCl (pH 7.5). Before application for biochemical analysis samples were heated at 80°C for 15 min to allow the protein complexes to adapt to their temperature optimum. For in vitro assembly under suboptimal temperatures using wild-type subunits this step was performed at 37°C.
Protein (4 mg) was fractionated by sucrose gradient ultracentrifugation from 10% to 40% as described (Frentzel et al., 1994) and equal amounts were separated on SDS-Laemmli gels. Native gel analysis of the proteins was performed using 10–15% precast gels in the PhastSystem (Amersham Pharmacia Biotech, Uppsala, Sweden) and Coomassie staining.
Chymotryptic activity of the proteasome was assayed using the synthetic peptide substrate Suc-Leu-Leu-Val-Tyr linked to the fluorometric reporter aminomethylcoumarin and quantitated using 360-nm excitation and 460-nm emission wavelengths.
Sample preparation
The specimen, in 100 μM Tris-HCl (pH 7.5), was incubated for 15 min at 80°C and cooled to ambient temperature before grid preparation. A 240-μl sample of the protein (10 μg/ml) in 0.25% methylamine tungstate stain was sprayed on to the Butvar 76 side of a carbon-coated grid (Stoops et al., 1991) and rapidly dried under a flush of N2 gas.
Data collection
Forty-six micrographs were collected on a JEOL 1200 electron microscope operating at 100 kV and a magnification of 50,000× using defocus settings from 0.5 to 2.6 μm. Micrographs were scanned on a Zeiss imaging scanner (Zeiss, Jena, Germany) with a step size of 14 μm, corresponding to a pixel size of 2.8 Å on a specimen scale.
Image processing and fitting of atomic structures
All image processing steps and fitting of x-ray structures were performed using SPIDER (Frank et al., 1996). The accurate micrograph defocus settings were estimated from the data using an automated procedure (Huang et al., 2003) and were used to correct for the effects of the contrast transfer function. This was done by inverting the sign of phases in the windowed particles according to the sign of the contrast transfer function. A total of 16,662 particles were manually selected and subjected to the reference-free alignment (Penczek et al., 1992) and K-means classification resulting in 500 classes. From this set, 394 class averages were calculated for the most populous classes. The class averages revealed two common views of the structure: a ring-like end view with a strong indication of sevenfold symmetry and a side view with two parallel bars implying the presence of two rings. To determine the three-dimensional (3-D) structure without any reference to the x-ray crystallographic structure of the 16S precursor complex, random Eulerian angles were assigned to the 394 class averages and an initial 3-D structure was calculated with additional enforcement of the sevenfold symmetry. This seed structure was subsequently used as a reference in 3-D projection matching procedure (Penczek et al., 1994), in which the Eulerian angles assigned to class averages were iteratively modified and which converged in 21 steps to a stable solution. The refinement of the structure was carried out independently using two random halves of the data set of individual particle views. This allowed adjustment of the refinement parameters to avoid overfitting of the data. After 15 steps of refinement using halves of the data set, four additional steps of 3-D projection alignment were performed using the whole set of particle views, yielding a 3-D structure of the 16S precursor complex at 16.2-Å resolution, as determined by the Fourier shell correlation with a 0.5 cutoff (Penczek, 1998). The handedness of the EM structure was set to the handedness of the x-ray structure of 20S proteasome from A. fulgidus. Similarly, the magnification of the EM structure was estimated by comparison with the x-ray structure and resulted in adjustment of the EM pixel size to 3.1 Å. (In this scale, the resolution of the EM map was 17.7 Å.) The relatively large magnification mismatch of 11% had to be attributed to the uniform shrinkage of the support Butvar film caused by exposure to the electron beam (Stoops et al., 1992). The Fourier amplitudes of the EM map were adjusted such that their rotational average matched the rotational average of the Fourier amplitudes of the x-ray structure and the EM map was low-pass filtered to its nominal resolution. To confirm the validity of the thus-determined structure, the appropriately filtered crystal structure of two rings of the 20S proteasome from A. fulgidus was used as the initial reference and refined with 394 class averages as described above. The result proved to be virtually identical to the structure determined ab initio. The docking of the x-ray structures into the EM map was done using dedicated SPIDER script. In this procedure, α- and β-rings were processed separately. Atomic coordinates of individual subunits from respective rings were extracted from the PDB coordinates of the 20S A. fulgidus and converted into electron density maps. The fitting was performed using an exhaustive search for Eulerian angles and for small values of possible translations. For each putative orientation of the β-subunit within the EM map, the two neighboring β-subunits were generated using known symmetry relations and if this resulted in spatial overlap between subunits, the position was excluded from further considerations; otherwise, the correlation coefficient between the two density maps was calculated. The final position of the β-subunits corresponded to the maximum correlation coefficient found. Visualizations were carried out in Web (Frank et al., 1996), IRIS Explorer, and RIBBONS (Carson, 1997).
RESULTS
Biochemical analysis of processing incompetent proteasome precursor intermediates
To structurally analyze 16S proteasome precursor intermediates we used the heat-stable 20S proteasome of the thermophile A. fulgidus. The structures of this 20S proteasome as well as the early (α-ring) and a late precursor complex (preholoproteasome) have been determined crystallographically (Groll et al., 2003). Therefore, we generated a number of constructs (Fig. 1 a) to capture 16S intermediates at stages where conformational changes may occur. First, we analyzed which proteasome complexes are formed using the wild-type α-subunit together with the β-subunit or their mutated derivatives, respectively (Fig. 1, a and b, 1–3). As expected, both wild-type subunits assembled into active 20S proteasomes with a size of ∼700 kDa (equivalent to 28 subunits). Elimination of the complete propeptide did not affect the biogenesis process and resulted in active 20S complexes analogous to the Thermoplasma 20S proteasome (Seemuller et al., 1996; Fig. 1 b). Mutation of the catalytic residue, threonine-1 to glycine (Thr-1/Gly), led to the formation of processing-incompetent and thereby inactive 20S complexes. These are composed of 28 subunits representing the state of preholoproteasomes as previously described (Groll and Huber, 2003; Fig.1 b). However, substitution of the wild-type propeptide for the human β5 propeptide resulted in proteolytically inactive complexes smaller than 20S with a size of ∼350 kDa (16S, equivalent to 14 subunits; Fig. 1 b, 4–6). Even after truncation of the human β5 prosequence from 59 to 14 or 11 residues, similar 16S complexes were obtained suggesting a species-specific sequence requirement for proper proteasome maturation (Fig. 1 b). Optimal assembly conditions for the A. fulgidus wild-type α- and β-subunits are at 80°C; however, the dynamics of this process would not allow us to capture short-lived 16S precursor complexes. More precursor complexes can be observed with wild-type subunits when following slow proteasome assembly under suboptimal temperatures (37°C). Nevertheless, the two wild-type subunits assembled mainly into complexes with sedimentation coefficients of 20S (Fig. 1 b, top panel) representing a mixture of active 20S proteasomes and preholoproteasomes (four rings). In addition, 16S precursor complexes were formed, which contain exclusively the β-subunit proprotein (pro-β) as a marker for precursor intermediates. Thus, 16S precursor intermediates naturally exist and can be observed in low quantities under appropriate conditions. The chimeric β-subunit construct, in which the wild-type 11 amino acid prosequence of A. fulgidus was exchanged with the 59 amino acid prosequence of the human β5 subunit, showed the same migration behavior on density gradients as the wild-type 16S precursor (Fig. 1 b, bottom panel). Therefore, we arrested proteasome biogenesis in its dynamic progression at the 16S state. This implies that the foreign prosequences of different length do not impair the folding and assembly of the subunits, but rather interfere with the final 20S proteasome formation. The captured 16S precursor complexes, complete with the 59 amino acid prosequence fused to the β-subunits, were used for the subsequent structural analysis.
FIGURE 1.

Biochemical analysis of proteasome precursor intermediates. Gels were stained with Coomassie blue and band identity was verified by mass spectrometry. (a) Various β-subunit constructs with the nomenclature on the right and the respective propeptide sequences on the left with the active sites underlined. (b) Sucrose gradient fractionation of in vitro assembled complexes of A. fulgidus wild-type proteasome (top panel) and that with the chimeric β-subunit (bottom panel). The 16S precursor complexes are in fractions 11 and 12, whereas fractions 14–16 contain 20S complexes (proteasomes and preholoproteasomes). Band identities and two- and four-ring structures are indicated. (c) Native gel analysis of the indicated proteasome and proteasome precursor complexes of A. fulgidus wild type and their mutated derivatives resulting in 20S complexes (constructs 1–3). The chimeric proteins bear the full length (59 aa) and truncated (14 and 11 aa) human β5 propeptides (constructs 4–6) and are arrested in the 16S stage. Two- and four-ring cartoons symbolize the structure of the complex.
EM structure of the 16S precursor complex
The initial analysis of two-dimensional averages of individual particle views confirmed the expected architecture of the 16S precursor complex, i.e., the presence of two sevenfold symmetrical rings (Fig. 2). The 3-D structure has been symmetrized accordingly. Although at the resolution achieved (17.7 Å) few internal details of the structure are resolved, both the configuration of the subunits and their shape are well defined (Fig. 3, a–d). The α-ring is made up of a central capped hub from which seven α-subunits radiate outwards as spokes. The α-subunit N-terminal domains form the hub region in the center of the ring with a curved arm extending out to the large globular domain. The β-subunits are kidney shaped with the midlobe indentation facing toward the inner surface of the β-ring. The lobe adjacent to the α-ring is larger and protrudes toward the center of the ring as a beak-like domain. The α-ring is capped, whereas the β-ring has a central pore (Fig. 3, c and d). The connections between the seven α-subunits are located at the center of the 16S structure, whereas those between the seven β-subunits lie between the spokes of the 16S β-ring. The α- and β-rings are fused by two connections between each α- and β-subunit.
FIGURE 2.
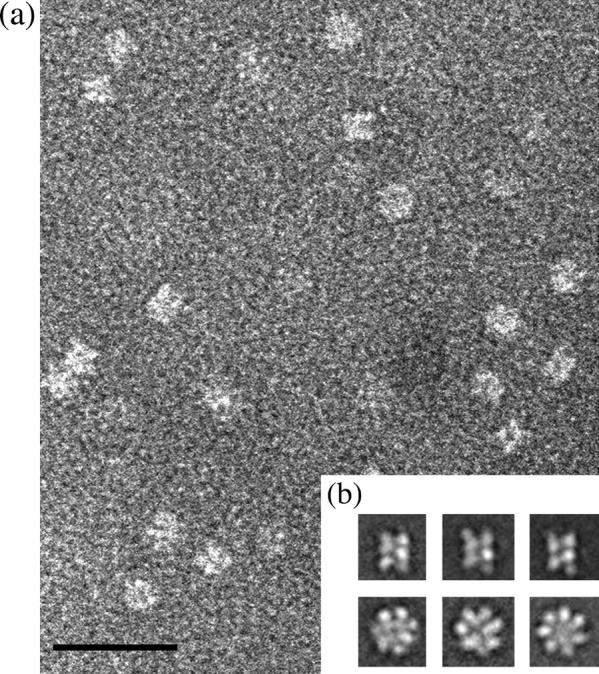
Electron micrograph and class averages of 16S particles. (a) Raw images of 16S proteasome precursor complex in negative stain. The scale bar represents 30 nm. (b) The class averages revealed two common views of the structure: three side-view averages with two parallel bars implying the presence of two rings (top panel) and three ring-like end-view averages (bottom panel) with a strong indication of sevenfold symmetry. The scale bar represents 30 nm.
FIGURE 3.

The 3-D EM structure of 16S from A. fulgidus. (a) Side view with the α-ring at the top and β-ring below. The two connections can be seen between each α- and β-subunit. (b) Vertical slice through 16S revealing the capped α-ring and open β-ring. (c) View from the α-ring side revealing the closed ring and no inter-α-subunit contacts between the spokes. (d) View from the β-ring side revealing the open pore and β-subunit connections. The scale bar represents 10 nm.
Fitting the 20S crystal structure into the EM density map
The coordinates from one asymmetric unit (one α-ring and one β-ring) of the 20S crystal structure from A. fulgidus (Groll et al., 2003) were used as an initial atomic model for the structure of the 16S precursor complex. The α-ring alone was also crystallized and the backbone tracing of the crystal structure of the α-ring alone was identical to the backbone tracing of the α-ring in the 20S proteasome crystal structure (Groll et al., 2003). The crystal structure of the 20S proteasome generally fits the EM density map of the 16S complex well; however, upon closer examination of the individual rings and subunits, differences were identified.
The α-ring from the crystal structure of 20S proteasome fits well into the α-ring of the EM density map of the 16S complex and only a few flexible loops, among them the C-terminus, extend outside the surface (Fig. 4 a). The EM map of the 16S α-ring is capped and contains sufficient volume to enable all the N-terminal residues of the α-subunits to fit into it. This is in contrast to the open 20S α-ring crystal structure in which the region of N-terminal residues 1–10 is undefined due to its flexibility (Fig. 4 a). Defined loops from residues 45–55 in the α-subunits of the crystal structure, which contribute to the major contact region between α-subunits within the crystal structure, protrude from the EM density map of the 16S. Because the 16S EM map lacks density between α-subunits in this region, the loop can presumably reside in an unassigned region of the map in each α-subunit spoke and does not contribute to inter-α-subunit contacts.
FIGURE 4.

The ribbon schematics of the α- and β-rings in the orientation of the crystal structure of 20S placed into the EM map of 16S. (a) A view from the α-ring end shows the protruding loop (residues 45–55), which is circled in red. (b) A view from the β-ring end shows the protruding regions Helix 2 and following loop (residues 84–96), and Helix 5 (residues 188–202), which are circled in red and black, respectively. The scale bar represents 10 nm.
Unlike the α-ring structure, the crystal structure of the β-ring from 20S exhibits more discrepancies in its fit with the EM density map of 16S. There are two large regions that do not satisfy the density map (Fig. 4 b). In the EM structure, the pore of the β-ring is both wider and lies closer to the α-ring when compared with the pore in the crystal structure. A beak-like domain in the EM structure protrudes toward the center of the pore and is positioned downwards, pointing toward the α-ring side of the β-ring. When compared with the crystal structure, this beak-like domain consists of Helix 2 and its ensuing loop, which are composed of residues 84–96. To fit the crystal structure of the β-subunit into the EM map it was necessary to position the beak-like domain lower in the EM density. This resulted in a rotation of the β-subunit by 28° about the inter-β-subunit contact point. This rotation of the β-subunit resulted in a shift of the beak region by 20 Å with respect to its original position in the crystal structure of the 20S proteasome (Fig. 5, a–c). The rotation also brought another previously exposed domain (Helix 5, residues 188–202) into the density map (Fig. 4 b). When the crystal structure of the β-ring was first positioned into the β-ring of the EM map, this helix was located outside the EM density of the β-subunits (Fig. 4 b). Hence, the rotation of the β-subunits satisfied not only the positioning of the beak-like domain into the density, but it also positioned the protruding helical structures of each β-subunit into the EM density map (Fig. 5 d).
FIGURE 5.

Comparison between the β-subunits in the EM map of 16S and in the crystal structure of the 20S proteasome. Fit of the β-subunits into the EM map showing the increased pore size in the EM structure. (a) A top view of the β-ring showing the fit of the β-subunits in the crystal structure orientation. All subunits are identical but each is shaded a different color. (b) A top view of the β-ring showing the fit of the β-subunits in the EM structure orientation. All subunits are identical but each is shaded a different color. (c) Helix 1 of the β-subunits makes contact with the α-subunits. β-Subunits in the crystal orientation shown in red and β-subunits in the EM orientation shown in blue. (d) Ribbon diagram of the β-ring in the orientation corresponding to the position of the EM map fitted into the β-ring map.
The contact regions between all β-subunits are similar in both the EM and crystal structures, as visualized in the comparison of the two density maps (Fig. 6). As viewed from the side of the structures, a contact is made from the end of Helix 1 (residues 52–70) of each β-subunit to a loop region (residues 115–120) of each adjacent β-subunit. This contact lies closest to the α-ring. The second contact is located slightly higher in the β-ring, toward the edge of the 16S precursor complex structure, and extends from a loop and β-strand (residues 20–28) of each β-subunit to a loop region of each adjacent β-subunit (residues 120–125).
FIGURE 6.

Inter-β-subunit connections. An overlay of the β-rings from the EM and crystal structure, shown in blue and red, respectively. The β-subunits are tilted and the view is from the α-side of the 16S structure. The two connections between two β-subunits can be seen with one connection positioned closer to the α-ring. A contact is made from the end of Helix 1 to a loop region of a neighboring β-subunit. This contact lies closest to the α-ring. The second contact is located slightly higher in the β-ring, toward the edge of the 16S precursor complex structure, and extends from a loop and β-strand of the β-subunit to a loop region of the neighboring β-subunit. The scale bar represents 10 nm.
The movement of the β-subunits to fit the beak-like domain into the EM density does not cause any significant alterations in the connections between the α-ring and the β-ring, nor does its movement disrupt the overall contacts between the β-subunits. A higher resolution structure will enable any movement in these connections to be studied in more detail during the conformational change of the β-subunit.
Conformational change within the β-subunits
The rotation of the β-subunits in the EM structure of the 16S precursor complex with respect to their positions in the crystal structure of the 20S proteasome suggests that a structural change occurs amongst the β-subunits during maturation into the 20S proteasome. In our work, we have rotated the β-subunits from the crystal structure orientation to satisfy the EM structure. However, the proteasome biogenesis pathway proceeds from the 16S precursor complex state to the 20S proteasome state and so we propose that the reverse rotation of the β-subunits will occur during 20S proteasome formation. Therefore, during proteasome biogenesis, the β-subunits in the ring of a 16S precursor complex move upwards and toward an approaching 16S precursor complex in which the β-subunits are undergoing the same conformational change. To form the 20S proteasome, the β-subunits must undergo a conformational change from their 16S orientation to form the necessary contacts.
We examined the inter-β-ring contacts in the 20S crystal structure and identified that the beak-like domain is the main contact point between the two β-rings. The beak-like domains in the β-subunits of one 16S precursor complex ring form contacts with β-turn domains (residues 25–30) in diagonally opposing β-subunits of the second 16S precursor complex (Fig. 7). The beak-like domains must adopt new orientations during 20S formation for these contacts to be formed and this is achieved by the conformational change of the β-subunits. The active catalytic residue, Thr-1, is in close proximity to the beak-like region and the conformational change in the β-subunits allows the catalytic residue to move from a protected region in the 16S precursor complex state to a location in the 20S proteasome state more accessible for its function (Fig. 7).
FIGURE 7.

Inter-β-ring subunit contacts and Thr-1 locations in the crystal structure of 20S. (a) The 20S structure from A. fulgidus. (b) Highlighted region of 20S shown as a ribbon diagram of four β-subunits, indicating the β-β-subunit interactions and the location of the catalytic residue, Thr-1. (c) Closeup of the ribbon diagram of four β-subunits with the beak-like domains circled. The beak-like domains (residues 84–96) in one β-ring interact with the β-turn domain (residues 25–30) of the β-subunits in the adjacent β-ring. Due to the arrangement of the rings, the β-subunits interact with each other across a diagonal. There are two regions of interaction between each pair of subunits. All interactions are identical, between the beak-like domain and β-turn domain. The catalytic residue (Thr-1) is shown. Thr-1 is in close proximity to the beak domain.
DISCUSSION
We have captured the proteasome in the two-ringed precursor complex state and have determined the 3-D structure using EM and single particle analysis. The α-ring from the 20S proteasome crystal structure fits the EM density map better than the corresponding fit to that of the β-ring. There was no density found for an α-α contact loop in the α-ring, which is present in the 20S α-ring crystal structure. This may indicate that the loop is more flexible in the 16S EM structure than in the 20S crystal structure and does not, therefore, necessarily have a role in the α-ring formation in the 16S precursor complex. In agreement with this idea, the size of the α-α contact with this loop has been seen to vary within other proteasome structures. The same loop is much longer in Thermoplasma and yeast than in Rhodococcus erythropolis (Kwon et al., 2004). The capping of the α-ring in the EM structure of the 16S precursor complex was also characterized in the EM tomographic reconstruction of the 20S proteasome from T. acidophilum (Koster et al., 1997). This capping has not been seen in the crystal structure of the 20S proteasome nor in the crystal structure of the α-ring alone (Groll et al., 2003). In the crystal structure of the α-ring alone, only part of this region is ordered as not all N-terminal residues were defined in the final structure. The crystal structure of the α-ring alone contained some density in the location of the N-terminal domains, whereas the crystal structure of 20S proteasome lacked density in this area. This indicates that the N-terminus of the α-ring is far more flexible in the 20S state than in both the single α-ring and the 16S precursor complex. The innate flexibility of this region allows the α-annulus to remain closed until the substrates are presented for degradation through the 20S proteasome (Groll et al., 1997; Lowe et al., 1995; Wenzel and Baumeister, 1995). This flexibility has also been thought to restrict the release of degradation products from the interior of the proteasome (Hill et al., 2002).
The crystal structure of the β-ring proved more challenging to fit into the EM density. Rotation of all seven β-subunits into the EM map allowed the crystal structure to fully fit within the EM map. In the crystal structure, one helical region (residues 188–202) does not have any major contacts between subunits or rings and so it is not significantly altered during the formation of 20S proteasomes. This is in agreement with Groll et al. (2003), who noted that the contact regions are not significantly different, between the crystal structures of free α- and the αβ-assembly in 20S, indicating a preexisting complementarity of the α-β contact surfaces.
It is not clear why the presence of the human β5 propeptides prevents 20S formation of the chimeric complexes. Most likely, however, it is due to a stabilization of the observed β-subunit conformation preventing the subunit rotation required for 20S formation. However, we cannot exclude that the foreign propeptide may result in a nonnative conformation of the 16S precursor complex or misfolded chimeric β-subunits. At the same time, propeptide substitution prevents final 20S formation independent of its length. Moreover, the estimated yields of soluble complexes from E. coli extracts were similar for wild-type and chimeric subunit combinations. The location of the seven human β5 propeptides attached to the N-terminus of each β-subunit cannot be seen in the EM density map. Due to the limited resolution of the structure, the location of the propeptides was not identified. We propose that they are positioned in the β-ring in a location protected from surface exposure to block access to the catalytic residue, Thr-1, until the conformational change of the β-subunits and propeptide cleavage occurs.
The conformational change in the second region, the beak-like domain, is the driving force in our proposed model for 20S proteasome formation. We postulate that the beak-like domain (residues 84–96) in each β-subunit moves up toward an approaching 16S precursor complex, in which the same conformational change is taking place in its β-ring. These beak-like domains allow residues in the β-ring to be exposed for contacts to be formed between two 16S precursor complexes to assemble into the 20S proteasome. These connections are between the residues of the beak-like domain of each β-subunit and the residues of the β-turn region of the diagonally opposing β-subunits.
The catalytic sites of the 20S proteasome are positioned within the β-subunits and consist of the catalytic residue, Thr-1, which becomes active only after the cleavage of the N-terminal propeptide. Additionally, the β-propeptides are thought to correctly position the 16S precursor complexes for the generation of 20S proteasomes in the maturation pathway (Groll et al., 2003). According to our model, during 20S proteasome formation, the catalytic residue, Thr-1, moves from being protected in the 16S structure to a more exposed position in the 20S structure as the β-subunits change conformation. Within the EM structure of 16S precursor complex, Thr-1 is positioned deep in the β-ring toward the inner walls, whereas in the crystal structure of the 20S proteasome Thr-1 is on the exposed surface of the β-subunits. The 16S precursor complexes with and without propeptides are inactive and this may be explained by the protected location of Thr-1 in the 16S structure (Schmidtke et al., 1997). It is the association and dimerization of two 16S precursor complexes into the preholoproteasome that leads to the final processing and autocatalytic removal of the β-subunit propeptides, resulting in proteasome activation (Chen and Hochstrasser, 1996; Heinemeyer et al., 1997).
We, therefore, propose a model for the final steps in proteasome assembly and maturation. During 16S precursor complex formation in Archaea, α-subunits oligomerize spontaneously into seven-membered rings, onto which β-subunits subsequently assemble. After formation of the 16S precursor complex, the β-subunits must undergo a conformational change and rotate upwards, toward the second 16S precursor complex in which the same conformational change is occurring. This rearrangement is induced either by the completely assembled β-ring or by an induced fit mechanism involving the second 16S precursor. One can speculate that in eukaryotes this mechanism is supported by maturation proteins (Krüger et al., 2003; Ramos et al., 1998; Witt et al., 2000). When the β-subunits have undergone their conformational change, they may fuse with the β-subunits in the second 16S precursor complex, through the interaction of the beak-like and the β-turn domains. Upon formation of these contacts, a 20S preholoproteasome is created. It remains inactive until the propeptide is cleaved off, thereby generating a catalytically active 20S proteasome.
Comparing the EM 16S precursor complex structure with that of the crystal structure of the 20S proteasome, we show that the structural changes within the β-subunit ring of the 16S precursor complex are essential for the formation of a proteolytically fully active 20S proteasome.
Acknowledgments
We thank Robert Huber for helpful discussion. We acknowledge Daniela Zantopf for her excellent technical assistance.
This work was supported in part by grants from the Welch Foundation (AU-1522) and National Institutes of Health (R01 NS 43258 and R01 GM 60635) to P.A.P. and from the Deutsche Forschungsgemeinschaft (KL 421/8-2) to P.M.K. and E.K.
Srinivas Mullapudi and Lee Pullan contributed equally to this work.
Ozlem T. Bishop's present address is University of the Western Cape, Biotechnology Department, Belville 7535, South Africa.
References
- Carson, M. 1997. Ribbons. Methods Enzymol. 277:493–505. [PubMed] [Google Scholar]
- Chen, P., and M. Hochstrasser. 1996. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell. 86:961–972. [DOI] [PubMed] [Google Scholar]
- Ditzel, L., R. Huber, K. Mann, W. Heinemeyer, D. H. Wolf, and M. Groll. 1998. Conformational constraints for protein self-cleavage in the proteasome. J. Mol. Biol. 279:1187–1191. [DOI] [PubMed] [Google Scholar]
- Frank, J., M. Radermacher, P. Penczek, J. Zhu, Y. Li, M. Ladjadj, and A. Leith. 1996. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116:190–199. [DOI] [PubMed] [Google Scholar]
- Frentzel, S., B. Pesold-Hurt, A. Seelig, and P. M. Kloetzel. 1994. 20S proteasomes are assembled via distinct precursor complexes. Processing of LMP2 and LMP7 proproteins takes place in 13–16S preproteasome complexes. J. Mol. Biol. 236:975–981. [DOI] [PubMed] [Google Scholar]
- Gerards, W. L., J. Enzlin, M. Haner, I. L. Hendriks, U. Aebi, H. Bloemendal, and W. Boelens. 1997. The human alpha-type proteasomal subunit HsC8 forms a double ringlike structure, but does not assemble into proteasome-like particles with the beta-type subunits HsDelta or HsBPROS26. J. Biol. Chem. 272:10080–10086. [DOI] [PubMed] [Google Scholar]
- Glickman, M. H., and A. Ciechanover. 2002. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82:373–428. [DOI] [PubMed] [Google Scholar]
- Groll, M., H. Brandstetter, H. Bartunik, G. Bourenkow, and R. Huber. 2003. Investigations on the maturation and regulation of archaebacterial proteasomes. J. Mol. Biol. 327:75–83. [DOI] [PubMed] [Google Scholar]
- Groll, M., L. Ditzel, J. Lowe, D. Stock, M. Bochtler, H. D. Bartunik, and R. Huber. 1997. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 386:463–471. [DOI] [PubMed] [Google Scholar]
- Groll, M., W. Heinemeyer, S. Jager, T. Ullrich, M. Bochtler, D. H. Wolf, and R. Huber. 1999. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc. Natl. Acad. Sci. USA. 96:10976–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll, M., and R. Huber. 2003. Substrate access and processing by the 20S proteasome core particle. Int. J. Biochem. Cell Biol. 35:606–616. [DOI] [PubMed] [Google Scholar]
- Heinemeyer, W., M. Fischer, T. Krimmer, U. Stachon, and D. H. Wolf. 1997. The active sites of the eukaryotic 20S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 272:25200–25209. [DOI] [PubMed] [Google Scholar]
- Hill, C. P., E. I. Masters, and F. G. Whitby. 2002. The 11S regulators of 20S proteasome activity. Curr. Top. Microbiol. Immunol. 268:73–89. [DOI] [PubMed] [Google Scholar]
- Huang, Z., P. R. Baldwin, S. Mullapudi, and P. A. Penczek. 2003. Automated determination of parameters describing power spectra of micrograph images in electron microscopy. J. Struct. Biol. 144:79–94. [DOI] [PubMed] [Google Scholar]
- Jager, S., M. Groll, R. Huber, D. H. Wolf, and W. Heinemeyer. 1999. Proteasome beta-type subunits: unequal roles of propeptides in core particle maturation and a hierarchy of active site function. J. Mol. Biol. 291:997–1013. [DOI] [PubMed] [Google Scholar]
- Kingsbury, D. J., T. A. Griffin, and R. A. Colbert. 2000. Novel propeptide function in 20S proteasome assembly influences beta subunit composition. J. Biol. Chem. 275:24156–24162. [DOI] [PubMed] [Google Scholar]
- Koster, A. J., R. Grimm, D. Typke, R. Hegerl, A. Stoschek, J. Walz, and W. Baumeister. 1997. Perspectives of molecular and cellular electron tomography. J. Struct. Biol. 120:276–308. [DOI] [PubMed] [Google Scholar]
- Krüger, E., U. Kuckelkorn, A. Sijts, and P. M. Kloetzel. 2003. The components of the proteasome system and their role in MHC class I antigen processing. Rev. Physiol. Biochem. Pharmacol. 148:81–104. [DOI] [PubMed] [Google Scholar]
- Kwon, Y. D., I. Nagy, P. D. Adams, W. Baumeister, and B. K. Jap. 2004. Crystal structures of the Rhodococcus proteasome with and without its pro-peptides: implications for the role of the pro-peptide in proteasome assembly. J. Mol. Biol. 335:233–245. [DOI] [PubMed] [Google Scholar]
- Lowe, J., D. Stock, B. Jap, P. Zwickl, W. Baumeister, and R. Huber. 1995. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science. 268:533–539. [DOI] [PubMed] [Google Scholar]
- Mayr, J., E. Seemuller, S. A. Muller, A. Engel, and W. Baumeister. 1998. Late events in the assembly of 20S proteasomes. J. Struct. Biol. 124:179–188. [DOI] [PubMed] [Google Scholar]
- Nandi, D., E. Woodward, D. B. Ginsburg, and J. J. Monaco. 1997. Intermediates in the formation of mouse 20S proteasomes: implications for the assembly of precursor beta subunits. EMBO J. 16:5363–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penczek, P. A. 1998. Measures of resolution using Fourier shell correlation. J. Mol. Biol. 280:115–116. [Google Scholar]
- Penczek, P. A., R. A. Grassucci, and J. Frank. 1994. The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy. 53:251–270. [DOI] [PubMed] [Google Scholar]
- Penczek, P. A., M. Radermacher, and J. Frank. 1992. Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy. 40:33–53. [PubMed] [Google Scholar]
- Ramos, P. C., J. Hockendorff, E. S. Johnson, A. Varshavsky, and R. J. Dohmen. 1998. Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell. 92:489–499. [DOI] [PubMed] [Google Scholar]
- Schmidt, M., and P. M. Kloetzel. 1997. Biogenesis of eukaryotic 20S proteasomes: the complex maturation pathway of a complex enzyme. FASEB J. 11:1235–1243. [DOI] [PubMed] [Google Scholar]
- Schmidtke, G., R. Kraft, S. Kostka, P. Henklein, C. Frommel, J. Lowe, R. Huber, P. M. Kloetzel, and M. Schmidt. 1996. Analysis of mammalian 20S proteasome biogenesis: the maturation of beta-subunits is an ordered two-step mechanism involving autocatalysis. EMBO J. 15:6887–6898. [PMC free article] [PubMed] [Google Scholar]
- Schmidtke, G., M. Schmidt, and P. M. Kloetzel. 1997. Maturation of mammalian 20S proteasome: purification and characterization of 13S and 16S proteasome precursor complexes. J. Mol. Biol. 268:95–106. [DOI] [PubMed] [Google Scholar]
- Seemuller, E., A. Lupas, and W. Baumeister. 1996. Autocatalytic processing of the 20S proteasome. Nature. 382:468–471. [DOI] [PubMed] [Google Scholar]
- Stoops, J. K., T. S. Baker, J. P. Schroeter, S. J. Kolodziej, X. D. Niu, and L. J. Reed. 1992. Three-dimensional structure of the truncated core of the Saccharomyces cerevisiae pyruvate dehydrogenase complex determined from negative stain and cryoelectron microscopy images. J. Biol. Chem. 267:24769–24775. [PMC free article] [PubMed] [Google Scholar]
- Stoops, J. K., J. P. Schroeter, J. P. Bretaudiere, N. H. Olson, T. S. Baker, and D. K. Strickland. 1991. Structural studies of human alpha 2-macroglobulin: concordance between projected views obtained by negative-stain and cryoelectron microscopy. J. Struct. Biol. 106:172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel, T., and W. Baumeister. 1995. Conformational constraints in protein degradation by the 20S proteasome. Nat. Struct. Biol. 2:199–204. [DOI] [PubMed] [Google Scholar]
- Witt, E., D. Zantopf, M. Schmidt, R. Kraft, P. M. Kloetzel, and E. Krüger. 2000. Characterisation of the newly identified human Ump1 homologue POMP and analysis of LMP7(beta 5i) incorporation into 20S proteasomes. J. Mol. Biol. 301:1–9. [DOI] [PubMed] [Google Scholar]
- Zuhl, F., E. Seemuller, R. Golbik, and W. Baumeister. 1997. Dissecting the assembly pathway of the 20S proteasome. FEBS Lett. 418:189–194. [DOI] [PubMed] [Google Scholar]
- Zwickl, P., J. Kleinz, and W. Baumeister. 1994. Critical elements in proteasome assembly. Nat. Struct. Biol. 11:765–770. [DOI] [PubMed] [Google Scholar]