

Abstract
Escherichia coli multidrug resistance protein E (EmrE) is an integral membrane protein spanning the inner membrane of Escherichia coli that is responsible for this organism's resistance to a variety of lipophilic cations such as quaternary ammonium compounds (QACs) and interchelating dyes. EmrE is a 12-kDa protein of four transmembrane helices considered to be functional as a multimer. It is an efflux transporter that can bind and transport cytoplasmic QACs into the periplasm using the energy of the proton gradient across the inner membrane. Isothermal titration calorimetry provides information about the stoichiometry and thermodynamic properties of protein-ligand interactions, and can be used to monitor the binding of QACs to EmrE in different membrane mimetic environments. In this study the ligand binding to EmrE solubilized in dodecyl maltoside, sodium dodecyl sulfate and reconstituted into small unilamellar vesicles is examined by isothermal titration calorimetry. The binding stoichiometry of EmrE to drug was found to be 1:1, demonstrating that oligomerization of EmrE is not necessary for binding to drug. The binding of EmrE to drug was observed with the dissociation constant (KD) in the micromolar range for each of the drugs in any of the membrane mimetic environments. Thermodynamic properties demonstrated this interaction to be enthalpy-driven with similar enthalpies of 8–12 kcal/mol for each of the drugs in any of the membrane mimetics.
INTRODUCTION
Multidrug transporters recognize and extrude a wide range of toxic compounds from the cell. These transporters have been divided into four major families based on sequence and substrate specificity: the major facilitator superfamily, ATP-binding cassette family, resistance/nodulation/cell division family, and the small multidrug resistance (SMR) family (Paulsen et al., 1996; Putman et al., 2000). The SMR family can be divided further into two categories: small multidrug efflux proteins and Sug proteins (Edwards and Turner, 1998; Paulsen et al., 1996). Members of the SMR family have ∼110 residues and are the smallest known functional unit of multidrug resistance. Studies have shown SMR transporters to be composed of a four-helix antiparallel bundle (Arkin et al., 1996; Edwards and Turner, 1998; Mordoch et al., 1999; Paulsen et al., 1995; Schwaiger et al., 1998).
Ethidium multidrug resistance protein (EmrE) is an SMR protein found in Escherichia coli, which extrudes lipophilic cations such as quaternary ammonium compounds from the cell resulting in bacterial resistance to these toxic compounds (Paulsen et al., 1996; Schuldiner et al., 1997; Yerushalmi and Schuldiner, 2000a,b). Using the energy of the proton gradient across the inner membrane of E. coli, EmrE transports two protons across the membrane into the cytoplasm while transporting drug from the cytoplasm into the periplasm, antiport to the proton translocation (Fig. 1) (Glaubitz et al., 2000; Lebendiker and Schuldiner, 1996; Muth and Schuldiner, 2000; Yerushalmi and Schuldiner, 2000a,b; Yerushalmi et al., 2001). The oligomerization of EmrE subunits has been suggested to be necessary for this transport to provide multiple Glu14 residues for both drug and proton binding (Koteiche et al., 2003; Ma and Chang, 2004). Whether or not the drug partitioning into the membrane is necessary to access the binding site of EmrE is still unknown.
FIGURE 1.

EmrE uses energy from the proton gradient across the inner membrane of Escherichia coli to efflux quaternary ammonium compounds from the cytoplasm to periplasm as shown. Extramembrane (EM) loops and N- and C-termini are labeled for clarity. The topology was established by Son et al. (2003).
The affinities of various lipophilic cations to EmrE in different membrane mimetics were examined using isothermal titration calorimetry (ITC) to determine the energetics and stoichiometry involved.
EXPERIMENTAL PROCEDURES
Purification of EmrE
EmrE protein was expressed from E. coli strain LE392Δunc containing the expression plasmid pMS119EH with the cloned emrE gene positioned behind a tac-promoter. Cells were grown to a density of 0.5 (A600) in 1 L Terrific Broth inoculated at 37°C and IPTG was added to a final concentration of 0.1 mM. The cells were then incubated for 3 h, harvested by centrifugation, and thoroughly washed with SMR A buffer (50 mM MOPS, 8% glycerol, 5 mM EDTA, 1 mM DTT, pH 7). Cells were resuspended in SMR A buffer with 100 μM PMSF and lysed by two passes through a French press (16,000 psi). A low speed centrifugation (9000 × g for 15 min) removed the heavy cellular constituents and unlysed cells, followed by a high-speed spin (110,000 × g for 1.5 h) to collect the membrane fraction. The membranes were resuspended in SMR A buffer and diluted to a final protein concentration of 10 mg/mL.
EmrE can be purified from membranes using organic solvent extraction as described previously (Winstone et al., 2002). Ten milliliters of the membrane fraction was extracted with 300 mL of 3:1 chloroform/methanol (CM). Fifty milliliters of double-distilled water was added to separate the water-soluble constituents from the organic phase. The organic phase was collected and briefly centrifuged to ensure complete phase separation of the aqueous layer. The organic phase was then evaporated below 6 mL using a rotovap. The protein was further purified and separated from any extracted lipid using Sephadex LH-20 hydrophobic chromatography (Sephadex, Amersham, Piscataway, NJ) with an isocratic elution in 1:1 CM solvent using an Aktä purifier (Aktä, Amersham). Solvent from the purified EmrE was removed under N2 gas and the dried protein was stored at −70°C until use. Protein purified in this fashion is easily reconstituted into small unilamellar vesicles (SUVs) and demonstrates transport activity (Winstone et al., 2002).
Solubilization of EmrE in detergent
Sodium dodecyl sulfate (SDS) (from BioRad, Mississauga, Ontario, Canada), and N-dodecyl-β-D-maltoside (DM) (Anatrace, Maumee, OH) were found to solubilize this protein at useful levels. Stock detergent solutions were made with 8% w/v SDS detergent in SMR B buffer (5 mM MOPS, 10 mM NaCl, 10 μM dithiothreitol (DTT), pH 7) and 2% w/v DM in SMR B buffer. Each tube of dried EmrE was exposed to 300 μL of the desired detergent solution. The EmrE suspension was vortexed for 2 h at room temperature followed by an overnight freeze-thaw cycle at −20°C. The sample was then centrifuged at 14,000 × g for 10 min to remove any insolubles. The pellet was discarded and the protein concentration of the supernatant was determined by a modified Lowry assay (Peterson, 1977), and diluted with its respective detergent solution in SMR B buffer so that the EmrE concentration was 2 mg/ml. This sample was stored at −70°C for later use.
Reconstitution of EmrE into small unilamellar vesicles
A tube of dried EmrE was resuspended in 110 μL of 3:1 CM. Of the 110 μL, 10 μL was set aside and dried under N2 gas. The dry pellet was resuspended in SDS solution as previously described and a modified Lowry assay (Peterson, 1977) was carried out on this sample to determine the protein content in the remaining 100 μL of EmrE solution. Two milligrams of EmrE was removed from this suspension, added to 1.5 mL of 25 mg/ml E. coli polar lipid extract (from Avanti, Alabaster, AL), and dried under N2 gas. One milliliter of SMR C buffer (0.5 mM MOPS, pH 7) was added to the dried pellet. The sample was vortexed for 20 min at room temperature to resuspend the pellet. Five freeze-thaw cycles were carried out on this sample at −70°C. Three cycles of sonication were carried out on the sample for 3 s each at 25% power (5 μA amplitude), and stored at −70°C for later use. SUVs were also constructed similarly in the absence of EmrE.
ITC calorimetry of EmrE
Prepared samples of EmrE in detergent or SUV solutions were thawed. To exchange into a buffer lacking salt and reducing agent, 0.5 mL of EmrE sample and 1.5 mL of SMR C buffer (0.5 mM MOPS, pH 7) with detergent or SUVs was loaded on a 5-mL Hi Trap Desalting Column (Phamacia, Amersham). The column had previously been equilibrated with six column volumes of SMR C buffer before loading of EmrE. The column was eluted with SMR C buffer, and the first 2 mL of elution containing the EmrE protein was collected. The eluted EmrE was diluted to a final concentration of 0.480 mg/ml (40 μM), and degassed in a thermovac at room temperature for 5 min. The degassed sample was injected into the sample cell of a MicroCal ITC calorimeter (MicroCal, Origin, Northampton, MA). Calorimetry trials were carried out at 25°C.
Ligand consisting of ethidium (Et), proflavin (Pro), cetylpyridinium (CTPC), methyl viologen (MV), or tetraphenylphosphonium (TPP) solubilized in SMR B buffer containing the same concentration and type of lipid (SDS, DM, or SUV) used in the protein sample was prepared as the titrant. Sixty injections of this titrant containing either 0.5 mM ligand for strong interactions or 2.0 mM ligand for weaker interactions were injected into the ITC sample cell.
Injections occurred at intervals of 240 s, and the duration time of each injection was 8 s. Heat transfer (μcal/s) was measured as a function of elapsed time (seconds). Heats of dilution were subtracted from the heats collected in the corresponding experiments. Independent preparations of EmrE were used in this experiment to verify that binding trends were consistent.
Calorimetry trials were also carried out in the absence of EmrE in the same experimental conditions as described above. No change in heat released was observed in the injections throughout the experiment.
ITC of drug binding to membrane mimetics
Each mimetic and drug was tested for binding. The titrant was made up of 40 mM drug in SMR B buffer (lacking detergent or SUVs), whereas the sample cell contained either 4 mM detergent in SMR B buffer, or SUVs in SMR B buffer. The SUVs were prepared as described previously, with the exception of 0.70 ml of 25 mg/ml E. coli polar lipid extract being used instead. Preparation of samples and calorimetry was carried out as previously described.
Thermodynamic calculations
From analysis of each generated thermogram, the heat of dilution (hdil) was used to determine the heat of reaction (δhi) for each injection by the equation
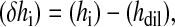 |
(1) |
where hi is the heat released at a given injection calculated by the integration of the respective injection peak. The integration of an injection peak was used to calculate hi. The reaction enthalpy (ΔH) can be calculated from the equation
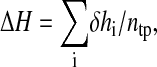 |
(2) |
where ntp is the total moles of protein in the cell. Plotting ΔH against the molecular ligand/protein ratio produces a binding isotherm.
Nonlinear regression fitting to the binding isotherm (ORIGIN software, MicroCal) gave the equilibrium dissociation constant of the ligand KD. From the value of KD, the free energy of binding (ΔG°) and entropy of binding (ΔS) can be calculated from the following relationships,
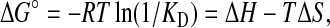 |
(3) |
where T is 273 K and R is 1.9872 cal/K per mol.
RESULTS
Isothermal calorimetry was employed to study the binding of lipophilic cations to EmrE. In the experiments, the ligand was in large excess over the protein which allowed the binding reaction to be driven near completion. However, before heats of binding could be detected at a sufficient signal/noise ratio, extensive exploration of experimental conditions was carried out.
For an acceptable signal/noise ratio and consistent baseline to be generated for analysis, many parameters of the procedure had to be tested and several optimization trials needed to be carried out. Preliminary trials (data not shown) consistently had too low a signal to be analyzed. A range of protein concentration from 0.05 to 0.5 mg/ml had been tested. Until the protein concentration was increased to a suitable level, little or no signal was detected. The ratio of the molecules of EmrE to the molecules of ligand introduced per injection had also been tested in the range of 0.1:10. It was found that a ratio in the range of 10:4 was suitable for measuring the heats evolved from EmrE binding to any of the substrates in any of the membrane mimetics. A modified solubilization protocol to achieve this concentration was employed as described above, after various parameters and methods of resolubilizing the dried protein had been tested. Such parameters included intensities, time intervals, and cycles of sonication; time and temperature of vortexing during resuspension; and concentrations of detergent or E. coli polar lipid extract present during resuspension. Much experimentation with these parameters had to be carried out before a suitable signal/noise ratio was achieved. The protocol described in the methods above consistently yields an EmrE concentration >2 mg/ml (166 μM) in each of the mimetic environments, which is necessary for a sufficient signal/noise ratio. It was also discovered that concentrations of NaCl and reducing agent DTT could be successfully removed or decreased in the EmrE samples using a size exclusion column for buffer exchange without affecting solubilization or functionality of the protein in the short term of the experiment. Both components add noise to the signal, especially DTT, which undergoes oxidation throughout the experiment. It was not until parameters of protein, NaCl, and DTT concentration were optimized that a sufficient signal was achieved.
The heats of binding are measured in a thermogram Fig. 2 A which demonstrates a titration of 30 injections containing 2 mM of ethidium into a solution of 40 μM EmrE reconstituted into SUVs. Each peak represents an injection of drug. Negative deflections from the baseline upon addition of drug indicate that heat was evolved during binding (an exothermic ligand binding event).
FIGURE 2.
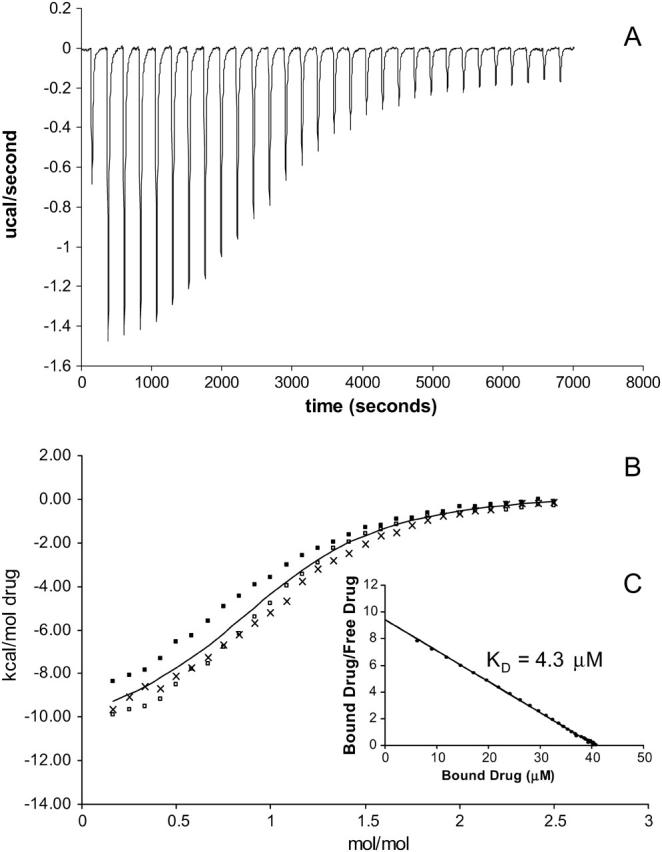
Representative titration calorimetry of EmrE in SUVs with ethidium. (A) Each peak corresponds to the injection of 8 μl of 0.5 mM ethidium in SUVs into the reaction cell containing 40 μM EmrE in SUVs. The concentration of E. coli polar lipid that formed SUVs in this experiment was 37.5 mg/ml. (B) Cumulative heat of reaction is displayed as a function of the injection number. The solid line is the least-squares fit to the experimental data of separate trials (indicated by symbols ×, ▪, and □). It corresponds with a KD of 5.5 μM. (C) Linearization of the data in a single trial in a Scatchard plot as an alternative way of measuring the KD.
Binding isotherms derived from multiple thermograms of ethidium-EmrE binding in SUVs are displayed in Fig. 2 B with a curve of best fit through the compiled data points. The stoichiometry and values of KD extracted from these curves are summarized in Table 1. Independent preparations of EmrE make up this data and demonstrate the reproducibility of the experiment.
TABLE 1.
Dissociation constants and thermodynamic data for binding of drug to EmrE in various mimetic environments at 25°C
| Environment | Drug | KD (μM) | ΔG (kcal/mol) | ΔH (kcal/mol) | ΔS (kcal/mol per K) |
|---|---|---|---|---|---|
| SUV | Ethidium | 5.5 ± 2.1 | −7.2 ± 0.2 | −10.7 ± 0.1 | −11.8 ± 1.0 |
| Methyl viologen | 38.2 ± 8.7 | −6.1 ± 0.1 | −7.5 ± 0.7 | −4.9 ± 2.7 | |
| Proflavin | 10.7 ± 2.8 | −6.8 ± 0.2 | −8.9 ± 0.2 | −7.1 ± 1.0 | |
| TPP | Could not be determined | ||||
| SDS | Ethidium | 5.2 ± 1.4 | −7.2 ± 0.2 | −11.4 ± 0.1 | −14.0 ± 0.6 |
| Methyl viologen | 5.4 ± 1.2 | −7.2 ± 0.1 | −9.7 ± 0.2 | −8.4 ± 0.6 | |
| Proflavin | 4.5 ± 0.8 | −7.3 ± 0.1 | −10.6 ± 0.2 | −11.1 ± 0.9 | |
| TPP | 4.8 ± 0.8 | −7.3 ± 0.1 | −12.1 ± 0.1 | −16.0 ± 0.5 | |
| DM | Ethidium | 6.3 ± 1.0 | −7.1 ± 0.1 | −10.9 ± 0.1 | −12.7 ± 0.2 |
| Methyl viologen | 46.2 ± 10.5 | −5.9 ± 0.1 | −7.7 ± 1.6 | −7.1 ± 4.7 | |
| Proflavin | 5.2 ± 0.9 | −7.2 ± 0.1 | −9.6 ± 0.1 | −7.8 ± 0.5 | |
| TPP | 25.5 ± 6.2 | −6.3 ± 0.1 | −9.9 ± 0.5 | −12.2 ± 2.2 | |
Each value represents three separate trials.
The values of KD were in the micromolar range which is similar to the dissociation constants reported for other multidrug-resistant proteins to these drugs (Lewinson and Bibi, 2001; Marham et al., 1996; Vázquez-Laslop et al., 1999). In comparing affinities of the various drugs to EmrE reconstituted in SUVs and in DM environments, the ethidium and proflavin ligands gave rise to stronger binding than tetraphenylphosphonium and methyl viologen in which binding was weaker and even undetected for tetraphenylphosphonium in the SUV mimetic (Table 1). The shift from a more sigmoidal plot of the binding isotherm in the case of ethidium (Fig. 2 B) toward a more hyperbolic plot in the case of methyl viologen (Fig. 3 B) demonstrates stronger versus weaker binding, respectively. In the SDS mimetic, each of the ligands bound with similar affinities (Table 1). In the case of using cetylpyridinium (CTPC) ligand, binding was not detected in any given membrane mimetic.
FIGURE 3.
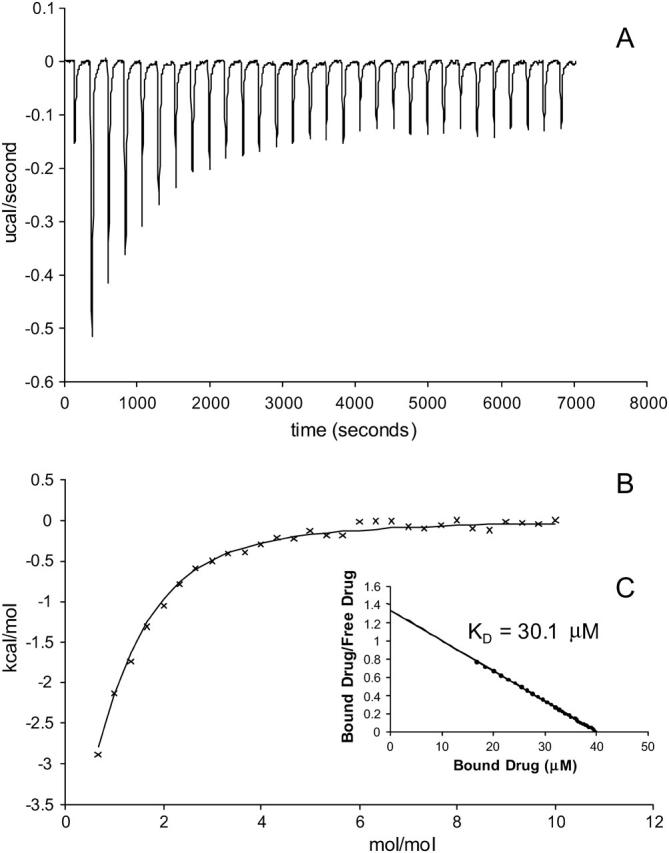
Representative titration calorimetry of EmrE in SUVs with methyl viologen. (A) Each peak corresponds to the injection of 8 μl of 0.500 mM methyl viologen in SUVs into the reaction cell containing 40 μM EmrE in SUVs. The concentration of E. coli polar lipid that formed SUVs in this experiment was 37.5 mg/ml. (B) Cumulative heat of reaction is displayed as a function of the injection number. The solid line is the least-squares fit to the experimental data of separate trials (indicated by symbols ×, ▪, and □). It corresponds with a KD of 38.2 μM. (C) Linearization of the data in a single trial in a Scatchard plot as an alternative way of measuring the KD.
In regard to the thermodynamic data, there did not appear to be any trends in the enthalpy or entropy of the reaction among the drugs in any given environment (Table 1). The inflection point of the isothermograms occurred at ∼1:1 (mol/mol) of drug to protein, indicating a stoichiometry of one molecule of drug bound to one molecule of protein.
Control experiments were performed in which each ligand in each of the membrane mimetics was injected into the ITC sample cell containing the same membrane mimetic in the absence of EmrE. These trials were carried out using the same concentrations and injection volumes as carried out previously in the presence of protein. Heat released during these trials was constant for each injection and could only be observed when higher volumes of injectant were used. It was measured that each injection contributed <0.9 μcal of heat for each ligand and membrane mimetic, well below the heat released upon drug-EmrE binding. These experiments demonstrated that the exothermic heats of drug partitioning from its membrane mimetic in the syringe into a greater amount of the membrane mimetic in the sample cell were negligible in contrast to drug-protein binding and could be disregarded.
To explore drug-lipid partitioning interactions, drug in the absence of each membrane mimetic was titrated into each of the membrane mimetic solutions. A sample thermogram of EtBr partitioning into DM is illustrated in Fig. 4. The interaction was too weak to determine the stoichiometry of drug binding to membrane mimetic and the confidence in the values is less because of the weaker binding, but the relative magnitude between experiments is still meaningful. It was observed that lipid partitioning of each drug into any of the membrane mimetics displayed similar weak binding (Table 2). The exception was CTPC, which bound to SUVs and SDS with dissociation constants in the high nanomolar range, whereas binding was undetected in DM. There were significant enthalpic and entropic contributions in the case of CTPC binding to membrane mimetic. In binding CTPC to SUVs, ΔG was observed to be −8.3 kcal/mol with ΔH and ΔS at −5.3 kcal/mol and 10.1 cal/mol K, respectively. In binding CTPC to SDS, ΔG was observed to be −9.1 kcal/mol with ΔH and ΔS at −6.4 kcal/mol and 8.9 cal/mol K, respectively.
FIGURE 4.
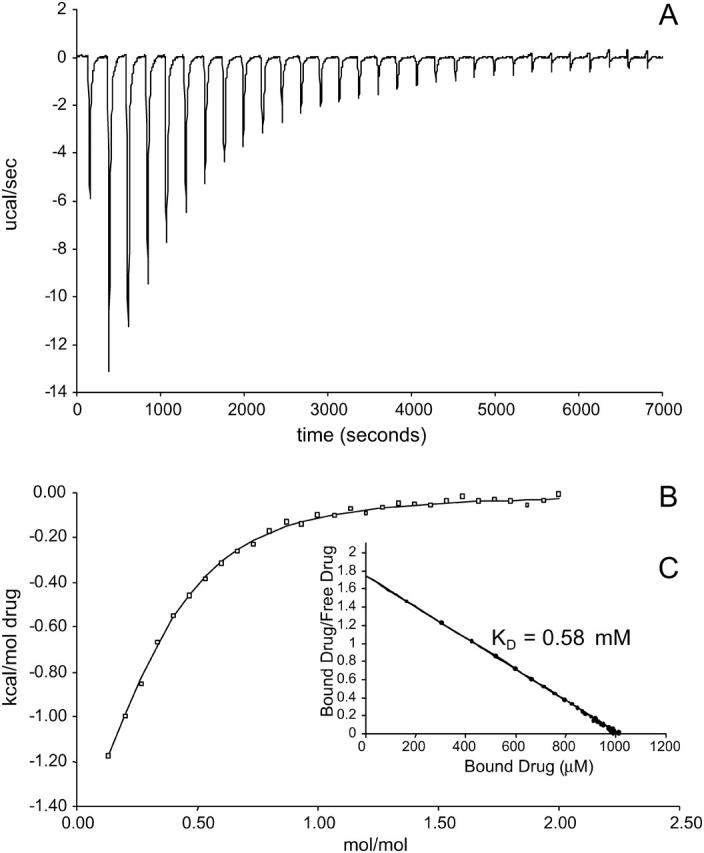
(A) Thermogram of EtBr binding to SUVs with a KD of 0.58 mmol. Each peak corresponds to the injection of 8 μl of 40 mM EtBr solution into the reaction cell containing SUVs. The concentration of E. coli polar lipid that formed SUVs in this experiment was 17.5 mg/ml. (B) Cumulative heat of reaction is displayed as a function of the injection number. The solid line is the least-squares fit to the experimental data points. It corresponds with a KD of 0.58 mM. (C) Linearization of the data in a single trial in a Scatchard plot as an alternative way of measuring the KD.
TABLE 2.
Binding constants for binding of drug to various mimetic environments at 25°C
| Environment | Drug | KD (mM) | ΔG (kcal/mol) | ΔH (kcal/mol) | ΔS (cal/mol per K) |
|---|---|---|---|---|---|
| SUV | Ethidium | 0.6 | −4.4 | −2.1 | 7.8 |
| Methyl viologen | 4.1 | −3.3 | −2.3 | 3.1 | |
| Proflavin | 2.1 | −3.7 | −2.0 | 5.6 | |
| TPP | 0.2 | −4.9 | −2.7 | 7.6 | |
| CTPC | 8.4 × 10−4 | −8.3 | −5.3 | 10.1 | |
| SDS | Ethidium | 0.3 | −4.9 | −2.6 | 7.7 |
| Methyl viologen | 1.8 | −3.8 | −2.6 | 3.8 | |
| Proflavin | 0.4 | −4.7 | −2.4 | 7.9 | |
| TPP | 0.4 | −4.6 | −2.0 | 8.7 | |
| CTPC | 2.4 × 10−4 | −9.1 | −7.4 | 5.6 | |
| DM | Ethidium | 5.2 | −3.1 | −1.5 | 5.4 |
| Methyl viologen | 11.8 | −2.6 | −1.0 | 5.4 | |
| Proflavin | 17.1 | −2.4 | −1.6 | 2.8 | |
| TPP | 10.8 | −2.7 | −1.3 | 4.8 | |
| CTPC | Could not be determined | ||||
The values in this table represents data from a single trial of three runs.
The concentration of detergents used were well above the critical micelle concentrations for SDS and DM. Aggregation numbers of 62–101 for SDS (Anatrace measurement) and 78–149 for DM (VanAken et al., 1986, Anatrace measurement) were used to determine the ratio of micelles/EmrE subunits. This ratio is on an order of 40 in SDS, and 4 in DM.
DISCUSSION
Studies of multidrug-resistant proteins have demonstrated two types of residues involved in binding to quaternary ammonium compounds. Structures of the compounds are shown in Fig. 5. The first essential residue is a negatively-charged acidic residue for binding to the positive charge of the ligand (Edgar and Bibi, 1999; Ekaterina et al., 2001; Muth and Schuldiner, 2000; Paulsen et al., 1996). This residue is highly conserved among multidrug resistant proteins (Paulsen et al., 1996). The Glu14 residue of EmrE serves such a purpose (Muth and Schuldiner, 2000). The second type of residue involved in ligand binding are aromatic residues (Dougherty, 1996; Zhong et al., 1998). Crystallographic studies of proteins involved in multidrug resistance have shown aromatic residues involved in van der Waal and π-π interactions with the aromatic rings of the ligand, as well as π-interactions with the positive charge on the ligand (Ekaterina et al., 2001; Schumacher and Brennan, 2003). EmrE contains several aromatic residues which may assist in this interaction. Several aromatic residues are highly conserved among SMR proteins (Putman et al., 2000), and one or more of these residues may aid in binding to the drug. Mutation of Y40, Y53, F44, Y60, or W63 to a cysteine residue results in a non-expressed or less-functional protein (Mordoch et al., 1999; Yerushalmi and Schuldiner, 2000a,b). This suggests that one or more of these residues may be involved in assembly or protein-ligand interactions.
FIGURE 5.

Structures of lipophilic cations used in this study.
Ligand interactions with the membrane have also been considered. It has been observed with other similar quaternary ammonium compounds such as anthracyclines, which interact with multidrug resistant protein P-glycoprotein partition into the headgroup/acyl chain interface of the inner leaflet of the membrane (Gallois et al., 1998). It was also observed that different headgroups (charged/zwitterionic/neutral) and differently charged drugs (monovalent/divalent) did not significantly contribute to partitioning of drug into the lipid (Gallois et al., 1998), which reflects the data collected in Table 2. Rather it is the hydrophobicity of the drug that is the major contributor of membrane partitioning (Gallois et al., 1998). This would make the binding interaction entropically favorable as well, since the water forming a clathrate cage around the hydrophobic regions of the drug are released upon the drug's partitioning into the lipid (Butler et al., 2004). Our goal was to examine whether or not drug partitioning into the membrane mimetic occurs to account for the possibility that EmrE recruits drug from the membrane. Drug binding to membrane mimetic was evaluated to asses this issue. Each drug demonstrated the ability to partition into various membrane mimetics (Table 2). It should be noted that drug-membrane mimetic interactions (Table 2) are still considerably weaker than drug binding to EmrE (Table 1), which is still the more energetically favorable reaction.
Under investigation is whether EmrE binds drug from the inner leaflet of the membrane or if drug is accessible to EmrE via the cytoplasm. Multidrug resistant proteins Lmr, MexA-MexB-OmpR, QacA, and P-glycoprotein have demonstrated interaction with drug solubilized in the inner leaflet of the membrane (Bolhuis, 1996; Bolhuis et al., 1996; Edgar and Bibi, 1999; Homolya, 1993; Mitchell et al., 1998; Ocaktan et al., 1997; Shapiro and Ling, 1997). A mechanism of multidrug resistance is the flipase activity of MDR proteins to relocate drugs from the inner leaflet to the outer leaflet of the membrane. This study has provided evidence of each drug having the capability to partition into different membrane mimetics (Fig. 2), so it is possible that EmrE may bind to drug solubilized in the membrane much like what is proposed for other multidrug resistant proteins. Crystallographic studies have suggested accessibility of drug to EmrE from both the membrane and cytoplasm (Ubarretxena-Belandia et al., 2003). However, it is still uncertain in what direction the drug enters the binding site of EmrE.
In comparing the chemical structures of the various ligands in this experiment to the calorimetric results obtained, certain postulations may be made. Firstly, the SUV and DM environments do not seem to alter the binding of EmrE to ligand in any way with the exception of TPP. TPP binding to EmrE was not observable by the method of ITC in the SUV mimetic but could be detected in the DM mimetic. It was shown that TPP still bound to each of these membrane mimetics (Table 2). Ethidium (Et) and proflavin (Pro) ligands showed stronger binding than the tetraphenylphosphonium (TPP) and methyl viologen (MV) ligands. Steric hindrance from the phenyl rings on the TPP substrate may make it less accessible to the binding site of EmrE. Likewise, the second positive charge on MV may interfere with its accessibility to the EmrE binding site.
SDS is an environment that denatures many soluble proteins; however, many membrane proteins retain their structure and have been studied in this mimetic (van de Ven et al., 1993; Mortishire-Smith et al., 1995; Lee et al., 2003; Sulistijo et al., 2003). Circular dichroism and fluorescence studies performed in SDS, DM, and SUVs have shown that EmrE in each of these environments has a similar structure (Federkeil et al., 2003). EmrE is in a slightly more open conformation in SDS than DM and SUVs (Federkeil et al., 2003), which could make its binding site more accessible to drug. The TPP and MV show binding with an affinity similar to that of Et and Pro ligands in this environment, perhaps as a result of better accessibility to the binding site for these ligands. It should also be noted that since the structure of EmrE in the different mimetics is similar as assessed by CD and fluorescence, each of the mimetics without protein bind drug similarly with dissociation constants in the millimolar range (Table 2), and the binding of EmrE to drug is also similar in each of these different mimetics (Table 1).
The ITC method was able to detect CTPC binding to the membrane mimetics but not to EmrE. The presence of the acyl chain of this molecule would interact with the lipid more favorably. Partitioning out of the lipid and into the binding site of EmrE may be energetically unfavorable for this drug.
EmrE is a protein known to form homo-oligomers (Ma and Chang, 2004; Muth and Schuldiner, 2000; Tate et al., 2001; Torres and Arkin, 2000; Rotem et al., 2001; Ubarretxena-Belandia et al., 2003; Yerushalmi et al., 1996). The most recent x-ray crystallographic study has reported a tetrameric EmrE (Ma and Chang, 2004). The results here suggest that oligomerization is not necessary for EmrE binding to drug. The binding stoichiometry of the EmrE binding drug in our study was observed to be 1:1. In the environments of SDS and DM, EmrE has been shown to exist as a monomer when purified using the organic solvent extraction methodology (Winstone et al., 2005). Both environments display a binding stoichiometry of 1:1 to each of the drugs, indicating that the monomer by itself is capable of binding drug. The stoichiometry of drug-EmrE binding in SUVs which better approximates the membrane of E. coli was also found to be 1:1. Considering that the micelle/EmrE ratio is 40 in SDS and 4 in DM and that one EmrE subunit occupies one DM micelle (Winstone et al., 2005), it can be concluded that the EmrE monomer can bind substrate. However, it is still possible that oligomerization is necessary for transport of the drug across the membrane as the other EmrE subunits are required to bind protons necessary for the antiport of drug with protons (Muth and Schuldiner, 2000; Yerushalmi and Schuldiner, 2000a,b). Additionally, dimerization could further stabilize the binding providing a second-half of a binding site, and this has been observed in studies in which a multimer of EmrE was present in which TPP dissociation constants in the nanomolar range have been reported (Rotem et al., 2001; Tate et al., 2003). Whether oligomerization occurs before or after drug binding to EmrE has yet to be determined, although these experiments demonstrate that oligomerization is not necessary for binding to drug substrate.
Crosslinking and crystallographic studies have shown that oligomeric EmrE has two Glu14 residues in close proximity with one another (Koteiche et al., 2003; Ma and Chang, 2004; Ubarretxena-Belandia et al., 2003). Although the EmrE monomer is capable of binding drug as shown in this and other studies (K. A. Duncalf and R. J. Turner, unpublished), it has been postulated that the formation of a dimer better stabilizes its interaction (Butler et al., 2004) with drug, since Glu14 residues from both subunits serve to stabilize the positive charge on the drug (as opposed to just one in the monomeric form). This interaction must still be weak enough to accommodate drug release during transport. Studying EmrE purified in different ways and solubilized in different membrane mimetics has resulted in different oligomeric states (Elbaz et al., 2004; Ma and Chang, 2004; Tate et al., 2003; Ubarretxena-Belandia et al., 2003; Winstone et al., 2005). This could account for higher binding affinities for TPP observed in other publications in the nanomolar range in which a dimer or higher order of oligomerization may be present (Elbaz et al., 2004; Muth and Schuldiner, 2000; Tate et al., 2003).
In conclusion, this study of isothermal titration calorimetry has demonstrated the weak, nonspecific binding of EmrE to a variety of lipophilic cationic drugs. It has demonstrated that this interaction is similar in various membrane mimetics. Also the binding stoichiometry of drug to EmrE has been shown to be 1:1. Although the oligomer can accommodate the protons required for transport as discussed, oligomerization is not required for the binding event as demonstrated by this study. Using ITC, the oligomerization state and thermodynamics of EmrE binding to lipophilic cationic drugs in different membrane mimetics is now understood.
Acknowledgments
We thank Tara Winstone and Karen Duncalf for useful discussions. We also thank Aaron Yamniuk and Weiguo Jing of Dr. H. Vogel's laboratory for training on the ITC.
This work was supported by a grant to R.J.T. from the National Sciences and Engineering Research Council of Canada.
References
- Arkin, I. T., W. P. Russ, M. Lebendiker, and S. Schuldiner. 1996. Determining the secondary structure and orientation of EmrE, a multi-drug transporter, indicates a transmembrane four-helix bundle. Biochemistry. 35:7233–7238. [DOI] [PubMed] [Google Scholar]
- Bolhuis, H. 1996. Multidrug resistance in Lactococcus lactis: evidence for ATP-dependent drug extrusion from the inner leaflet of the cytoplasmic membrane. EMBO J. 15:4239–4245. [PMC free article] [PubMed] [Google Scholar]
- Bolhuis, H., H. W. van Veen, J. R. Brands, M. Putman, B. Poolman, A. J. M. Driessen, and W. N. Konings. 1996. Energetics and mechanism of drug transport mediated by the lactococcal multidrug transporter LmrP. J. Biol. Chem. 271:24123–24128. [DOI] [PubMed] [Google Scholar]
- Butler, P., I. Ubarretxena-Belandia, T. Warne, and C. Tate. 2004. The Escherichia coli multidrug transporter EmrE is a dimer in the detergent-solubilised state. J. Mol. Biol. 340:797–808. [DOI] [PubMed] [Google Scholar]
- Dougherty, D. A. 1996. Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 271:163–168. [DOI] [PubMed] [Google Scholar]
- Edwards, R. A., and R. J. Turner. 1998. Alpha-periodicity analysis of small multidrug resistance (SMR) efflux transporters. Biochem. Cell Biol. 76:791–797. [DOI] [PubMed] [Google Scholar]
- Edgar, R., and E. Bibi. 1999. MdfA, an Escherichia coli multidrug resistance protein with an extraordinary broad spectrum of drug recognition. EMBO J. 15:822–832. [Google Scholar]
- Ekaterina, E., Z. Heldwein, and R. Brennan. 2001. Crystal structure of the transcription activator BmrR bound to DNA and a drug. Nature. 409:378–382. [DOI] [PubMed] [Google Scholar]
- Elbaz, E., S. Steiner-Murdoch, T. Danieli, and S. Schuldiner. 2004. In vitro synthesis of fully functional EmrE, a multidrug transporter, and study of its oligomeric state. Proc. Natl. Acad. Sci. USA. 101:1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federkeil, S., T. Winstone, G. Jickling, and R. J. Turner. 2003. Examination of EmrE conformational differences in various membrane mimetic environments. Biochem. Cell Biol. 81:61–70. [DOI] [PubMed] [Google Scholar]
- Gallois, L., M. Fiallo, A. Laigle, W. Priebe, and A. Garnier-Suillerot. 1998. The overall partitioning of anthracyclines into phosphatidyl-containing model membranes depends neither on the drug charge nor the presence of anionic phospholipids. Eur. J. Biochem. 241:879–887. [DOI] [PubMed] [Google Scholar]
- Glaubitz, C., A. Gröger, K. Gottschalk, P. Spooner, A. Watts, S. Schuldiner, and H. Kessler. 2000. 31P-CP-MAS NMR studies on TPP+ bound to the ion-coupled multidrug transport protein EmrE. FEBS Lett. 480:127–131. [DOI] [PubMed] [Google Scholar]
- Homolya, L. 1993. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J. Biol. Chem. 268:21493–21496. [PubMed] [Google Scholar]
- Koteiche, H. A., M. D. Reeves, and H. S. McHaourab. 2003. Structure of the substrate binding pocket of the multidrug transporter EmrE: site-directed spin labeling of transmembrane segment 1. Biochemistry. 42:6099–6105. [DOI] [PubMed] [Google Scholar]
- Lebendiker, M., and S. Schuldiner. 1996. Identification of residues in the translocation pathway of EmrE, a multidrug antiporter from Escherichia coli. J. Biol. Chem. 271:21193–21199. [DOI] [PubMed] [Google Scholar]
- Lee, K., S. Shin, J. Hong, S. Yang, J. Kim, K. Hahm, and Y. Kim. 2003. Solution structure of termite-derived antimicrobial peptide, spinigerin, as determined in SDS micelle by NMR spectroscopy. Biochem. Biophys. Res. Commun. 309:591–597. [DOI] [PubMed] [Google Scholar]
- Lewinson, O., and E. Bibi. 2001. Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli multidrug transporter MdfA. Biochemistry. 40:12612–12618. [DOI] [PubMed] [Google Scholar]
- Ma, C., and G. Chang. 2004. Structure of the multidrug resistance efflux transporter EmrE from Escherichia coli. Proc. Natl. Acad. Sci. USA. 101:2852–2857. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Markham, P., M. Ahmed, and A. Neyfakh. 1996. The drug-binding activity of the multidrug-responding transcriptional regulator BmrR resides in its C-terminal domain. J. Bacteriol. 178:473–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, B. A., M. H. Brown, and R. A. Skurray. 1998. QacA multidrug efflux pump from Staphylococcus aureus: comparative analysis of resistance to diamidines, biguanidines, and guanylhydrazones. Antimicrob. Agents Chemother. 42:475–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordoch, S. S., D. Granot, M. Lebendiker, and S. Schuldiner. 1999. Scanning cysteine accessibility of EmrE, an H+-coupled multidrug transporter from Escherichia coli, reveals a hydrophobic pathway for solutes. J. Biol. Chem. 274:19480–19486. [DOI] [PubMed] [Google Scholar]
- Mortishire-Smith, R., S. Pitzenberger, C. Burke, C. Middaugh, V. Garsky, and R. Johnson. 1995. Solution structure of the cytoplasmic domain of phospholamban: phosphorylation leads to a local perturbation in secondary structure. Biochemistry. 34:7603–7613. [DOI] [PubMed] [Google Scholar]
- Muth, T. R., and S. Schuldiner. 2000. A membrane-embedded glutamate is required for ligand binding to the multidrug transporter EmrE. EMBO J. 19:234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocaktan, A., H. Yoneyama, and T. Nakae. 1997. Use of fluorescence probes to monitor function of the subunit proteins of the MexA-MexB-OprM drug extrusion machinery in Pseudomonas aeruginosa. J. Biol. Chem. 272:21964–21969. [DOI] [PubMed] [Google Scholar]
- Paulsen, I. T., M. H. Brown, S. J. Dunstan, and R. A. Skurray. 1995. Proton-dependent multidrug efflux systems. J. Bacteriol. 177:2827–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen, I. T., R. A. Skurry, R. Tam, M. H. Saier, Jr., R. J. Turner, J. H. Weiner, E. G. Goldberg, and L. L. Grinius. 1996. The SMR family: a novel family of multidrug efflux proteins involved with the efflux of lipophilic drugs. Mol. Microbiol. 19:1167–1175. [DOI] [PubMed] [Google Scholar]
- Peterson, G. L. 1977. A simplification of the protein assay method of Lowry et al., which is more generally applicable. Anal. Biochem. 83:346–356. [DOI] [PubMed] [Google Scholar]
- Putman, M., H. W. Van Veen, and W. N. Konings. 2000. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 64:672–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem, D., N. Salman, and S. Schuldiner. 2001. In vitro monomer swapping in EmrE, a multidrug transporter from Escherichia coli, reveals that the oligomer is the functional unit. J. Biol. Chem. 276:48243–48249. [DOI] [PubMed] [Google Scholar]
- Son, M. S., C. Del Castilho, K. Duncalf, D. Carney, J. H. Weiner, and R. J. Turner. 2003. Mutagenesis of SugE, a small multidrug resistance protein (SMR), suggests an antiporter activity. Biochem. Biophys. Res. Commun. 312:914–992. [DOI] [PubMed] [Google Scholar]
- Schuldiner, S., M. Lebendiker, and H. Yerushalmi. 1997. EmrE, the smallest ion-coupled transporter, provides a unique paradigm for structure-function studies. J. Exp. Biol. 200:335–341. [DOI] [PubMed] [Google Scholar]
- Schumacher, M., and R. Brennan. 2003. Deciphering the molecular basis of multidrug recognition: crystal structures of the Staphylococcus aureus multidrug binding transcription regulator QacR. Res. Microbiol. 154:69–77. [DOI] [PubMed] [Google Scholar]
- Schwaiger, M., M. Lebendiker, H. Yerushalmi, M. Coles, A. Groger, C. Schwarz, S. Schuldiner, and H. Kessler. 1998. NMR investigation of the multidrug transporter EmrE, an integral membrane protein. Eur. J. Biochem. 254:610–619. [DOI] [PubMed] [Google Scholar]
- Shapiro, A., and V. Ling. 1997. Extraction of Hoechst 33342 from the cytoplasmic leaflet of the plasma membrane by P-glycoprotein. Eur. J. Biochem. 250:115–121. [DOI] [PubMed] [Google Scholar]
- Sulistijo, E., T. Jaszewski, and K. MacKenzie. 2003. Sequence-specific dimerization of the transmembrane domain of the “BH3-only” protein BNIP3 in membranes and detergent. J. Biol. Chem. 278:51950–51956. [DOI] [PubMed] [Google Scholar]
- Tate, C. G., E. R. S. Kunji, M. Lebendiker, and S. Schuldiner. 2001. The projection structure of EmrE, a proton-linked multidrug transporter from Escherichia coli, at 7 Å. EMBO J. 20:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate, C., I. Ubarretxena-Belandia, and J. Baldwin. 2003. Conformational changes in the multidrug transporter EmrE associated with substrate binding. J. Mol. Biol. 332:229–242. [DOI] [PubMed] [Google Scholar]
- Torres, J., and I. T. Arkin. 2000. Recursive use of evolutionary conservation data in molecular modeling of membrane proteins. Eur. J. Biochem. 267:3422–3431. [DOI] [PubMed] [Google Scholar]
- Ubarretxena-Belandia, I., J. Baldwin, S. Schuldiner, and C. Tate. 2003. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 22:6175–6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Ven, F., J. van Os, J. Aelen, S. Wymenga, M. Remerowski, R. Konings, and C. Hilbers. 1993. Assignment of 1H, 15N, and backbone 13C resonances in detergent-solubilized M13 coat protein via multinuclear multidimensional NMR: a model for the coat protein monomer. Biochemistry. 32:8322–8328. [DOI] [PubMed] [Google Scholar]
- VanAken, T., S. Foxall-VanAken, S. Castlemen, and S. Ferguson-Miller. 1986. Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins. Methods Enzymol. 125:27–35. [DOI] [PubMed] [Google Scholar]
- Vázquez-Laslop, N., P. Markham, and A. Neyfakh. 1999. Mechanism of ligand recognition by BmrR, the multidrug-responding transcriptional regulator: mutational analysis of the ligand-binding site. Biochemistry. 38:16925–16931. [DOI] [PubMed] [Google Scholar]
- Winstone, T., K. Duncalf, and R. J. Turner. 2002. Optimization of expression and the purification by organic extraction of the integral membrane protein EmrE. Protein Expr. Purif. 26:111–121. [DOI] [PubMed] [Google Scholar]
- Winstone, T. L., M. Jidenko, M. le Claire, C. Ebel, K. A. Duncalf, and R. J. Turner. 2005. Organic solvent extracted EmrE solubilized in dodecyl maltoside is monomeric and binds drug ligand. Biochem. Biophys. Res. Comm. In press. [DOI] [PubMed]
- Yerushalmi, H., M. Lebendiker, and S. Schuldiner. 1996. Negative dominance studies demonstrate the oligomeric structure of EmrE, a multidrug antiporter for Escherichia coli. J. Biol. Chem. 271:31044–31048. [DOI] [PubMed] [Google Scholar]
- Yerushalmi, H., S. S. Mordoch, and S. Schuldiner. 2001. A model for coupling of H+ and substrate fluxes based on “time-sharing” of a common binding site. J. Biol. Chem. 276:12744–12748.11278804 [Google Scholar]
- Yerushalmi, H., and S. Schuldiner. 2000a. A common binding site for substrates and protons in EmrE, an ion-coupled multidrug transporter. FEBS Lett. 476:93–97. [DOI] [PubMed] [Google Scholar]
- Yerushalmi, H., and S. Schuldiner. 2000b. An essential residue in EmrE, a multidrug antiporter from Escherichia coli. Biochemistry. 39:14711–14719. [DOI] [PubMed] [Google Scholar]
- Zhong, W., J. P. Gallivan, Y. Zhang, L. Li, H. A. Lister, and D. A. Dougherty. 1998. From ab initio quantum mechanics to molecular neurobiology: a cation-π binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. USA. 95:12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]