

Abstract
The molecular interactions on a protein-resistant surface coated with low-molecular-weight poly(ethylene glycol) (PEG) copolymer brushes are investigated using the extended surface forces apparatus. The observed interaction force is predominantly repulsive and nearly elastic. The chains are extended with respect to the Flory radius, which is in agreement with qualitative predictions of scaling theory. Comparison with theory allows the determination of relevant quantities such as brush length and adsorbed mass. Based on these results, we propose a molecular model for the adsorbed copolymer morphology. Surface-force isotherms measured at high resolution allow distinctive structural forces to be detected, suggesting the existence of a weak equilibrium network between poly(ethylene glycol) and water—a finding in accordance with the remarkable solution properties of PEG. The occurrence of a fine structure is interpreted as a water-induced restriction of the polymer's conformational space. This restriction is highly relevant for the phenomenon of PEG protein resistance. Protein adsorption requires conformational transitions, both in the protein as well as in the PEG layer, which are energetically and kinetically unfavorable.
INTRODUCTION
Poly(ethylene glycol) (PEG) surface grafts have gained considerable attention as being stable films that provide resistance to nonspecific protein adsorption (Elbert and Hubbell, 1996; Harris and Zalipsky, 1997). This protein resistance is of practical importance for a number of surface-engineering applications including biomaterials, drug delivery, or biosensors.
The molecular mechanisms underlying protein resistance of grafted PEG have not yet been fully identified. Two different aspects of the problem are usually considered: first, the brush-induced “steric” repulsion that is thought to prevent direct contact between proteins and the underlying surface (Szleifer, 1997); and second, hydration shells, which energetically suppress adsorption of proteins onto the PEG layer. Recently, it has been argued that the protein resistance also observed on well-ordered model systems of self-assembled monolayers of oligo(ethylene glycol) may alternatively be due to charging effects (Chan et al., 2003; Herrwerth et al., 2003; Kreuzer et al., 2003).
The PEG-water interaction is truly remarkable, as one can see from its unusual and well-documented solution properties. It is commonly understood that PEG is an amphiphilic polymer, which is only structurally soluble in water, i.e., hydrated PEG preferentially resides in polar gauche-conformations (Björling et al., 1991) that closely match the structure of water. It is widely recognized that there must be a particularly enhanced water structure around PEG chains (Müller and Rasmussen, 1991) that gives rise to a solubility gap (phase separation) at higher solution temperatures (Karlström, 1985; Saeki et al., 1976). Based on thermodynamic considerations, a widely cited hydration model has been proposed (Kjellander and Florin, 1981), which portrays a helical PEG conformation encaged into a surrounding water structure. Indeed, the existence of extensive equilibrium hydration structures was postulated based on heat-capacity measurements (Kingman et al., 1990). A minimum of ∼2–3 water molecules per PEG monomer seems to be required to complete basic hydration (Maisano et al., 1993).
Low-molecular-weight PEG does not readily adsorb onto surfaces. It must be grafted to the surface to form a brush layer of sufficiently high surface density to achieve protein resistance. Common grafting strategies include the use of block copolymers (Costello et al., 1993), electrostatic adsorption (Claesson and Gölander, 1987), chemical functionalization (Himmelhaus et al., 2003; Prime and Whitesides, 1993; Raviv et al., 2002), surface-induced polymerization (Ma et al., 2004), cross-linking of star-shaped PEG (Groll et al., 2004), or covalent grafting of PEG chains to phospholipids (Kuhl et al., 1994; Tirosh et al., 1998).
In this work, poly(ethylene glycol) was grafted onto a cationic poly-l-lysine backbone, forming the comb-like polymer, poly(l-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG). The PLL backbone acts as an anchor onto anionic surfaces such as mica (Fig. 1 a). This type of copolymer exhibits an extraordinarily strong adsorption onto many different surfaces. Different PLL-g-PEG architectures are known to exhibit different degrees of protein resistance (Kenausis et al., 2000; Pasche et al., 2003). In this article, we have focused on the most efficient architecture for protein resistance.
FIGURE 1.

(a) Chemical structure of PLL-g-PEG used in this study. Before the grafting reaction in solution, the molecular weight of PLL was 12 kDa (polymerization: ξPLL = 96, rF ≈ 5.4 nm) and for PEG is was 2 kDa (polymerization: ξPEG = 45 and rF ≈ 3.4 nm). The PEG was then grafted to one lysine out of g = 3.5. (b) Schematic setup of the extended surface forces apparatus used to measure the surface force isotherms. The surface separation, D, is measured using thin-film interferometry in transmission (see interference pattern on left) while a calibrated motor, M, approaches and compresses the surfaces via a compliant spring, k. The mica surfaces are in crossed-cylinder geometry and optionally coated with the PLL-g-PEG copolymer.
Our motivation in studying these PEG films in the extended surface-forces apparatus (eSFA) is to obtain additional insights into the molecular morphology and interactions of the adsorbed copolymer film and to identify the molecular mechanisms responsible for its excellent protein resistance.
MATERIALS AND METHODS
We have adsorbed PLL-g-PEG onto clean mica surfaces and measured the surface-force isotherms between them in aqueous solutions using the eSFA. Additionally we have measured the surface forces in the case where only one mica surface was covered with the adsorbed polymer.
Copolymer synthesis and chemical structure
The PLL-g-PEG used for this study was synthesized and characterized in our group according to procedures previously described in great detail (Huang et al., 2000; Pasche et al., 2003). The molecular weight of the poly(l-lysine) was 12 kDa, corresponding to 20 kDa PLL-HBr used for synthesis, and the polydispersity was MW/Mn = 1.2. The molecular weight of the poly(ethylene glycol) was 2 kDa and the polydispersity Mw/Mn = 1.1. The structure of this copolymer is illustrated schematically in Fig. 1 a. The lysine side chains are positively charged in aqueous solutions, with a pK = 10.5. The grafting ratio, g, is defined as the total number of lysine units divided by the number of PEG-modified units. The grafting ratio of the PLL-g-PEG copolymer used in this study was g = 3.5. The degree of polymerization of PLL is ξPLL = 96 and the polymerization of PEG ξPEG = 45. Therefore, each PLL-g-PEG copolymer carries an average number of 27 grafted PEG side chains and 69 positively charged lysine side chains. The average distance between PEG grafting points along the PLL backbone is d0 ≈ 1.2 nm. The total molar mass of one PLL-g-PEG molecule is 67 kDa. This particular PLL-g-PEG architecture (i.e., grafting ratio and molecular weight) was chosen because it results in a highly protein-resistant adsorbed layer (Kenausis et al., 2000; Pasche et al., 2003).
Copolymer adsorption
We used the cleavage plane of mica crystals as a model substrate in this work. Ruby mica is known to expose negative surface sites on the basal cleavage plane that are neutralized by K+ ions in the bulk crystal. The remaining K+ ions can dissociate in an aqueous environment and a double layer is formed in the vicinity of this negatively charged surface. The surface density of such ion sites (0.48 nm2)−1 is determined by the crystal structure.
The adsorption process of PLL-g-PEG on mica is controlled by electrostatic forces, Van der Waals attraction, and steric contributions that originate from the grafted PEG side chains. PEG2000 itself is of too low a molecular weight to adsorb efficiently on mica. The adsorption kinetics of the PLL-g-PEG copolymer have been investigated on metal oxide substrates in great detail and were found to be very fast and highly irreversible (Kenausis et al., 2000).
In our study, PLL(12 k)-g(3.5)-PEG(2 k) was adsorbed onto the mica substrates from a 1 mg/ml aqueous PLL-g-PEG solution ex situ with an incubation time of 30 min at room temperature. The substrates were thoroughly rinsed with ultrapure water to remove any possible excess copolymer or ions. After PLL-g-PEG adsorption, the substrates were kept wet at all times during handling.
Aqueous solutions used
All experiments were performed using ultrapure water (puriss. p.a., 0.2 μm membrane filtered; Fluka, Milwaukee, WI) as received. The fluid cell of the eSFA has a volume of ∼80 ml. For the addition of salt, 10 ml of the water was drained from the SFA and replaced by 10 ml of a 0.2-μm-filtered solution of KCl (ultra, >99.0%; Sigma, St. Louis, MO) at the appropriate concentration.
Mica surface preparation
Thin mica sheets were prepared by manual cleavage of optical-quality ruby mica blocks in a class-100 laminar-flow cabinet. Sets of thin mica pieces of uniform thickness (in the range of 1.5–4.5 μm) were obtained at an approximate size of 20 × 20 mm. A number of smaller (≈8 × 8 mm) rectangular pieces were cut from the larger sheet.
To avoid potential complications due to the recently discovered nanoparticles on conventionally cut mica (Heuberger and Zäch, 2003; Kohonen et al., 2003; Ohnishi et al., 1999), we used surgical scissors for cutting. The cut mica sheets were deposited on a thicker, freshly cleaved mica block for intermediate storage and further handling. One edge of each mica piece was overlapped with a clean Teflon tape to facilitate subsequent liftoff.
The mica sheets were then coated with a silver film of 40 nm thickness by means of thermal evaporation in vacuum (Pbase < 5 × 10−6 mbar). The silvered mica sheets were finally lifted off and glued onto cylindrical lenses using epoxy resin glue. After gluing, the samples were immediately (≈5 min) inserted into the sealed measurement apparatus. The fluid cell was then purged with dry nitrogen and the mica thickness determined using thin-film interferometry.
The extended surface-forces apparatus
The experimental data were obtained using the eSFA (Fig. 1 b), which is an enhanced and automated version of the SFA 3 (Surforce, Santa Barbara, CA) (Israelachvili and McGuiggan, 1990). The eSFA is located inside a thermally insulated enclosure at a controlled temperature at 25.00(2)°C (Heuberger et al., 2001). The mica surfaces are arranged in crossed-cylinder geometry inside a fluid cell. The optical measurement of surface separation is based on thin-film interferometry (Israelachvili, 1973). The eSFA uses fully automated spectrum acquisition and evaluation, which permits measurements to be carried out rapidly and with great precision. Interference spectra are evaluated by means of the numerical fast spectral correlation algorithm (Heuberger, 2001), allowing the determination of both film thickness, D, and refractive index, n, at high acquisition rates (1–10 Hz). The instrumental precision of film-thickness measurement is typically σD = ±25 pm, and for the refractive index it is σn < ±0.05 (for D > 1 nm). A detailed error calculation is given elsewhere (Heuberger, 2001). In a typical experiment, the optical thickness of the mica substrates is first determined in a dry N2 atmosphere while the mica surfaces are in unloaded adhesive contact.
All surface-force isotherms shown here were measured by approaching the surfaces toward each other with an actuator at a constant velocity of v = 1.00(5) nm/s. Surface forces, F(D), are calculated based on the difference between the fine-calibrated actuator movement, M(t), and the optically measured film thickness, D(t), measured in the point of closest approach. To obtain the normalized surface force, one can use the simple relationship
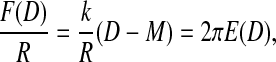 |
(1) |
where R = (Rx × Ry)0.5 is the effective radius representing the local surface curvature and k = 1002(23) N/m is the compliance of the force-measuring spring (Fig. 1 b).
The calculated force, F(D), is normalized by the effective local radius, R ≈ 18(2) mm of curvature. The mica surfaces showed only minor elastic flattening during compression of the polymer film, as seen from an analysis of the interference fringes. Using the Derjaguin approximation, one can thus estimate the free energy per unit area, F/R ≈ 2πE(D), which allows easy comparison between data and theory (Derjaguin, 1934; Israelachvili, 1991).
The calculation of this free energy (i.e., F/R) is subject to instrumental errors that are discussed in detail elsewhere (Zäch and Heuberger, 2000). Typical drift rates of the eSFA are 20 pm/min (Heuberger et al., 2001) and the cylinder radii, Ri, can be determined with <5% relative error using lateral scanning (Heuberger, 2001). In this work, the accuracy of F(D)/R was typically ±0.1 mN/m. The error bars shown in Figs. 2, 3, and 5 correspond to this instrumental error. Errors due to variations in sample preparation are discussed in the text.
FIGURE 2.

Surface-force isotherms (black is loading; gray is unloading) measured in two situations: (a) where only one, or (b) where both mica surfaces were covered with the PLL-g-PEG copolymer. The data points shown here were obtained in 1 mM KCl solution at 25°C. The data points were continuously acquired while the motor (c.f. Fig. 1 b) was operated at constant velocity, dM/dt = −1 nm/s; this corresponds to a compression rate of d(F/R)/dt ≈ 55 μNm−1/s. The marks, LA and LB, designate the PEG brush length obtained from a fit of the data (dashed gray lines) to the de Gennes scaling theory. Error bars represent instrumental errors, as discussed in the Methods section.
FIGURE 3.

Refractive index measured simultaneously with the surface-force isotherms for the two separate situations: (a) where only one, or (b) where both mica surfaces were covered with the PLL-g-PEG copolymer. The medium was ultrapure water in equilibrium with atmospheric CO2 at a temperature of 25°C. The solid gray lines correspond to the refractive index envelope predicted by the simple model described in Eq. 2. The error bars shown along the solid lines represent the estimated statistical errors based on the standard deviation of ± σλ = 10 pm for interference peak detection, which is typical for this type of fast spectral correlation setup (Heuberger, 2001). The inset shows the difference between the simple model and the measurement; a significant depression of refractive index is visible at a surface separation of 20 nm. This n-depression is reproducible between different samples and in quantitative agreement with a recently reported ≈10% density decrease found in water adjacent to a PEG-ylated surface (Schwendel et al., 2002). Error bars represent instrumental errors, as discussed in the Methods section.
FIGURE 5.

Magnification of the surface-force isotherm between two PLL-g-PEG covered surfaces in pure water. Discrete film-thickness transitions with a step size 125 ± 25 pm (statistics based on 40 isotherms) reveal the presence of long-lived, load-bearing equilibrium structures in the PLL-g-PEG film. Similar film-thickness transitions are also observed in the case where only one surface is decorated with PEG. The transitions are observed in sequences of 6–10 individual transitions, separated by regions of similar extension (≈1–1.5 nm) without clear transitions. The loading speed was held constant at 1 nm/s using a closed feedback loop. For the sake of simplicity, only one error bar is shown, which represents the instrumental precision of this measurement.
RESULTS
Surface-force isotherms
Typical surface-force isotherms, F(D)/R, for adsorbed PLL-g-PEG copolymer films measured in 1 mM KCl aqueous solution are shown in Fig. 2. The value D = 0 nm indicates the absolute location of the mica surfaces (±0.2 nm, absolute). Under compression, D represents the polymer film thickness. Two curves are shown: in curve a the adsorbed copolymer covers only one mica surface; and in curve b both surfaces are covered by the adsorbed copolymer film. The measured force is chiefly repulsive, i.e., F/R > 0. The high-load regime can be well described by an exponential function. The film thickness of curve b is slightly less than twice that of curve a for a comparable compression. The exponential decay length is larger for the symmetric case. We note that the highly compressed copolymer film (F/R > 1 mN/m) deforms virtually free of hysteresis, which suggests an equilibrium measurement. The absence of hysteresis under compression is in agreement with previous force measurements of surface-grafted PEG chains (Efremova et al., 2001; Sheth and Leckband, 1997). In the highly compressed region, we found that the repulsive force was independent of the salt concentrations used here. This behavior is expected because of the nonionic nature of PEG and is in agreement with previous studies on grafted PEG brushes (Raviv et al., 2002). Only in the low-force regime, where the polymer is barely compressed, did we observe weak attractive forces (see below).
Refractive index and adsorbed mass
Simultaneously with the interferometric measurement of the surface separation, D, we have determined the refractive index, n(D) (Fig. 3). This is a measure of the average density in a volume of thickness D and diameter Ø ≈ 1 μm. Because the refractive index of the adsorbed copolymer is higher than that of water, we expect to observe an increase of n as the surface separation is decreased. To estimate the total amount of PEG between the surfaces from the refractive index we can adopt a simple model (Raviv et al., 2002)
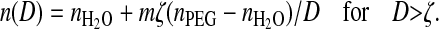 |
(2) |
ζ = Γ/ρ is the equivalent thickness of the dried PLL-g-PEG layer; Γ is the adsorbed PLL-g-PEG mass per surface area, and ρ ≈ 1.12 g/cm3 is the dry polymer density. If we further assume two identical copolymer layers on both surfaces, we can set m = 1 for the asymmetric case and m = 2 for the symmetric case.
Fitting Eq. 2 to our data yields an adsorbed polymer mass Γ ≈ 200 ± 100 ng/cm2, which is in good agreement with the typical adsorbed mass of this PLL-g-PEG architecture on various metal oxide substrates, as determined from an optical waveguide technique (180 ± 20 ng/cm2) (Kenausis et al., 2000; Pasche et al., 2003).
The inset of Fig. 3 shows the difference between the model described by Eq. 2 and the measurement. We observe a significant depression of the refractive index at a separation of Dmin = 21 ± 1 nm. If interpreted in terms of water density, this is equivalent to a zone of 5–10% reduced density 5–10 nm above the PEG layers. We note that a quantitatively similarly density depression was recently suggested based on neutron reflectivity measurements (Schwendel et al., 2002).
Comparison with theory
To compare the surface-force isotherms with theory, we have to consider several different surface-force contributions. Van der Waals forces between mica surfaces can be neglected here because the closest mica separations are beyond the range of detectable force. Due to the charged nature of both mica and poly-lysine, we need to consider the presence of double-layer forces. We invoke the well-known double-layer model to describe the exponential force profiles at large separations (i.e., outside the polymer layer) with the characteristic Debye length, κ−1, which depends on the ionic composition of the solution:
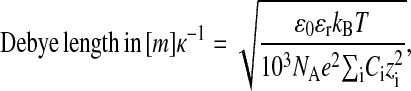 |
(3) |
where ɛ0 is the dielectric field constant, ɛr the relative dielectric constant of the aqueous solution, kB the Boltzmann constant, T = 298.15 K the absolute temperature, NA the Avogadro number, e the electronic charge, Ci the molar concentration, and zi the valency, of each ionic species, i, in the solution.
Assuming a constant dielectric permittivity for the aqueous solutions, ɛr ≈ 80, the Debye lengths for ultrapure water (pH = 5.6, dissolved CO2) and 1 mM KCl solution are κ−1(H2O) ≈ 190 nm and κ−1(1 mM KCl) ≈ 9.7 nm, respectively. The measured surface-force isotherms were fitted in the regime of sufficient surface separation, i.e., 50 nm < D < 2.5 μm, using the well-known expression of double-layer forces between curved surfaces (Verwey and Overbeek, 1945):
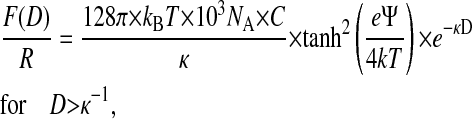 |
(4) |
where C is the molar concentration of the salt solution and Ψ the surface potential in Volts.
There is no significant long-range force in 1 mM KCl (Ψ = 20 ± 20 mV). In pure water the measurement was of similar magnitude, but less reliable due to the accumulation of small nonlinearities in the actuator over the longer distance range.
The surface-force isotherms presented in Fig. 2 reveal a strong exponential repulsion at small surface separations. This force is commonly assigned to the steric repulsion known for polymer-bearing surfaces. If the number of available conformers is restricted by an opposing surface, the associated entropy reduction gives rise to a repulsive force. Depending on the average distance, d, between PEG grafting sites, one can invoke different theoretical models to describe this steric force. For low-molecular-weight PEG, in particular, there is some controversy in the literature about the correct assignment of these regimes (Hansen et al., 2002). The so-called “brush” regime is often invoked for d < 2rF, and the so-called “mushroom” regime for d > 2rF. The quantity rF is the Flory radius of the polymer random coil in a good solvent,
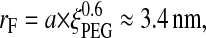 |
(5) |
where a = 0.35 nm is the PEG monomer size (Hansen et al., 2002), and ξPEG = 45 the degree of polymerization of PEG2000.
The average distance, d, between PEG surface grafting sites determined from the optically measured adsorbed mass (200 ± 100 ng/cm2) is dn = 1.4 ± 0.4 nm. Because this optically determined dn is clearly smaller than the random coil size, 2rF = 6.8 nm, we conclude that the chains are extended with respect to their Gaussian dimension. The length, L, of such a brush can be predicted qualitatively from this grafting distance and the known number of monomer units. Using the scaling relation proposed by Alexander (1977) we have:
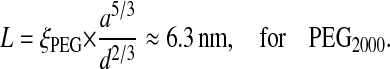 |
(6) |
Scaling theory strictly applies to highly extended (i.e., high lateral compression), end-grafted polymers of high molecular weight, so it can only be considered as an approximate guide here. We can also attempt to fit the parameters of the de Gennes scaling theory for brushes (deGennes, 1987) to reproduce the measured surface-force isotherm. After integration of the brush-induced pressure (Eq. 17 in deGennes, 1987), scaling theory predicts a surface force of the form:
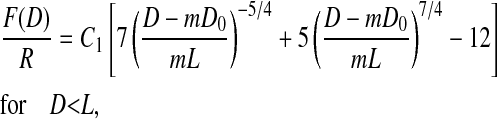 |
(7a) |
where
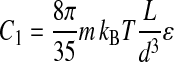 |
(7b) |
with ɛ ≈ 1 a dimensionless prefactor and m = 1 for the asymmetric experiment (Eq. 7a) and m = 2 for the symmetric experiment (Eq. 7b). The parameter D0 is an offset that accounts for the finite thickness of the underlying PLL anchor.
Fitting of the surface-force isotherm (Fig. 2) gives a reasonable quantitative agreement in the regime of strong compression. Fixing the offset to D0 = 1 nm gives the following brush lengths: LA = 8.2 nm and LB = 6.3 nm. The result, Lb < La, indicates the presence of interdigitation effects in case b as opposed to case a. Interdigitation is not included in this simple model and will be discussed in more detail in a later publication.
Weak attractive forces
A magnification of the weak-force regime reveals rather small attractive forces. In Fig. 4 a, two different isotherms are displayed for the case of polymer adsorbed onto one surface only. One curve is measured in pure water (triangles) and one is measured in 1 mM KCl solution (squares). The weak double-layer force contribution has been mathematically subtracted for better illustration of the hysteresis. The isotherms were fitted to Eq. 4 outside the range of the steric repulsion (i.e., >30 nm) and then extrapolated into the range shown here. In the pure water, the Debye length is sufficiently large to justify a linear approximation of Eq. 4. The loading branch in pure water (black triangles) revealed a small attractive force that is strongest at F/R(D = 16 ± 2 nm) = −0.18 ± 0.05 mN/m. The maximum range of these small attractive forces is ∼48 ± 5 nm. These small attractive forces are not observed during approach in salt solution (black squares). The gray curves show the corresponding unloading branches. The adhesive minima are located in the same range of surface separation, i.e., D = 16 ± 2 nm. The difference of the force between loading and unloading branches was comparable in both solutions. For comparison, the length of a fully stretched PLL backbone is ΛPLL ≈ 34 nm and a fully stretched PEG chain is ΛPEG ≈ 17 nm.
FIGURE 4.

Magnification of surface-force isotherms in the low-force regime. The weak double-layer repulsion forces have been subtracted to illustrate the small hysteresis. Black symbols are loading cycles and gray symbols are unloading cycles; squares stand for 1 mM KCl solution and triangles for water; panel a illustrates the attractive forces in the asymmetric case, and panel b shows those observed in the symmetric case. The loading/unloading speed was 1 nm/s. The absolute errors in this graph are dominated by uncertainties of fitting (and subtracting) the weak double-layer force. For the sake of simplicity the estimated error bar is shown exemplarily for one point only.
The curves shown in Fig. 4 b illustrate the weak-force regime for the case of polymer adsorbed onto both surfaces. As described above (Fig. 4 a), the weak double-layer force contribution has been subtracted. In both solutions we observed no attractive forces during loading and a weak attractive force during unloading at a surface separation of D = 21 ± 1 nm. The small repulsion observed in pure water is at the significance limit of this measurement.
Embedded fine structure
When we magnify the D axis of the surface-force isotherms in the repulsive regime, as exemplified in Fig. 5, we find a ubiquitous fine structure in the form of distinctive film-thickness transitions. These transitions are embedded into the known steric repulsion curve described above (Fig. 1). The characteristic step size is 125 ± 25 pm. The data points were acquired at ≈1.9-s intervals, which means that some of the observed features effectively have a lifetime of at least 30 s. A similar fine structure could be observed for all salt concentrations used here, both for the symmetric and the asymmetric case, and, in a temperature range between 1 and 40°C.
Manual analysis of ∼40 surface-force isotherms taken at 25°C reveals that these steps tend to occur in groups of 6–10 at diverse film compressions, starting around the nominal brush length. Similar groups of steps are also observed during unloading—often at comparable film compressions to those observed during loading. The surface-force isotherms are free of hysteresis in this load regime. This suggests that these fine transitions are caused by equilibrium structures that exist within this hydrated PLL-g-PEG layer.
The fine structure is not an instrumental artifact. The precision of the interferometric distance measurements used here is ±25 pm, as previously shown in detail (Heuberger, 2001). The actuator did not induce the steps because it moved at constant velocity (relative error <5%), as opposed to the stepwise measurement of force isotherms often used in other studies. More generally, we can exclude any instrumental instabilities (e.g., drift, vibrations, actuator irregularities) as the origin of the fine structure by the following argument. The sample surfaces are mechanically coupled to the eSFA body via the force-measuring spring (Fig. 1 b). The contact stiffness of the polymer film can be seen as a nonlinear spring in series with this force-measuring spring and the other much stiffer springs in the apparatus. At the compression where steps were observed, the contact stiffness varies from ∼100 kN/m to 1000 kN/m (c.f. slope of isotherm in Fig. 2). This is 1000 times higher than the spring constant, k = 1 kN/m, of the force-measuring spring. To induce film transitions of the observed size in the contact zone, the eSFA frame would thus have to perform quasiperiodic jumps over distances of 125 nm. Careful analysis of high-resolution data both with and without actuator action at various surface separations and actuator speeds reveals that the largest instrumental instabilities are three orders of magnitude too small to produce such an artifact (Heuberger et al., 2000).
It is also important to note that the observed step size does not vary systematically with the amount of overall film compression. Because the effective stiffness of the polymer film varies over a wide range in this experiment, we can conclude that the transitions are indeed an intrinsic property of the polymer layer and reflect a characteristic structural length scale.
DISCUSSION
Based on the surface force and refractive index measurements, we propose the model of a well-organized film (Fig. 6). The PLL backbone is adsorbed to the mica surface. The in-plane order of the adsorbed PLL backbones cannot be determined here, but the high effective grafting density (dn ≈ 1.4 nm) compared to the PEG grafting distance along the PLL backbone, d0 ≈ 1.2 nm, indicates that the PEG side chains are indeed standing up and that the PLL backbones cover most of the available surface area. The PEG chains are in a brush-like configuration. For an adsorbed mass of 200 ng/cm2 and a film thickness of 8.2 nm, the volume fraction of water in the film is 82%, which corresponds to a number of 10 water molecules per ethylene-oxide (ethylene oxide)-mer present in the film.
FIGURE 6.

Schematic model exemplifying the molecular morphology of one PLL-g-PEG film adsorbed to mica based on the compression isotherms discussed in the text. The copolymer film has a high degree of molecular organization. The PLL backbone is strongly adsorbed to the mica surface and the PEG side chains extend away from the surface in a brush-like configuration. The proposed polymer density profile, ρ(D), is shown on the left. The gray dashed lines in the background denote the underlying hydration structure, which gives rise to a quantized fine structure in the compression isotherm (c.f. Fig. 5).
A significant amount of PLL on the outside of the polymer film can be excluded due to the absence of strong electrostatic and adhesive surface forces against clean mica. However, the weak attractive forces shown in Fig. 4 a indicate that a very small number of PLL chain segments can still directly interact with the opposing surface. The differences of the weak attraction seen with different ionic concentrations can be explained as follows. The Debye length in 1 mM KCl aqueous solution is κ−1 ≈ 9.6 nm, as opposed to κ−1 ≈ 190 nm in pure water (pH 5.6 due to dissolved CO2). As a consequence, extending poly-cationic PLL segments are expected to contract in salt solution. Additionally, electrostatic anchoring to the opposing negatively charged mica surface is less likely due to the enhanced double layer.
We have shown that the short-range repulsion can be reasonably well described by theories of grafted flexible polymer chains in a brush-like configuration (Alexander, 1977; deGennes, 1987). Analysis of the surface-force isotherms in terms of scaling theory at low forces indicate that the density profile is not rectangular, but must exhibit a finite polymer density beyond the length of the de Gennes brush (Fig. 2) (Hansen et al., 2002; Milner and Witten, 1988; Zhulina et al., 1989). Comparison of de Gennes brush lengths between the asymmetric (a) and symmetric (b) experiments yields Lb < La. This finding indicates that there is excess entropy of interdigitation in case b in contrast to case a. Interdigitation reduces the apparent brush length. This effect is not accounted for in the theoretical model, c.f. Eq. 7 and is sometimes neglected in comparable experimental studies.
The steric repulsion exhibited by the PLL-g-PEG film is in good qualitative agreement with previous direct force measurements reported for comparable grafted PEG systems (Claesson and Gölander, 1987; Costello et al., 1993; Efremova et al., 2001; Kenworthy et al., 1995; Kuhl et al., 1994; Needham et al., 1992; Raviv et al., 2002). However, the observation of fine film-thickness transitions is unprecedented. The question is, thus, how can the PEG layer consist of flexible chains and still exhibit quantized structural forces?
At a sufficiently high density, a surface-adsorbed polymer might be in a glass-like state (Kremer, 1986). It has been shown that many water-soluble polymers can exhibit a small but finite yield strength when spread and compressed at the water/air interface. However, in these studies, PEG was found to exhibit only a vanishingly small such effect, if any (Cohen-Stuart et al., 1986). Furthermore, the glass-transition temperature of PEG2000 is rather low, Tg ≈ −40°C, and the PLL-g-PEG layer investigated in this study consists of 83% water. A glass-like polymer film is expected to exhibit a finite yield stress and plasticity over a wide temperature range, which is in contrast with the hysteresis-free (i.e., elastic) compression isotherm observed here. Some kind of glassy state as the source of the observed transitions can thus be excluded.
The characteristic step size of 125 ± 25 pm reported here is smaller than any molecular unit present in this system and thus cannot be attributed to a confinement-induced dynamic “layering” effect (Israelachvili and Pashley, 1983).
The change of free energy between two consecutive transitions can be estimated according to Eq. 1, ΔE ≈ ΔF/2πR ≈ 0.4 ± 0.2 mJ/m2. Assuming that all PEG chains contribute to the transitions and taking the area per PEG chain determined above (aPEG ≈ 2.0 × 10−18 m2), we obtain a change of free energy per PEG chain ΔG/PEG = 0.2 ± 0.1 kT. The change of free energy is small compared to kT and thus compatible with the chain flexibility paradigm. It is interesting to note that the quantum mechanically calculated change of free energy for conformational transitions in hydrated PEG is of comparable magnitude (Wang et al., 2000). The step size observed here (Fig. 5) is also comparable to the change of the oxygen-oxygen distance during a trans-gauche transition around the C-C bond in PEG.
The force isotherms measure the change of free energy upon compression. The observation of the fine structure in the absence of hysteresis thus implies two things: first, that the derivative of the entropy versus surface separation, dS/dD, is no longer a monotonous function; and secondly, a considerable fraction of the molecules in the contact region undergo the transition simultaneously, hence, there must be a molecular mechanism synchronizing these events.
To illustrate the first point, we note that a PEG chain can be in ∼1062 different conformations (random walk model). In the contact zone there are ∼108 PEG chains, which results in a total of 1070 molecular configurations contributing to the eSFA measurement. Under confinement, the number of possible conformations is reduced continuously, and, due to the high number of possible states, no noticeable transitions are expected. The presence of fine structures must therefore be understood as a significant restriction or degeneration of the polymer conformational space.
Regarding the second point we note that the high degree of molecular synchronization is linked to the nonmonotonous change of free energy. It can be understood in terms of a critical stress that is locally needed to induce the transition. As soon as one PEG chain undergoes the transition, the load on the neighboring chains will increase above the critical stress, thus, triggering a cascade of similar transitions in its neighborhood. Considering the curved contact geometry in this experiment, one can readily show that a region of a few micrometers diameter can undergo the same transition simultaneously.
It is important to note that the transition discussed here is not an individual change of conformation, but rather a confinement-induced critical transition of the conformational space in a thermodynamic sense (i.e., a phase transition). Therefore, the measured barriers in the system free energy are not expected to produce appreciable loading speed dependence in this quasiequilibrium experiment. Indeed, an experimental variation of actuator speed in the range from 0.1 to 5 nm/s (data not shown) revealed no dependence on this parameter.
To this end, a comparison with the literature is made to identify possible molecular mechanisms that give rise to the observed fine structure. The existence of an extensive equilibrium hydration structure was postulated in numerous studies, namely in aqueous PEG solution experiments (Bieze et al., 1994; Janelli et al., 1994; Karlström, 1985; Kjellander and Florin, 1981), single-molecule force measurements (Oesterhelt et al., 1999), and recent surface-monolayer studies (Grunze and Pertsin, 2000) as well as theory (Wang et al., 1997, 2000). The extraordinary solution properties of PEG are reviewed in the introduction above. It is known that gauche conformers are more polar and offer two hydrogen bond acceptor sites with an oxygen-oxygen distance (2.88 Å) that is very close to that of bulk water (2.85 Å). For example, H-bonding with two successive oxygen atoms in PEG by one single water molecule forms a pentagonal bridge structure (Bandyopadhyay et al., 2000) reminiscent of the pentagonal equilibrium structure predominant in bulk water (Dougherty and Howard, 1998). Namely, it was shown that the gauche conformers are overpopulated in PEG in contact with water (Björling et al., 1991). Similarly, the existence of conformational substates and bound water beyond the first hydration shell was detected as an anomalous excess heat capacity (Kingman et al., 1990).
Although the influence of water on the PEG conformational structure is widely accepted, it is less clear to what extent the water structure is modified by the presence of PEG. The density depression illustrated in Fig. 3 would suggest that a modified water structure potentially exists several nanometers beyond the nominal brush length. This is comparable to recent neutron-scattering observations (Schwendel et al., 2002) on ethylene-oxide self-assembled monolayers. The general idea is that this effect be linked to the hydrophobic effect in the vicinity of the amphiphilic PEG layer.
The PLL(12 k)-g(3.5)-PEG(2 k) copolymer studied here is used as a very effective protein-resistant coating. It is thus interesting to discuss the role of the embedded fine structure in terms of protein adsorption. It is generally accepted that in a wide range of grafting densities, direct adsorption to the underlying substrate is prevented by the osmotic pressure (i.e., steric repulsion of the polymer film). Another important ingredient for protein resistance is that the proteins do not stick to the PEG. Recent SFA measurements suggest that there may be a rather high activation barrier of up to 44 kT, which kinetically hinders the proteins (e.g., streptdavidin) from adsorbing (Efremova et al., 2001). If correct, then protein resistance is ultimately a nonequilibrium effect. Indeed, adhesion energies on the order of ≈−2 kT per protein were reported between strepdavidin and PEG brushes after external compression (Efremova et al., 2001; Sheth and Leckband, 1997).
Considering the size of a protein (footprint ≈ 20 nm2) and the fact that proteins adsorb via multiple sites, we estimate that at least 10 PEG chains directly contribute to the adsorption barrier of a single protein. Therefore, it takes ∼2 kT per protein to induce one transition of the kind observed here. It is important to note that to adsorb to PEG, a protein will restrict the conformational space of the PEG chains, even in the absence of a net compression. A simple thermodynamical analysis of this quasiequilibrium situation reveals that ∼99.8% of the proteins are incapable of inducing more than three conformational transitions in PEG and thus will be repelled within the first 0.4 nm (i.e., three or less transitions).
With the possibility at hand to directly detect transitions in the conformational space of the hydrated PEG, it is now planned to apply this new tool to various other polymer architectures, particularly those that are known to be less protein resistant.
CONCLUSION
In quantitative agreement with previous studies, steric forces are found during compression of surfaces exposing grafted PEG2000 brushes on mica substrates. The compression isotherms can be fitted using a combination of double-layer repulsion and polymer-brush compression. The effective surface potential is small, 20 ± 20 mV, and the obtained polymer brush lengths agree with theoretical predictions based on chemical structure. The grafting density and film thickness were determined from refractive index and force measurements, respectively. A morphological model for the structure of the adsorbed copolymer film is presented. A significant density depression above the PEG layer is found from an analysis of the refractive index envelope. Multiple, partially coherent conformational transitions are resolved that are embedded into the well-known steric repulsion. The conformational space of PEG is significantly frustrated and quantized as a function of film thickness. This is in agreement with the presence of an equilibrium hydration structure, also postulated in the literature. The nonmonotonous change of free energy during each structural transition is ∼0.2 kT per PEG chain. The resulting energy barrier is capable of inhibiting the adsorption of proteins within the first 0.4 nm of the polymer film.
Acknowledgments
We thank N. P. Huang for the synthesis of the PLL-g-PEG and S. Tosatti for providing unpublished data on refractive index for PEG400 solutions. Furthermore, we acknowledge invaluable discussions and advice from O. Borisov, J. Vörös, and M. A. Cohen-Stuart. This work profited from technical assistance provided by M. Elsener and J. Vanicek.
Financial support was provided by the TopNano21 Program of the Council of the Swiss Federal Institutes of Technology (ETH-Rat) as well as the US Air Force Office of Scientific Research under contract No. F49620-02-1-0346.
References
- Alexander, S. 1977. Adsorption of chain molecules with a polar head a scaling description. Le Journal de Physique. 38:983–987. [Google Scholar]
- Bandyopadhyay, S., M. Tarek, M. L. Lynch, and M. L. Klein. 2000. Molecular dynamics study of the poly(oxyethylene) surfactant C12E2 and water. Langmuir. 16:2000. [Google Scholar]
- Bieze, T. W. N., A. C. Barnes, C. J. M. Huige, J. E. Enderby, and J. C. Leyte. 1994. Distribution of water around poly(ethylene oxide): a neutron diffraction study. J. Phys. Chem. 98:6568–6576. [Google Scholar]
- Björling, M., G. Karlström, and P. Linse. 1991. Conformational adaptation of poly(ethylene oxide). J. Phys. Chem. 95:6706–6709. [Google Scholar]
- Chan, Y.-H. M., R. Schweiss, C. Werner, and M. Grunze. 2003. Electrokinetic characterization of oligo- and poly(ethylene glycol)-terminated self-assembled monolayers on gold and glass surfaces. Langmuir. 19:7380–7385. [Google Scholar]
- Claesson, P. M., and C.-G. Gölander. 1987. Direct measurements of steric interactions between mica surfaces covered with electrostatically bound low-molecular-weight polyethylene oxide. J. Colloid Interface Sci. 117:366–374. [Google Scholar]
- Cohen-Stuart, M. A., J. T. F. Keurentjes, B. C. Bonekamp, and J. G. E. M. Fraaye. 1986. Gelation of polymers adsorbed at a water-air interface. Colloids and Surfaces. 17:91–102. [Google Scholar]
- Costello, B. A. De L., P. F. Luckham, and T. F. Tadros. 1993. Forces between adsorbed low molecular weight graft copolymers. J. Colloid Interface Sci. 156:72–77. [Google Scholar]
- deGennes, P. G. 1987. Polymers at an interface: a simplified view. Adv. Colloid Interface Sci. 27:189–209. [Google Scholar]
- Derjaguin, B. V. 1934. Untersuchungen über die Reibung und Adhäsion. IV. Kolloid Zeitschrift. 69:155–164. [in German]. [Google Scholar]
- Dougherty, R. C., and L. N. Howard. 1998. Equilibrium structural model of liquid water: evidence from heat capacity, spectra, density, and other properties. J. Chem. Phys. 109:7379–7393. [Google Scholar]
- Efremova, N. V., S. R. Sheth, and D. E. Leckband. 2001. Protein-induced changes in poly(ethylene glycol) brushes: molecular weight and temperature dependence. Langmuir. 17:7628–7636. [Google Scholar]
- Elbert, D. L., and J. Hubbell. 1996. Surface treatments of polymers for biocompatibility. Annual Review of Materials Science. 26:365–370. [Google Scholar]
- Groll, J., E. V. Amirgoulova, T. Ameringer, C. D. Heyes, C. Röcker, G. U. Nienhaus, and M. Möller. 2004. Biofunctionalized, ultrathin coatings of cross-linked star-shaped poly(ethylene oxide) allow reversible folding of immobilized proteins. J. Am. Chem. Soc. 126:4234–4239. [DOI] [PubMed] [Google Scholar]
- Grunze, M., and A. Pertsin. 2000. Molecular Conformations in Organic Monolayers Affect their Ability to Resist Protein Adsorption. L. Fabbrizzi and A. Poggi, editors. Springer-Verlag, Berlin/Heidelberg, Germany.
- Hansen, P. L., J. A. Cohen, R. Podgomik, and V. A. Parsegian. 2002. Osmotic properties of poly(ethylene glycols): quantitative features of brush and bulk scaling laws. Biophys. J. 84:350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, J. M., and S. Zalipsky. 1997. Poly(ethylene glycol). J. M. Harris and S. Zalipsky, editors. American Chemical Society, Washington, DC.
- Herrwerth, S., W. Eck, S. Reinhardt, and M. Grunze. 2003. Factors that determine the protein resistance of oligoether self-assembled monolayers: internal hydrophilicity, terminal hydrophilicity, and lateral packing density. J. Am. Chem. Soc. 125:9359–9366. [DOI] [PubMed] [Google Scholar]
- Heuberger, M. 2001. The extended surface forces apparatus. Part I. Fast spectral correlation spectroscopy. Rev. Sci. Instrum. 72:1700–1707. [Google Scholar]
- Heuberger, M., J. Vanicek, and M. Zäch. 2001. The extended surface forces apparatus. Part II. Precision temperature control. Rev. Sci. Instrum. 72:3556–3560. [DOI] [PubMed] [Google Scholar]
- Heuberger, M., and M. Zäch. 2003. Nano-fluidics: structural forces, density anomalies and the pivotal role of nano-particles. Langmuir. 19:1943–1947. [Google Scholar]
- Heuberger, M., M. Zäch, and N. D. Spencer. 2000. Sources and control of instrumental drift in the surface forces apparatus. Rev. Sci. Instrum. 71:4502–4508. [Google Scholar]
- Himmelhaus, M., T. Bastuck, S. Tokumitsu, M. Grunze, L. Livadaru, and H. J. Kreuzer. 2003. Growth of a dense polymer brush layer from solution. Europhys. Lett. 64:378–384. [Google Scholar]
- Huang, N.-P., R. Michel, J. Voros, M. Textor, R. Hofer, A. Rossi, L. Elbert, J. Hubbell, and N. D. Spencer. 2000. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: surface-analytical characterization and resistance to serum and fibrinogen adsorption. Langmuir. 17:489–498. [Google Scholar]
- Israelachvili, J. N. 1973. Thin film studies using multiple beam interferometry. J. Colloid Interface Sci. 44:259–271. [Google Scholar]
- Israelachvili, J. N. 1991. Intermolecular & Surface Forces. Academic Press, London, UK.
- Israelachvili, J. N., and P. M. McGuiggan. 1990. Adhesion and short-range forces between surfaces. I. New apparatus for surface force measurements. J. Mater. Res. 5:2223–2231. [Google Scholar]
- Israelachvili, J. N., and R. M. Pashley. 1983. Molecular layering of water at surfaces and origin of repulsive hydration forces. Nature. 306:249–250. [Google Scholar]
- Janelli, M. P., S. Magazu, G. Maisano, D. Majolino, and P. Migliardo. 1994. Non-ideal compressibility in poly(ethylene oxide)-water solutions induced by H-bond interactions. J. Mol. Struct. 322:337–343. [Google Scholar]
- Karlström, G. 1985. A new model for upper and lower critical solution temperatures in poly(ethylene oxide) solutions. J. Phys. Chem. 89:4962–4964. [Google Scholar]
- Kenausis, G., J. Voros, D. L. Elbert, N.-P. Huang, R. Hofer, L. Ruiz-Taylor, M. Textor, J. A. Hubbell, and N. D. Spencer. 2000. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: attachment mechanism and effects of polymer architecture on resistance to protein adsorption. J. Phys. Chem. B. 104:3298–3309. [Google Scholar]
- Kenworthy, A. K., K. Hristova, D. Needham, and T. J. McIntosh. 1995. Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly(ethylene glycol). Biophys. J. 68:1921–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, N. G., A. Rosenberg, M. Bastos, and I. Wadsö. 1990. Heat capacity of poly(ethylene glycol)-water mixtures: poly(ethylene glycol)-water interactions. Thermochimica Acta. 169:339–346. [Google Scholar]
- Kjellander, R., and E. Florin. 1981. Water structure and changes in thermal stability of the system poly(ethylene oxide)-water. J. Chem. Soc. Faraday Trans. I. 77:2053–2077. [Google Scholar]
- Kohonen, M. M., F. C. Meldrum, and H. K. Christenson. 2003. Particles on melt-cut mica sheets are platinum. Langmuir. 19:975–976. [Google Scholar]
- Kremer, K. 1986. Glassy states of adsorbed flexible polymers and spread polymer “monolayers”. Le Journal de Physique. 47:1269–1271. [Google Scholar]
- Kreuzer, H. J., R. L. C. Wang, and M. Grunze. 2003. Hydroxide ion adsorption on self-assembled monolayers. J. Am. Chem. Soc. 125:8384–8389. [DOI] [PubMed] [Google Scholar]
- Kuhl, T. L., D. E. Leckband, D. D. Lasic, and J. N. Israelachvili. 1994. Modulation of interaction forces between bilayers exposing short-chained ethylene oxide headgroups. Biophys. J. 66:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H., J. Hyun, P. Stiller, and A. Chilkoti. 2004. “Non-fouling” Oligo(ethylene glycol)- functionalized polymer brushes synthesized by surface-initiated atom transfer radical polymerization. Adv. Mater. 16:338–341. [Google Scholar]
- Maisano, G., D. Majolino, P. Migliardo, S. Venuto, F. Aliotta, and S. Magazu. 1993. Sound velocity and hydration phenomena in aqueous polymeric solutions. Mol. Phys. 78:421–435. [Google Scholar]
- Milner, S. T., and T. A. Witten. 1988. Theory of the grafted polymer brush. Macromolecules. 21:2610–2619. [Google Scholar]
- Müller, E. A., and P. Rasmussen. 1991. Densities and excess volumes in aqueous poly(ethylene glycol) solutions. Journal of Chemical Engineering Data. 36:214–217. [Google Scholar]
- Needham, D., K. Hristova, T. J. McIntosh, M. Dewhirst, N. Wu, and D. D. Lasic. 1992. Polymer-grafted liposomes: physical basis for the “stealth” property. J. Liposome Res. 2:411–430. [Google Scholar]
- Oesterhelt, F., M. Rief, H. E. Gaub. 1999. Single molecule force spectroscopy by AFM indicates helical structure of poly(ethylene-glycol) in water. New J. Phys. 1:6.1–6.11. [Google Scholar]
- Ohnishi, S., M. Hato, T. Tamada, and H. K. Christenson. 1999. Presence of particles on melt-cut mica sheets. Langmuir. 15:3312–3316. [Google Scholar]
- Pasche, S., S. M. De Paul, J. Voros, N. D. Spencer, and M. Textor. 2003. Poly(L-lysine-graft-poly(ethylene glycol) assembled monolayers on niobium oxide surfaces: a quantitative study of the influence of polymer interfacial architecture on resistance to protein adsorption by ToF-SIMS and in situ OWLS. Langmuir. 19:9216–9225. [Google Scholar]
- Prime, K. L., and G. M. Whitesides. 1993. Adsorption of proteins onto surfaces containing end-attached oligo(ethylene oxide): a model system using self-assembled monolayers. J. Am. Chem. Soc. 115:10714–10721. [Google Scholar]
- Raviv, U., J. Frey, R. Sak, P. Laurat, R. Tadmore, and J. Klein. 2002. Properties and interactions of physigrafted end-functionalized poly(ethylene glycol) layers. Langmuir. 18:7482–7495. [Google Scholar]
- Saeki, S., K. Nobuhiro, M. Nakata, and M. Kaneko. 1976. Upper and lower critical solution temperatures in poly (ethylene glycol) solutions. Polymer. 17:685–689. [Google Scholar]
- Schwendel, D., T. Hayashi, R. Dahint, A. Pertsin, M. Grunze, R. Steitz, and F. Schreiber. 2002. Interaction of water with self-assembled monolayers: neutron reflectivity measurements of the water density in the interface region. Langmuir. 19:2284–2293. [Google Scholar]
- Sheth, S. R., and D. Leckband. 1997. Measurements of attractive forces between proteins and end-grafted poly(ethylene glycol) chains. Proc. Natl. Acad. Sci. USA. 94:8399–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szleifer, I. 1997. Polymers and proteins: interactions at interfaces. Curr. Opin. Colloid Interface Sci. 2:337–344. [Google Scholar]
- Tirosh, O., Y. Barenholz, J. Katzhendler, and A. Priev. 1998. Hydration of polyethylene glycol-grafted liposomes. Biophys. J. 74:1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verwey, E. J. W., and J. T. G. Overbeek. 1945. Theory of Stability of Lyophobic Colloids. Elsevier, Amsterdam, The Netherlands.
- Wang, R. L. C., H. J. Kreuzer, and M. Grunze. 1997. Molecular conformation and solvation of oligo(ethylene glycol)-terminated self-assembled monolayers and their resistance to protein adsorption. J. Phys. Chem. B. 101:9767–9773. [Google Scholar]
- Wang, R. L. C., H. J. Kreuzer, and M. Grunze. 2000. The interaction of oligo(ethylene oxide) with water: a quantum mechanical study. Phys. Chem. Chem. Phys. 2:3613–3622. [Google Scholar]
- Zäch, M., and M. Heuberger. 2000. Statistical and systematic errors of the surface forces apparatus. Langmuir. 16:7309–7314. [Google Scholar]
- Zhulina, E. B., O. V. Borisov, and V. A. Priamitsyn. 1989. Theory of steric stabilization of colloid dispersions by grafted polymers. J. Colloid Interface Sci. 137:495–511. [Google Scholar]