

Abstract
Although the addition of just the excitation light field at the focus, or of just the fluorescence field at the detector is sufficient for a three- to fivefold resolution increase in 4Pi-fluorescence microscopy, substantial improvements of its optical properties are achieved by exploiting both effects simultaneously. They encompass not only an additional expansion of the optical bandwidth, but also an amplified transfer of the newly gained spatial frequencies to the image. Here we report on the realization and the imaging properties of this 4Pi microscopy mode of type C that also is the far-field microscope with the hitherto largest aperture. We show that in conjunction with two-photon excitation, the resulting optical transfer function displays a sevenfold improvement of axial three-dimensional resolution over confocal microscopy in aqueous samples, and more importantly, a marked transfer of all frequencies within its inner region of support. The latter is present also without the confocal pinhole. Thus, linear image deconvolution is possible both for confocalized and nonconfocalized live-cell 4Pi imaging. Realized in a state-of-the-art scanning microscope, this approach enables robust three-dimensional imaging of fixed and live cells at ∼80 nm axial resolution.
INTRODUCTION
Improving the axial resolution is a rewarding but challenging current endeavor in fluorescence microscopy (Diaspro, 2002; Hell, 2003). For the largest aperture available, the axial extent of the main focal spot is ∼2λ/n, where λ is the wavelength of light and n the refractive index of the mounting medium. This is about four times larger than the ∼λ/(2n) value found in the transverse direction. The interference of the focused wavefronts of two opposing objective lenses readily produces a focal axial width of ∼λ/(4n). However, with current apertures, the focal maximum is also accompanied by interference side lobes (Hell and Stelzer, 1992a; Hell et al., 1997). The periodic repetition of peaks in the point-spread function (PSF) of a microscope signifies that the associated spatial frequency is weak or nonexistent in its optical transfer function (OTF) (Gu and Sheppard, 1994; Schrader et al., 1997; Gustafsson, 1999; Nagorni and Hell, 2001a). Thus, the side lobe height crucially determines the viability of an interference-based approach to axial superresolution. As a rule of thumb, the first-order side lobes should be <∼50% of the main focal maximum (Hänninen et al., 1995; Nagorni and Hell, 2001a).
The height and number of the side lobes decrease dramatically with the aperture angle of the lenses, but with current lenses, adding two wavefronts entails primary lobe heights of ∼70% of the maximum (Hell and Stelzer, 1992a). Effective for the lobe suppression are the mechanism of two-photon fluorescence excitation (Denk et al., 1990) of the fluorophore and confocal detection (Hell and Stelzer, 1992b). The third option is the simultaneous constructive interference of the excitation and the detection wavefront pairs (Hell and Stelzer, 1992a,b; Gustafsson et al., 1995; Nagorni and Hell, 2001a). The latter is efficient if the actual wavelengths are significantly different, because wavelength disparity leads to different axial locations of the side lobes. As a result, fluorescence at the side lobes of the excitation spot is weakened by incomplete detection. In I5M microscopy (Gustafsson et al., 1995, 1999; Nagorni and Hell, 2001a), the joint action of illumination and detection interference is mandatory because other mechanisms of lobe suppression are absent here. Still, the OTF of the I5M features periodic regions of minor transfer (Nagorni and Hell, 2001a) (see also Appendix).
The approach of pairing coherent illumination with coherent detection through both lenses (see Fig. 1) has been introduced in the framework of 4Pi microscopy where it is referred to as “4Pi microscopy of type C” (Hell and Stelzer, 1992a). Although “4Pi type C” has been shown for light scattering (Hell et al., 1994), it has not been explored in 4Pi imaging of fluorescent samples to date. This is because unlike in I5M, other less-demanding lobe suppression mechanisms were available. However, current ambitions of 4Pi microscopy with regard to live-cell imaging (Egner et al., 2002, 2004) render OTF improvement desirable, especially in light of the fact that live-cell imaging requires water-immersion lenses. Because they have a smaller aperture angle than their oil- or glycerol-immersion counterparts, water-immersion lenses are less suitable for this type of axial resolution improvement. In combination with two-photon excitation, theory (Hell and Stelzer, 1992b) predicts side lobes of <15%, resulting in an OTF with comparatively low intraband modulation (Nagorni and Hell, 2001a). The latter is invaluable for axial superresolution imaging of live cells where optical conditions are imperfect (Egner et al., 2002). These arguments provided strong incentives for us to address both the physical and technical challenges of fluorescence 4Pi microscopy of type C. Here we report on the realization of this imaging mode along with detailed studies of its optical sectioning property under aqueous conditions. First imaging applications display an axial resolution of 80–100 nm in live cells.
FIGURE 1.

Sketch of the 4Pi microscope of type C. Excitation light originating from the microscope stand is divided by the beam splitter BS and focused onto the same spot by the opposing objective lenses O1 and O2. The lenses L1, L2, and L3 and the mirrors M1, M2, and M3 form the intermediate optical system, ensuring that the two scanning pivotal points coincide with the entrance pupils of the two objective lenses. Fluorescence is collected by both lenses, recombined at BS, and directed toward the microscope stand. The pathlength difference between the two interferometric arms is smaller than the coherence length of the fluorescence light, so that fluorescence interferes at the detector as well (i.e., type C). Dispersion compensation over a large wavelength range is ensured by movable optical wedges in the lower interferometric arm whose thickness is compensated by a glass window in the upper arm. Scanning is performed by a standard confocal microscope unit.
THE INSTRUMENT
The 4Pi microscope consists of a 4Pi unit and a standard confocal scanning microscope (TCS SP2, Leica Microsystems Heidelberg GmbH, Mannheim, Germany). The 4Pi unit is tightly mounted to the microscope turret to maintain the scanning and spectral detection capabilities of the confocal system (Fig. 1). Although the microscope is able to adopt both single- and two-photon excitation, in these studies we refrained to the latter to optimize the OTF. For this purpose, the beam of a mode-locked Ti:Sapphire laser (Mira, Coherent, Palo Alto, CA) was coupled into the optical train of the scanner. The 4Pi unit was stabilized against thermal distortions through a symmetrical design of the mechanical components. The exit pupil of the confocal microscope is imaged into the entrance pupil of the objective lenses by the lens L1, and the identical lenses L2 and L3. The excitation laser beam is split into two beams of similar intensity and focused into the sample by the objective lenses. The pupils of the objective lenses are uniformly illuminated by the laser beams to ensure optimal performance of the system. The objective lenses O1 and O2 are mounted on a precision mechanical and a piezo-driven translation stage, respectively. This allows the alignment of the lenses along a common axis. Adjusting the mirrors M1, M2, and M3, and the beam splitter BS enables the alignment of the excitation beams with respect to the entrance pupil of the objective lenses.
Fast lateral scanning is performed by the beam scanner of the system, i.e., by tilting the two beams in the respective pupils of the objective lenses. Axial scanning is accomplished by translating the sample along the optic axis (z) with a piezo-scanning stage. The stage is horizontally oriented like in a standard confocal microscope. The fluorescence is collected through both lenses, merged at the beam splitter, and reflected backward to the confocal microscope. It is fed into the standard spectral detection unit of the confocal scanner.
Compared with the reported 4Pi system using just illumination interference, i.e., 4Pi microscopy of type A (Egner et al., 2002), where the phase can simply be adjusted by changing the length of one interferometric arm, the phase modification is now less straightforward. This stems from the fact that small differences between the two optical arms necessitate the observance of dispersion. Achromatic constructive interference is only possible if each beam passes identical optical material. Although both arms feature identical types of components, minute differences in glass thickness arising from the individual (sub)lenses compromise achromatic phase adjustment. To compensate for this, a pair of movable glass wedges has been inserted in the lower interferometric arm, and a glass window with the mean wedge thickness has been placed in its upper counterpart. This one-time adjustment enables the adaptation of the dispersion of the two interferometric arms. In daily operation, for an optimal PSF, the phase is selected such that both the excitation as well as the fluorescence interfere constructively. The individual arm lengths are adjusted by the mirrors M2 and M3, whereby M2 is mounted on a mechanical translation stage and M3 is controlled piezoelectrically. The optical design ensures invariance of the 4Pi PSF across a field of view of 50 μm × 50 μm.
The axial resolution
The effective point-spread function (PSF)  of a two-photon excitation 4Pi microscope of type C is given by the product of the illumination and the detection PSF:
of a two-photon excitation 4Pi microscope of type C is given by the product of the illumination and the detection PSF:
 |
with  and
and 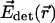 denoting the illumination and detection focal fields of the lenses and
denoting the illumination and detection focal fields of the lenses and 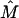 being a transformation matrix accounting for the counterpropagation of the vector fields (Egner et al., 2002). The function
being a transformation matrix accounting for the counterpropagation of the vector fields (Egner et al., 2002). The function 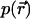 describes the image of the detection pinhole in the focal region.
describes the image of the detection pinhole in the focal region.
The most stringent test for the axial resolution of a three-dimensional (3D) microscope is that of the ability to distinguish infinitely extended transverse (x,y) planes (Wilson and Sheppard, 1984). It is described by the microscope's z-response to a monomolecular fluorescent plane:  Therefore the z-response was measured and compared with the theoretical predictions. For this purpose, we placed ultrathin fluorescent layers into the common focal region of the lenses and adjusted the illumination and fluorescence wavefront pairs for constructive interference at the focus and the detection pinhole, respectively. The resulting z-responses are shown in Fig. 2 along with their theoretical counterparts, both for water (Leica PL APO 63×/1.20 W CORR) and glycerol-immersion lenses (Leica HCX PL APO 100×/1.35 GLYC CORR). The z-response of the glycerol-immersion lens was obtained with a polydiacetylene layer (Schrader et al., 1998b) excited at the wavelength λ = 840 nm. The water-immersion lenses were measured using a layer of the fluorophore Oregon Green (Molecular Probes, Eugene, OR) excited at λ = 880 nm. In both cases, the detection pinhole size corresponded to the size of the back-projected Airy disk. The laser power was chosen to be well below 1 mW, i.e., significantly below the onset of saturation or photobleaching. The fluorescence emission was centered at 565 nm for the polydiacetylene layer and at 526 nm for the Oregon Green layer. The calculations were performed for these conditions by applying a vectorial theory (Richards and Wolf, 1959; Wolf, 1959). To comply with the apodization at the periphery of the aperture, we assumed an effective numerical aperture of 1.15 (rather than 1.2) for the water lenses; similarly, 1.30 was assumed for the glycerol lenses. The measured full-width at half-maximum (FWHM) of the z-response for the glycerol- and water-immersion cases was 105 nm and 112 nm, with lobe heights of 16 and 24%, respectively (Fig. 2). In a 4Pi microscope of type C, the height and shape of the lobes of the effective PSF
Therefore the z-response was measured and compared with the theoretical predictions. For this purpose, we placed ultrathin fluorescent layers into the common focal region of the lenses and adjusted the illumination and fluorescence wavefront pairs for constructive interference at the focus and the detection pinhole, respectively. The resulting z-responses are shown in Fig. 2 along with their theoretical counterparts, both for water (Leica PL APO 63×/1.20 W CORR) and glycerol-immersion lenses (Leica HCX PL APO 100×/1.35 GLYC CORR). The z-response of the glycerol-immersion lens was obtained with a polydiacetylene layer (Schrader et al., 1998b) excited at the wavelength λ = 840 nm. The water-immersion lenses were measured using a layer of the fluorophore Oregon Green (Molecular Probes, Eugene, OR) excited at λ = 880 nm. In both cases, the detection pinhole size corresponded to the size of the back-projected Airy disk. The laser power was chosen to be well below 1 mW, i.e., significantly below the onset of saturation or photobleaching. The fluorescence emission was centered at 565 nm for the polydiacetylene layer and at 526 nm for the Oregon Green layer. The calculations were performed for these conditions by applying a vectorial theory (Richards and Wolf, 1959; Wolf, 1959). To comply with the apodization at the periphery of the aperture, we assumed an effective numerical aperture of 1.15 (rather than 1.2) for the water lenses; similarly, 1.30 was assumed for the glycerol lenses. The measured full-width at half-maximum (FWHM) of the z-response for the glycerol- and water-immersion cases was 105 nm and 112 nm, with lobe heights of 16 and 24%, respectively (Fig. 2). In a 4Pi microscope of type C, the height and shape of the lobes of the effective PSF  are strongly determined by the ratio between the excitation and the detection wavelengths. In most cases, the lobes of
are strongly determined by the ratio between the excitation and the detection wavelengths. In most cases, the lobes of 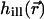 do not coincide in space with those of
do not coincide in space with those of  thereby suppressing each other. The z-response of the glycerol lens (Fig. 2, bottom) features lower lobes in this experiment, because of the slightly more suitable wavelength ratio, but also because the glycerol-immersion lenses feature a semiaperture angle that is larger by 4° than that of their water-immersion counterparts.
thereby suppressing each other. The z-response of the glycerol lens (Fig. 2, bottom) features lower lobes in this experiment, because of the slightly more suitable wavelength ratio, but also because the glycerol-immersion lenses feature a semiaperture angle that is larger by 4° than that of their water-immersion counterparts.
FIGURE 2.

Axial resolution of the 4Pi microscope of type C using two-photon excitation for water immersion (top row) and glycerol immersion (bottom row). (A) xz-image of an ultrathin extended fluorescent plane. (B) Measured and calculated z-responses featuring a FWHM of 112 nm and 105 nm, in the water- and glycerol-immersion case, respectively. (C) Modulus of the optical transfer functions along the optic axis (z-OTF) exhibiting an approximately sevenfold-enlarged passband over that of a confocal microscope (shaded), with strong values across the full region of support. The comparatively low side lobes of 24% and 16% in the water and glycerol case (B) are equivalent to a comparatively weak modulation of the z-OTF, which is essential for avoiding interference-induced artifacts. Note the good agreement between experiment and theory.
Fourier transformation of the z-responses readily yields the OTF along the reciprocal optic axis, 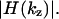 Referred to as the z-OTF,
Referred to as the z-OTF,  quantifies the ability to distinguish planes at a given spatial frequency along the optic axis. The z-OTFs in Fig. 2 are normalized to the central zero-frequency peak which, unlike in I5M, is not a singularity. Fig. 2 reveals that the z-OTF of the two-photon excitation 4Pi microscope of type C is contiguous, featuring a comparatively low intraband modulation. In fact it is the best-filled OTF support reported so far in 4Pi and related imaging (Nagorni and Hell, 2001a). The first minimum in the glycerol z-OTF is 0.4, whereas in the water-immersion case it is 0.22. The largest transmitted object frequencies range between 45 μm−1 and 50 μm−1; the range stems from the slightly different wavelengths used in the measurements, as well as from the different objective lenses, i.e., water and glycerol. The gain in the spatial bandwidth represents a sevenfold enlargement of the OTF as compared with a standard (two-photon) confocal microscope (7 μm−1). A comparison of the measured axial responses with the theory shows remarkable agreement.
quantifies the ability to distinguish planes at a given spatial frequency along the optic axis. The z-OTFs in Fig. 2 are normalized to the central zero-frequency peak which, unlike in I5M, is not a singularity. Fig. 2 reveals that the z-OTF of the two-photon excitation 4Pi microscope of type C is contiguous, featuring a comparatively low intraband modulation. In fact it is the best-filled OTF support reported so far in 4Pi and related imaging (Nagorni and Hell, 2001a). The first minimum in the glycerol z-OTF is 0.4, whereas in the water-immersion case it is 0.22. The largest transmitted object frequencies range between 45 μm−1 and 50 μm−1; the range stems from the slightly different wavelengths used in the measurements, as well as from the different objective lenses, i.e., water and glycerol. The gain in the spatial bandwidth represents a sevenfold enlargement of the OTF as compared with a standard (two-photon) confocal microscope (7 μm−1). A comparison of the measured axial responses with the theory shows remarkable agreement.
Imaging live yeast cells with two-photon 4Pi microscopy of type C
To explore the imaging capability of the system, we recorded the mitochondrial network of live Saccharomyces cerevisiae cells (strain BY4743), whose mitochondrial matrix was labeled with green fluorescent protein (GFP) (Westermann and Neupert, 2000). The cells were embedded in a physiological buffer solution with 1% low-melting agarose; they were subsequently mounted between two coverslips. Apart from immobilizing the cells, the agarose slightly increases the average refractive index of the buffer medium from 1.33 to 1.34, thus reducing minute but accumulative refractive index changes in the sample (Egner et al., 2002). The small linear phase shift arising from the refractive index mismatch between the buffer and the immersion liquid can be actively compensated (Schrader et al., 1998a). An evidence for the applicability of 4Pi microscopy of type C to cells is the fact that the side lobes in the yeast xz-images are only slightly larger than those found with the fluorescent layer, i.e., <30% of the main peak and therefore much below 50% (Fig. 3). We also note that the sample need not be placed between two coverslips, because one of the objective lenses can be a dipping lens.
FIGURE 3.

(A) xz-Section of a raw (not deconvolved) 4Pi-image data stack of the mitochondrial network of a live yeast cell, featuring the hallmarks of the main maximum and of the two axial side lobes. (B) The raw intensity profile integrated along the x axis from the data within the box shows that the lobes are <30% in the live cell. The cell was mounted in a physiological buffer and the data were recorded with water-immersion lenses.
The imaging was performed in a bidirectional scanning mode with a line frequency of 400 Hz. The imaging speed was not limited by the scanning technology but by the inherent photon flux. A typical mitochondrial stack of 6.25 × 6.25 × 4.5 μm3 (x × y × z) in size, featuring pixels of 12 nm (x,y) and 41 nm (z) was recorded within 1.5 min. During this time span, the mitochondrial structure is not expected to change significantly. The initial phase adjustment was carried out with a fluorescent layer before recording. The random phase variations were smaller than π/6 throughout the whole stack, proving that the sample-induced aberrations were rather weak (Egner et al., 2002).
Still, the presence of the lobes (Fig. 3) requires deconvolution of the raw data with the aim of removing the lobe-induced replication artifacts. A single-step linear deconvolution, such as Wiener filtering, has several advantages over its nonlinear positivity-constrained counterparts. It is much faster compared with iterative techniques, the selection of a regularization parameter is less critical, no iteration stopping criterion is needed, the deconvolution process is more transparent, and the deconvolved image still is a convolution of a (lobe-free) PSF with the object to be imaged. The latter is important because it enables quantitative analysis of the deconvolved image data. Finally, a linear deconvolution is mathematically reversible.
However, linear deconvolution is possible only if the modulus of the OTF is well above the noise floor. In single-lens microscopes, the OTF is weak only at its periphery, but in 4Pi and related systems, weak OTF values are also encountered at the “critical frequency” representing the lobe distance. In the 4Pi imaging (of type A) of live cells, linear deconvolution has so far been impossible due to the deep OTF minima stemming from the rather low-aperture angle of water-immersion lenses (Egner et al., 2002). The improved OTF of the 4Pi microscope of type C (Fig. 2) over that of reported 4Pi systems has now led us to investigate whether the removal of the side lobes by linear Wiener filtering is possible in live-cell data. In fact, we compared the results obtained by Wiener filtering with those obtained through an iterative positivity-constrained nonlinear Richardson-Lucy algorithm (Richardson, 1972; Nagorni and Hell, 2001b). In both cases we employed PSFs or OTFs calculated for the applied experimental conditions. The deconvolved 3D data were surface rendered with a 3D-visualization software (Amira, Konrad-Zuse-Zentrum, Berlin, Germany).
Fig. 4 shows a typical surface-rendered reconstruction of the mitochondrial network of a live yeast cell, as obtained by nonlinear (A) and linear (B) deconvolution. The linear Wiener filter not only removed the effects of the side lobes, but also increased the axial resolution to slightly better than 80 nm. The comparison of the results demonstrates good agreement between the two deconvolution methods and, importantly, the applicability of fast linear filtering in 4Pi imaging of type C.
FIGURE 4.

Mitochondrial network of a live yeast cell recorded with a two-photon excitation 4Pi microscope of type C. The displayed 3D image was obtained after nonlinear (A) and linear (B) deconvolution and subsequent surface rendering of the raw 3D data. Note the good agreement of the two images indicating the efficiency of the mathematically simpler and more reliable linear deconvolution. The mitochondrial matrix was labeled with EGFP.
Nonconfocalized 4Pi imaging of type C
The measurement of the z-OTF (Fig. 2) has shown that the joint use of coherent illumination and fluorescence wavefront pairs improves the transfer of the critical spatial frequencies. This raises the question as to the contribution of the confocal pinhole to this improvement. Potential removal of the pinhole may relax design constraints. In fact, nonconfocal 4Pi imaging of type A has been reported for beads (Hell et al., 1995), but the ∼70% lobe height and the image data recorded in that work indicated that this mode was not applicable to the 3D imaging of cells at all.
Therefore we recorded the z-responses of the water-immersion lens for various pinhole diameters. The corresponding z-OTFs 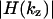 are displayed in Fig. 5 A. To assess the loss of the signal at the pinhole, we also displayed the relative change of the peak height of the z-response (Fig. 5 B). The latter was normalized to the signal obtained with a pinhole diameter of one Airy disk. The curves in Fig. 5 demonstrate that the local minima of the z-OTFs become more pronounced with increasing pinhole diameter, as expected. However, even for a rather ineffective pinhole diameter of size 2.5 times the Airy disk, the local minima exceed the relative value of 0.13.
are displayed in Fig. 5 A. To assess the loss of the signal at the pinhole, we also displayed the relative change of the peak height of the z-response (Fig. 5 B). The latter was normalized to the signal obtained with a pinhole diameter of one Airy disk. The curves in Fig. 5 demonstrate that the local minima of the z-OTFs become more pronounced with increasing pinhole diameter, as expected. However, even for a rather ineffective pinhole diameter of size 2.5 times the Airy disk, the local minima exceed the relative value of 0.13.
FIGURE 5.

(A) Modulus of the optical transfer function along the optic axis (z-OTF) of a two-photon excitation 4Pi microscope of type C as a function of the pinhole size given in Airy disk units. The undesired periodic modulation of the z-OTF decreases with increasing degree of confocalization. In this unaberrated case, the comparatively large pinhole size of 2.5 times the Airy disk still entails local minima of acceptably high levels of >0.13. (B) The increase of the pinhole diameter is accompanied by an increase in fluorescence signal. The data of Fig. 4 were recorded with a pinhole diameter of 1 Airy disk.
Several conclusions can be drawn from this experiment. First, the fluorescence loss through confocalization is limited to at most 40% of the total signal at the peak. Second, in two-photon excitation 4Pi microscopy of type C, one can refrain from using a confocal pinhole provided that the signal is still strong enough at the local minima of the OTF. These findings are corroborated by the images in Fig. 6 showing mitochondria of yeast cells recorded with the detection pinhole virtually open (2.5 times the Airy disk). Owing to the contiguous well-filled OTF, the 3D images are virtually free of artifacts both for the nonlinear (A) and linear (B) deconvolution of the raw data. Again, the resulting axial resolution is of the order of 80 nm.
FIGURE 6.

Mitochondrial network of a live yeast cell recorded with a nonconfocalized two-photon excitation 4Pi microscope of type C, again for nonlinear (A) and linear (B) deconvolution and subsequent surface rendering of the raw 3D data. The success of the linear deconvolution of the nonconfocally recorded data underscores the resilience of this novel form of 4Pi microscopy with respect to interference-induced artifacts. This is due to the fact that in a coherently illuminating and detecting two-lens system, the disparity between the two-photon excitation and the fluorescence wavelength leads to low interference side lobes or a strongly filled OTF. This applies also in the absence of a confocal pinhole and to the water-immersion lenses required for high-resolution imaging of objects mounted in aqueous media. The axial resolution in the live cell is 80 nm.
CONCLUSION AND OUTLOOK
The coherent superposition of the two-photon excitation spherical wavefront pairs of two opposing lenses in conjunction with that of their fluorescence emission counterparts, i.e., two-photon 4Pi microscope of type C, leads to an axial resolution of ∼80 nm in 3D imaging of aqueous samples. We have demonstrated that this imaging mode can be realized in an optical setup that is applicable for live cells. Compared with other related imaging modes, the reported 4Pi microscope of type C is by far more resilient to periodic artifacts induced by interference. This is due to the rather low interference side lobes of the PSF, which is equivalent to a contiguous and well-filled OTF. The quadratic intensity dependence and strong excitation/detection wavelength disparity brought about by two-photon excitation considerably contributes to this improvement in optical performance. Although multiphoton excitation is straightforward in use, the associated lower signal level and the significant costs still allow interesting possibilities to explore whether novel combinations of coherent detection and single-photon excitation can be found, which may lead to acceptable conditions for the imaging of aqueous samples in the future. As of now, with the approach reported herein, a sevenfold-improved axial resolution over that of confocal microscopy has now become possible in the 3D imaging of live cells.
Acknowledgments
The authors thank Jay Jethwa for careful reading of the manuscript and Andreas Schönle for help with his image analysis software ImSpector.
This work was supported by the German Ministry of Education and Research.
APPENDIX
Our motivation for exploring two-photon excitation 4Pi microscopy of type C stems from a theoretical analysis of the OTFs of the various options of using two lenses coherently. A comparison of 4Pi microscopy of type A, type C, and of I5M (see Fig. 7) shows that the OTF of the I5M is much more structured than that of the two-photon excitation 4Pi microscope of type A and C. Moreover, within its region of support, the I5M features areas in which the newly gained spatial frequencies are transferred only weakly. If the transferred signal at these frequencies is close to or below the noise level, periodic artifacts arise in the image. Of the explored options (Fig. 7, A–C), the two-photon excitation 4Pi microscope of type C promises the best performance.
FIGURE 7.

OTFs of various techniques using two opposing lenses coherently. (A) Two-photon excitation 4Pi microscope of type A, i.e., interference of just the illumination wavefronts. (B) Two-photon 4Pi microscope of type C, i.e., simultaneous interference of both the illumination and the fluorescence wavefronts. (C) OTF of the I5M microscope that uses single-photon wide-field standing wave excitation combined with the interference of the fluorescence light at a camera. The white contour line in panels A–C corresponds to the 2% amplitude of the OTF of a standard confocal microscope. In panels A and B, the part of the OTF that lies within this area is largely equivalent to the OTF of the confocal microscope and the regions outside represent the spatial frequencies that are gained over confocal microscopy by the respective method. The coverage of the newly gained regions is significantly stronger in 4Pi microscopy of type C than in I5M. The insert in panel C shows the same I5M-OTF, but normalized to 10%, to highlight weak regions of the OTF.
The OTFs were calculated according to a vectorial theory (Schönle and Hell, 2002), under the assumption of a half-aperture angle of 1.1 rad (i.e., the value of a water-immersion lens of a numerical aperture of 1.2). For the 4Pi microscope of type A and C, the detection pinhole diameter corresponded to the size of the Airy disk at the medium fluorescence wavelength. The very center of the I5M-OTF is a singularity; the height of the central peak in the I5M-OTF in our calculations stems from an assumed volume of signal collection of (10 μm)3. For larger volumes, the weakly transferred amplitudes in the I5M-OTF fall back in relative height with regard to the central peak. The calculations were performed with the following wavelengths: two-photon excitation at 870 nm, fluorescence at 510 nm, and single-photon excitation at 480 nm.
Hilmar Gugel and Jörg Bewersdorf contributed equally to this work.
References
- Denk, W., J. H. Strickler, and W. W. Webb. 1990. Two-photon laser scanning fluorescence microscopy. Science. 248:73–76. [DOI] [PubMed] [Google Scholar]
- Diaspro, A. 2002. Confocal and Two-Photon Microscopy. A. Diaspro, editor. Wiley-Liss, New York.
- Egner, A., S. Jakobs, and S. W. Hell. 2002. Fast 100-nm resolution 3D-microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA. 99:3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner, A., S. Verrier, A. Goroshkov, H.-D. Söling, and S. W. Hell. 2004. 4Pi-microscopy of the Golgi apparatus in live mammalian cells. J. Struct. Biol. 147:70–76. [DOI] [PubMed] [Google Scholar]
- Gu, M., and C. J. R. Sheppard. 1994. Three-dimensional transfer functions in 4Pi confocal microscopes. J. Opt. Soc. Am. A. 11:1619–1627. [Google Scholar]
- Gustafsson, M. G. L. 1999. Extended resolution fluorescence microscopy. Curr. Opin. Struct. Biol. 9:627–634. [DOI] [PubMed] [Google Scholar]
- Gustafsson, M. G. L., D. A. Agard, and J. W. Sedat. 1995. Sevenfold improvement of axial resolution in 3D widefield microscopy using two objective lenses. Proc. SPIE. 2412:147–156. [Google Scholar]
- Gustafsson, M. G. L., D. A. Agard, and J. W. Sedat. 1999. I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J. Microsc. 195:10–16. [DOI] [PubMed] [Google Scholar]
- Hänninen, P. E., S. W. Hell, J. Salo, E. Soini, and C. Cremer. 1995. Two-photon excitation 4Pi confocal microscope: enhanced axial resolution microscope for biological research. Appl. Phys. Lett. 66:1698–1700. [Google Scholar]
- Hell, S. W. 2003. Toward fluorescence nanoscopy. Nat. Biotechnol. 21:1347–1355. [DOI] [PubMed] [Google Scholar]
- Hell, S. W., S. Lindek, C. Cremer, and E. H. K. Stelzer. 1994. Measurement of the 4Pi-confocal point spread function proves 75 nm resolution. Appl. Phys. Lett. 64:1335–1338. [Google Scholar]
- Hell, S. W., M. Schrader, P. E. Hänninen, and E. Soini. 1995. Resolving fluorescence beads at 100–200 nm distance with a two-photon 4Pi-microscope working in the near infrared. Opt. Commun. 117:20–24. [Google Scholar]
- Hell, S. W., M. Schrader, and H. T. M. van der Voort. 1997. Far-field fluorescence microscopy with three-dimensional resolution in the 100 nm range. J. Microsc. 185:1–5. [DOI] [PubMed] [Google Scholar]
- Hell, S. W., and E. H. K. Stelzer. 1992a. Properties of a 4Pi-confocal fluorescence microscope. J. Opt. Soc. Am. A. 9:2159–2166. [Google Scholar]
- Hell, S. W., and E. H. K. Stelzer. 1992b. Fundamental improvement of resolution with a 4Pi-confocal fluorescence microscope using two-photon excitation. Opt. Commun. 93:277–282. [Google Scholar]
- Nagorni, M., and S. W. Hell. 2001a. Coherent use of opposing lenses for axial resolution increase in fluorescence microscopy. I. Comparative study of concepts. J. Opt. Soc. Am. A. 18:36–48. [DOI] [PubMed] [Google Scholar]
- Nagorni, M., and S. W. Hell. 2001b. Coherent use of opposing lenses for axial resolution increase in fluorescence microscopy. II. Power and limitation of nonlinear image restoration. J. Opt. Soc. Am. A. 18:49–54. [DOI] [PubMed] [Google Scholar]
- Richards, B., and E. Wolf. 1959. Electromagnetic diffraction in optical systems II. Structure of the image field in an aplanatic system. Proc. R. Soc. Lond. A. 253:358–379. [Google Scholar]
- Richardson, W. H. 1972. Bayesian-based iterative method of image restoration. J. Opt. Soc. Am. 62:55–59. [Google Scholar]
- Schönle, A., and S. W. Hell. 2002. Calculation of vectorial three-dimensional transfer functions in large-angle focusing systems. J. Opt. Soc. Am. A. 19:2121–2126. [DOI] [PubMed] [Google Scholar]
- Schrader, M., K. Bahlmann, G. Giese, and S. W. Hell. 1998a. 4Pi-confocal imaging in fixed biological specimens. Biophys. J. 75:1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader, M., U. G. Hofmann, and S. W. Hell. 1998b. Ultrathin fluorescent layers for monitoring the axial resolution in confocal and two-photon fluorescence microscopy. J. Microsc. 191:135–140. [DOI] [PubMed] [Google Scholar]
- Schrader, M., M. Kozubek, S. W. Hell, and T. Wilson. 1997. Optical transfer functions of 4Pi confocal microscopes: theory and experiment. Opt. Lett. 22:436–438. [DOI] [PubMed] [Google Scholar]
- Westermann, B., and W. Neupert. 2000. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 16:1421–1427. [DOI] [PubMed] [Google Scholar]
- Wilson, T., and C. J. R. Sheppard. 1984. Theory and Practice of Scanning Optical Microscopy. Academic Press, New York.
- Wolf, E. 1959. Electromagnetic diffraction in optical systems I. An integral representation of the image field. Proc. R. Soc. Lond. A. 253:349–357. [Google Scholar]