

Abstract
The orientation and motion of a model lysine-terminated transmembrane polypeptide were investigated by molecular dynamics simulation. Recent 2H NMR studies of synthetic polypeptides with deuterated alanine side chains suggest that such transmembrane polypeptides undergo fast, axially symmetric reorientation about the bilayer normal but have a preferred average azimuthal orientation about the helix axis. In this work, interactions that might contribute to this behavior were investigated in a simulated system consisting of 64 molecules of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and one α-helical polypeptide with the sequence acetyl-KK-(LA)11-KK-amide. In one simulation, initiated with the peptide oriented along the bilayer normal, the system was allowed to evolve for 8.5 ns at 1 atm of pressure and a temperature of 55°C. A second simulation was initiated with the peptide orientation chosen to match a set of experimentally observed alanine methyl deuteron quadrupole splittings and allowed to proceed for 10 ns. Simulated alanine methyl group orientations were found to be inequivalent, a result that is consistent with 2H NMR observations of specifically labeled polypeptides in POPC bilayers. Helix tilt varied substantially over the durations of both simulations. In the first simulation, the peptide tended toward an orientation about the helix axis similar to that suggested by experiment. In the second simulation, orientation about the helix axis tended to return to this value after an excursion. These results provide some insight into how interactions at the bilayer surface can constrain reorientation about the helix axis while accommodating large changes in helix tilt.
INTRODUCTION
The orientations and dynamics of various transmembrane polypeptides and transmembrane segments of membrane-associated proteins have been investigated using both 15N and 2H solid-state NMR techniques (Jones et al., 1998; Morrow and Grant, 2000; Marassi et al., 2000; Marassi and Opella, 2000; Wang et al., 2000; Harzer and Bechinger, 2000; Sharpe et al., 2001, 2002; van der Wel et al., 2002). In 2H NMR observations of particular polypeptides modeled on the transmembrane segment of epidermal growth factor receptor (Jones et al., 1998, Morrow and Grant, 2000; Sharpe et al., 2001), the magnitudes and inequivalence of splittings from methyl group deuterons on alanines at different positions along the helix showed that the polypeptides were tilted and undergoing rapid axially symmetric reorientation about the bilayer normal while also maintaining a preferred orientation about the helix axis.
Similar results were obtained for polypeptides having a more uniform hydrophobic sequence consisting of alternating alanines and leucines terminated by two lysines at each end (Sharpe et al., 2002). Polypeptides of this type have been labeled as KALP polypeptides by Killian and co-workers (de Planque et al., 1999, 2003; Strandberg et al., 2002; Morein et al., 2002; de Planque and Killian, 2003; Killian, 2003). 2H NMR observations of labeled alanine methyl groups on these polypeptides were consistent with reorientation of the tilted helix about the bilayer normal with a correlation time of ∼10−7 s (Sharpe et al., 2002). The existence of a preferred average orientation about the helix long axis appears to be consistent with observations on other transmembrane polypeptides (Jones et al., 1998; Marassi et al., 2000; Marassi and Opella, 2000; Wang et al., 2000). The fact that each deuterated alanine residue gave rise to a unique splitting ruled out the possibility of slow exchange among two or more orientations of the polypeptide about its helix axis, a situation that would have given rise to a superposition of doublets with different splittings. NMR observations of deuterated alanines along a tryptophan-anchored α-helix of alternating alanines and leucines (WALP 19) have also indicated an intrinsic tilt and preferred orientation about the helix axis (van der Wel et al., 2002).
Although NMR experiments can indicate the average orientation and dynamics of the polypeptide over the timescale characteristic of the 2H experiment, they do not provide much information concerning fluctuations about the average orientation on shorter timescales. For tilted polypeptides with a preferred orientation about the helix axis, the amplitude of motions that allow the polypeptide to sample a range of orientations about a preferred orientation might have a significant effect on the accessibility of particular regions of the polypeptide surface to other polypeptides in the membrane. Knowledge of transmembrane polypeptide dynamics may thus have some general relevance to understanding of helix-helix interaction.
The 2H NMR observations of Sharpe et al. (2002) show that axially symmetric reorientation of the tilted polypeptide about the bilayer normal is fast on the ∼10−5 s characteristic time of the 2H NMR experiment. Molecular dynamics (MD) simulations are not typically of sufficient duration to show the kind of motion that is presumed to be responsible for axially symmetric averaging of the quadrupole interaction on this timescale. Such simulations can, however, display rapid small amplitude motions that modulate the quadrupole interaction about its average. Although these may have little direct effect on the observed quadrupole splittings, such motions can give insight into interactions that are relevant to the adoption of a preferred tilt and azimuthal orientation. They can also give some insight into whether such motions might affect the accessibility of particular portions of the helix surface to peptide-peptide interaction.
Petrache et al. (2002) used molecular dynamics to study five tryptophan-terminated alternating alanine-leucine polypeptides (WALP polypeptides) of different lengths in dipalmitoyl phosphatidylcholine and dimyristoyl phosphatidylcholine bilayers. They found helix tilts ranging from 10 to 20°. The tilt obtained in the simulation depended on the arrangement of the tryptophan pairs about the helix axis and did not vary monotonically with helix length. They also reported large variations in instantaneous tilt over the durations of the simulations.
We have carried out molecular dynamics simulations of the polypeptide Ac-KK-(LA)11-KK-amide in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer using two different starting conditions. This peptide is one of those for which quadrupole splittings of selectively deuterated alanine methyl groups indicated orientational inequivalence of alanine residues along the helix backbone within the bilayer interior (Sharpe et al., 2002). The first simulation starts with the polypeptide aligned along the bilayer normal and follows the development of helix tilt with respect to the bilayer normal as the simulation proceeds for 8.5 ns. This simulation provides some insight into how alanine methyl group orientation, with respect to the bilayer normal, couples to helix tilt and polypeptide orientation about the helix axis.
The 8.5-ns duration of the first simulation may not have been sufficient for the polypeptide to relax to the orientation implied by the experimentally observed alanine methyl deuteron quadrupole splittings. For this reason, a second simulation was run with the initial polypeptide tilt and orientation about the helix axis chosen to match the experimentally observed splittings given the assumption of fast reorientation about the bilayer normal. The way in which the simulated polypeptide departed from this initial orientation over the course of the subsequent 10-ns simulation provides further insight into the interactions that give rise to the adoption of the experimentally observed polypeptide orientation.
METHODS
Initial bilayer preparation
An Lα-phase POPC membrane structure was obtained from previously reported molecular dynamics results for runs at constant number, pressure, area, and temperature (NPAT) on a bilayer composed of 72 POPC lipids (36 in each monolayer) at low hydration (∼13.5 waters per lipid) (Feller et al., 1997; Armen et al., 1998). The system contained ∼970 water molecules in total and the simulation cell dimensions were 48 × 48 × 51.58 Å. The water layer thickness required for full hydration is ∼13 Å. To achieve this in this work, patches of TIP3 water (Jorgensen et al., 1983) were placed directly above and below the bilayer. A spacing of 3 Å was initially left between the existing water and the patches to ensure no bad contacts between the added patches of water molecules and the existing layer. An X-plor format psf file (Brünger, 1992) of the system was then built using the Psfgen construction tool included with the NAMD2 (Nelson et al., 1996; Kalé et al., 1999) MD simulation package. An all-atom model was employed in accordance with the CHARMM 27 topology and parameter files (Mackerell et al., 1998; Schlenkrich et al., 1996). The final system contained 37.5 waters/lipid headgroup corresponding to a water concentration of 52% w/w. At this level of hydration the bilayer is expected to be well beyond the Lβ phase at the target temperature (328 K) and ambient pressure.
Simulations were carried out on an IBM RS/6000 SP1 and a SGI Onyx 3400. The NAMD2 package (Nelson et al., 1996; Kalé et al., 1999) was used for simulations whereas MD trajectory progress, visualization, and analysis were carried out using visual molecular dynamics (Humphrey et al., 1996) on a Pentium class computer running Linux 2.4 and an SGI Onyx 3400 (Mountain View, CA). Simulation conditions were as follows. To satisfy the condition of constant pressure, the system was coupled, at all times, to a 1-atm pressure bath using the Nose-Hoover Langevin piston method (Feller et al., 1995). A splitting function with a cutoff of 12 Å was used for separation of long- and short-range electrostatic interactions. A cutoff of 12 Å was also employed for calculating Van der Waals interactions and a switching function was used to relax the van der Waals potential to zero over a distance of 2 Å. Pair list distance was set at 13.5 Å and a hydrogen group based cutoff was set at 3 Å. Periodic boundary conditions were employed in all cell dimensions and the particle mesh Ewald (PME) method (Darden et al., 1993) was used for full evaluation of electrostatic interactions. Typically eight processors were used during MD simulations. The SHAKE algorithm (Ryckaert et al., 1977) was used to constrain all bonds between hydrogens and heavy atoms with a tolerance of 10−5 Å.
The bilayer system was prepared by first coupling it to a thermal bath at 100 K (Brünger, 1992) and allowing the system cell to fluctuate in all dimensions. The physical gaps between the original and added water layers disappeared after 50 ps of simulation. The system was then subjected to 13,000 steps of conjugate gradient minimization with a tolerance of 0.0005. The equilibration process was then initiated by weak coupling to a thermal bath at 328 K. After 600 ps of simulation, a variety of bilayer properties were measured to ensure that the system had approached equilibrium.
Polypeptide construction
The polypeptide CH3CO-KK-(LA)11-KK-CO-NH2 is expected to be α-helical in the membrane environment and peptide was prepared with this initial conformation. Observations reported by Zhang et al. (1995) confirm α-helical geometry for a 12-subunit LA segment embedded in a bilayer membrane. The helical peptide used in the simulation was constructed using MOLDEN (Schaftenaar and Noordik, 2000). All side-chain dihedral angles were chosen from the backbone dependent rotamer library for proteins (Dunbrack and Karplus, 1993). After construction, the polypeptide was subjected to 3500 steps of conjugate gradient energy minimization. Minimization of the protein conformational energy was completed in four stages similar to those described by Belohorcová et al. (1997). In the first 500 steps, dihedrals were restrained using a restraint potential with an energy constant of 50 kcal/mol. The Cβ positions of the alanine residues and the Cβ and Cγ positions of the leucine and lysine residues were initially held fixed. In the next 500 steps, dihedrals were restrained using a restraint potential with an energy constant of 25 kcal/mol. The Cβ positions of the alanine residues and the Cβ and Cγ positions of the leucine and lysine residues were harmonically restrained with a force constant of 25 kcal/(mol Å2). In the next 500 steps, dihedrals were restrained using a restraint potential with an energy constant of 5 kcal/mol. The Cβ positions of the alanine residues and the Cβ and Cγ positions of the leucine and lysine residues were harmonically restrained with a force constant of 5 kcal/(mol Å2). Over the final 2000 steps of the protein minimization procedure, there were no restraints on side-chain dihedrals nor harmonic restraints on atomic positions.
These measures were used to ensure that the peptide remained α-helical during energy minimization. The polypeptide was then aligned with its long axis oriented along the z-direction by diagonalization of the moment of inertia tensor followed by application of appropriate rotations.
Lipid-peptide system preparation and construction
After equilibration of the bilayer, four lipid molecules were removed from the upper and lower leaflets, respectively. The system was then coupled to a weak repulsive cylindrical potential that forced lipids outward to allow the bilayer to accommodate the polypeptide. The simulation parameters were as described above. The PME technique was used for evaluating electrostatic forces without truncating long-range forces. The PME technique requires that the periodic cell charge be zero. To counteract the four charged lysine side chains, Cl− ions were added to the system by randomly removing two water molecules from the upper and lower water layers (Pandit and Berkowitz, 2002). The helix was initially held fixed while the cylindrical potential was relaxed. Over a period of 208 ps the lipids completely enclosed the polypeptide. After this preparation, the simulation was allowed to proceed for a total duration of 8.5 ns including equilibration and dynamics.
A second simulation was run with the initial orientation of the polypeptide chosen to match that inferred from NMR observations of the corresponding polypeptide deuterated on selected alanines. This orientation was found by optimizing the fit to quadrupole splittings observed for methyl deuterons on alanine residues 14 and 16 and to the set of splittings observed from the same polypeptide simultaneously deuterated on the alanine methyls of residues 8, 10, 12, 14, 16, 18, and 20. The association of splittings with a particular orientation was done under the assumption of rapid axially symmetric reorientation of the polypeptide about the bilayer normal. The initial polypeptide conformation and preparation were as for the first simulation. The polypeptide, with its orientation matched to the experimentally observed quadrupole splittings, was inserted into the equilibrated bilayer as described previously. After the lipids had again enclosed the polypeptide, the simulation proceeded in the same way as for the first simulation. The total time for the second simulation was ∼10 ns.
RESULTS AND DISCUSSION
Bilayer preparation
The root mean squared displacement (RMSD) of molecular coordinates with respect to the starting configuration is given by
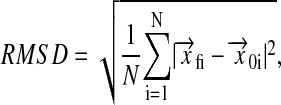 |
(1) |
where the vectors 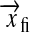 and
and 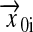 describe the position of atom i and their difference is the displacement of atom i from its position at the beginning of the simulation. The evolution of RMSD for the lipids over an initial 600-ps bilayer preparation period is shown in Fig. 1. This quantity increases sharply during the first 100 ps. This rapid initial increase reflects changes in lipid conformation and intermolecular spacing as the bilayer relaxes from its initial state toward lower energy configurations. After this rapid initial adjustment, the RMSD continues to change at a slower rate as molecules reorient and diffuse within the bilayer. Shinoda et al. (1997) suggested that trans-gauche transition rates for torsional motion of the lipid acyl chains might provide some indication of equilibration time needed in MD simulations of pure lipid bilayers. They suggested that characteristic times for torsional motions could range from ∼10 ps for carbons in the acyl chains to as long as 400–700 ps for torsions at the base of the lipid headgroup.
describe the position of atom i and their difference is the displacement of atom i from its position at the beginning of the simulation. The evolution of RMSD for the lipids over an initial 600-ps bilayer preparation period is shown in Fig. 1. This quantity increases sharply during the first 100 ps. This rapid initial increase reflects changes in lipid conformation and intermolecular spacing as the bilayer relaxes from its initial state toward lower energy configurations. After this rapid initial adjustment, the RMSD continues to change at a slower rate as molecules reorient and diffuse within the bilayer. Shinoda et al. (1997) suggested that trans-gauche transition rates for torsional motion of the lipid acyl chains might provide some indication of equilibration time needed in MD simulations of pure lipid bilayers. They suggested that characteristic times for torsional motions could range from ∼10 ps for carbons in the acyl chains to as long as 400–700 ps for torsions at the base of the lipid headgroup.
FIGURE 1.

Root mean squared displacement (RMSD) of bilayer coordinates during the initial 600-ps bilayer simulation before insertion of the peptide.
The initial bilayer simulation corresponds to conditions of 55°C and 50% water by weight. The properties of the simulated POPC bilayer after the 600-ps preparation period are consistent with previous simulations of POPC in the liquid crystalline phase (Heller et al., 1993; Pasenkiewicz-Gierula et al., 2000). The area/lipid headgroup slowly increases over the 600-ps preparation period as lipids redistribute and the simulation cell fluctuates under the influence of applied external pressure. The average area per lipid headgroup during this period is ∼63 Å2. This is similar to areas per lipid obtained in previous simulations of POPC in the liquid crystalline phase (Pasenkiewicz-Gierula et al., 2000). For comparison, average interfacial areas/lipid in the liquid crystalline phase, obtained from recent analyses of diffraction data, are reported to be 64 Å2 for DPPC at 50°C and 72.5 Å2 for DOPC at 30°C (Nagle and Tristram-Nagle, 2000).
The state of a bilayer is also reflected by the orientational order parameters of the lipid acyl chain methylene segments. Experimentally, such order parameters can be obtained from 2H NMR quadrupole splittings of deuterium labeled segments. The orientational order parameter is then defined as 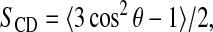 where θ is the angle between the CD bond and the bilayer normal and the average is over motions of the chain. Fig. 2 shows orientational order parameter profiles for the palmitoyl and oleoyl chains obtained by averaging over the simulation trajectory before addition of the polypeptide and over the trajectory of Simulation 1 with the polypeptide present. Order parameters in the absence of the peptide are typical of liquid crystalline POPC bilayers and are in good agreement with results of Seelig and Waespe-Sarcevic (1978). In particular, the characteristic dip in order parameter near the oleoyl double bond was successfully reproduced by the simulation.
where θ is the angle between the CD bond and the bilayer normal and the average is over motions of the chain. Fig. 2 shows orientational order parameter profiles for the palmitoyl and oleoyl chains obtained by averaging over the simulation trajectory before addition of the polypeptide and over the trajectory of Simulation 1 with the polypeptide present. Order parameters in the absence of the peptide are typical of liquid crystalline POPC bilayers and are in good agreement with results of Seelig and Waespe-Sarcevic (1978). In particular, the characteristic dip in order parameter near the oleoyl double bond was successfully reproduced by the simulation.
FIGURE 2.

(A) Average orientational order parameter profile for the POPC saturated chain in Simulation 1 with (•) and without (○) the polypeptide present. (B) Average orientational order parameter profile for the POPC unsaturated chain in Simulation 1 with (•) and without (○) the polypeptide present.
Before insertion of the peptide, the average distance between phosphates on opposite sides of the bilayer fluctuated slightly around ∼39.5 Å over the course of the simulation. Comparison with observations of liquid crystalline DOPC and DPPC (Weiner and White, 1992; Nagle and Tristram-Nagle, 2000) suggests that the simulation generates a reasonable bilayer thickness for peptide-free POPC bilayers.
Simulation 1: polypeptide initially oriented along the bilayer normal
After the bilayer was prepared as described above, the polypeptide was first inserted into the bilayer with its helix axis oriented along the bilayer normal. The trajectory following from this initial polypeptide orientation will be referred to as Simulation 1. The solid symbols in Fig. 2 show that addition of the transmembrane polypeptide segment induced a small but significant increase in the average orientational order of both lipid chains. This peptide-induced increase in acyl chain orientational order is consistent with the results of previous experimental and simulation studies of bilayers containing helical polypeptides (Subczynski et al., 1998; Belohorcová et al., 1997). During the first 7 ns after insertion of the peptide, the average phosphate-phosphate separation increased from ∼39.5 to ∼43 Å and then decreased to ∼41 Å over the next 1.5 ns. The duration of Simulation 1 was not sufficient to determine whether this reflected a transient response to peptide insertion or a more persistent peptide-induced enhancement of bilayer thickness fluctuations.
Fig. 3 illustrates the polypeptide dihedral angles ϕ and ψ for each residue, other than terminal lysines, averaged over the course of the 8.5-ns trajectory of Simulation 1. The overall average dihedral angles are ψ = −42.2° and ϕ = −63.6°, which is within the α-helical range. The helix is stable over the course of the simulation and there does not appear to be significant unraveling toward the ends of the helix. These results are consistent with the report, based on Fourier transform infrared spectroscopy observations, that proteins composed of 12 LA subunits are transmembrane and retain α-helical conformation when incorporated in bilayers of phosphatidylcholines with varying acyl chain lengths (Zhang et al., 1995).
FIGURE 3.

Peptide dihedral angles ψ (○) and ϕ (•) versus residue position along the helix backbone averaged over the trajectory for the simulation in which the peptide is initially oriented along the bilayer normal.
The orientation of the simulated polypeptide was characterized by the tilt angle, τ, of the helix axis with respect to the bilayer normal and the azimuthal orientation, ρ, of the polypeptide about its axis. To define the azimuthal angle, a reference direction in the bilayer was obtained by taking the cross product of the helix axis and the bilayer normal. This direction is well defined when the helix is tilted but more ambiguous as the helix orientation approaches the bilayer normal. A marker direction attached to the helix was obtained by projecting the alanine 14 methyl axis onto the plane perpendicular to the helix axis. The azimuthal angle ρ was then defined as the angle between the helix-fixed marker direction and the reference direction. Fig. 4 illustrates the characterization of helix orientation using these angles.
FIGURE 4.

Axes used to define polypeptide tilt and azimuthal orientation. The polypeptide backbone is represented as a ribbon with the side chain of alanine 14 shown explicitly. Axis 1 is the bilayer normal. Axis 2 is the helix axis. The tilt, τ, is the angle between Axis 1 and Axis 2. Axis 3, the cross product of the bilayer normal and helix axis, provides a reference direction for the azimuthal orientation. Axis 4 is directed along the methyl group symmetry axis of alanine 14. Axis 5 is the projection of Axis 4 onto the plane perpendicular to the helix axis. The azimuthal orientation, ρ, is measured between Axis 5 and Axis 3. The polypeptide is shown in the orientation 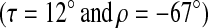 that gives the best fit to observed methyl deuteron splittings for alanines 8, 10, 12, 14, 16, 18, and 20. This is also the starting orientation for Simulation 2.
that gives the best fit to observed methyl deuteron splittings for alanines 8, 10, 12, 14, 16, 18, and 20. This is also the starting orientation for Simulation 2.
Fig. 5 shows the helix tilt and azimuthal orientation along with the angles between the bilayer normal and the methyl axes of two alanines, 12 and 14, over the course of Simulation 1, in which the initial orientation of the polypeptide is along the bilayer normal. Fig. 5 A shows that after constraints on the polypeptide are lifted, at ∼100 ps, the helix begins to tilt to accommodate the hydrophobic length of the transmembrane segment, which is greater than the hydrophobic thickness of the bilayer interior. After an initial period of rapid reorientation, the tilt of the helix axis with respect to the bilayer normal continues to fluctuate slowly over the next 8.5 ns.
FIGURE 5.

The time dependence of selected polypeptide orientational parameters obtained from the trajectory of Simulation 1 (peptide helix axis initially oriented along the bilayer normal). Angles shown are (A) the tilt (τ) of the helix axis with respect to the bilayer normal, (B) the azimuthal orientation (ρ) about the helix axis, (C) shows the angle (θ12) between the C-CH3 bond axis of alanine residue 12 and the bilayer normal, and (D) the angle (θ14) between the C-CH3 bond axis of alanine residue 14 and the bilayer normal.
Fig. 5 B presents the time dependence of the polypeptide orientation about the helix axis over the course of the simulation. Changes in the angle ρ between the reference direction and the helix-fixed marker direction reflect rotation of the tilted helix about its axis. For a tilted helix, such rotations will change the portions of the helix surface that are oriented perpendicular to the bilayer normal and, in doing so, change the portions of the surface expected to be most accessible for interaction with neighboring polypeptides. In Fig. 5 B, this angle is not well defined during the intervals when the orientation of the helix axis approaches the bilayer normal, but otherwise this angle fluctuates rapidly with an amplitude of ∼20° about an average orientation that appears to remain stable for periods of a few nanoseconds.
As the helix tilt grows and the orientation about the helix becomes clearly defined, the trend displayed in Fig. 5 B is toward an angle of ∼−50° between the helix fixed reference and the bilayer fixed reference directions. As noted below, this is close to the orientation inferred from observed alanine methyl deuteron splittings.
Fig. 5, C and D, show the angles between the alanine methyl axes and the bilayer normal for alanine residues 12 and 14. If the polypeptide reorients rapidly, on the deuterium NMR timescale, about the bilayer normal, the observed alanine methyl deuteron quadrupole splittings are determined by these angles. For an ideal α-helix having 3.6 residues per turn, one would expect the projections of two C-CH3 bond axes (on two alanines separated by a single leucine) onto a plane perpendicular to the helix axis to be separated by ∼160°. Over the course of the simulation, the average separation of the projections of the alanine 12 and alanine 14 methyl axes is ∼159°. The correlation between the orientations of the two alanine methyl axes with respect to the bilayer normal, shown in Fig. 5, C and D, reflects this fixed geometric relationship between the two alanine side groups. Fig. 5 also suggests some correlation between the helix axis tilt and the orientation of each methyl axis with respect to the bilayer normal. An increase in the tilt of the helix axis leads to an increase in angular displacement of the C-CH3 bond axis for the alanine at position 12 on the polypeptide. Depending on the direction of helix tilt and the initial orientation of the alanine C-CH3 bond, this behavior might be expected even if there were no rotation about the helix axis. Interestingly, the C-CH3 bond on the next alanine, at position 14, displays orientational fluctuations in the opposite direction. An increase in angular displacement of the helix axis from the bilayer normal leads to a decrease in angular displacement of the C-CH3 bond axis from the bilayer normal for the alanine at position 14 of the polypeptide. This behavior is a consequence of the direction of the bilayer normal falling roughly between the directions the two alanine methyl axes and remaining close to the plane defined by the two methyl axis directions as the tilt fluctuates.
The methyl axis orientations plotted in Fig. 5, C and D, illustrate differences between the long and short timescale motions of the helix. Over short times, the methyl axis orientations fluctuate, with amplitudes of ∼5–8° about short-term averages that, in turn, vary along with the helix tilt over a longer timescale. This observation suggests that for a given tilt of the helix axis, there is a preferred average orientation of the helix about that axis. Fluctuations about that preferred orientation are rapid but constrained in amplitude.
As noted above, the observation that different alanine residues are spectrally inequivalent implies a tilt of the polypeptide helix axis with respect to the bilayer normal, the adoption of a preferred polypeptide orientation about its helix axis and rapid rotation of the tilted helix about the bilayer normal with a correlation time of ∼10−7 s (Sharpe et al., 2002). To obtain splittings from the simulation that can be compared to experimental observations, it is necessary to average the quadrupole interaction over timescales that are comparable to the characteristic timescale (∼10−5 s) of the quadrupole echo experiment and thus much longer than the duration of the simulation. We can estimate the average quadrupole interaction by assuming that the polypeptide undergoes axially symmetric reorientation about the bilayer normal. The expected splitting for deuterons on a given alanine methyl then reflects the average, over the simulation trajectory, of the angle between that methyl axis and the bilayer normal.
If the small asymmetry parameter for a CD bond is neglected, the 2H NMR spectrum for a CD bond on a static, rigid molecule is a doublet with prominent edges split by
 |
(2) |
where β is the angle between the CD bond and the applied magnetic field. If the molecule undergoes axially symmetric reorientation with a correlation time that is short with respect to the ∼10−5 s timescale characteristic of the 2H NMR experiment, the splitting is reduced by an additional factor, 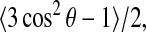 where θ is the angle between the CD bond and the axis about which the molecule is rotating. The average in this factor is over any internal and/or wobbling motions of the molecule that modulate the interaction over the characteristic timescale of the experiment. For the case of axially symmetric reorientation, the angle β is redefined as the angle between the applied magnetic field and the axis about which the molecule reorients. For the deuterated methyl group on a labeled alanine, fast rotation of the CD bonds about the symmetry axis of the methyl group introduces an additional factor of 1/3 and θ can then be redefined as the angle between the methyl group symmetry axis and the axis about which the molecule is reorienting. For peptides incorporated into multilamellar vesicle bilayers, the spectrum is a superposition of doublets corresponding to a spherical distribution of β. The resulting powder pattern or Pake doublet, characteristic of axially symmetric peptide reorientation, has prominent edges (90° edges) corresponding to molecules reorienting about axes perpendicular to the applied field. The prominent 90° edges for a methyl-deuterated alanine on a transmembrane polypeptide segment will then be split by
where θ is the angle between the CD bond and the axis about which the molecule is rotating. The average in this factor is over any internal and/or wobbling motions of the molecule that modulate the interaction over the characteristic timescale of the experiment. For the case of axially symmetric reorientation, the angle β is redefined as the angle between the applied magnetic field and the axis about which the molecule reorients. For the deuterated methyl group on a labeled alanine, fast rotation of the CD bonds about the symmetry axis of the methyl group introduces an additional factor of 1/3 and θ can then be redefined as the angle between the methyl group symmetry axis and the axis about which the molecule is reorienting. For peptides incorporated into multilamellar vesicle bilayers, the spectrum is a superposition of doublets corresponding to a spherical distribution of β. The resulting powder pattern or Pake doublet, characteristic of axially symmetric peptide reorientation, has prominent edges (90° edges) corresponding to molecules reorienting about axes perpendicular to the applied field. The prominent 90° edges for a methyl-deuterated alanine on a transmembrane polypeptide segment will then be split by
 |
(3) |
where θ is now the angle between the alanine methyl symmetry axis and the axis about which the peptide is reorienting. The quantity in the angular brackets is the second Legendre polynomial, 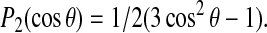
Experimentally, the splittings for a polypeptide simultaneously deuterated on the methyls of alanine residues 8, 10, 12, 14, 16, 18, and 20 were found to be clustered within two ranges, 4–5 kHz and 11–14 kHz (Sharpe et al., 2002). Use of selectively deuterated polypeptides allowed the splittings for residues 14 and 16 to be assigned as ∼14 kHz and ∼11 kHz, respectively (Sharpe et al., 2002). Alanine methyl splittings for residues 8, 10, 12, 18, and 20 were thus known to fall within one of the 4–5 kHz or 11–14 kHz ranges but could not be assigned more specifically.
As noted above, the possibility of rapid reorientation about the polypeptide helix axis was ruled out by both the magnitude and the inequivalence of these splittings. If the motion responsible for averaging of the quadrupole interaction were rotation about the polypeptide helix axis, θ in Eq. 2 would represent the angle between the C-CH3 bond axis and the long axis of the transmembrane segment and the average would be over the simulation trajectory. For an ideal α-helix, the orientation of the C-CH3 bond axis with respect to the molecular long axis is ∼56°. This is very close to the magic angle (54.7° ), so that averaging of the quadrupole interaction by rapid reorientation about the helix axis would result in quadrupolar splittings being close to zero. In the simulation, the angles between the alanine C-CH3 bond axes and the peptide long axis varied between 54° and 59°. If the polypeptide were assumed to rotate rapidly about the helix long axis, the splittings for the seven alanine residues that were simultaneously deuterated in the experiments of Sharpe et al. (2002) would be expected to range from 0.16 to 3.42 kHz. This spread reflects small departures from ideal α-helical geometry in the simulation. The splittings obtained from the simulation by assuming rapid reorientation about the helix axis are thus smaller and more uniform than the experimentally observed set of splittings, and fast rotation about the helix axis can be ruled out. The dominant axially symmetric reorientation must then be about the bilayer normal (Sharpe et al., 2002).
For the case of rapid axially symmetric reorientation about the bilayer normal, the angle 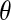 in Eq. 3 is defined to be between the bilayer normal and the C-CH3 bond axis. For alanine residues 12 and 14, it is this angle that is plotted in Fig. 5, C and D. Fig. 6, B and C, show corresponding values of P2(cos θ) over the trajectory of simulation 1. Fig. 6 A shows the corresponding helix tilt angle for comparison. Taking into account that
in Eq. 3 is defined to be between the bilayer normal and the C-CH3 bond axis. For alanine residues 12 and 14, it is this angle that is plotted in Fig. 5, C and D. Fig. 6, B and C, show corresponding values of P2(cos θ) over the trajectory of simulation 1. Fig. 6 A shows the corresponding helix tilt angle for comparison. Taking into account that  ranges from −0.5 to 1.0, it is apparent that the ±10° fluctuations shown in Fig. 5, C and D, translate into substantial fluctuations in
ranges from −0.5 to 1.0, it is apparent that the ±10° fluctuations shown in Fig. 5, C and D, translate into substantial fluctuations in  . The observed quadrupole splitting is proportional to the average of this quantity over the ∼10−5 s timescale that is characteristic of the deuterium NMR experiment. Thus whereas the standard deviation in
. The observed quadrupole splitting is proportional to the average of this quantity over the ∼10−5 s timescale that is characteristic of the deuterium NMR experiment. Thus whereas the standard deviation in  is comparable in magnitude to that of
is comparable in magnitude to that of 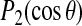 itself, it is the more precisely determined average of this value that is experimentally meaningful.
itself, it is the more precisely determined average of this value that is experimentally meaningful.
FIGURE 6.

(A) The time dependence of the helix tilt (τ) derived from the trajectory of Simulation 1 (peptide helix axis initially oriented along the bilayer normal). (B) The time dependence of 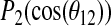 for Simulation 1. (C) The time dependence of
for Simulation 1. (C) The time dependence of 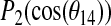 for Simulation 1. Here θn is the instantaneous angle between the bilayer normal and the methyl group axis for the alanine residue n and
for Simulation 1. Here θn is the instantaneous angle between the bilayer normal and the methyl group axis for the alanine residue n and  .
.
Table 1 shows quadrupolar splittings calculated for alanine methyl deuterons on residues 8, 10, 12, 14, 16, 18, and 20 by averaging 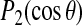 over the trajectory of Simulation 1 and using Eq. 3 to estimate the corresponding splitting. This presumes axially symmetric reorientation of the polypeptide about the bilayer normal with a correlation time longer than the duration of the simulation but shorter than the characteristic deuterium NMR timescale of ∼10−5 s. Also tabulated for comparison are the experimentally observed splittings and the splittings corresponding to the helix orientation that yields the best fit to known splittings. For convenience, this is the orientation that is used, in Fig. 4, to illustrate the definitions of tilt and azimuthal orientation about the helix axis.
over the trajectory of Simulation 1 and using Eq. 3 to estimate the corresponding splitting. This presumes axially symmetric reorientation of the polypeptide about the bilayer normal with a correlation time longer than the duration of the simulation but shorter than the characteristic deuterium NMR timescale of ∼10−5 s. Also tabulated for comparison are the experimentally observed splittings and the splittings corresponding to the helix orientation that yields the best fit to known splittings. For convenience, this is the orientation that is used, in Fig. 4, to illustrate the definitions of tilt and azimuthal orientation about the helix axis.
TABLE 1.
Quadrupole splittings (|ΔνQ|) in kHz for selected alanine methyl deuterons
| Alanine | Experimentally observed splittings* | Peptide oriented for best fit to observed splittings† | Average over Simulation 1‡ | Average over Simulation 2‡ |
|---|---|---|---|---|
| 8 | 4–5 or 11–14 | 5.1 | 6.0 | 6.7 |
| 10 | 4–5 or 11–14 | 13.4 | 9.2 | 15.9 |
| 12 | 4–5 or 11–14 | 11.3 | 6.9 | 18.9 |
| 14 | 14 | 14.9 | 8.3 | 18.2 |
| 16 | 11 | 11.2 | 3.3 | 22.9 |
| 18 | 4–5 or 11–14 | 13.3 | 4.2 | 17.4 |
| 20 | 4–5 or 11–14 | 4.9 | 2.8 | 14.9 |
From Sharp et al. (2002). Experimentally observed values were obtained from a polypeptide simultaneously deuterated on alanines 8, 10, 12, 14, 16, 18, and 20, a polypeptide deuterated on alanine 14, and a polypeptide deuterated on alanine 16. Splittings for the multiply deuterated polypeptide fell into two ranges (4–5 kHz or 11–14 kHz) and could not be further resolved.
Splittings obtained by choosing polypeptide tilt (τ = 12°) and azimuthal orientation (ρ = −67°) that gives the best fit to experimentally observed splittings under assumption of axially symmetric rotation about bilayer normal.
Splittings calculated by assuming rotation about the bilayer normal and averaging 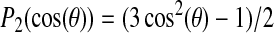 over simulation trajectories. For Simulation 1, the helix axis is initially oriented along the bilayer normal. For Simulation 2, the initial orientation of the polypeptide
over simulation trajectories. For Simulation 1, the helix axis is initially oriented along the bilayer normal. For Simulation 2, the initial orientation of the polypeptide 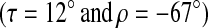 corresponds to the best fit to experimentally observed alanine methyl deuteron quadrupole splittings.
corresponds to the best fit to experimentally observed alanine methyl deuteron quadrupole splittings.
The splittings obtained by averaging over the trajectory of Simulation 1 capture some of the inequivalence observed experimentally but do not correctly reproduce the detailed distribution of observed splittings. Given the sensitivity of  to fluctuations in the methyl axis orientation, this is not surprising. Although the maximum splitting obtained by assuming axially symmetric reorientation about the bilayer normal is less than that observed experimentally, the distributions of simulated and experimental splittings span similar ranges. The maximum alanine deuteron splitting obtainable by assuming rotation of the tilted helix about the bilayer normal is constrained by the peptide tilt (Sharpe et al., 2002). The duration of the simulation may be too short to capture slow variations in the helix tilt which, over the correlation time of the reorientation, might give rise to a larger average tilt. Another possibility is that the duration of Simulation 1 was not sufficient to show structural alterations, such as partial relaxation of the helix near the bilayer surface, that might allow the lysines to maintain contact with the bilayer surface for larger helix tilt angles and possibly different orientations of the peptide about the helix axis. This particular effect has been referred to as “snorkeling” (Segrest et al., 1990; Killian and von Heijne, 2000). The specific orientation adopted by the helix in a particular environment may be quite sensitive to details of the peptide-membrane interaction.
to fluctuations in the methyl axis orientation, this is not surprising. Although the maximum splitting obtained by assuming axially symmetric reorientation about the bilayer normal is less than that observed experimentally, the distributions of simulated and experimental splittings span similar ranges. The maximum alanine deuteron splitting obtainable by assuming rotation of the tilted helix about the bilayer normal is constrained by the peptide tilt (Sharpe et al., 2002). The duration of the simulation may be too short to capture slow variations in the helix tilt which, over the correlation time of the reorientation, might give rise to a larger average tilt. Another possibility is that the duration of Simulation 1 was not sufficient to show structural alterations, such as partial relaxation of the helix near the bilayer surface, that might allow the lysines to maintain contact with the bilayer surface for larger helix tilt angles and possibly different orientations of the peptide about the helix axis. This particular effect has been referred to as “snorkeling” (Segrest et al., 1990; Killian and von Heijne, 2000). The specific orientation adopted by the helix in a particular environment may be quite sensitive to details of the peptide-membrane interaction.
Simulation 2: polypeptide initially tilted
A second simulation was started by returning to the state of the bilayer at the end of the peptide-free equilibration and then inserting the polypeptide with an initial orientation obtained by fitting to experimentally observed alanine methyl deuteron quadrupole splittings of the corresponding polypeptide. As noted above, it is this orientation that is shown in Fig. 4. As the second simulation proceeded, the bilayer again accommodated itself to the presence of the polypeptide but, in this case, with an initial conformation more closely approximating experimental observations.
Fig. 7 A shows the evolution of the helix tilt over the 10-ns simulation trajectory. At ∼2 ns, the tilt increased from ∼14 to ∼22° with an excursion to >30° between 7 and 8 ns. Fig. 7 B shows the orientation of the polypeptide about its helix axis over the same period. It is interesting that even while the tilt departs significantly from its initial value in this simulation, the orientation about the helix axis during the last 2 ns of the simulation is similar to its value during the first 4 ns. It is also interesting that the overall trend in Simulation 1, as shown in Fig. 5 B, is toward a similar azimuthal orientation. This may reflect interactions of the peptide with the bilayer that can impose an azimuthal restoring torque about the preferred polypeptide orientation.
FIGURE 7.

The time dependence of selected polypeptide orientational parameters obtained from the trajectory of Simulation 2 (initial orientation of the peptide corresponds to the best fit to observed alanine methyl deuteron quadrupole splittings). Angles shown are (A) the tilt (τ) of the helix axis with respect to the bilayer normal, (B) the azimuthal orientation (ρ) about the helix axis, (C) shows the angle (θ12) between the C-CH3 bond axis of alanine residue 12 and the bilayer normal and (D) the angle (θ14) between the C-CH3 bond axis of alanine residue 14 and the bilayer normal.
The orientations of the methyl axes of alanines 12 and 14 are shown in Fig. 7, C and D, respectively. Over the course of the 10-ns simulation, both methyl orientations move by ∼10°, in opposite directions, from their initial orientations. The magnitude of the tilt changes by a comparable amount over the same period. The abrupt changes in methyl orientation between 7 and 8 ns are coincident with a change in orientation of the polypeptide about its helix axis at the same time, as seen in Fig. 7 B. Fig. 8, B and C, show the corresponding values of 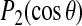 . For the methyl group of alanine 12,
. For the methyl group of alanine 12,  samples more than one-third of its allowed range. For alanine 14, the angle between the methyl axis and the bilayer normal is close to 90° and this is reflected by the saturation of
samples more than one-third of its allowed range. For alanine 14, the angle between the methyl axis and the bilayer normal is close to 90° and this is reflected by the saturation of 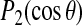 near its lower bound of −0.5.
near its lower bound of −0.5.
FIGURE 8.

(A) The time dependence of the helix tilt (τ) derived from the trajectory of Simulation 2 (initial orientation of the peptide corresponds to the best fit to observed alanine methyl deuteron quadrupole splittings). (B) The time dependence of  for Simulation 2. (C) The time dependence of
for Simulation 2. (C) The time dependence of 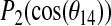 for Simulation 2. Here θn is the instantaneous angle between the bilayer normal and the methyl group axis for the alanine residue n and
for Simulation 2. Here θn is the instantaneous angle between the bilayer normal and the methyl group axis for the alanine residue n and 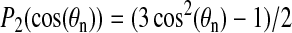 .
.
Simulated quadrupole splittings for the methyl deuterons of alanines 8, 10, 12, 14, 16, 18, and 20 were again calculated by averaging  over the trajectory, of Simulation 2 in this case, and assuming axially symmetric rotation about the bilayer normal. These are shown in the last column of Table 1. Although the width of this distribution, relative to the largest splitting, is again comparable to that of the observed distribution, all of the splittings obtained from Simulation 2 are larger than observed experimentally. This reflects the increase, by ∼10°, in the helix axis tilt angle away from its initial value over the course of the simulation.
over the trajectory, of Simulation 2 in this case, and assuming axially symmetric rotation about the bilayer normal. These are shown in the last column of Table 1. Although the width of this distribution, relative to the largest splitting, is again comparable to that of the observed distribution, all of the splittings obtained from Simulation 2 are larger than observed experimentally. This reflects the increase, by ∼10°, in the helix axis tilt angle away from its initial value over the course of the simulation.
The tilt of the polypeptide away from an initial orientation chosen to match observed alanine methyl deuteron splittings suggests that large excursions from the preferred average tilt are easily accommodated. The initial orientation of the polypeptide in Simulation 2 was chosen to match experimental observations that reflect the average of the orientation-dependent quadrupole interaction over the characteristic experimental timescale. The simulated bilayer, however, must adapt to the peptide insertion, and some initial perturbation of the tilt is expected. Some adjustment from the initial polypeptide conformation, particularly near the bilayer surface, is also expected. Accordingly, the extent to which transient orientations correspond to observed alanine methyl deuteron splittings during a short simulation may not be as instructive as a consideration of relationships between different orientational parameters as the system evolves.
It is interesting that the orientation of the polypeptide about its helix axis, as shown in Fig. 7 B, returns to its initial value even as the tilt continues to move away from its initial value. The orientation about the helix axis is likely determined by interactions at the membrane surface and thus sensitive to the positions of the terminal lysines about the helix axis. Once these residues have been optimally positioned to interact with the surface, rotation about the helix axis may be significantly constrained. This would be consistent with experimental observations. This would also be consistent with the observation, in the first simulation, that orientation about the helix axis is tending toward the orientation seen through most of the second simulation.
The helix tilt appears to be less constrained. This may reflect the abilities of both the bilayer and the polypeptide to accommodate large fluctuations in polypeptide tilt. The bilayer can do so by local deformation of the surface. The flexibility of the lysine side chains also allows their charged groups to remain close to the bilayer surface whereas the polypeptide tilt varies.
Fig. 9 shows the positions of the four lysine α-carbons at either end of the polypeptide for both simulations. The average positions of the lipid phosphorus atoms are shown for comparison. Fig. 10 shows corresponding positions for the charged nitrogens at the ends of the lysine side chains. It should be noted that the phosphorus positions are averages over all lipids and that any local deformation of the bilayer surface near the polypeptide end would be masked in this representation. In Simulation 1, the α-carbons are generally closer to the bilayer center than the average phosphorus positions. The penultimate lysine residues 2 and 25 are, on average, deeper than the terminal residues 1 and 26. The charged nitrogens at the ends of the lysine side chains are consistently closer to the bilayer surface. The corresponding relationships from simulation 2 are similar. The flexibility of the lysine side chain is reflected by the observation that charged nitrogens on the side chains of lysine residues 2 and 25 are sometimes farther from the bilayer center than those of the terminal lysine resides 1 and 26. The positions of the lysine side-chain nitrogens, relative to the average phosphorus position, are not very sensitive to fluctuations in helix tilt. This is consistent with the observation that tilt is less constrained than orientation about the helix axis in Simulation 2.
FIGURE 9.

Comparison of lysine α-carbon positions with average phosphorus positions along the z axis from the two molecular dynamics trajectories. Top panels show α-carbon positions for lysine residues 1 and 26 obtained from the trajectories of Simulation 1 (A) and Simulation 2 (C). Bottom panels show the α-carbon positions for lysine residues 2 and 25 obtained from the trajectories of Simulation 1 (B) and Simulation 2 (D). In all panels, the finer traces show the average positions of the phosphorus atoms on either side of the bilayer.
FIGURE 10.

Comparison of lysine side-chain nitrogen positions with average phosphorus positions along the z axis from the two molecular dynamics trajectories. Top panels show side-chain nitrogen positions for lysine residues 1 and 26 obtained from the trajectories of Simulation 1 (A) and Simulation 2 (C). Bottom panels show side-chain nitrogen positions for lysine residues 2 and 25 obtained from the trajectories of Simulation 1 (B) and Simulation 2 (D). In all panels, the finer traces show the average positions of the phosphorus atoms on either side of the bilayer.
Table 2 summarizes the average differences, Δz, in the projection of position along the bilayer normal between the lysine 1 and lysine 26 α-carbons, the lysine 1 and lysine 26 side-chain nitrogens, the lysine 2 and lysine 25 α-carbons, and the lysine 2 and lysine 25 side-chain nitrogens for both simulations. The separations between average lipid phosphorus positions across the bilayer are included for comparison. These separations provide some information regarding how the larger average helix tilt seen in Simulation 2 is accommodated by the lysines at either end of the peptide. Not surprisingly, Δz for the α-carbons on the terminal lysines (1 and 26) is larger in Simulation 1 than in Simulation 2 for which the helix tilt is larger. The same difference between Δz from simulation 1 and Δz from simulation 2 is seen for the α-carbons on the penultimate lysines (2 and 25) and the side-chain nitrogens of the terminal lysines (1 and 26). In contrast, Δz for the side-chain nitrogens on lysines 2 and 25 has similar values in the two simulations. Taken together, this implies that the side chains on the penultimate lysines are more perturbed by the increased tilt than those on the terminal lysines. Table 2 also shows the differences between the average α-carbon separation and the average nitrogen separation for each pair of lysines. For the terminal lysines, this difference does not change between the two simulations. A similar difference is seen for the penultimate lysines in Simulation 1. The difference is significantly larger, though, for the penultimate lysines in Simulation 2. Although the greater average helix tilt in Simulation 2 results in the lysine α-carbons being, on average, deeper in the bilayer, the side-chain nitrogens remain close to the bilayer surface. The results in Table 2 suggest that this requires more extension of the lysine 2 and lysine 25 side chains than of the lysine 1 and lysine 26 side chains.
TABLE 2.
Projections along the bilayer normal of the average separations between lipid phosphorus atoms on opposite sides of the bilayer and between lysine 1 and lysine 26 α-carbons, lysine 1 and lysine 26 side-chain nitrogens, lysine 2 and lysine 25 α-carbons, and lysine 2 and lysine 25 side-chain nitrogens
| Simulation 1 | Simulation 2 | |
|---|---|---|
| Average POPC phosphorus Δz | 41.5 Å | 41.1 Å |
α-carbons: 
|
36.8 Å | 35.1 Å |
Side-chain nitrogens: 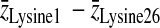
|
42.7 Å | 40.9 Å |
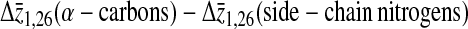 |
5.9 Å | 5.8 Å |
α-carbons: 
|
34.0 Å | 32.2 Å |
Side-chain nitrogens: 
|
39.3 Å | 39.3 Å |
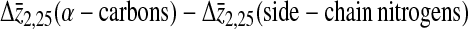 |
5.3 Å | 7.1 Å |
Also shown are the differences between the α-carbon and nitrogen separations for lysine 1 and lysine 26 and the differences between the α-carbon and nitrogen separations for lysine 2 and lysine 25.
SUMMARY
The simulations described here provide insights into how interactions at the membrane surface can constrain rotation about the helix axis while allowing fluctuations in helix tilt. They are relevant to understanding 2H NMR observations of alanine methyl deuterons on lysine-terminated transmembrane polypeptides. Such experiments show that the lysine-terminated transmembrane polypeptides exhibit fast axially symmetric reorientation about the bilayer normal while adopting stable preferred orientations about the helix axis. Although large excursions in helix tilt may be accommodated by extension of the terminal lysine side chains, a situation often referred to as “snorkeling,” the localization of lysine side-chain charges at the bilayer surface appears to contribute to the adoption of a preferred orientation of the tilted helix about its axis. When the simulation is started with the helix oriented along the bilayer normal, the polypeptide tends toward the preferred azimuthal orientation as the simulation proceeds. If the simulation is started with the polypeptide close to the orientation that gives the best fit to observed alanine methyl deuteron splittings, the orientation about the helix axis appears to be maintained even as the helix tilt deviates significantly from the experimentally observed average.
These observations suggest that in studies of transmembrane orientation using alanine methyl deuteron splittings, the tilt of lysine-terminated polypeptides may fluctuate substantially over timescales shorter than the characteristic time of the deuterium NMR experiment. Orientation about the tilted helix axis appears to be more tightly constrained by interactions at the bilayer surface.
Although this study focused on lysine-terminated polypeptides, the constraint of orientation about the helix axis of a tilted polypeptide by interactions involving polar or charged residues near the membrane surface may be more general. Because this may affect the mutual accessibility of particular residues on adjacent polypeptides, studies of peptide-peptide interaction using model polypeptides may be sensitive to the axial arrangement of charged and polar residues at the peptide ends.
Acknowledgments
The authors are grateful to Memorial University of Newfoundland and the University of New Brunswick for access to computing resources, the Advanced Computation and Visualization Centre and the Advanced Computational Research Laboratory, respectively, made available via C3.ca. The authors are also grateful to the Theoretical and Computational Biophysics Group (University of Illinois at Urbana Champaign, Beckman Institute) for making available the NAMD2 molecular dynamics code and to James Polson for helpful discussions.
This research was supported by grants from the Natural Science and Engineering Research Council of Canada and the Canadian Institutes of Health Research.
References
- Armen, R. S., O. D. Uitto, and S. E. Feller. 1998. Phosopholipid component volumes: determination and application to bilayer structure calculations. Biophys. J. 75:734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belohorcová, K., J. H. Davis, T. B. Woolf, and B. Roux. 1997. Structure and dynamics of an amphiphilic peptide in a lipid bilayer. Biophys. J. 73:3039–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T. 1992. X-plor, Version 3.1: A System for X-Ray Crystallography and NMR. Yale University, New Haven, CT.
- Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- de Planque, M. R. R., B. B. Bonev, J. A. A. Demmers, D. V. Greathouse, R. E. Koeppe II, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring (review). Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe II, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane α-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- Dunbrack, R. L., and M. Karplus. 1993. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 230:543–574. [DOI] [PubMed] [Google Scholar]
- Feller, S. E., D. Yin, R. W. Pastor, and A. D. MacKerell. 1997. Molecular dynamics simulation of unsaturated lipid bilayers at low hydration: parameterization and comparison with diffraction studies. Biophys. J. 73:2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, S. E., Y. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- Harzer, U., and B. Bechinger. 2000. Alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: a CD, 15N, and 31P solid-state NMR spectroscopy investigation. Biochemistry. 39:13106–13114. [DOI] [PubMed] [Google Scholar]
- Heller, H., M. Shaefer, and K. Schulten. 1993. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid-crystal phases. J. Phys. Chem. 97:8343–8360. [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Jones, D. H., K. R. Barber, E. W. VanDerLoo, and C. W. M. Grant. 1998. The EGF receptor transmembrane domain: 2H NMR implications for orientation and motion in a bilayer environment. Biochemistry. 37:16780–16787. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2 Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- Killian, J. A. 2003. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 555:134–138. [DOI] [PubMed] [Google Scholar]
- Killian, J. A., and G. Von Heijne. 2000. How proteins adapt to a membrane-water interface. Trends in Biological Sciences. 25:429–434. [DOI] [PubMed] [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Marassi, F. M., C. Ma, J. J. Gessell, and S. J. Opella. 2000. Three-dimensional solid-state NMR spectroscopy is essential for resolution of resonance from in-plane residues in uniformly 15N-labeled helical membrane proteins in oriented lipid bilayers. J. Magn. Reson. 144:156–161. [DOI] [PubMed] [Google Scholar]
- Marassi, F. M., and S. J. Opella. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morein, S., J. A. Killian, and M. M. Sperotto. 2002. Characterization of the thermotropic behavior and lateral organization of lipid-peptide mixtures by a combined experimental and theoretical approach: effects of hydrophobic mismatch and role of flanking residues. Biophys. J. 82:1405–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow, M. R., and C. W. M. Grant. 2000. The EGF receptor transmembrane domain: peptide-peptide interactions in fluid membranes. Biophys. J. 79:2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M. T., W. Humphrey, A. Gursoy, A. Dalke, L. V. Kale, R. D. Skeel, and K. Schulten. 1996. NAMD: a parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. 10:251–268. [Google Scholar]
- Pandit, S. A., and M. L. Berkowitz. 2002. Molecular dynamics simulation of dipalmitoylphosphatidylserine bilayer with Na+ counterions. Biophys. J. 82:1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula, M., K. Murzyn, T. Rog, and C. Czaplewski. 2000. Molecular dynamics simulation studies of lipid bilayer systems. Acta Biochim. Pol. 47:601–611. [PubMed] [Google Scholar]
- Petrache, H. I., D. M. Zuckerman, J. N. Sachs, J. A. Killian, R. E. Koeppe II, and T. B. Woolf. 2002. Hydrophobic matching mechanism investigated by molecular dynamics simulations. Langmuir. 18:1340–1351. [Google Scholar]
- Ryckaert, J.-P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- Schaftenaar, G., and J. H. Noordik. 2000. Molden: a pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 14:123–134. [DOI] [PubMed] [Google Scholar]
- Schlenkrich, M., J. Brickmann, A. D. MacKerell, Jr., and M. Karplus. 1996. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhauser, Boston, MA. 31–81.
- Seelig, J., and N. Waespe-Sarcevic. 1978. Molecular order in cis and trans unsaturated phospholipids bilayers. Biochemistry. 17:3310–3315. [DOI] [PubMed] [Google Scholar]
- Segrest, J. P., H. De Loof, J. G. Dohlman, C. G. Brouillette, and G. M. Anantharamaiah. 1990. Amphipathic helix motif: classes and properties. Proteins. 8:103–117. [DOI] [PubMed] [Google Scholar]
- Sharpe, S., K. R. Barber, C. W. M. Grant, D. Goodyear, and M. R. Morrow. 2002. Organization of model helical peptides in lipid bilayers: insight into the behavior of single-span protein transmembrane domains. Biophys. J. 83:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe, S., C. W. M. Grant, K. R. Barber, J. Giusti, and M. R. Morrow. 2001. Structural implications of a Val→Glu mutation in transmembrane peptides from the EGF receptor. Biophys. J. 81:3231–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda, W., N. Namiki, and S. Okazaki. 1997. Molecular dynamics study of a lipid bilayer: convergence, structure, and long time dynamics. J. Chem. Phys. 106:5731–5743. [Google Scholar]
- Strandberg, E., S. Morein, D. T. S. Rijkers, R. M. J. Liskamp, P. C. A. van der Well, and J. A. Killian. 2002. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry. 41:7190–7198. [DOI] [PubMed] [Google Scholar]
- Subczynski, W. K., R. N. A. H. Lewis, R. N. McElhaney, R. S. Hodges, J. S. Hyde, and A. Kusumi. 1998. Molecular organization and dynamics of 1-palmitoyl-2-oleoylphosphatidylcholine bilayers containing a transmembrane α-helical peptide. Biochemistry. 37:3156–3164. [DOI] [PubMed] [Google Scholar]
- van der Wel, P. C. A., E. Strandberg, J. A. Killian, and R. E. Koeppe, II. 2002. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 83:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K. Nishimura, Z. Gan, R. Fu, J. R. Quine, and T. A. Cross. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144:162–167. [DOI] [PubMed] [Google Scholar]
- Weiner, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.-P., R. N. A. H. Lewis, G. D. Henry, B. D. Sykes, R. S. Hodges, and R. N. McElhaney. 1995. Peptide models of helical hydrophobic transmembrane segments of membrane proteins. 1. Studies of the conformation, intrabilayer orientation, and amide hydrogen exchangeability of Ac-K2-(LA)12-K2-amide. Biochemistry. 34:2348–2361. [DOI] [PubMed] [Google Scholar]