

Abstract
The structure of individual nucleosomes organized within reconstituted 208-12 arrays at different levels of compaction was examined by tapping mode atomic force microscopy in air and liquid. Reconstitution at lower histone octamer to DNA weight ratios showed an extended beads-on-a-string morphology with less than the expected maximum of 12 nucleosome core particles per array, each particle located in the most favored positioning site. A correlation of the contour lengths of these arrays with the number of observed particles revealed two distinct populations of particles, one with ∼50 nm of bound DNA and a second population with ∼25 nm. The measured nucleosome center-to-center distances indicate that this ∼25 nm is not necessarily symmetrically bound about the dyad axis, but can also correspond to DNA bound from either the entry or exit point of the particle to a location at or close to the dyad axis. An assessment of particle heights suggests that particles wrapping ∼25 nm of DNA are most likely to be subnucleosomal particles, which lack either one or both H2A-H2B dimers. At a higher reconstitution ratio, folded compact arrays fully populated with 12 nucleosome core particles, were observed. Liquid measurements demonstrated dynamic movements of DNA loops protruding from these folded arrays.
INTRODUCTION
DNA in the eukaryotic nucleus is densely packaged into chromatin, of which the basic structural repeat unit is the nucleosome (Kornberg, 1974). This consists of a nucleosome core particle (NCP) along with linker DNA that connects successive particles. Recently, the structure of the NCP has been resolved by x-ray crystallography to a resolution of 1.9 Å (Davey et al., 2002). It is defined by ∼146 basepairs (bp) of DNA, which wrap in ∼1.65 left-handed superhelical turns around a core histone octamer, composed of a histone (H3-H4)2 tetramer and two histone H2A-H2B dimers. These NCPs are positioned every ∼200 bp throughout the eukaryotic genome, forming long arrays that are considered as the first level of DNA compaction. Further compaction of the nucleosomal array in the presence of linker histones results, at least in vitro, in a higher order chromatin structure that is termed the 30-nm fiber (Finch and Klug, 1976; van Holde, 1988). The structure of the higher order chromatin fiber remains a controversial issue. The two main competing models, the solenoid and zig-zag models, describe different linker DNA arrangement within the fiber (reviewed in Widom, 1998; Woodcock and Dimitrov, 2001).
Importantly, hierarchical levels of chromatin compaction play a crucial role not only in maintaining an orderly packaging of the DNA, but also in the regulation of DNA template-dependent processes, by controlling the access to the genetic code. This is relevant to the vital processes of DNA replication, recombination, transcription, and repair. Mechanisms of transitions between different chromatin structures remain to be fully characterized (reviewed in Horn and Peterson, 2002); however, a number of proposals have been made. For example, access of DNA binding proteins may involve ATP-dependent remodeling processes (for a detailed review see Becker and Horz (2002)) or covalent histone modifications (Jenuwein and Allis, 2001). Another interesting mechanism was proposed in the “site exposure model” (Polach and Widom, 1995, 1996). This mechanism involves access to the DNA template via spontaneous thermal fluctuations that result in an unpeeling of the DNA in a stepwise manner starting from either the entry or exit DNA point toward the dyad axis position (superhelical locations (SHLs) ±7–0 (Fig. 1)). Whether these mechanisms occur independently or as a concerted effort has still to be investigated for all the different processes involving access to the genetic code. Current chromatin research is greatly focused on the elucidation of these processes, and slowly a more complete picture is beginning to emerge.
FIGURE 1.

(Upper) A simple schematic of the direct DNA - histone hydrogen bonds determined from high-resolution x-ray crystallography studies of the NCP (Luger and Richmond, 1998; Davey et al., 2002). Interactions are formed predominantly at sites where the DNA minor groove faces inward to the histone core and these have been labeled with SHLs from −7 to +7. For clarity, interactions are illustrated only for one superhelical turn (eight of the fourteen regions) of the NCP. (Lower) A stretch of nucleosomal DNA, where all fourteen SHLs are indicated. The greatest numbers of interactions are located at the central bound DNA region, SHLs −0.5 and 0.5 (at the dyad axis). The least number of interactions are found at the entry-exit regions, SHLs ±5.5 and ±6.5. The other SHLs have an intermediate number of direct hydrogen bonds. An estimate of the binding strength at each SHL is shown in the figure (in terms of kBT) and was calculated from the known binding free energy, ΔG of −13.83 kcal/mol, of a NCP positioned on the same 208 sequence (Gottesfeld and Luger, 2001). This clearly does not take into account entropic considerations but does give a rough indication of possible strength at each site. Distances in terms of both bp and nm are indicated; the zero position is taken as the most centrally bound basepair at the location of the dyad axis. Possible binding motifs for a 25-nm length of DNA are indicated.
Here, we used atomic force microscopy (AFM) to examine in detail the arrangement and structure of NCPs positioned along 208-12 DNA templates at different core histone saturation levels. In contrast to previous AFM studies (Allen et al., 1993; Leuba et al., 1998; d'Erme et al., 2001; Schnitzler et al., 2001; Tomschik et al., 2001; Yodh et al., 2002), the samples used here were not fixed. We believe that this is of relevance to the results we present here. In particular, it is known that glutaraldehyde fixation has serious consequences in terms of internucleosomal interactions (Yodh et al., 1999). A comparison between our own results and those of other groups further demonstrates that a long fixation procedure may capture individual NCPs with binding motifs that are different from those in nonfixative conditions. In this study AFM imaging in air was used to provide high-resolution structural data of the arrays. This information was subsequently used for compaction analysis and accurate determination of NCP number and positioning. AFM imaging in liquid is ideal for studying the dynamic behavior of the molecules (Bennink et al., 2003). However, difficulties arose when trying to image the naked linker DNA along with the individual particles and hence could not be used for precise quantitative analysis of contour lengths and internucleosomal distances. Despite this, results of liquid measurements were used to ensure that no drying artifacts were present in the arrays imaged in air, and furthermore revealed for the first time that the DNA within compact arrays is not tightly wrapped but has rather dynamic behavior.
MATERIALS AND METHODS
Purification of histone octamers and nucleosomal array reconstitution
Native histone octamers were prepared from chicken erythrocytes (Simon and Felsenfeld, 1979). The histone octamer purity and stoichiometry were analyzed on 15% SDS-PAGE gels and it was found that all four core histones, H2A, H2B, H3, and H4, were present in equal stoichiometry and H1 was completely absent. The DNA template was a linear fragment containing 12 tandem repeats of a 208-bp nucleosome positioning sequence from 5S rDNA from the sea urchin Lytechinus variegates (Simpson et al., 1985). Nucleosomal arrays were reconstituted with purified histone octamers on 208-12 DNA templates at various saturation levels using stepwise salt dialysis (Hansen et al., 1989). Different saturation levels were achieved by mixing purified histone octamers with linear 208-12 DNA templates for octamer to DNA weight ratios of 0.75:1, 1:1, 1.25:1, and 1.5:1 (the concentration of DNA (190 ng/μl) was kept constant, and the reactions were carried out on ice, all were at 2 M NaCl). The samples were incubated at 37°C for 30 min to promote binding at the positioning sites. Stepwise salt dialysis was next performed; reaction mixtures were successively dialyzed against 1 M, 0.75 M, and 0 M NaCl solutions buffered with 10 mM TE (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) at 4°C. Each dialysis step was carried out for 4 h and the last step was overnight. Aliquots were taken from each sample after the last dialysis step were analyzed on a 1.6% agarose gel for 4 h and 45 min at 60 V in 1× TAE buffer (40 mM Tris - HCl, pH 8.0, 40 mM acetic acid, 1 mM EDTA).
(H3-H4)2 tetramer reconstitution was performed by reconstituting octamers and then by washing the octamer reconstitute with 1 M NaCl, 10 mM TEA-HCl (pH 7.5), followed by washing with 10 mM TEA-HCl (pH 7.5). This was done to make sure that the tetrasome-fibers contained only one tetramer per particle, and not two (Tomschik et al., 2001).
AFM methodology
Imaging was performed on a custom-built AFM (van der Werf et al., 1993), using silicon nitride cantilevers with spring constants of ∼0.5 N/m (Veeco Metrology, Sunnyvale, CA). Data was collected either in tapping mode in air (resonant frequency of 100–110 kHz) at ambient temperature and humidity, or in liquid (resonant frequency of 30–33 kHz) using an imaging buffer of 10 mM TE, 10 mM NaCl, 2 mM MgCl2, pH 7.5. The samples for imaging in air were prepared by diluting the stock reconstituted samples 10- to 20-fold in 10 mM TE, 5 mM NaCl, 2 mM MgCl2, pH 7.5. Next 5 μl was deposited onto freshly cleaved mica and left to incubate for 5 min in a humid environment, and then the samples were rinsed with ultrapure water and very gently dried with nitrogen gas. The samples for imaging in liquid were prepared by taking 5 μl of the stock reconstituted samples and diluting 10- to 20-fold in the same buffer as for the air samples, but containing 10 mM NaCl. These salt conditions were found to be optimal for immobilization to the mica surface. Five microliters of this diluted sample was deposited onto freshly cleaved mica and left to incubate for 30 s in a humid environment, and then 200 μl of the same dilution buffer was added on top.
RESULTS
Gel electrophoresis
An initial assessment of the reconstituted samples with different octamer/DNA weight ratios (Rw) was made using gel electrophoresis. The results are shown in Fig. 2 a. Variations in template saturation levels are clearly evident in the gel band shifts; different migration distances represent different levels of compaction. It was observed that with increasing amounts of histone octamer the migration band through the gel became more compact. This indicated an increase in the homogeneity in saturation of the 208-12 templates with NCPs.
FIGURE 2.

(a) Reconstituted nucleosomal arrays analyzed on 1.6% agarose gels for Rw of 0.75:1 (lane 1), 1:1 (lane 2), 1.25:1 (lane 3), and 1.5:1 (lane 4). Lane 5 is the 208-12 DNA fragment alone. Lane 6 is a Lambda/BstEII marker. (b) An AFM image of arrays reconstituted at Rw of 0.75:1 showing the procedure used to determine center-to-center distances for consecutive NCPs. The center of each NCP in an array is manually marked by the computer mouse pointer using custom designed software which then records x, y position and height z. x and y are determined with an accuracy of ∼2 nm and z is determined with an accuracy of ∼0.5 nm. The distance between the centers is then determined either by tracing the contour length of the DNA between them, if visible, or by estimating the shortest distance between the two x, y locations. (c) AFM image taken in liquid from the same sample as in b to confirm that the drying procedure does not mechanically disrupt the samples. This control was carried out for all samples as an independent check. (Bar, 50 nm; z range, 6 nm).
AFM image analysis
AFM images in air were used to determine distances between consecutive NCPs within the nucleosomal arrays. This analysis was carried out using custom-designed software. Distance errors due to the broadening effect of the AFM tip were avoided by measuring center-to-center distances of consecutive NCPs (Fig. 2 b). The contour lengths of individual arrays were measured only for those with clearly distinguishable ends. Fig. 2 c is the same Rw of 0.75:1 as shown in Fig. 2 b; however, this is a measurement performed in liquid. Problems associated with making accurate length measurements on molecules in motion, such as center-to-center distances and contour lengths, are clear. The liquid measurements, however, are very useful to verify that there are no drying artifacts in the arrays imaged in air. A visual comparison between the images taken in air and in liquid showed that the arrays maintained similar morphologies, indicating that the samples were not mechanically damaged by the deposition procedure.
Core histone octamer/DNA Rw of 1:1
An example of an AFM image from nucleosomal arrays reconstituted at an Rw of 1:1 is shown in Fig. 3 b. The arrays exhibited the typical beads-on-a-string morphology, populated with an average number of 10 NCPs, regularly spaced along the DNA template. A Gaussian fit to a histogram of the center-to-center distances (Fig. 3 a) determined a discrete peak at 22 nm. This result is in agreement with previously published data, in which NCP center-to-center distances of ∼20 nm are reported for the same tandemly repeated 208 DNA templates with extended linker DNA (Garcia-Ramirez et al., 1992; Hammermann et al., 2000; Yodh et al., 2002). It is interesting that the relationship between the center-to-center distance and the length of linker DNA depends only slightly on relative orientations of NCPs (for 146 bp of NCP-bound DNA, 62 bp remain as linker DNA, equivalent to ∼20 nm). For example it has been shown that linker DNA of 62 bp results in NCP center-to-center distances ranging from 21 to 23 nm (van Holde and Zlatanova, 1996).
FIGURE 3.

(a) A histogram of center-to-center distances for arrays at Rw of 1:1, taken from a total of 54 individual molecules. (b) A typical AFM image in air of an array reconstituted at this Rw (bar, 50 nm; z range, 6 nm). (c) A histogram of NCP heights reveals a single peak at 3.3 nm. (d) A histogram of center-to-center distances for arrays at Rw of 0.75:1, taken from a total of 111 individual molecules, reveals a multipeak distribution. (e) A typical AFM image in air of an array reconstituted at Rw of 0.75:1 (bar, 50 nm; z range, 6 nm). (f) A histogram of particle heights reveals three distinct peaks at 1.3, 2.4, and 3.5 nm.
The narrow histogram distribution clearly indicates that NCPs are located within the preferred positioning sites. A minor peak at ∼40 nm was also observed (see next section). Height measurements of the NCPs (Fig. 3 c) reveal a single peak at 3.3 nm. Caution must be taken when interpreting height measurements due to artifacts associated with different tapping conditions; this is particularly a problem when comparing separate images (van Noort et al., 1997; Rossell et al., 2003; Moreno-Herrero et al., 2003). It should be noted that all of our imaging conditions, such as tapping amplitude, setpoint, and tip-sample adhesion, were kept as constant as possible and the variation in height from one image to the next is estimated to be <0.5 nm.
Core histone octamer/DNA Rw of 0.75:1
Fig. 3 e shows an AFM image of a nucleosomal array reconstituted at Rw of 0.75:1. In contrast to the 1:1 Rw samples, visual inspection of the AFM images surprisingly shows particles of different size bound to the DNA templates (see also Fig. 2, b and c). Plotting the height measurements of these particles in a histogram reveals three discrete peaks (Fig. 3 f). This suggests that the particles observed here are likely to be not only histone octamers but also subnucleosomal particles, which lack either one or both H2A-H2B dimers. It is believed that the formation of a NCP is a process that first involves the binding of a (H3-H4)2 tetramer, followed by the binding of two separate H2A-H2B dimers (Smith et al., 1984).
To identify the observed particles, a control experiment was carried out where the 208-12 templates were reconstituted with (H3-H4)2 tetramers. A typical AFM image of a tetramer-DNA fiber is shown in Fig. 4 a. The height measurements of the tetrameric histone-DNA complexes reveal a single peak at 2.3 nm (Fig. 4 b). A comparison between the results obtained from the octameric and from the tetrameric fibers indicates that the middle-height particles observed at the 0.75:1 octameric reconstitution are histone tetramer-DNA complexes.
FIGURE 4.

(a) A typical AFM image of a tetramer-DNA array (bar, 100 nm; z range, 3 nm). (b) A histogram of particle heights reveals a single peak at 2.3 nm.
The particles bound to the DNA templates with heights of: 1.3 nm, 2.4 nm, and 3.5 nm were classified as H2A-H2B dimers, subnucleosomal particles (DNA-histone tetramer particles or DNA-hexamer particles), and complete NCPs. Since dimers bind nonspecifically to the DNA and are known to bind a DNA length that is too short to result in compaction (Luger et al., 1997), these small particles were not considered in any of the further analyses.
Thus, excluding the ∼1.3 nm high particles, it was obtained that DNA templates are populated with an average number of six particles. Center-to-center distance analysis resulted in a histogram with a number of discrete peaks (Fig. 3 d), which was fitted using a multiple Gaussian fit. In addition to a peak at 21 nm, similar to the 1:1 Rw (Fig. 3 a) and consistent with particles located at two adjacent nucleosome positioning sites, many other peaks were observed. This suggested a more complex histone binding pattern along the DNA templates.
Additionally the data was considered in terms of center-to-center distances for particles classified on a height basis (Fig. 5). The histograms are of center-to-center distances between two high 3.5-nm particles (H-H), between two medium 2.4-nm particles (M-M), and between a high and a medium-sized particle (H-M). Comparing these plots to that of all the center-to-center distances combined (Fig. 3 d) shows that in the case of the H-H measurements the peak at ∼100 nm has completely disappeared, H-M measurements show significant peaks at ∼45, ∼60, and ∼100 nm and the M-M measurements show peaks at ∼40 and ∼115 nm.
FIGURE 5.

Center-to-center distances of arrays reconstituted at Rw of 0.75:1, in which the particles are classified on a height basis, between two high 3.5-nm particles (H-H; shaded), a high and a medium-sized particle (H-M; white), and two medium 2.4-nm particles (M-M; black).
“25-nm wrap” model
To elucidate these results a model was developed which assumed that the particles did not always have the expected 50 nm (146 bp) of DNA bound, but also 25 nm. This model was derived based on evidence in the literature that the central region of the 146 bp of NCP DNA is bound most tightly with a gradation of affinities from the end (Polach and Widom, 1995, 1996). It is also evident from AFM and optical tweezers studies that NCPs can tightly wrap a 25-nm length of DNA (Brower-Toland et al., 2002; Sato et al., 1999; L. H. Pope, unpublished). To explain the data, it was required to includ in the model not only a 25-nm DNA binding motif symmetrically positioned about the dyad axis (from SHL −3.5 to 3.5), but also another which could be from either the entry or exit point of the DNA up to the dyad position, i.e., from SHLs ±7 to 0 (Fig. 1).
Taking into account these binding motifs, some of the possible arrangements of particles within an array are illustrated by Fig. 6 a. An overview of the model of all possible binding motifs and the expected center-to-center distances between adjacent particles is given in Fig. 6 b. The peaks in the experimental histogram data (Fig. 3 d) from 21 to 72 nm are in agreement with this model. Furthermore, the model assumes that not all of the nucleosome positioning sites are occupied and takes the maximum number of unoccupied binding sites between two particles as 2. One unoccupied site gives a distance increase of 70 nm (50 nm nucleosomal DNA + 20 nm linker DNA). All other experimental distances >72 nm can be explained by adding 70 nm to the model distances presented in Fig. 6 b.
FIGURE 6.

(a) Schematic of some of the possible arrangements of binding motifs, along a nucleosomal array, that could be adopted by particles wrapping either 50 nm or 25 nm of DNA. The distances, indicated by the arrows, can be calculated from the known 70-nm long 208-bp DNA sequence. (Inset) For simplicity the DNA involved in the NCP is presented as 50-nm nucleosomal DNA (dark shaded) and 10-nm linker DNA (light shaded) from either side. (b) Distances (nm) obtained by combining four different binding motifs, assumed by the model: (1) 50 nm of bound DNA from SHL −7 to +7 (see Fig. 1 for clarity), providing a DNA length for connection to the next particle of 10 nm for both n1 and n2; (2) 25 nm of DNA bound from SHL −7 to 0, providing 35 nm for n1 and 10 nm for n2; (3) 25 nm of DNA bound from SHL 0 to +7, providing 10 nm for n1 and 35 nm for n2; and (4) 25 nm of DNA bound from SHL −3.5 to +3.5, providing 22.5 nm for both n1 and n2. n1 and n2 are the first and the second positions, respectively, of consecutive particles along an array. The interacting DNA of each binding motif is indicated in black. In addition, the model assumes a maximum separation of two missing NCPs, where one missing NCP gives a distance of 70 nm. (c) A bar chart representing the comparison between the model and the experimental data normalized to the first peak. The model assumes that all motifs are adopted with equal probability and a maximum separation of two missing NCPs is possible, where one missing NCP gives a distance of 70 nm. The bar chart from the experimental data is calculated from the areas of the multi-Gaussian fitted peaks.
A consideration of the model binding motifs that the distances in Fig. 5 possibly correspond to supports the idea that the majority of particles that bind just ∼25 nm of DNA are likely to be subnucleosomal particles. For example, the most possible candidates for M-M distances of ∼40 and ∼115 (i.e., 45 + 70) nm are combinations, (2)-(2), (3)-(3), (4)-(4), which contain 25 nm of bound DNA and have ∼2.4 nm height. A contribution of (1) in those combinations cannot be completely ruled out. The fact that distances of ∼100 (i.e., ∼32.5 + 70) nm were not observed for H-H group indicates that mainly M particles, or subnucleosomal particles, contributed to these values.
In addition, the model shows that different combinations of binding motifs can result in the same distance. Hence, some center-to-center distances are expected with a higher frequency than others, if each motif is adopted with equal probability. It is clear that if certain binding motifs are more favorable than others this will be apparent in the measured data. A direct comparison of the model with the experimental data is given by the bar chart shown in Fig. 6 c. In agreement with the model, the most frequently observed distances were ∼20 and ∼45 nm. However, it should be noted that there are some inconsistencies between the model and the experimental results. For example, the model distances of 32.5 nm and 127.5 nm were not observed in the experimental data. It is possible that the histogram peak at ∼30 nm may be obscured by the peaks at 21 nm and 43 nm (Fig. 3 d). However, the peak at ∼128 nm is clearly not in the measured data and may reflect an unfavorable binding motif. Finally, the data clearly suggest that for these saturation levels there are typically one or less unoccupied sites between neighboring particles; two unoccupied sites is observed much less frequently (peak at 160 nm). A shift in center-to-center distance versus frequency plots is to be expected for different reconstitution conditions.
Contour length analysis for Rw of 0.75:1 and 1:1
Further insight into the amount of DNA bound within the particles was obtained from analysis of array contour lengths. For individual arrays, reconstituted at Rw of 0.75:1 and 1:1, both contour length and number of particles per array were determined (Fig. 7 a). This plot shows that DNA compaction increases with an increasing number of particles. A decrease in contour length of ∼50 nm is expected for each NCP containing 146 bp of DNA. In this case the contour length of the nucleosomal array (L) can be described as
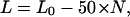 |
(1) |
where L0 is the length of the naked 208-12 DNA (2 500 bp, ∼850 nm), and N is the number of observed NCPs per array. Eq. 1 yields a single straight line for a plot of array length L versus N (line P0 in Fig. 7 a).
FIGURE 7.

(a) A plot of the number of particles per array versus its contour length for molecules reconstituted at Rw of 0.75:1 (○) or at Rw of 1:1 (•). The data was collected with an accuracy of ±5 nm. The fit to the Eq. 2 is given by the lines P0 to P12, which represent the number of particles with 25 nm of DNA bound from 0 to 12, respectively. (b) A plot of the percentage of particles with 25 nm of bound DNA as a function of number of particles per array. Error bars represent the mean error of the standard deviation.
Clearly this equation, however, does not hold for the 0.75:1 data. The data fall along a number of lines that deviate from P0 by a change in the slope (lines P1 to P12), where the length of the arrays differs by ∼25 nm. Eq. 1 can now be rewritten as
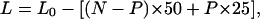 |
(2) |
where P is the number of particles with 25 nm of bound DNA and (N−P) is the number of particles containing 50 nm DNA. This supports our model that the particles do not necessarily have 50 nm of bound DNA, but can bind also 25 nm of DNA and form a stable particle. Using Eq. 2 it was then possible to determine the percentage of particles containing 25 nm of bound DNA for each nucleosomal array. This calculation was carried out for each value of N (Fig. 7 b) and showed that for reconstitutions at Rw of 0.75:1, with increasing N the percentage of particles with ∼25 nm DNA decreased to a minimum of ∼50%. For arrays reconstituted at Rw of 1:1, at values of N ≥ 9, the percentage decreased to ∼0%. From these results it is clear that the 0.75:1 Rw reconstitution conditions result in the formation of nucleosomal arrays that contain more particles with ∼25 nm, rather than ∼50 nm, of bound DNA. In contrast, arrays at the Rw of 1:1 typically contained particles with ∼50 nm DNA.
These results suggest that more saturated DNA templates, formed at higher histone octamer concentrations, allow the formation of NCPs with the expected 50-nm length of bound DNA. The difference in percentage P for the two different samples containing arrays with the same number of particles also supports the height data showing that the particle compositions are different. If arrays with the same number of particles were of the same composition, they would be expected to have the same number of 25-nm and 50-nm DNA-wrapped particles. The only reasonable explanation that can be given of this observation is that at the lower Rw of 0.75:1, more of the particles are not complete NCPs but subnucleosomal particles, likely to contain either just the (H3-H4)2 tetramer or a hexamer of (H3-H4)2 plus one H2A-H2B dimer. It seems logical to expect that missing H2A-H2B dimers from the particle could result in a reduced ability to stably wrap 50 nm of DNA.
To examine the effects of fixation on the composition of the NCPs, a control experiment was undertaken where 208-12 nucleosomal arrays reconstituted at Rw of 0.75:1 were fixed with glutaraldehyde. The contour length of fixed nucleosomal arrays (typical AFM image is shown in Fig. 8 a) as a function of the number of particles is shown in Fig. 8 b. Eq. 1 was used to fit the data where the decrease in contour length was ∼50 nm per particle. The analysis indicated that the NCPs in the fixed arrays contain 50 nm bound DNA.
FIGURE 8.

(a) An AFM image in air of a fixed array reconstituted at Rw of 0.75:1 (bar, 50 nm; z range, 4 nm). (b) A plot of the number of particles per array versus its contour length. Eq. 1 was used to fit the data.
Core histone octamer to DNA Rw of 1.5:1
Fig. 9 a shows nucleosomal arrays reconstituted at Rw of 1.5:1. It is interesting to see that under these conditions the arrays no longer exhibit an extended beads-on-a-string structure but are much more compact. These compact structures resemble that of a “bunch-of-grapes” morphology. Measurement of the heights of the NCPs in these compact structures of 3.4 (± 1.1) nm (data not shown) reveals a value close to that measured for the extended beads-on-a-string structures (Fig. 3 c). These results indicate that the compact nucleosomal arrays are flat on the surface, where all particles are clearly visible. Simple counting of the number of NCPs per structure revealed a value of 12, consistent with fully saturated DNA templates.
FIGURE 9.

(a) An AFM image in air of compact arrays reconstituted at Rw of 1.5:1 (bar, 50 nm; z range, 6 nm). (b-d) Consecutive AFM images in liquid, focusing on one individual array reconstituted at Rw of 1.5:1 (bar, 50 nm; z range, 6 nm). The time between successive images is 30 s. The arrows indicate the dynamic movement of DNA loops protruding from the array.
Furthermore, AFM measurements in liquid (Fig. 9, b–d) show consecutive images of a dynamic movement of DNA loops protruding from one of these compact structures. It should be noted that in contrast to the images taken in air, the poor resolution here is a result of imaging molecules in motion. From AFM measurements in both air and liquid, it was not possible to determine the order of the NCPs along the DNA.
DISCUSSION
At Rw of 0.75:1 and 1:1 it was observed that the histone particle arrays had extended beads-on-a-string conformations. In both cases the average number of particles was less than the maximum expected number of 12, and it was also evident that the number of particles increased with increasing octamer concentration in the reconstitution protocol. For arrays reconstituted at Rw of 1:1, the particles were located in the most favored positioning site along the template and were of similar dimensions (height of ∼3.3 nm). The most probable center-to-center distance was found to be 22 nm, in excellent agreement with previous studies of the same 208-12 template with extended linker DNA lengths of ∼20 nm and particles that wrap ∼50 nm of DNA. These results are consistent with DNA wrapped around a complete histone octamer (forming a NCP) and bound at the preferred positioning site.
For arrays reconstituted at Rw of 0.75:1 differently sized particles were distributed along the DNA template. A histogram of particle heights showed three different distributions centered at 1.3 nm, 2.4 nm, and 3.5 nm that most likely correspond to H2A-H2B dimers, subnucleosomal particles and complete nucleosome core particles. Assuming that the dimers do not compact the DNA template in length, analysis of center-to-center distances of particles with heights of ∼2.4 and ∼3.5 nm revealed a number of other discrete values in addition to the expected peak at 21 nm. These discrete distances suggested that the particles were accurately positioned within the binding sites, however, in contrast to the 1:1 reconstitution, with different DNA-binding motifs. The existence of two stable well-positioned particle populations, as the model describes, one with ∼50 nm of bound DNA and a second with ∼25 nm, could be explained in a number of different ways. Considering the results of the Rw of 1:1, it is likely that the particles wrapping ∼50 nm of DNA are complete NCPs containing the histone octamer. Particles wrapping only ∼25 nm of DNA could be histone octamers, hexamers, or tetramers. We can consider the results here in terms of all these different possibilities for particle constitution:
Wrapping of DNA around nucleosomal particles
Firstly, if the 25-nm DNA-wrapped particles contain a histone octamer then these results support the view that specific regions of the DNA are able to unbind without dissociation of histones from the template. This behavior is consistent with the site exposure model where nucleosomes as dynamic structures, transiently expose stretches of their DNA with decreasing tendencies from the ends to the central region (Polach and Widom, 1995, 1996). This would provide evidence that NCPs exist as different populations of stable particles, wrapping, for example, either ∼50 or ∼25 nm of DNA.
Another explanation is that in the subsaturated array, the ∼25 nm binding motif depends on the conformations of adjacent particles and the stability that they confer. For example, if positioning site n − 2 is occupied, site n − 1 is unoccupied, and site n + 1 is occupied, then internucleosomal interactions between n and n − 2 are expected to be weaker than those between n and n + 1, due to an increased interparticle distance. This type of particle environment may subsequently induce nonsymmetrical particle stability about the dyad position.
In contrast, from the assessment of H-bond interactions, it would be intuitive to expect that in the case of an intact NCP this ∼25 nm of DNA would be bound symmetrically about the dyad axis between SHLs −3.5 and 3.5 where the greatest number of direct H-bond interactions exist (Fig. 1). This is clearly not the sole 25-nm binding motif from the results presented here.
Wrapping of DNA around subnucleosomal particles
However, the height analysis of the particles suggests that the subsaturated arrays contain both NCPs and subnucleosomal particles. Evidence in the literature that supports our data comes from micrococcal nuclease digestion of tetrasomes, in which 25-nm rather than 50-nm binding motifs are observed (Dong and van Holde, 1991; Hansen and Wolffe, 1994; Alilat et al., 1999). Hence from both our own data and previously published data it appears most likely that full NCPs wrap ∼50 nm of DNA, whereas subnucleosomal particles wrap ∼25 nm of DNA.
The different 25-nm binding motifs of subnucleosomal particles could be considered as a result of both intra- and internucleosomal interactions that are lost when H2A-H2B dimers are absent from the structure (Luger et al., 1997). For example, if a particle that binds ∼25 nm of DNA consists of a histone hexamer or tetramer rather than an octamer, then tails that are usually available to participate in interparticle interactions will be absent. It may in such a way bias the stability of DNA bound to a neighboring particle in a nonsymmetrical manner. If we consider intranucleosomal stability through examining the crystal structure of the NCP, it is clear that the absence of a single H2A-H2B dimer would result in the loss of interactions only at one side of the particle. Hence particles containing histone hexamers could be responsible for the nonsymmetric DNA binding motif that is observed here. It should be noted that loss of a single dimer is, however, more likely to result in a particle with >25 nm of DNA, rather ∼30 nm. This exact amount is difficult to determine from the technique that we describe here. However, in the case where both H2A-H2B dimers are absent, DNA-histone interactions would be broken, resulting in a 25-nm binding motif symmetrical about the dyad axis. All of the above-mentioned explanations are consistent with our experimental observations.
Effect of glutaraldehyde fixation and surface forces
The samples used here containing NCPs with 25 nm of wrapped DNA were not fixed with glutaraldehyde or any other fixatives. We believe that glutaraldehyde fixation has serious consequences in terms of intra- and internucleosomal interactions. Analysis of the effects of glutaraldehyde fixation on the composition of 208-12 nucleosomal arrays showed that NCPs in fixed arrays contain 50 nm bound DNA. The cross-linking mechanism of glutaraldehyde is thought to act through lysine groups of the histone tails (Sewell et al., 1984), and hence might alternate tail-tail interactions. This fixation process also could capture individual NCPs in more static conformations that are different from those we observe in nonfixative conditions.
Another reason for observing the partially unwrapped NCPs is that surface forces encountered in the AFM technique could enhance intrinsic NCP lability in nonfixed arrays. Unfixed samples show a lower average NCP occupancy than fixed samples (Leuba et al., 1994; Yodh et al., 1999). Thus, NCPs are apparently somewhat unstable when they are not fixed and surface effects could alter the structure by depleting the histone H2A-H2B dimers. It should be mentioned here that in solution, elevated salt (∼100 mM) is required to achieve significant H2A-H2B depletion from nucleosomes. Since the samples are in low salt, they should not be significantly depleted in H2A-H2B. Nevertheless, mica surface can generate high local salt concentrations and thus could be the cause of the depletion.
However, the same AFM procedure used in this work is gentle enough to visualize the more highly loaded nucleosomal arrays (Rw of 1:1) without causing almost any disruption, based on the percentage of particles wrapping ∼25 nm DNA (Fig. 7 b). Here in the saturated samples possible enhanced internucleosomal contacts could stabilize those arrays against surface-induced depletion.
Furthermore, dynamic AFM imaging of nonfixed nucleosomal arrays in liquid, which reflects more solution conformation conditions, revealed quantized behavior of wrapping/unwrapping events with the same step sizes of 50 nm and 25 nm DNA as in the experiments performed under air conditions (D. N. Nikova, unpublished). This is an indication that the partial wrapping is not an AFM artifact caused by the drying procedure but is rather an intrinsic property of the NCP.
“Structural snapshots” of a nonfixed nucleosomal array
Since the samples we studied were not fixed, we may consider the AFM images taken in air as structural “snapshots” of dynamic nucleosomal arrays in which DNA is able to wrap/unwrap from the core histone surface. Although it is likely that different structural intermediates are transiently visited by a “dynamic” particle, our data provide evidence that specific binding motifs are preferred, and that these are likely to reflect the most energetically favorable conformations. These stable conformations were classified as follows: 1), ∼50 nm of DNA bound from SHLs −7 to +7; 2), ∼25 nm of DNA bound from either the entry or exit point to the dyad axis, i.e., from SHLs ±7 to 0; and 3), ∼25 nm of DNA symmetrically bound at the dyad axis from SHLs −3.5 to +3.5. Since from the discussion above it seems most intuitive that these ∼25-nm particles are subnucleosomal particles, we can further extend the site exposure model to suggest that removal of a H2A-H2B dimer may be sufficient to unpeel the DNA from one end of the histone core particle, in a process that results in a stable particle but with altered accessibility to nucleosomal DNA. It is known that transcriptionally active chromatin contains NCPs depleted in H2A and H2B (Hansen and Wolffe, 1994; Wolffe, 1994). Furthermore, a recent report suggests that RNA Pol II can displace H2A-H2B dimers (Kireeva et al., 2002).
Folded nucleosomal arrays
Earlier AFM studies of condensed chromatin fibers isolated from chicken erythrocytes and Tetrahymena thermophila, respectively, revealed irregular and blobby structure, where individual nucleosomes are no longer distinct (Martin et al., 1995; Zlatanova et al., 1998). In contrast, the high-resolution images obtained here at Rw of 1.5:1 showed that the individual NCPs are clearly discernible and the number of NCPs per structure can be attained. It was interesting to see that at this reconstitution ratio, fully populated DNA templates containing 12 NCPs were capable of folding from the beads-on-a-string structure to a higher order of compaction (Fig. 9 a). It is known that DNA charge neutralization plays a major role in chromatin compaction (Schwarz and Hansen, 1994; Schwarz et al., 1996). However, although the presence of polyvalent cations is absolutely necessary for chromatin compaction, these ionic factors alone cannot induce folding if a NCP is missing from the fiber. Although in our study MgCl2 was present at a concentration of 2 mM in all samples, arrays containing 10 or 11 NCPs did not undergo compaction. These observations imply that a structural factor such as NCP positioning and occupancy play an important role in chromatin folding. This is consistent with earlier studies suggesting that compaction in 1–2 mM MgCl2 is only possible when all sites in a DNA template are occupied; if just one NCP of 208-12 array is missing, folding is inhibited (Fletcher et al., 1994; Schwarz and Hansen, 1994).
Furthermore, our results demonstrate that the NCP-NCP interactions that are required here for the formation of a secondary compact structure are likely to be between nonneighboring particles. The nonneighbor interactions are too few to hold the folded structure together when a run of <12 consecutive NCPs is present. It is likely that when there are no closely spaced NCPs, the attraction between them decreases, resulting in unfolded structures. We note that at Rw of 1:1 typically the 12-mer arrays contain 10 NCPs (Fig. 3 b) and it is possible that the structures formed here represent an intermediate between the fully extended and compact states. It would be interesting at this Rw of 1.5:1 to study longer nucleosomal arrays to observe the compaction process in more detail.
Additionally, the consecutive dynamic AFM images in buffer (Fig. 9, b–d) indicated that the folded nucleosomal arrays were intrinsically stable, at least on the surface, in solution. However, stretches of the DNA were highly dynamic, loops clearly protruded from the folded structure and then disappeared again. The results suggest that the DNA within folded arrays is not tightly bound but is able to wrap/unwrap from the structure. Such behavior is relevant to processes where proteins gain access to the genetic code without dramatic changes in the compaction level.
CONCLUSIONS
The results from this study clearly demonstrate that simply through variation of reconstitution conditions, different structural forms of chromatin can be generated. These range from an extended array that may represent an “active” chromatin structure, where the absence of H2A-H2B dimers is of particular significance, to a more stable beads-on-a-string array, which could provide information about the importance of nearest neighbor internucleosomal interactions. Finally, the preparation of folded arrays may provide valuable information about higher-order chromatin structure, but also could allow studies of the dynamics between the extended and compact forms of chromatin through the alteration of internucleosomal interactions. Additionally the data support the view that consecutive runs of NCPs are required for folding to higher-order levels of compaction from which it is evident that internucleosomal interactions between nonneighboring NCPs play a central role. Clearly AFM studies of nucleosomal arrays, in different structural forms, in both liquid and air conditions will provide vital clues toward the dynamics of individual nucleosomes and the effect of the environment in which these structures are situated. This research is of particular relevance to processes that require mechanisms for the release of DNA from the NCP in a regulated manner. Future work will ideally focus more on liquid measurements to study the dynamics of these particles in greater detail.
Acknowledgments
We are grateful to Dr. S. Leuba (University of Pittsburgh) for the gift of 208-12 DNA. The schematic of the NCP in Fig. 1 was kindly provided by Anthony de Vries (University of Twente, The Netherlands).
Research is supported by the Netherlands Organization for Scientific Research (NWO), division Earth and Life Sciences (ALW). L.H.P. was supported by The Dutch Foundation for Fundamental Research on Matter.
References
- Alilat, M., A. Sivolob, B. Révet, and A. Prunell. 1999. Nucleosome dynamics. Protein and DNA contributions in the chiral transition of the tetrasome, the histone (H3–H4)2 tetramer-DNA Particle. J. Mol. Biol. 291:815–841. [DOI] [PubMed] [Google Scholar]
- Allen, M. J., X. F. Dong, T. E. O'Neill, P. Yau, S. C. Kowalczykowski, J. Gatewood, R. Balhorn, and E. M. Bradbury. 1993. Atomic force microscope measurements of nucleosome cores assembled along defined DNA sequences. Biochemistry. 32:8390–8396. [DOI] [PubMed] [Google Scholar]
- Becker, P. B., and W. Horz. 2002. ATP-dependent nucleosome remodeling. Annu. Rev. Biochem. 71:247–273. [DOI] [PubMed] [Google Scholar]
- Bennink, M., D. Nikova, K. van der Werf, and J. Greve. 2003. Dynamic imaging of single DNA-protein interactions using atomic force microscopy. Anal. Chim. Acta. 479:3–15. [Google Scholar]
- Brower-Toland, B. D., C. L. Smith, R. C. Yeh, J. T. Lis, C. L. Peterson, and M. D. Wang. 2002. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA. 99:1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey, C. A., D. F. Sargent, K. Luger, A. W. Maeder, and T. J. Richmond. 2002. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- d'Erme, M., G. Yang, E. Sheagly, F. Palitti, and C. Bustamante. 2001. Effect of poly(ADP-ribosyl)ation and Mg2+ ions on chromatin structure revealed by scanning force microscopy. Biochemistry. 40:10947–10955. [DOI] [PubMed] [Google Scholar]
- Dong, F., and K. E. van Holde. 1991. Nucleosome positioning is determined by the (H3–H4)2 tetramer. Proc. Natl. Acad. Sci. USA. 88:10596–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch, J. T., and A. Klug. 1976. Solenoidal model for superstructure in chromatin. Proc. Natl. Acad. Sci. USA. 73:1897–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, T. M., P. Serwer, and J. C. Hansen. 1994. Quantitative analysis of macromolecular conformational changes using agarose gel electrophoresis: application to chromatin folding. Biochemistry. 33:10859–10863. [DOI] [PubMed] [Google Scholar]
- Garcia-Ramirez, M., F. Dong, and J. Ausio. 1992. Role of the histone “tails” in the folding of oligonucleosomes depleted of histone H1. J. Biol. Chem. 267:19587–19595. [PubMed] [Google Scholar]
- Gottesfeld, J. M., and K. Luger. 2001. Energetics and affinity of the histone octamer for defined DNA sequences. Biochemistry. 40:10927–10933. [DOI] [PubMed] [Google Scholar]
- Hammermann, M., K. Toth, C. Rodemer, W. Waldeck, R. P. May, and J. Langowski. 2000. Salt-dependent compaction of di- and trinucleosomes studied by small-angle neutron scattering. Biophys. J. 79:584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J. C., J. Ausio, V. H. Stanik, and K. E. van Holde. 1989. Homogeneous reconstituted oligonucleosomes, evidence for salt-dependent folding in the absence of histone H1. Biochemistry. 28:9129–9136. [DOI] [PubMed] [Google Scholar]
- Hansen, J. C., and A. P. Wolffe. 1994. A role for histones H2A/H2B in chromatin folding and transcriptional repression. Proc. Natl. Acad. Sci. USA. 91:2339–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, P. J., and C. L. Peterson. 2002. Molecular biology. Chromatin higher order folding—wrapping up transcription. Science. 297:1824–1827. [DOI] [PubMed] [Google Scholar]
- Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science. 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- Kireeva, M. L., W. Walter, V. Tchernajenko, V. Bondarenko, M. Kashlev, and V. M. Studitsky. 2002. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol. Cell. 9:541–552. [DOI] [PubMed] [Google Scholar]
- Kornberg, R. D. 1974. Chromatin structure: a repeating unit of histones and DNA. Science. 184:868–871. [DOI] [PubMed] [Google Scholar]
- Leuba, S. H., G. Yang, C. Robert, B. Samori, K. van Holde, J. Zlatanova, and C. Bustamante. 1994. Three-dimensional structure of extended chromatin fibers as revealed by tapping-mode scanning force microscopy. Proc. Natl. Acad. Sci. USA. 91:11621–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba, S. H., C. Bustamante, K. van Holde, and J. Zlatanova. 1998. Linker histone tails and N-tails of histone H3 are redundant: scanning force microscopy studies of reconstituted fibers. Biophys. J. 74:2830–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389:251–260. [DOI] [PubMed] [Google Scholar]
- Luger, K., and T. J. Richmond. 1998. DNA binding within the nucleosome core. Curr. Opin. Struct. Biol. 8:33–40. [DOI] [PubMed] [Google Scholar]
- Martin, L. D., J. P. Vesenka, E. Henderson, and D. L. Dobbs. 1995. Visualization of nucleosomal substructure in native chromatin by atomic force microscopy. Biochemistry. 34:4610–4616. [DOI] [PubMed] [Google Scholar]
- Moreno-Herrero, V., J. Colchero, and A. M. Baro. 2003. DNA height in scanning force microscopy. Ultramicroscopy. 96:167–174. [DOI] [PubMed] [Google Scholar]
- Polach, K. J., and J. Widom. 1995. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 254:130–149. [DOI] [PubMed] [Google Scholar]
- Polach, K. J., and J. Widom. 1996. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol. 258:800–812. [DOI] [PubMed] [Google Scholar]
- Rossell, J. P., S. Allen, M. C. Davies, C. J. Roberts, S. J. B. Tendler, and P. M. Williams. 2003. Electrostatic interactions observed when imaging proteins with the atomic force microscope. Ultramicroscopy. 96:37–46. [DOI] [PubMed] [Google Scholar]
- Sato, M. H., K. Ura, K. I. Hohmura, F. Tokumasu, S. H. Yoshimura, F. Hanaoka, and K. Takeyasu. 1999. Atomic force microscopy sees nucleosome positioning and histone H1-induced compaction in reconstituted chromatin. FEBS Lett. 452:267–271. [DOI] [PubMed] [Google Scholar]
- Schnitzler, G. R., C. L. Cheung, J. H. Hafner, A. J. Saurin, R. E. Kingston, and C. M. Lieber. 2001. Direct imaging of human SWI/SNF-remodeled mono- and polynucleosomes by atomic force microscopy employing carbon nanotube tips. Mol. Cell. Biol. 21:8504–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, P. M., A. Felthauser, T. M. Fletcher, and J. C. Hansen. 1996. Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry. 35:4009–4015. [DOI] [PubMed] [Google Scholar]
- Schwarz, P. M., and J. C. Hansen. 1994. Formation and stability of higher order chromatin structures. Contributions of the histone octamer. J. Biol. Chem. 269:16284–16289. [PubMed] [Google Scholar]
- Sewell, B., C. Bouloukos, and C. von Holt. 1984. Formaldehyde and glutaraldehyde in the fixation of chromatin for electron microscopy. J. Microsc. 136:103–112. [DOI] [PubMed] [Google Scholar]
- Simon, R. H., and G. Felsenfeld. 1979. A new procedure for purifying histone pairs H2A + H2B and H3 + H4 from chromatin using hydroxylapatite. Nucleic Acids Res. 6:689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, R. T., F. Thoma, and J. M. Brubaker. 1985. Chromatin reconstituted from tandemly repeated cloned DNA fragments and core histones: a model system for study of higher order structure. Cell. 42:799–808. [DOI] [PubMed] [Google Scholar]
- Smith, D. R., I. J. Jackson, and D. D. Brown. 1984. Domains of the positive transcription factor specific for the Xenopus 5S RNA gene. Cell. 37:645–652. [DOI] [PubMed] [Google Scholar]
- Tomschik, M., M. A. Karymov, J. Zlatanova, and S. H. Leuba. 2001. The archaeal histone-fold protein HMf organizes DNA into bona fide chromatin fibers. Structure (Camb). 9:1201–1211. [DOI] [PubMed] [Google Scholar]
- van der Werf, K. O., C. Putman, B. G. de Grooth, F. Segerink, E. Schipper, N. F. van Hulst, and J. Greve. 1993. Compact stand-alone atomic force microscope. Rev. Sci. Instrum. 64:2892–2897. [Google Scholar]
- van Holde, K., and J. Zlatanova. 1996. What determines the folding of the chromatin fiber? Proc. Natl. Acad. Sci. USA. 93:10548–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde, K. E. 1988. In Chromatin. A. Rich, editor. Springer, New York. 111–148.
- van Noort, J., K. O. van der Werf, B. G. de Grooth, N. F. van Hulst, and J. Greve. 1997. Height anomalies in tapping mode atomic force microscopy in air caused by adhesion. Ultramicroscopy. 69:117–127. [Google Scholar]
- Widom, J. 1998. Structure, dynamics, and function of chromatin in vitro. Annu. Rev. Biophys. Biomol. Struct. 27:285–327. [DOI] [PubMed] [Google Scholar]
- Wolffe, A. P. 1994. Transcription: in tune with the histones. Cell. 77:13–16. [DOI] [PubMed] [Google Scholar]
- Woodcock, C. L., and S. Dimitrov. 2001. Higher-order structure of chromatin and chromosomes. Curr. Opin. Genet. Dev. 11:130–135. [DOI] [PubMed] [Google Scholar]
- Yodh, J. G., Y. L. Lyubchenko, L. S. Shlyakhtenko, N. Woodbury, and D. Lohr. 1999. Evidence for nonrandom behavior in 208–12 subsaturated nucleosomal array populations analyzed by AFM. Biochemistry. 38:15756–15763. [DOI] [PubMed] [Google Scholar]
- Yodh, J. G., N. Woodbury, L. S. Shlyakhtenko, Y. L. Lyubchenko, and D. Lohr. 2002. Mapping nucleosome locations on the 208–12 by AFM provides clear evidence for cooperativity in array occupation. Biochemistry. 41:3565–3574. [DOI] [PubMed] [Google Scholar]
- Zlatanova, J., S. H. Leuba, and K. van Holde. 1998. Chromatin fiber structure: morphology, molecular determinants, structural transitions. Biophys. J. 74:2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]