

Abstract
The magnetic relaxation dispersion profiles for formate, acetate, and water protons are reported for aqueous solutions of hemoglobin singly and doubly labeled with a nitroxide and mercury(II) ion at cysteines at β-93. Using two spin labels, one nuclear and one electron spin, a long intramolecular vector is defined between the two β-93 positions in the protein. The paramagnetic contributions to the observed 1H spin-lattice relaxation rate constant are isolated from the magnetic relaxation dispersion profiles obtained on a dual-magnet apparatus that provides spectral density functions characterizing fluctuations sensed by intermoment dipolar interactions in the time range from the tens of microseconds to ∼1 ps. Both formate and acetate ions are found to bind specifically within 5 Å of the β-93 spin-label position and the relaxation dispersion has inflection points corresponding to correlation times of 30 ps and 4 ns for both ions. The 4-ns motion is identified with exchange of the anions from the site, whereas the 30-ps correlation time is identified with relative motions of the spin label and the bound anion in the protein environment close to β-93. The magnetic field dependence of the paramagnetic contributions in both cases is well described by a simple Lorentzian spectral density function; no peaks in the spectral density function are observed. Therefore, the high frequency motions of the protein monitored by the intramolecular vector defined by the electron and nuclear spin are well characterized by a stationary random function of time. Attempts to examine long vector fluctuations by employing electron spin and nuclear spin double-labeling techniques did not yield unambiguous characterization of the high frequency motions of the vector between β-93 positions on different chains.
INTRODUCTION
The global and local dynamics of folded macromolecular structures, such as proteins, are fundamentally important to gain understanding of both the kinetic and thermodynamic aspects of macromolecular function. The folded structure of these macromolecules contains catalytic sites and internal cavities that, though essential for proper protein function, are improperly configured or inaccessible if the protein is treated as a rigid, static structure. However, because the energies required for the protein to sample other structures near the energy minimum of its folding pathway are not large, the protein structure is reasonably represented by distribution of conformational substates, the components of which may be sampled transiently to modulate function.
Hemoglobin and myoglobin have been extensively studied and provide a model for how molecular dynamics couples the structure of a protein with its biological function. Hemoglobin (Hb) is a multimeric, noncovalent aggregation of four distinct polypeptide chains—a dimer of heterodimers, typically denoted as α2β2, that pack together into a nearly spherical array ∼55 Å in diameter. Although each α-chain contacts both β-chains, there are few interactions between either the two α- or β-chains. Each subunit contains a heme prosthetic group located in a crevice near the exterior of the protein that is responsible for the functional ligand binding of the protein. Although the heme groups are ∼25 Å apart, hemoglobin has long been considered the paradigm for allosteric cooperativity in proteins (Silva et al., 1992; Mueser et al., 2000; Lukin et al., 2003).
The specific molecular motions required for hemoglobin allosteric effects have been studied with a variety of computational (Mouawad et al., 2002) and experimental techniques (Yuan et al., 2002; Acharya et al., 2003; Tsai et al., 2003; Chan et al., 2004). The original Perutz mechanism, based on the initial crystal structures of the tense and relaxed states, incorporates a positional change of a histidine with respect to the heme of an α1 subunit upon binding of a ligand (Perutz, 1970; Perutz et al., 1998). This movement induces a relative shift between the corresponding α1 and β2 subunits that modifies the network of salt bridges and hydrogen bonds at the α1–β2 interface and, thereby, changes the ligand accessibility to the heme in the β2 subunit. The structural details of these conformational transitions and their time dependence are still debated. However, local as well as longer range conformational fluctuations remain as an important aspect of hemoglobin function.
The idea that structural fluctuations are crucial is not new. For both myoglobin (Bourgeois et al., 2003) and cytochrome c oxidase (Koutsoupakis et al., 2003), large-scale asynchronous internal motions of specific structural entities were reported over a broad time range extending from picoseconds to milliseconds. These motions generate transient barriers and channels within the protein for internal ligand migration. Another example is the phosphorylation of the allosteric enzyme aspartate transcarbamoylase which activates the protein by stabilizing a pre-existing structure instead of inducing a new structure (Beernink et al., 1999; Volkman et al., 2001). Taken together, these results strongly suggest that protein crystal structures represent the selective detection of one structure from a distribution of conformers in the same manner as a protein effector stabilizing a particular active conformer. With this perspective, the efficiency of the protein function may be strongly dependent on the amplitudes and rate constants for the intrinsic structural fluctuations of the macromolecule.
The objective of this work is to develop experimental methods for observing the intramolecular dynamics of a protein to elucidate potential contributions to function. Hemoglobin provides an ideal candidate because its structure is well defined, it has been extensively studied, it is readily obtainable, and dynamics play an essential role in function in that high frequency motions of the protein are required to permit oxygen accessibility to the iron atoms of the embedded heme groups. Nuclear magnetic resonance spectroscopy provides an excellent approach to this problem because various aspects of the spectroscopy are sensitive to a wide range of frequencies from kHz to THz. Measurement of the spin-lattice relaxation rate constants generally provides a dynamical report at approximately the nuclear Larmor frequency employed because the relaxation must be stimulated by the magnetic noise near this frequency. Because the Larmor frequency is linear in the applied magnetic field strength, the inter- and intramolecular dynamics may be examined by recording the dependence of the spin-lattice relaxation rate constant on the magnetic field strength or the nuclear Larmor frequency. The magnetic field dependence of the spin-lattice relaxation rate constant is called the magnetic relaxation dispersion (MRD) profile. For diamagnetic systems, the maximum frequency is limited by the availability of very high magnetic field strengths to the range below 5 × 109 s−1, or correlation time constants greater than several hundred picoseconds. However, the time or frequency range may be extended significantly over that usually accessible in nuclear magnetic resonance experiments by exploiting nuclear spin-lattice relaxation induced by electron spins because the paramagnetic contributions to the nuclear relaxation equation include terms involving the electron Larmor frequency. As a consequence, the sensitivity to short time dynamics is extended to frequencies of the order of 3 × 1012 s−1, or correlation times on the order of several tenths of a picosecond. The low end of the accessible frequency range is limited by the practical factors of eliminating stray magnetic fields in a magnetic resonance laboratory. For example, the magnetic field of the earth corresponds to a proton Larmor frequency of ∼104 s−1.
We report here high-resolution magnetic relaxation dispersion experiments on hemoglobin solutions, which characterize internal fluctuations in the range from tens of nanoseconds to picoseconds, by exploiting paramagnetic contributions to proton spin-lattice relaxation rates that are measured over the range of magnetic field strengths from just above the earth field to 7 T. We present a strategy for defining and monitoring the fluctuations of specific intramolecular vectors in proteins, as well as a strategy for controlling the magnitudes of the nuclear spin relaxation rates, using a spin dilution technique to make the data acquisition technically feasible over the whole range of magnetic field strengths.
BACKGROUND AND THEORY
The experimental apparatus employed for this work obtains the magnetic relaxation dispersion profile by employing two isolated magnets (Wagner et al., 1999) although fringe field experiments are also useful in this context (Redfield, 2003; Roberts et al., 2004). The nuclear spins of the sample are first polarized in a highly homogeneous 300 MHz superconducting magnet. The sample is then rapidly moved to a satellite electromagnet where its nuclear spins are allowed to relax for a variable amount of time before the sample is returned to the high magnetic field where the residual magnetization is detected. Because the detection field is constant, this field cycling strategy has the advantage that the relaxation dispersion profile may be recorded for a wide range of magnetic fields with nearly constant sensitivity. Not only does this circumvent the usual B7/4 field dependence of the signal/noise ratio, but also high resolution spectroscopy is possible over the complete range of evolution field values. Unfortunately, the finite transit time between the two magnets creates two experimental constraints:
The highest observable relaxation rate constants are <10 s−1.
The sample transit through the low field region between the magnets eliminates magnetization from spins with large low-field relaxation rates, such as protein protons.
The problem of the second constraint, however, may be controlled by using the technique known as chemical-exchange dilution. This technique exploits the effects of chemical exchange on the observed spin of a labile solute that samples both protein and nonprotein sites. The relaxation rate for the labile ligand spin may be adjusted by changing the relative populations of protein and nonprotein environments, thus scaling the whole relaxation dispersion profile.
In the presence of exchange between protein and nonprotein environments, the observed proton spin-lattice relaxation rate constant for the particular solute, R1observed, is a weighted average of the relaxation rate constant of the solute in the bulk, free environment and the protein-associated, bound environment,
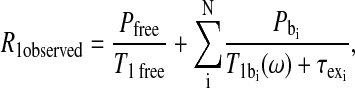 |
(1) |
where T1 free and 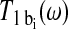 are the relaxation times of the ligand in the free and bound environments, respectively, and
are the relaxation times of the ligand in the free and bound environments, respectively, and 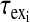 is the mean residence time in the ith bound environment. The expressions
is the mean residence time in the ith bound environment. The expressions  and
and 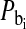 constitute the probability that the observed solute associates with the protein at the ith binding site. If the protein concentration is low compared with the ligand concentration, the bound probabilities may be written in terms of the protein and the ligand concentrations as
constitute the probability that the observed solute associates with the protein at the ith binding site. If the protein concentration is low compared with the ligand concentration, the bound probabilities may be written in terms of the protein and the ligand concentrations as
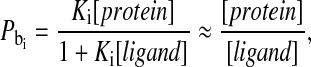 |
(2) |
where Ki is the association constant between the macromolecule and the solute at the ith binding site position and the simplification is valid for sufficiently large Ki.
In general, the mean residence times, 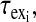 may have a distribution of values that reflect their different chemical and steric environments. If the mean residence times are small compared with the bound-site relaxation times, the observed relaxation rate constant is simply the weighted average of the rate constants in all environments. Eqs. 1 and 2 show that the magnetic field dependence of the relaxation rate constant of the bound environment is mapped onto the observed relaxation rate constant of the labile ligand resonance and that magnitude of this contribution is scaled by the concentration ratio between the protein and the labile ligand. Hence, this ratio may be adjusted to place the relaxation rate constant into a range that is readily measured in the field cycling spectrometer.
may have a distribution of values that reflect their different chemical and steric environments. If the mean residence times are small compared with the bound-site relaxation times, the observed relaxation rate constant is simply the weighted average of the rate constants in all environments. Eqs. 1 and 2 show that the magnetic field dependence of the relaxation rate constant of the bound environment is mapped onto the observed relaxation rate constant of the labile ligand resonance and that magnitude of this contribution is scaled by the concentration ratio between the protein and the labile ligand. Hence, this ratio may be adjusted to place the relaxation rate constant into a range that is readily measured in the field cycling spectrometer.
The observed 1H spin-lattice relaxation rate constants contain contributions from intra- and intermolecular dipolar interactions of both a homonuclear and heteronuclear nature. All of the data reported here were obtained in D2O solutions to minimize the dynamic range problem of a large water proton signal and to minimize the solvent contribution to the proton relaxation rates of the observed labile ligands, i.e., formate ion or acetate ion. The substitution of D2O for H2O reduces the proton relaxation rate of formate ion to ∼0.03 s−1 and increases the sensitivity to macromolecule sites by approximately a factor of 10 compared with using H2O. Consequently, the observed relaxation rate constants for both the formate and residual water proton in the diamagnetic samples are dominated by contributions from intermolecular dipole-dipole interactions between the observed ligand proton and the protein protons. Because this mutual dipolar coupling involves protons that are unlike, and is modulated by random rotational motions of their intermoment vector, it can be described by the expression
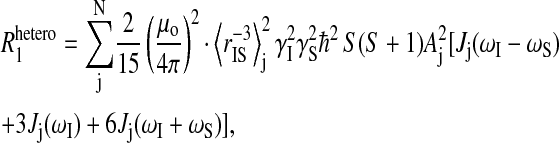 |
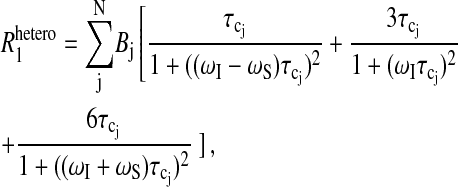 |
(3) |
where N is the number of sites; μo = 4π × 10−7Tm A−1 is the free space permeability; I is the observed nuclear (proton) spin quantum number interacting with a nucleus with spin quantum number S; γI and γS are their corresponding magnetogyric ratio; ℏ is Planck's constant divided by 2π; rIS is the distance between the two spins; 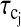 is the correlation time for the vector fluctuations between the jth pair; and J(ω) is the spectral density function that characterizes the frequency dependence of magnetic noise created by the molecular motion of the system that induces relaxation. The order parameter A scales the apparent strength of the coupling if there are local high frequency motions present in the protein binding site (Lipari and Szabo, 1982a,b; Tugarinov et al., 2001). Though the sum includes all significant interproton couplings between the bound nuclear spin and the protein protons in the vicinity, typically only a few terms will dominate because of the strong distance dependence. Furthermore, since the resonance frequency of the two unlike protons will differ only by their chemical shift, then ωI ≈ ωS, to within a few parts per million. With this simplification, Eq. 3 reduces to
is the correlation time for the vector fluctuations between the jth pair; and J(ω) is the spectral density function that characterizes the frequency dependence of magnetic noise created by the molecular motion of the system that induces relaxation. The order parameter A scales the apparent strength of the coupling if there are local high frequency motions present in the protein binding site (Lipari and Szabo, 1982a,b; Tugarinov et al., 2001). Though the sum includes all significant interproton couplings between the bound nuclear spin and the protein protons in the vicinity, typically only a few terms will dominate because of the strong distance dependence. Furthermore, since the resonance frequency of the two unlike protons will differ only by their chemical shift, then ωI ≈ ωS, to within a few parts per million. With this simplification, Eq. 3 reduces to
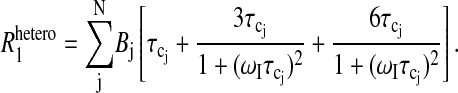 |
(4) |
The experimental strategy to be described is to introduce a paramagnetic center at a specific position in the protein using a nitroxide-spin label, which has an electron spin-lattice relaxation time longer than the rotational correlation time of the protein. For the hemoglobin samples containing this nitroxide spin label, the paramagnetic contributions to the observed 1H spin-lattice relaxation rate constants represent heteronuclear dipole-dipole interactions between the observed protons and the electron of the nitroxide that again can be expressed with Eq. 3, but now with S = ½ for the spin quantum number for the electron, and with ωS ≈ 658ωI for its corresponding Larmor frequency.
The chemical exchange rate of the observed ligand species enters the relaxation equations in two ways. The first is through Eq. 1 or the mixing equation. The second results if the exchange rate of the observed ligand is comparable to or more rapid than the rotational correlation time. Then the effective correlation time associated with the relaxation rate constant of the bound ligand is determined by both the rotation and exchange events,
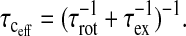 |
(5) |
The signature of this situation is that the measured correlation time, 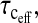 is significantly shorter than the global correlation time for the reorientation of the protein, a situation that is obvious from the relaxation dispersion profile.
is significantly shorter than the global correlation time for the reorientation of the protein, a situation that is obvious from the relaxation dispersion profile.
Although it is sometimes possible to isolate a unique ligand-binding site on a protein without specific modification, a general problem is unambiguous identification of the positions of the electron moment relative to the detected bound site(s) of the labile nuclear moment. Consequently, a chemical labeling strategy was employed to provide a specific site for the observed ligands, formate and acetate. Cysteine residues provide excellent thermodynamic selectivity for introducing both electron-spin labels and sites for labile ligand exchange. The cysteine spin-labeling chemistry is well known and widely used in the context of site-directed spin-labeling experiments (Hubbell et al., 1996). The free sulfhydryl group also reacts quantitatively with mercury(II) ion, thus providing a metal site for exchange of labile ligands such as formate or acetate if the metal-ligand association constants are sufficiently large. Hence, the observed ligand relaxation rate constant of Eq. 1 may be written in terms of several contributions,
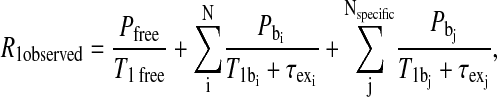 |
(6) |
where the weak sites are distinguished from the specific mercury-labeled sites or other high affinity sites. Both terms on the right side of Eq. 6 may have diamagnetic and paramagnetic contributions.
To isolate the relaxation contributions of the specific vector defined by the nuclear and electron spin-label site pair, four experiments are required: 1), diamagnetic unlabeled protein, Hb; 2), diamagnetic mercury(II)-labeled protein, HbHg; 3), paramagnetic nitroxide-labeled protein, HbSL; and 4), paramagnetic protein that is labeled with both a nitroxide and mercury(II), HbSL_Hg. The relaxation contributions for each case are summarized in Eq. 7 as
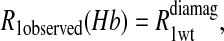 |
(7a) |
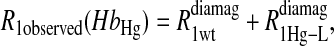 |
(7b) |
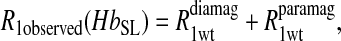 |
(7c) |
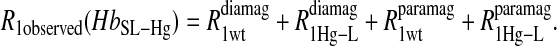 |
(7d) |
The 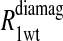 term is the relaxation rate constant observed for the wild-type reduced cyanohemoglobin and represents the sum of all of the nonspecific contributions of the observed ligand interacting with the protein.
term is the relaxation rate constant observed for the wild-type reduced cyanohemoglobin and represents the sum of all of the nonspecific contributions of the observed ligand interacting with the protein. 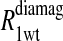 includes the first two terms on the right-hand side of Eq. 6. The
includes the first two terms on the right-hand side of Eq. 6. The 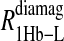 term of Eq. 7b represents the specific contribution from the observed labile ligand interacting with the mercury(II) ions bound at the two β-93 cysteine sites of reduced cyanohemoglobin; i.e., the last term of the right-hand side of Eq. 6. In Eq. 7c, the
term of Eq. 7b represents the specific contribution from the observed labile ligand interacting with the mercury(II) ions bound at the two β-93 cysteine sites of reduced cyanohemoglobin; i.e., the last term of the right-hand side of Eq. 6. In Eq. 7c, the 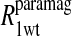 term represents the increase in the observed relaxation rate constant that is generated by labeling the β-93 cysteine sites of reduced cyanohemoglobin with the nitroxide-spin label instead of the mercury(II) ion. This term includes the interaction of the paramagnetic spin label with the protons of all observed labile ligand ions diffusing nearby or interacting with the protein. Finally, Eq. 7d represents the relaxation rate constant observed for reduced cyanohemoglobin that has been labeled with one mercury(II) and one nitroxide spin label in a statistical mixture. The
term represents the increase in the observed relaxation rate constant that is generated by labeling the β-93 cysteine sites of reduced cyanohemoglobin with the nitroxide-spin label instead of the mercury(II) ion. This term includes the interaction of the paramagnetic spin label with the protons of all observed labile ligand ions diffusing nearby or interacting with the protein. Finally, Eq. 7d represents the relaxation rate constant observed for reduced cyanohemoglobin that has been labeled with one mercury(II) and one nitroxide spin label in a statistical mixture. The 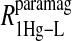 term represents the contribution to the observed relaxation rate constant generated by the specific interaction between the electron of the nitroxide spin label and the proton of the unique formate ion associated with the sulfur-bound mercury(II). Because the location of both of these interacting spins is well defined with respect to the protein structure, the dynamics of a specific vector within a protein can be defined and characterized quantitatively.
term represents the contribution to the observed relaxation rate constant generated by the specific interaction between the electron of the nitroxide spin label and the proton of the unique formate ion associated with the sulfur-bound mercury(II). Because the location of both of these interacting spins is well defined with respect to the protein structure, the dynamics of a specific vector within a protein can be defined and characterized quantitatively.
EXPERIMENTAL
The nuclear spin-lattice relaxation rate constants reported here were measured as a function of the magnetic field strength using an NMR spectrometer assembled in this laboratory and described previously (Wagner et al., 1999). The spectrometer employs two magnetically isolated independent magnets separated by 82 cm. The high field magnet was constructed by Magnex Scientific (Oxford, UK) and provides a high resolution magnetic field of 7.05 T. The second magnet, positioned below the superconducting magnet in an iron shield, is a variable gap, iron-core 4" electromagnet made by GMW Associates (San Carlos, CA). The magnetization decay measured as a function of the residence time in the satellite relaxation field is characterized by the spin-lattice time constant, T1, appropriate to the value of the satellite relaxation field. The magnetic relaxation dispersion profile is obtained by varying satellite field strength over the range of proton Larmor frequencies from 0.01 to 70 MHz. The relaxation rate constant of the 300-MHz superconducting polarization/detection field provides the high field limit. Data acquisition and sample movement are controlled by an Apollo system (Tecmag, Houston, TX), which drives the NVS 4214 double-solenoid air valve (SMC Pneumatics, Indianapolis, IN) used to direct the pressurized air and vacuum that moves the sample between the two magnet systems. The NMR probe built in this laboratory includes a standard linear resonance compressor single resonance circuit with an unload Q of ∼150. The high field is typically shimmed to a 1H line width of 10 Hz or better that is stable to the pounding of the sample excursions. All experiments were conducted at ambient temperature of the driving gas supply, which was ∼293 K.
Sample preparation
All samples were prepared from lyophilized bovine methemoglobin (M-9250; 61.95 kDa, Sigma Chemical, St. Louis, MO). A 500 μM methemoglobin solution was prepared by adding a buffer of 100 mM sodium formate (S-2140; Sigma) and 10 mM total of sodium phosphate monobasic (S-9638; Sigma) and dibasic (S-7907; Sigma) at pH 6.8 that used deionized 17 MΩ water obtained from an Ultrapure system (Barnsted, Dubuque, IA) using house deionized water as the feed. The hemoglobin was treated with a 10-fold molar excess of sodium hydrosulfite, or sodium dithionite (S-1256; Sigma) to produce the iron(II); then a 10-fold molar excess of potassium cyanide (3080-1; Baker, Phillipsburg, NJ) was added to stabilize the low spin, diamagnetic state of hemoglobin and prevent the observed ligands from binding at the iron. After allowing several hours, excess dithionite was removed by fast phase liquid chromatography size-exclusion gel filtration utilizing an AKTA purifier system (Amersham Biosciences, Piscataway, NJ) employing a prepacked HiPrep 26/10 desalting column (Code No. 17-5087-01) using Sephadex G-25 Fine with a 53 mL bed volume. The purified hemoglobin solution was concentrated by using Centricon YM-10 centrifugal filters with a molecular cutoff of 10,000 (No. 4205 Millipore), all obtained from Fisher Scientific (Pittsburgh, PA). An IEC Centra MP4 (International Equipment Company, Needham Heights, MA) was employed running at 8400 rpm for 100 min to reduce a 2-mL sample volume to ∼200 μL. A buffer exchange was then performed using the same Centricon filter by bringing the reduced solution volume back to 2 mL with a deuterium oxide buffer at 99.9% deuterium (Cambridge Isotope Laboratories, Andover, MA) that contained 100 mM sodium formate and 10 mM sodium phosphate buffered at pH 6.8. This buffer exchange process was repeated four times to reduce the water proton concentration of the MRD sample significantly. For the hemoglobin samples containing only the bound mercury label and not the nitroxide, two equivalents of mercuric acetate (M-6279; Sigma) were added to the hemoglobin-cyanide solution after the buffer exchange process to bind quantitatively to both (β-93) cysteine residues.
The nitroxide-labeled hemoglobin samples were prepared as previously described (Ogawa and McConnell, 1967). A 10-fold molar excess of the spin label 3-(2-iodoacetamido)-PROXYL (I-1259; Sigma) dissolved in acetonitrile was combined with the purified hemoglobin-cyanide solution. Under these conditions, the iodoacetamide spin label reacts slowly but specifically with the two β-93 cysteine sulfhydryl groups of the hemoglobin protein. The spin-labeling reaction was allowed to proceed for 2 h at room temperature, and then at 4°C, to allow the reaction to continue with minimal protein degradation. The progress of the spin-labeling reaction was monitored by electron paramagnetic resonance spectroscopy. The 10-μL aliquots of the hemoglobin-spin-labeling solution were transferred into a round (0.60 mm I.D. × 0.84 mm O.D.) quartz capillary tube (CV6084; VitroCom, Mountain Lakes, NJ). Using a Varian E-line 102 Centuries series spectrometer (Varian, Palo Alto, CA) and an X-band loop-gap resonator (Medical Advances, Milwaukee WI) with a standard two-loop, one-gap configuration, progress of the reaction could be observed and compared with methane thiosulfonate spin-label standard solutions made by weight. After ∼3 days when the spin-labeling reaction was approximately half completed, the AKTA purification system with an HiPrep 26/10 desalting column was again employed to remove the excess spin label and, thereby, terminate the reaction. The collected hemoglobin, with approximately one cysteine sulfhydryl group spin-labeled, was then divided in half, concentrated, and the buffer exchanged in the same manner as described previously for the non-spin-labeled hemoglobin samples. To one of these two samples, one equivalent of mercuric acetate (M-6279; Sigma) was added to occupy the remaining β-93 sulfhydryl group.
The final protein concentration of each sample was determined by using a Bradford assay protocol with Coomassie blue reagent (1856209; Pierce, Rockford, IL) and bovine serum albumin standards (23209; Pierce). The optical absorbance at 595 nm was recorded on a Varian Cary 4 UV-Visible spectrophotometer (Varian Cary, Palo Alto, CA). Using electron paramagnetic resonance spectroscopy, the nitroxide concentration for the two sample preparations containing the iodoacetamide spin label was determined by comparing the calculated second integral of the measured derivative spectrum with a calibration curve developed using various free nitroxides in solution.
Each sample was purged of oxygen by placing it in a gas-tight glove box that was purged with nitrogen gas after being sealed. To facilitate the elimination of the paramagnetic oxygen molecules dissolved in the sample solvent, carbon monoxide gas was slowly bubbled into the sample mixture for 40 min. The resulting 500 μL of sample was then loaded into the sample container consisting a 4.0 mm I.D. × 6.0 mm O.D. glass tube (Wilmad, Buena, NJ) mounted in a cylindrical Delrin barrel (15.7 mm O.D., Delrin, McMaster-Carr, New Brunswick, NJ). Because it does not leak oxygen and is relatively chemically inert, this glass design provides significant advantages over the earlier Delrin shuttles.
RESULTS AND DISCUSSION
Formate and acetate sites near β-93
The proton spin-lattice relaxation rates plotted as a function of the Larmor frequency are shown in Fig. 1 for the formate proton in aqueous hemoglobin solutions containing 100 mM formate ion for cyanohemoglobin (Hb) and cyanohemoglobin that has two equivalents of mercury(II) ion added (HbHg). Fig. 2 reports the MRD profiles in the same solutions for the residual water proton in the D2O. The low-field inflection of the water dispersion corresponds to a rotational correlation time of 50–60 ns that is consistent with other measurements of proteins this size (Denisov and Halle, 1996; Kiihne and Bryant, 2000; Van-Quynh et al., 2003). The corresponding inflections for the formate ion report time constants of 70 and 110 ns. The difference between these two is not significant because the small amplitude of the dispersion from global rotation in the Hb sample makes the uncertainty in the inflection point larger than that in the HbHg sample, which provides an additional specific formate ion binding site at both β-93 cysteine residues. The small amplitude of the relaxation dispersion is consistent with a small number of formate ion-protein interactions. There is also a weak, but experimentally significant, high frequency dispersion corresponding to Larmor frequencies of hundreds of MHz for both the formate and water protons. The significant field jump between 70 MHz, the maximum field of the variable low field magnet, and the detection field of 300 MHz precludes an accurate estimate of the correlation time. However, the step implies that the observed labile molecules suffer a correlation with hemoglobin protons that is interrupted in the time range of several nanoseconds. One possible source for this high frequency dispersion could be trace amounts of adventitious oxygen or contributions from iron. However, the iron contributions, if any, are eliminated by subtraction when specific terms in the relaxation equations are isolated and the sealed bubble-free tubes that were used in these experiments are not pervious to oxygen. The approximate magnitude and frequency of the high field decrease, based on Eqs. 3 and 4, is consistent with the formate proton interacting with 1–2 protons at van der Waals contact with a correlation time of ∼2 ns. The paramagnetically labeled protein variants to be discussed shortly define this interaction more clearly. The relaxation profiles of Fig. 1 demonstrate that addition of mercury(II) ion enhances the formate proton relaxation rate by creating a high affinity binding site for the formate ion or acetate ion. To characterize these effects more quantitatively, the data were fit to Eq. 4 with the parameters summarized in Table 1.
FIGURE 1.

The proton spin-lattice relaxation rate constant as a function of the magnetic field strength plotted as the proton Larmor frequency for the formate proton measured in the presence of reduced cyanohemoglobin (▴) and reduced cyanohemoglobin with a mercury(II) ion bound at each of the β-93 cysteine sites (♦). As Eq. 2 indicates, the observed relaxation rate constants are linear with respect to the protein concentration and the data shown have been normalized to represent 50 μM hemoglobin. These protein samples were in a D2O buffer with 100 mM sodium formate and 10 mM sodium phosphate at pH 6.8. The dashed line is the difference between the fits obtained for the two samples and represents the specific contribution from the formate at the β-93-mercury(II) sites. The three solid lines represent the least-square fits to the two data sets, and their difference (dashed line) using Eq. 4, and the parameters are listed in Table 1.
FIGURE 2.

The proton spin-lattice relaxation rate constant, as a function of the magnetic field strength plotted as the proton Larmor frequency for the residual water proton, measured in the presence of reduced cyanohemoglobin (▴) and reduced cyanohemoglobin, with a mercury(II) ion bound at each of the β-93 cysteine sites (♦) obtained from the same samples described in the caption for Fig 1. The solid lines represent the least-squares fits to the data using Eq. 4 with the parameters listed in Table 1.
TABLE 1.
Parameters determined from least-square fits to the hemoglobin MRD data
| A′ | B1 | τ1 | B2 | τ1 | |
|---|---|---|---|---|---|
| Formate | |||||
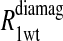
|
0.02 | 2.8E + 04 | 7.6E − 08 | 1.8E + 06 | 1.2E − 09 |
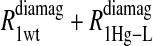
|
0.01 | 7.7E + 04 | 1.2E − 07 | 1.1E + 06 | 2.1E − 09 |
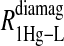
|
0.00 | 5.8E + 04 | 1.3E − 07 | ||
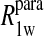
|
−0.27 | 3.4E + 09 | 2.9E − 11 | 3.0E + 07 | 1.7E − 09 |
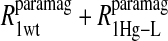
|
−0.23 | 3.3E + 09 | 3.2E − 11 | 4.0E + 07 | 1.9E − 09 |
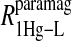
|
0.02 | 1.6E + 08 | 5.3E − 11 | 1.1E + 07 | 2.1E − 09 |
| Acetate | |||||
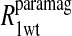
|
−0.03 | 3.5E + 08 | 3.4E − 11 | 4.3E + 06 | 4.2E − 09 |
| Water | |||||
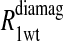
|
0.02 | 4.5E + 04 | 5.4E − 08 | 2.0E + 06 | 8.8E − 10 |
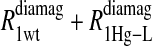
|
0.02 | 4.6E + 04 | 6.2E − 08 | 1.8E + 06 | 1.1E − 09 |
Eq. 4 with N = 2 was applied to the 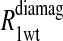 and the
and the 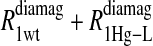 data for both water and formate protons. The constant A′ represents the residual relaxation rate constant remaining at the high field end for all of the fits tabulated here. The data for
data for both water and formate protons. The constant A′ represents the residual relaxation rate constant remaining at the high field end for all of the fits tabulated here. The data for 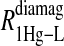 of the formate proton was fit in a similar fashion except with N = 1. Eq. 3 with N = 2 and physical constants corresponding to an electron was applied to the
of the formate proton was fit in a similar fashion except with N = 1. Eq. 3 with N = 2 and physical constants corresponding to an electron was applied to the 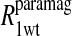 and the
and the 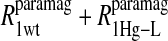 data for the formate protons and to the
data for the formate protons and to the 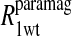 data for the acetate protons. The units of the correlation times are in seconds (s) and for the relaxation rates are s−1. Errors for the fitted parameters are estimated at 20%.
data for the acetate protons. The units of the correlation times are in seconds (s) and for the relaxation rates are s−1. Errors for the fitted parameters are estimated at 20%.
In D2O solutions containing hemoglobin, both the residual protons in water and the formate proton will suffer relaxation contributions from protons as well as deuterons. However, the deuteron relaxation contribution is much smaller than the proton-proton coupling. In addition, the concentration of protein-bound deuterons is limited to exchangeable proton sites, which are relatively rare. Although the deuteron concentration is large in the solvent, the correlation time for the deuteron-proton coupling is in the tens-of-picoseconds range, which is orders-of-magnitude shorter than that for protein-bound couplings. Thus, the detected proton relaxation dispersion profiles for the diamagnetic cases were fit to a sum of two Lorentzian functions representative of heteronuclear interactions according to Eq. 4 with N = 2.
The paramagnetic contributions to relaxation dispersion profiles were isolated by subtracting the fitted curves of the appropriate diamagnetic data from the data obtained from the paramagnetic samples. Three labile ligands were examined: formate ion, acetate ion, and water. For the formate ion, a doubly-labeled hemoglobin sample was also studied in an attempt to isolate the relaxation dispersion profile for a specific long vector in the protein between the two cysteine residues. The paramagnetic contributions to formate and acetate ion relaxation are shown in Figs. 3 and 4, respectively. The data are fit with two relaxation rate contributions of the form shown in Eq. 3. Inflection points at 2 ns and 30 ps associated with the electron Larmor frequency are well defined and demonstrate that the labile ligand samples a unique binding site in the vicinity of the paramagnet at the β-93 cysteine. The small high-field drop in the relaxation rate is consistent with the 2 ns correlation time inflection in the nuclear Larmor frequency term, 3J(ωI), and sensed as the low field inflection of both Figs. 3 and 4 caused by the electron Larmor frequency term, 7J(ωS), of Eq. 3.
FIGURE 3.

The proton spin-lattice relaxation rate constant as a function of the magnetic field strength plotted as the proton Larmor frequency for the formate proton measured in the presence of reduced cyanohemoglobin labeled with iodoacetic acid spin label (•) and reduced cyanohemoglobin labeled with one mercury(II) and one iodoacetic spin label in a statistical mixture (▾). As Eq. 2 indicates, the observed relaxation rate constants are linear with respect to the protein concentration and the data shown have been normalized to represent 50 μM hemoglobin. These protein samples were in a D2O buffer with 100 mM sodium formate and 10 mM sodium phosphate at pH 6.8. The corresponding unfilled symbols, (○) and (▿), represent the paramagnetic contribution to the respective MRD profile after subtraction of the diamagnetic contribution. The solid lines represent the least-square fits of the paramagnetic contributions using Eq. 3 with the parameters listed in Table 1. The dot-dashed line represents the difference between the fits obtained for the two samples, and represents the specific paramagnetic contribution detected by the formate proton at the β-93-mercury(II) site.
FIGURE 4.

The proton spin-lattice relaxation rate constant as a function of the magnetic field strength plotted as the proton Larmor frequency for acetate (⋄) and residual water protons (○) obtained with methemoglobin samples that are labeled at the β-93 sites with two equivalents of iodoacetic acid spin label. The acetate data were acquired with 100 μM methemoglobin in D2O with 200 mM sodium acetate. The residual water data was acquired with 200 μM methemoglobin in D2O with 100 mM potassium chloride and no sodium acetate. The solid lines represent the least-square fits using Eq. 10 (Table 2) for the residual water data and Eq. 3 (Table 1) for the acetate data.
There is no dispersion in the vicinity of 50 ns for global rotational reorientation observed in the paramagnetic contribution. The frequency of the low field inflection corresponds to a correlation time of 2 ns, which is ∼25 times shorter than the global rotational correlation time of the protein under these conditions. Thus the electron-nuclear correlation is interrupted by events that are faster than global reorientation of the protein. This result is consistent with the formate or acetate ion binding to the vicinity of the nitroxide label with a mean residence time of 2 ns. This correlation time is also consistent with the small drop in the diamagnetic relaxation rate at the highest Larmor frequencies shown in Fig. 2, but it is much more apparent in the paramagnetic protein data.
We note that the paramagnetic relaxation contributions to the formate ion and acetate ion relaxation dispersion profiles may have contributions from the relative translational diffusion of these observed species in the vicinity of the paramagnetic center on the protein. The diffusional contribution will generally have relaxation rates in the range of 1 s−1 mM−1 for nitroxides (Polnaszek and Bryant, 1984). However, several factors affect this initial estimate with respect to the data of this study:
The protein-bound nitroxide concentration is a factor-of-10 smaller than that for the cases studied in that work.
The estimate of 1 s−1 mM−1 presumes essentially uniform contact between the diffusing observed spin and the nitroxide, which is not possible when the nitroxide is bound to a protein residue that is sterically constrained. Thus, a reduction by a factor between 2 and 4 or more is appropriate because of limited steric access.
The estimate presumes that the distance of closest approach of the observed spin to the nitroxide is limited only by van der Waals contact, which may not be possible because of the protein getting in the way.
The local translational diffusion coefficient in the vicinity of the nitroxide may be different from that in the simpler solutions. Measurements of the change in the dynamics of water at the protein interface suggest that the diffusion coefficient is smaller by a factor of ∼3 at the protein-water interface (Polnaszek and Bryant, 1984; Polnaszek et al., 1987; Hodges et al., 1997), which would increase the size of the diffusive contribution.
Nevertheless, taken together these factors will limit the magnitude of the low field diffusional contribution to the order of 0.05 s−1 or less for the conditions of these experiments. Furthermore, the relaxation dispersion profiles do not require a large diffusive contribution because the Lorentzian functions account for the data well; it is not included in the least-squares fits displayed.
The 30-ps correlation times that appear in Figs. 3 and 4 report local relative motion of the bound formate and acetate ions adjacent to the nitroxide label. The inflection is well defined and reproducible. Because the same inflection frequency is observed for both the formate and acetate ion, the high frequency motion reported in this experiment cannot reflect simply rotation of the acetate ion methyl group. Instead, the inflection reports relative motion of the acetate or formate ion proton spins with respect to the nitroxide covalently linked to the β-93 cysteine. Assuming that the order parameter is unity and a single site, Eq. 3 implies that the magnitude of the relaxation rate change through the 30-ps inflection region is consistent with a distance between the electron and nuclear moments of ∼5 Å. This result is consistent with the identification of an anion binding site in close proximity to β-93 (Chiancone et al., 1975a,b, 1976; Norne et al., 1978).
The observed formate or acetate ions are not covalently bound to the protein but the diamagnetic contributions reported in Fig. 2 show that there are several weak binding interactions with the protein. However, the paramagnetic contribution to the proton relaxation of both formate and acetate ion are well accounted for by a single bound site close to the paramagnet. The 30-ps correlation time is of order 1000 times shorter than the global rotational correlation time of the protein and it is tempting to describe it as characterizing rapid internal breathing motions of the local structure. The convenient mental picture of such motions is some sort of structural vibration. If the motion that modulates this electron-nuclear coupling were periodic, then the Fourier transform of the periodic correlation function, i.e., the spectral density function, would have a peak. However, the experimental MRD profiles that provide direct measure of this spectral density function exhibit a monotonic decrease with increasing Larmor frequency. Thus the MRD profile reports that the motions of the intermoment vector between the nitroxide and the formate or acetate protons are interrupted randomly and characterized by a 30-ps correlation time.
The β-93–β-93 vector
The use of deuterium oxide as the solvent, as well as dilute observation ligands that exchange with specific protein binding sites, permit detection of weak or long-range interactions. Even though the β-93 SH groups are ∼20 Å apart, it may be possible to detect the electron-nuclear coupling at this distance. Fig. 5 shows the low field paramagnetic contribution to the proton spin-lattice relaxation rate divided by the order parameter as a function of distance for choices of the correlation time from 30 ps to 50 ns computed from Eq. 3 assuming a bound ligand probability of 0.001. In protein D2O solutions, the formate proton relaxation is limited by the residual proton concentration in the water and is typically ∼0.03 s−1. Therefore, a relaxation rate change of 0.02 s−1 represents a 66% change and is readily detectable. The calculations summarized in Fig. 5 show that when the correlation time is close to the global reorientation time, a vector as long as 20 Å may be easily detected; however, for shorter correlation times, the magnitude of the paramagnetic contribution may drop dramatically. These calculations are oversimplified in that they presume an effective global correlation time; nevertheless, they suggest that it is reasonable to look for dynamics associated with the mercury-nitroxide double label that define a unique and long intermoment vector between these two positions in the protein.
FIGURE 5.

Computed paramagnetic contributions to the proton spin-lattice relaxation rate constant as a function of the intermoment distance, assuming a bound state labile ligand probability of 0.001, an order parameter of 1, and correlation times of 50 ns (top), 10 ns, 1 ns, 500 ps, 100 ps, and 30 ps (bottom).
Indeed, the addition of mercury to the protein labeled with 50% nitroxide yields a measurable increase in the relaxation rate constant. The difference between the two curves fitted to the relaxation rates for the singly- and doubly-labeled protein is shown in Fig. 3. The inflection frequencies correspond to correlation times of 2 ns and 50 ps. Inspection of Fig. 5 implies that the correlation time of 50 ps is too small to provide a significant contribution from fluctuations in the Hg-nitroxide vector because the distance is of order 20 Å (Fermi et al., 1984). Further, because the rotational correlation is interrupted by faster events, the global rotational motion of the protein is not detected in this contribution. The 2-ns inflection is again assigned to the exchange event at the nitroxide site. The mercury-formate ion exchange rate has been measured in other systems and is slower than 107 s−1 (K. Victor, unpublished). Thus, because the effects of limited access to the metal site should slow this rate further, the observed 2-ns exchange event is not associated with the mercury-ligand exchange. Thus, for the doubly-labeled hemoglobin sample, it is unreasonable to ascribe the observed increase in the relaxation rate constant as being caused by the motions of the long vector defined by the double mercury-nitroxide label.
There are changes, however. The fact that the long correlation time is 2 ns, which is the same as in the protein labeled only with the nitroxide, is consistent with the detected change arising from the nitroxide site modified somewhat by the binding of mercury. The short correlation time changes by a factor of 2.5, although the uncertainty is significant because the relaxation rate changes are small. The change suggests that the fluctuations of the formate-nitroxide intermoment vector at the nitroxide β-93 site are slowed somewhat by modification of the remote β-93 site.
High frequency water dynamics
The diamagnetic contributions to the water spin-lattice relaxation rate shown in Fig. 2 are well understood in the context of the labile exchange of relatively few water molecules and labile protein hydrogens that bind to unique sites on the protein with lifetimes that are long compared with the rotational correlation time of the protein (Koenig and Schillinger, 1969a,b; Bryant, 1996; Halle et al., 1999). The vast majority of water molecules in the vicinity of the protein sample the protein surface in times that are short compared with the rotational correlation time of the protein. The well-known consequences are that there are few intermolecular nuclear-Overhauser-effect crosspeaks between water and protein protons at the surface of the protein (Wuthrich et al., 1992; Halle, 2003; Modig et al., 2004). Water molecule correlation times for motions at the protein interface are generally placed in the range of tens to hundreds of picoseconds.
The paramagnetic contributions to the water spin-lattice relaxation are shown in Fig. 6. Water relaxation rates in nitroxide-labeled proteins have been reported before, and successfully analyzed in terms of the translational mobility of the water at the nitroxide site (Polnaszek and Bryant, 1984). The electron-nuclear coupling provides a means for localizing the diffusive contributions to the nuclear spin relaxation to those regions very close to the paramagnetic center, and this class of measurements has been useful in defining interfacial translational diffusion coefficients. The relaxation equations for translational modulation of the electron-nuclear coupling have been developed by Freed and Hwang as well as by Ayant and co-workers (Ayant et al., 1975; Hwang and Freed, 1975; Freed, 1978). Fig. 6 shows a relaxation dispersion profile computed based on the Freed-Hwang model for the conditions of the experiments conducted on spin-labeled hemoglobin using the assumption that the effective translational diffusion constant is three times smaller than that for bulk water and that the distance of closest approach between the water proton and the nitroxide is the sum of the van der Waals radii or 2.5 Å. The computed diffusional relaxation rates based on the diffusional motions of the water proton in the vicinity of the paramagnetic center clearly does not match the experimental paramagnetic contribution. The relaxation rates are too small and the shape of the dispersion curve is poorly reproduced.
FIGURE 6.

The proton spin-lattice relaxation rate constant as a function of the magnetic field strength plotted as the proton Larmor frequency for the residual water proton obtained from the same samples described in the caption for Fig. 3. The corresponding open symbols, (○) and (▿), again represent the paramagnetic contribution to the respective MRD profile after subtraction of the diamagnetic contribution. The solid lines represent the least-square fits of the paramagnetic contribution to Eq. 10 resulting in the parameters summarized in Table 2. The dotted line represents an approximation to the translational contribution due to diffusion of bulk water near the nitroxide label, computed using the Freed equation and assuming a distance of closest approach of 2.6 Å and a relative diffusion constant of 6 × 10−10 m2/s. The result of the Freed calculation was used for the calculated parameters listed in Table 3.
A number of factors may cause this discrepancy:
The nitroxide site may be protected by its location in the interstrand crevice of the Hb molecule, which may change the effective distance of closest approach to the paramagnet by the observed ligand. Steric hindrance to ligand access would reduce the measurable relaxation rate; however, the parameters used in the calculation assumed van der Waals contact, and the computed relaxation rate is still less than the observed rate.
Generally, steric access from solution to a protein-bound spin label is reduced by the order of approximately a factor of 2 from that appropriate to an unbound free nitroxide, even if the nitroxide is bound at the surface because the large protein effectively screens the radical from solvent or small solute collisions on the protein side of the interface. In this case where the nitroxide is in a crevice between chains, the steric exclusion factor will be larger. Thus, the computed diffusional contribution is overestimated.
The water molecule motion in the vicinity of the β-93 nitroxide may not be diffusive; the water may stick for a variety of residence times. Indeed, the water molecules that bind to hemoglobin for times that are long in comparison to the rotational correlation time that creates the relaxation dispersion in the diamagnetic protein solutions are present in the spin-labeled protein, and each of these water molecules may have a unique paramagnetic relaxation contribution to the observed water proton relaxation rate. Were there only one such water molecule site close to the spin label, the relaxation dispersion would be modeled well by the sum of a diffusional contribution and a Lorentzian contribution like that observed for the acetate and formate ions. However, the paramagnetic contribution to the water relaxation dispersion profile is not described by the effects of a single binding site like that for the formate and acetate ions.
Although diffusive motion of the water protons in the neighborhood of the protein must make at least small contributions to the observed paramagnetic relaxation profile, the shape of the profile suggests that an alternate approach is justified. If we assume that the water suffers several binding events in the vicinity of the nitroxide position, there must be a distribution of them. We presently have no way to count them, but the shape of the MRD profile implies that there is a distribution of effective correlation times that characterizes the electron-nuclear coupling to water protons.
The paramagnetic contribution of water molecules that exchange with the protein in times that are long compared with the global rotational correlation time of hemoglobin will have correlation times for the electron-nuclear coupling that will be equal to the rotational correlation time of the protein, or of the order of 50 ns, provided that the electron spin-lattice relaxation time is long compared with 50 ns. Both the electron and nuclear Larmor frequency terms contribute to the observed paramagnetic relaxation and the data may be analyzed by incorporating both terms simultaneously, because the correlation times for each are not independent. With the rotational correlation time as the longest correlation time, the data shown in Fig. 6 were fit to a discrete sum of paramagnetic contributions using the well-known Solomon, Bloembergen, Morgan equations (Solomon, 1955; Bloembergen and Morgan, 1961), which may be written
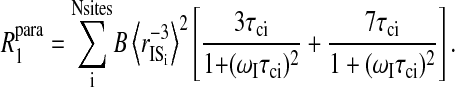 |
(8) |
The sum runs over binding sites each characterized by a correlation time and an intermoment distance to the paramagnet site, S, and the nuclear spin position, i. Equation 8 suggests that the analysis in terms of a distribution of correlation times is intractable because of simultaneous distributions of distance and correlation times. Although this is true in detail, the exercise holds some merit for the following reason. Each term of Eq. 8 may be written
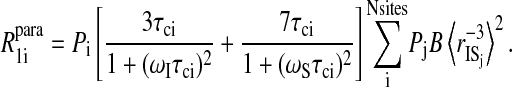 |
(9) |
If one assumes that the water binding sites are uniformly distributed through the protein at some uniform density per unit volume, then the sum over j-sites characterized by correlation time 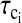 may be replaced by an integral over the volume of the protein from the distance of closest approach to the paramagnet to the diameter of the protein. In this case, the integral is a constant presuming that the sites for each correlation time are randomly distributed in the protein, which of course makes a possibly unwarranted assumption about the cooperativity of local high frequency protein dynamics. However, in this simplified limit, Eq. 8 may now be written
may be replaced by an integral over the volume of the protein from the distance of closest approach to the paramagnet to the diameter of the protein. In this case, the integral is a constant presuming that the sites for each correlation time are randomly distributed in the protein, which of course makes a possibly unwarranted assumption about the cooperativity of local high frequency protein dynamics. However, in this simplified limit, Eq. 8 may now be written
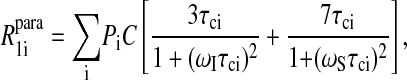 |
(10) |
where the sum now is over the correlation time set with each contribution of weight Pi and C is a constant. Presuming that there is a distribution of water molecules interacting with the protein, Eq. 10 is then applied to the data using a set of six correlation times to represent motions ranging from the rotational mobility of the protein and shorter correlation times that characterize different mean residence times in the vicinity of the paramagnetic center. It should be emphasized that these six fixed correlation times, tabulated in Table 2, are an arbitrary selection simply representing motions of different orders of magnitude. Although certainly not justifiable in detail, this analysis of the paramagnetic contributions to the water MRD data provides a semiquantitative view of the water molecule lifetime distribution in the vicinity of the electron spin label at the protein-water interface.
TABLE 2.
Pi parameters determined from least-square fits to the hemoglobin MRD data using a series of six terms of the heteronuclear interaction of Eq. 10 with fixed correlation times of 5 × 10−8, 5 × 10−10, 1 × 10−10, 5 × 10−11, 1 × 10−11, and 5 × 10−12 s
| Fractional contribution of corresponding τc term |
||||||
|---|---|---|---|---|---|---|
| τc | 5.0E − 08 | 5.0E − 10 | 1.0E − 10 | 5.0E − 11 | 1.0E − 11 | 5.0E − 12 |
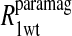 |
2.0E − 04 | 2.7E − 02 | 0.0E + 00 | 1.2E − 01 | 8.0E − 01 | 5.4E − 02 |
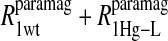 |
1.0E − 04 | 1.7E − 02 | 0.0E + 00 | 8.0E − 02 | 3.6E − 01 | 5.4E − 01 |
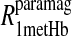 |
1.0E − 04 | 9.9E − 03 | 0.0E + 00 | 3.3E − 02 | 3.5E − 01 | 6.1E − 01 |
The tabulated Pi parameters represent the normalized fractional contribution to the total paramagnetic MRD profile for its corresponding fixed correlation time. The results of the analysis are illustrated in Figs. 4 and 5 for the residual water in the methemoglobin and cyanohemoglobin samples, respectively.
Table 2 summarizes the results of applying Eq. 10 to the isolated paramagnetic contributions of the water spin-lattice relaxation rate constants, in the presence of cyano- and methemoglobin, which are shown in Figs. 6 and 4, respectively. An alternative approach was also taken by first including a specific calculation of the diffusional contribution to the relaxation rate and subtracting that from the data before fitting the remainder to the sum of the correlation time distribution. Note that by using the Freed model with the assumptions that van der Waals contact is the distance of closest approach and the diffusion constant is that for water divided by 3, the diffusional contribution is very likely overestimated, because no account is taken of the steric exclusion of the water from some sides of the spin label by the steric constraints of the spin-label position. Nevertheless, the result of this calculation is shown in Fig. 6 and the analysis results using this calculation are listed in Table 3.
TABLE 3.
Pi parameters determined from least-square analysis of MRD data using a series of six terms of the heteronuclear interaction of Eq. 10 with fixed correlation times of 5 × 10−8, 5 × 10−10, 1 × 10−10, 5 × 10−11, 1 × 10−11, and 5 × 10−12 s
| Fractional contribution of corresponding τc term |
||||||
|---|---|---|---|---|---|---|
| τc | 5.0E − 08 | 5.0E − 10 | 1.0E − 10 | 5.0E − 11 | 1.0E − 11 | 5.0E − 12 |
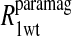 |
4.0E − 04 | 4.5E − 02 | 7.7E + 00 | 1.2E − 01 | 8.3E − 01 | 0.0E + 02 |
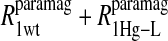 |
1.0E − 04 | 2.2E − 02 | 1.0E + 00 | 7.5E − 02 | 1.7E − 01 | 7.4E − 01 |
The analysis is identical to that in Table 2 for the residual water in the cyanohemoglobin samples except that an approximation to the translational contribution due to diffusion of bulk water near the nitroxide label, computed using the Freed equation with a distance of closest approach of 2.6 Å and a relative diffusion constant of 6 × 10−10 m2/s, has been first removed from the paramagnetic data displayed in Fig. 5 before the analysis.
In this context we note that the diffusional contribution to the formate and acetate relaxation dispersion was not detected as a distortion to the Lorentzian relaxation profile. If diffusional access to the nitroxide region of the protein were similarly restricted for the water, the diffusional contribution would also be negligible and the simple sum of Lorentzian functions would provide a more logical approach. However, the results of the two approaches are similar and should be regarded as essentially the same to the extent that either is justified. Although the numbers are slightly different, it is clear that the probability of long-lived water molecules in the vicinity of the paramagnetic center is very low and that the predominant correlation times for the water-protein interaction in the vicinity of the paramagnetic center are very short, in the range of tens of picoseconds.
The conclusion that the water molecule dynamics at the protein interface is dominated by rapid motions is not new (Denisov et al., 1995, 1999; Bryant, 1996; Denisov and Halle, 1996; Halle et al., 1999). However, the present data represents a measurement that spans the frequency range to times <1 ps and provides a reasonable approximation to a map of the water lifetime distribution on this region of the protein interface. The distribution is heavily weighted by correlation times in the vicinity of 10 ps, which is essentially what has been measured for any solute in water. If this region of the protein surface is representative, water molecule dynamics at the protein interface cannot pose a dynamical barrier to protein conformational rearrangements that may be important for function.
SUMMARY
The magnetic relaxation dispersion measurements made at high resolution and utilizing an exchange dilution protocol provide a means for defining specific intramolecular vectors in macromolecular systems and mapping the spectral density profiles that characterize the relative motions of the two positions labeled in space. In the present case, utilizing an organic radical characterized by a long electron spin-lattice relaxation time provides a direct approach to characterizing internal dynamics ranging between the rotational correlation time of the protein and ∼1 ps. Two high frequency events were identified by acetate and formate ions on hemoglobin in the proximity of β-93. One is a 2-ns exchange event between the ion and the protein surface. The other is an internal local motion of the electron-nuclear vector defined by the relative positions of the nitroxide and the observed nuclear spin of the anion. This motion is characterized by a 50-ps correlation time that originates from random, not periodic, fluctuations. The residual water protons interacting with the protein report a distribution of correlation times that are dominated by events on the order of 10 ps.
References
- Acharya, S. A., A. Malavalli, E. Peterson, P. D. Sun, C. Ho, M. Prabhakaran, A. Arnone, B. N. Manjula, and J. M. Friedman. 2003. Probing the conformation of hemoglobin Presbyterian in the R-state. J. Protein Chem. 22:221–230. [DOI] [PubMed] [Google Scholar]
- Ayant, Y., E. Belorizky, J. Alizon, and J. Gallice. 1975. Calculation of spectral density resulting from random translational movement with relaxation by magnetic dipolar interaction in liquids. J. Phys. I. 36:991–1004. [Google Scholar]
- Beernink, P. T., J. A. Endrizzi, T. Alber, and H. K. Schachman. 1999. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc. Natl. Acad. Sci. USA. 96:5388–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloembergen, N., and L. O. Morgan. 1961. Proton relaxation times in paramagnetic solutions. Effects of electron spin relaxation. J. Chem. Phys. 34:842–850. [Google Scholar]
- Bourgeois, D., B. Vallone, F. Schotte, A. Arcovito, A. E. Miele, G. Sciara, M. Wulff, P. Anfinrud, and M. Brunori. 2003. Complex landscape of protein structural dynamics unveiled by nanosecond Laue crystallography. Proc. Natl. Acad. Sci. USA. 100:8704–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, R. G. 1996. The dynamics of water-protein interactions. Annu. Rev. Biophys. Biomol. Struct. 25:29–53. [DOI] [PubMed] [Google Scholar]
- Chan, N.-L., J. S. Kavanaugh, P. H. Rogers, and A. Arnone. 2004. Crystallographic analysis of the interaction of nitric oxide with quaternary-T human hemoglobin. Biochemistry. 43:118–132. [DOI] [PubMed] [Google Scholar]
- Chiancone, E., J. E. Norne, S. Forsen, J. Bonaventura, M. Brunori, E. Antonini, and J. Wyman. 1975a. Identification of chloride-binding sites in hemoglobin by nuclear-magnetic-resonance quadrupole-relaxation studies of hemoglobin digests. Eur. J. Biochem. 55:385–390. [DOI] [PubMed] [Google Scholar]
- Chiancone, E., J. E. Norne, S. Forsen, M. Brunori, and E. Antonini. 1975b. Nuclear magnetic resonance quadrupole relaxation studies of chloride binding to the isolated hemoglobins from trout (Salmo irideus). Biophys. Chem. 3:56–65. [DOI] [PubMed] [Google Scholar]
- Chiancone, E., J. E. Norne, S. Forsen, A. Mansouri, and K. H. Winterhalter. 1976. Anion binding to proteins. NMR quadrupole relaxation study of chloride binding to various human hemoglobins. FEBS Lett. 63:309–312. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., and B. Halle. 1996. Protein hydration dynamics in aqueous solution. Faraday Discuss. 103:227–244. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., B. Halle, J. Peters, and H. D. Horlein. 1995. Residence times of buried water molecules in bovine pancreatic trypsin inhibitor and its G36S mutant. Biochemistry. 34:9046–9051. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., B. H. Jonsson, and B. Halle. 1999. Hydration of denatured and molten globule proteins. Nature Struct. Biol. 6:253–260. [DOI] [PubMed] [Google Scholar]
- Fermi, G., M. F. Perutz, B. Shaanan, and R. Fourme. 1984. The crystal structure of human deoxyhaemoglobin at 1.74 Å resolution. J. Mol. Biol. 175:159–174. [DOI] [PubMed] [Google Scholar]
- Freed, J. H. 1978. Dynamic effects of correlation functions on spin relaxation by diffusion in liquids. II. Finite jumps and independent T1 processes. J. Chem. Phys. 94:2843–2847. [Google Scholar]
- Halle, B. 2003. Cross-relaxation between macromolecular and solvent spins: the role of long-range dipole couplings. J. Chem. Phys. 119:12372–12385. [Google Scholar]
- Halle, B., V. P. Denisov, and K. Venu. 1999. Multinuclear relaxation dispersion studies of protein hydration. In Biological Magnetic Resonance. N.R. Krishna, editor. Kluwer Academic/Plenum, New York. 419–484.
- Hodges, M. W., D. S. Cafiso, C. F. Polnaszek, C. C. Lester, and R. G. Bryant. 1997. Water translational motion at the bilayer interface: an NMR relaxation dispersion measurement. Biophys. J. 73:2575–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell, W. L., H. S. McHaourab, C. Altenbach, and M. A. Lietzow. 1996. Watching proteins move using site-directed spin labeling. Structure. 4:779–783. [DOI] [PubMed] [Google Scholar]
- Hwang, L.-P., and J. H. Freed. 1975. Dynamic effects of pair correlation functions on spin relaxation by diffusion in liquids. J. Chem. Phys. 63:4017–4025. [Google Scholar]
- Kiihne, S., and R. G. Bryant. 2000. Protein-bound water molecule counting by resolution of SUP 1/H spin-lattice relaxation mechanisms. Biophys. J. 78:2163–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, S. H., and W. E. Schillinger. 1969a. Nuclear magnetic relaxation dispersion in protein solutions. I. Apotransferrin. J. Biol. Chem. 244:3283–3289. [PubMed] [Google Scholar]
- Koenig, S. H., and W. E. Schillinger. 1969b. Nuclear magnetic relaxation dispersion in protein solutions. II. Transferrin. J. Biol. Chem. 244:6520–6526. [PubMed] [Google Scholar]
- Koutsoupakis, C., T. Soulimane, and C. Varotsis. 2003. Ligand binding in a docking site of cytochrome c oxidase: a time-resolved step-scan Fourier transform infrared study. J. Am. Chem. Soc. 125:14728–14732. [DOI] [PubMed] [Google Scholar]
- Lipari, G., and A. Szabo. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104:4546–4559. [Google Scholar]
- Lipari, G., and A. Szabo. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104:4559–4570. [Google Scholar]
- Lukin, J. A., G. Kontaxis, V. Simplaceanu, Y. Yuan, A. Bax, and C. Ho. 2003. Quaternary structure of hemoglobin in solution. Proc. Natl. Acad. Sci. USA. 100:517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modig, K., E. Liepinsh, G. Otting, and B. Halle. 2004. Dynamics of protein and peptide hydration. J. Am. Chem. Soc. 126:102–114. [DOI] [PubMed] [Google Scholar]
- Mouawad, L., D. Perahia, C. H. Robert, and C. Guilbert. 2002. New insights into the allosteric mechanism of human hemoglobin from molecular dynamics simulations. Biophys. J. 82:3224–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueser, T. C., P. H. Rogers, and A. Arnone. 2000. Interface sliding as illustrated by the multiple quaternary structures of liganded hemoglobin. Biochemistry. 39:15353–15364. [DOI] [PubMed] [Google Scholar]
- Norne, J. E., E. Chiancone, S. Forsen, E. Antonini, and J. Wyman. 1978. 35cl NMR study of the release of chloride on oxygen binding to human hemoglobin. FEBS Lett. 94:410–412. [DOI] [PubMed] [Google Scholar]
- Ogawa, S., and H. M. McConnell. 1967. Spin-label study of hemoglobin conformations in solution. Proc. Nat. Acad. Sci. USA. 58:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz, M. F. 1970. Stereochemistry of cooperative effects in hemoglobin. Nature. 228:726–739. [DOI] [PubMed] [Google Scholar]
- Perutz, M. F., A. J. Wilkinson, M. Paoli, and G. G. Dodson. 1998. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu. Rev. Biophys. Biomol. Struct. 27:1–34. [DOI] [PubMed] [Google Scholar]
- Polnaszek, C. F., and R. G. Bryant. 1984. Nitroxide radical induced solvent proton relaxation: measurement of localized translational diffusion. J. Chem. Phys. 81:4038–4045. [Google Scholar]
- Polnaszek, C. F., D. Hanggi, P. W. Carr, and R. G. Bryant. 1987. Nuclear magnetic relaxation dispersion measurement of water mobility at a silica surface. Anal. Chem. Acta. 194:311–316. [Google Scholar]
- Redfield, A. G. 2003. Shuttling device for high-resolution measurements of relaxation and related phenomena in solution at low field, using a shared commercial 500 mHz NMR instrument. Magn. Res. Chem. 41:753–768. [Google Scholar]
- Roberts, M. F., Q. Cui, C. J. Turner, D. A. Case, and A. G. Redfield. 2004. High-resolution field-cycling NMR studies of a DNA octamer as a probe of phosphodiester dynamics and comparison with computer simulation. Biochemistry. 43:3637–3650. [DOI] [PubMed] [Google Scholar]
- Silva, M. M., P. H. Rogers, and A. Arnone. 1992. A third quaternary structure of human hemoglobin A at 1.7-Å resolution. J. Biol. Chem. 267:17248–17256. [PubMed] [Google Scholar]
- Solomon, I. 1955. Relaxation processes in a system of two spins. Phys. Rev. 99:559–565. [Google Scholar]
- Tsai, C.-H., V. Simplaceanu, N. T. Ho, T.-J. Shen, D. Wang, T. G. Spiro, and C. Ho. 2003. Site mutations disrupt interhelical H-bonds (A14W·A67T and B15W·B72S) involved in kinetic steps in the hemoglobin R to T transition without altering the free energies of oxygenation. Biophys. Chem. 100:131–142. [DOI] [PubMed] [Google Scholar]
- Tugarinov, V., Z. Liang, Y. E. Shapiro, J. H. Freed, and E. Meirovitch. 2001. A structural mode-coupling approach to 15N NMR relaxation in proteins. J. Am. Chem. Soc. 123:3055–3063. [DOI] [PubMed] [Google Scholar]
- Van-Quynh, A., S. Willson, and R. G. Bryant. 2003. Protein reorientation and bound water molecules measured by 1H magnetic spin-lattice relaxation. Biophys. J. 84:558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman, B. F., D. Lipson, D. E. Wemmer, and D. Kern. 2001. Two-state allosteric behavior in a single-domain signaling protein. Science. 291:2429–2433. [DOI] [PubMed] [Google Scholar]
- Wagner, S., T. R. J. Dinesen, T. Rayner, and R. G. Bryant. 1999. High-resolution magnetic relaxation dispersion measurements of solute spin probes using a dual-magnet system. J. Magn. Reson. 140:172–178. [DOI] [PubMed] [Google Scholar]
- Wuthrich, K., G. Otting, and E. Liepinsh. 1992. Protein hydration in aqueous solution. Faraday Discuss. 93:35–45. [DOI] [PubMed] [Google Scholar]
- Yuan, Y., V. Simplaceanu, J. A. Lukin, and C. Ho. 2002. NMR investigation of the dynamics of tryptophan side-chains in hemoglobins. J. Mol. Biol. 321:863–878. [DOI] [PubMed] [Google Scholar]