

Abstract
Phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) are the main lipid components of the inner bacterial membrane. A computer model for such a membrane was built of palmitoyloleoyl PE (POPE) and palmitoyloleoyl PG (POPG) in the proportion 3:1, and sodium ions (Na+) to neutralize the net negative charge on each POPG (POPE-POPG bilayer). The bilayer was simulated for 25 ns. A final 10-ns trajectory fragment was used for analyses. In the bilayer interfacial region, POPEs and POPGs interact readily with one another via intermolecular hydrogen (H) bonds and water bridges. POPE is the main H-bond donor in either PE⋯PE or PE⋯PG H-bonds; PG⋯PG H-bonds are rarely formed. Almost all POPEs are H-bonded and/or water bridged to either POPE or POPG but PE-PG links are favored. In effect, the atom packing in the near-the-interface regions of the bilayer core is tight. Na+ does not bind readily to lipids, and interlipid links via Na+ are not numerous. Although POPG and POPE comprise one bilayer, their bilayer properties differ. The average surface area per POPG is larger and the average vertical location of the POPG phosphate group is lower than those of POPE. Also, the alkyl chains of POPG are more ordered and less densely packed than the POPE chains. The main conclusion of this study is that in the PE-PG bilayer PE interacts more strongly with PG than with PE. This is a likely molecular-level event behind a regulating mechanism developed by the bacteria to control its membrane permeability and stability consisting in changes of the relative PG/PE concentration in the membrane.
INTRODUCTION
Phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) are the major lipid components of bacterial membranes. For instance, the inner membrane of Escherichia coli contains 70–80% PE, 20–25% PG, and <5% cardiolipin (Dowhan, 1997). In contrast, concentrations of PE and PG in the animal cell membrane are low. In the blood cell, PE and PG constitute 6% and 2%, respectively, of all the membrane phospholipids (Uran et al., 2001). The major lipid component of the animal cell membrane is phosphatidylcholine (PC) (Uran et al., 2001), which is rarely present in the bacterial membrane (Dowhan, 1997).
Comparative studies on PE and PC bilayers revealed that the hydrocarbon chain order is higher in PE than in PC bilayers (Urbina et al., 1998; Thurmond et al., 1991) due to a smaller cross sectional area of the PE molecule (Thurmond et al., 1991). In effect, the main-phase transition temperature of bilayers composed of PEs is higher than that of equivalent PCs (Huang and Li, 1999). The smaller cross sectional area of PE compared to PC originates from a smaller headgroup (the ammonium group is smaller than the choline group) and a smaller number of bound water molecules (McIntosh and Simon, 1986). The latter stems from the ability of the PE ammonium group, in contrast to the PC choline group, to form hydrogen (H) bonds with phosphate and carbonyl oxygen atoms of adjacent PE molecules. These intermolecular H-bonds replace some of the PE-water H-bonds and significantly strengthen interlipid contacts. Thus, the differences between PE and PC bilayers can be directly related to intermolecular H-bonding that is present in the PE bilayer but absent in the PC bilayer. PE⋯PE intermolecular H-bonds were observed both experimentally (Boggs, 1987; Hubner and Blume, 1998; and references cited therein) and in molecular dynamics (MD) simulations (Pink et al., 1998; Damodaran and Merz, 1994). In PC bilayers and monolayers, positively charged choline groups only weakly interact with negatively charged phosphate or carbonyl groups of adjacent PC molecules by forming charge pairs (Yeagle et al., 1977; Pasenkiewicz-Gierula et al., 1999; Kaznessis et al., 2002). The Berkowitz group identified these interactions as weak H-bonding (Pandit et al., 2003a).
Similarly to the ammonium group of PE, the hydroxyl group of PG has a potential to form intermolecular H-bonds. The occurrence of PG⋯PG H-bonds in a PG monolayer (Dicko et al., 1998) and bilayer (Zhang et al., 1997) was suggested by experimental studies. Limited formation of such bonds was demonstrated in MD simulation of a PG monolayer (Kaznessis et al., 2002). These interlipid interactions via H-bonding are evidently weakened by electrostatic repulsion of negatively charged PGs, since only at high ionic strength is the main-phase transition temperature of a PG bilayer similar to that of an equivalent PC bilayer (Huang and Li, 1999; Lamy-Freund and Riske, 2003). At low ionic strength, the gel-liquid transition displays a complex thermal behavior (Lamy-Freund and Riske, 2003).
Studies on the toxic effects of organic solvents on bacteria revealed that in an adaptive mechanism of solvent-tolerant bacteria the amount of PG relative to PE in the membrane changes; depending on the chemical character of the solvent molecules it increases or decreases (Weber and de Bont, 1996; Isken and de Bont, 1998). This alteration in the headgroup composition is the way of preserving the stability and low permeability of the membrane by increasing the average phospholipid headgroup area and presumably the chain order. The way in which a negatively charged PG headgroup could promote a higher order of chains in the membrane is not that obvious. Nevertheless, a higher order of PG than PC chains was observed in a MD simulation study of mixed lipid PC-PG monolayers (Kaznessis et al., 2002).
In this study, a MD simulation was applied to investigate the properties of a bilayer built of PE and PG molecules in the proportion 3:1. Mixed phospholipid-charged bilayers, which provide a good model for bacterial membranes, are studied much less frequently than bilayers made of zwitterionic phospholipids that constitute models of eukaryotic membranes. Indeed, the majority of lipid bilayers studied by MD simulation are uncharged single-component bilayers built predominantly of PC (cf. Scott, 2002) with only a few built of PE (Zhou and Schulten, 1995; Damodaran and Merz, 1994; Damodaran et al., 1992; Raghavan et al., 1992). Only recently the bilayers (Mukhopadhyay et al., 2004; Pandit and Berkowitz, 2002; Pandit et al., 2003a; Gurtovenko et al., 2004; Cascales et al., 1996; Cascales and de la Torre, 1997) and monolayers (Kaznessis et al., 2002) composed of charged or mixtures of uncharged and charged lipids have also been simulated. Those simulations demonstrated that the properties of such bilayers are dominated by electrostatic interactions among lipid headgroups, ions, and water at the membrane/water interface. Our study provides some new insight into the organization of the mixed phospholipid-charged bilayer, but it concerns predominantly the atomic-level mechanisms in such a bilayer that may be utilized by bacteria when adapting to environmental changes.
METHOD
Simulation systems
The POPE-POPG bilayer used in this study consisted of 54 palmitoyloleoyl-phosphatidylethanolamine (POPE), 18 palmitoyloleoylphosphatidylglycerol (POPG) (25 mol % POPG), and 1955 water molecules. Eighteen sodium ions (Na+) were added to neutralize the net charge of −1.0 in units of electronic charge (e) on each POPG molecule. The bilayer was constructed by replacing the PC choline moiety in the palmitoyloleoylphosphatidylcholine (POPC) bilayer equilibrated in 6-ns MD simulation (Murzyn et al., 2001) with ammonium and glycerol groups to make POPE and POPG, respectively. Details concerning the membrane construction are given in Murzyn and Pasenkiewicz-Gierula (1999). An initial 11-ns simulation of the POPE-POPG bilayer was carried out using AMBER 4.0 (Pearlman et al., 1991). In this part of the simulation, a residue-based cut-off distance of 12 Å was used to evaluate the nonbonded interactions. The simulation was continued for another 14 ns using AMBER 5.0 (Case et al., 1997). In this part of the simulation, the electrostatic interactions were evaluated using the particle mesh Ewald (PME) summation method (Essmann et al., 1995). Since only the PME simulation of a bilayer containing charged molecules generates reliable results (Róg et al., 2003), the initial 11-ns trajectory was ignored in the following analyses. The assessment of the POPE-POPG bilayer equilibration is thus limited to the 14-ns trajectory generated in PME simulation and is discussed in the Results section. Some results of the POPE-POPG bilayer analyses were compared to those of a POPC bilayer. The POPC bilayer was simulated for 17 ns; an initial 11-ns simulation was carried out using AMBER 4.0 and a 12-Å cut-off (Murzyn et al., 2001), then using AMBER 5.0 and a PME summation (Essmann et al., 1995) with the same parameters as in the case of the POPE-POPG bilayer (see below). The last 4-ns trajectory fragment was used for analyses (Róg et al., 2003). Fig. 1 shows the structure and numbering of atoms and torsion angles in POPC, POPE, and POPG molecules.
FIGURE 1.

Molecular structures with numbering of atoms and torsion angles of POPC (a), POPE (b), and POPG (c) (chemical symbol for carbon atoms, C, is omitted).
Simulation parameters
For phospholipids, OPLS parameters (Jorgensen and Tirado-Rives, 1988), for water, TIP3P parameters (Jorgensen et al., 1983), and for sodium ions, Aqvist's parameters (Aqvist, 1990) were used. The united-atom approximation was applied to the CH, CH2, and CH3 groups of the phospholipids. The numerical values for the atomic charges for POPE and POPG are given in Murzyn and Pasenkiewicz-Gierula (1999) and for POPC follow those in Charifson et al. (1990). Other details concerning the POPE-POPG bilayer can be found in Murzyn and Pasenkiewicz-Gierula (1999) and the POPC bilayer in Murzyn et al. (2001). Force field parameters, energy function, simulation protocol, and simulation parameters used in this study generated a bilayer that compares well with relevant experimental models (a validation of the POPE-POPG bilayer is given in the Discussion section and that of the POPC bilayer in Murzyn et al., 2001).
Simulation conditions
Three-dimensional periodic boundary conditions with the usual minimum image convention were used. The SHAKE algorithm (Ryckaert et al., 1977) was used to preserve the bond lengths of the OH and NH3 groups of water, POPG, and POPE molecules, and the time step was set at 2 fs (Egberts et al., 1994). To evaluate the nonbonded interactions, a PME summation (Essmann et al., 1995) with a real cut-off of 12 Å, β-spline interpolation order of 5, and direct sum tolerance of 10−6 was used. The list of nonbonded pairs was updated every 25 steps. Restraints of a flat-bottom harmonic potential as defined and implemented in the AMBER package (Case et al., 1997) were imposed on the double bond to prevent cis-trans isomerization.
The MD simulation was carried out at constant pressure (1 atm) and at the temperature of 310 K (37°C), which is above the main-phase transition temperature for POPC (−5°C) (Seelig and Waespe-Šarčević, 1978), pure POPE (26.1°C) (Huang and Li, 1999), and pure POPG (−4°C) (Boggs and Tümmler, 1993) bilayers. The temperatures of the solute and solvent were controlled independently. Both the temperature and pressure of the system were controlled by the Berendsen method (Berendsen et al., 1984). The relaxation times for temperatures and pressure were set at 0.4 and 0.6 ps, respectively. The applied pressure was controlled anisotropically, each direction being treated independently with the trace of the pressure tensor kept constant at 1 atm.
RESULTS
Equilibration of the membrane system
To evaluate the process of the POPE-POPG bilayer equilibration, time profiles of several parameters were recorded from the onset of PME simulation until 25 ns (14-ns time span) (Fig. 2). These parameters include the system temperature (Fig. 2 a) and potential energy (Fig. 2 b), the surface area per lipid (Fig. 2 c), the number of interlipid H-bonds (for explanation, see the Water/membrane interface section; Fig. 2 d), and the number of neighbors (for explanation, see the Membrane core section; Fig. 2 e). The surface area per lipid is a slowly converging parameter of MD simulation, but its average value, as well as that of the system temperature, was stable over this portion of the simulation time. The number of neighbors and the number of interlipid H-bonds are the key parameters of this study. They give an account of the bilayer core and the bilayer interface, respectively. Stabilization of their average values in time is necessary to carry out meaningful analyses of the MD trajectory. As Fig. 2 shows, the values as well as that of the system potential energy changed initially but became stable within 4 ns of PME simulation. Therefore, we concluded that the POPE-POPG bilayer had reached a steady state after 4 ns of PME simulation and the last 10-ns trajectory fragment generated between 15 and 25 ns of MD simulation was used for analyses. In a recent article, Falck et al. (2004) evaluated the equilibration process of PC bilayers containing varying amounts of cholesterol by monitoring the time profile of the average area per lipid in 100-ns MD simulations. The equilibration time increased with an increasing cholesterol concentration and was related to the rate of the PC lateral diffusion. The lateral diffusion coefficient for PC is 10-fold larger in the bilayer containing 0% than in that containing 50% cholesterol. As the bilayer containing 50% cholesterol equilibrated within 20 ns (Falck et al., 2004), the equilibration time for the pure PC bilayer must be significantly shorter. The average values given below are ensemble and time averages obtained from the block averaging procedure. Errors in the derived average values are standard error estimates.
FIGURE 2.

Diagrams showing the time development, from the onset of PME simulation (11 ns) until 25 ns, of the temperature (a), potential energy (b), surface area per lipid (c) (the equilibrium average surface area is 61.5 ± 0.2 Å2), number of intermolecular H-bonds (d), and number of neighbors (e) for the POPG-POPE bilayer. The thin line in c indicates the average value of the surface area in the time range between 15 and 25 ns.
Spatial properties of the membrane
A two-dimensional Voronoi tessellation method (Shinoda and Okazaki, 1998) allows one to compute the surface area of a single molecule in the bilayer made of lipids of a single type (cf. Murzyn et al., 2001). In the case of a mixed lipid bilayer, the method is less precise; nevertheless, it was applied to estimate the surface area of each POPE and POPG molecule in the POPE-POPG bilayer. From the estimated single molecular areas, the average values for POPE and POPG of 61.0 ± 0.2 and 62.8 ± 0.3 Å2, respectively, were obtained (Table 1). The averaged overall lipid-molecule surface area in the POPE-POPG bilayer is 61.5 ± 0.2 Å2 and in the POPC bilayer is 63.5 ± 0.2 Å2 (Róg et al., 2003). The average surface area per POPC agrees well with the values of 63 Å2 (Smaby et al., 1997) and 66 Å2 (Hyslop et al., 1990) obtained from POPC monolayer studies at surface pressures of 30 mN/m and 20 mN/m, respectively. As one would expect, the area per POPC is greater than the area per POPE; however, there is no experimental data concerning the area per POPE or POPG in the mixed lipid POPE-POPG bilayer.
TABLE 1.
Average values of parameters
| POPE | POPG | POPC | |
|---|---|---|---|
| Area (Å2) | 61.0 ± 0.2 | 62.8 ± 0.3 | 63.5 ± 0.2 |
| P-P distance (Å) | 36.4 ± 0.1 | 35.6 ± 0.1 | 35.5 ± 0.1 |
| P-C vector inclination (°) | 24 ± 1 | 36 ± 1 | 30 ± 1 |
| C-C vector inclination (°) | 36 ± 1 | 40 ± 1 | 44 ± 1 |
| No. of neighbors | 41.6 ± 0.05 | 40.3 ± 0.05 | 39.9 ± 0.05 |
| Diffusion coefficient (cm2/s × 10−7) | 3.5 ± 0.1 | 3.5 ± 0.1 | 4.5 ± 0.1 |
The surface area per lipid (Area), distance between average positions of phosphorus (P) atoms in the two leaflets of a bilayer (P-P distance), estimated orientation of the lipid α-chain (P-C vector inclination) and glycerol backbone (C-C vector inclination), number of neighbors, and short timescale diffusion coefficient of a lipid molecule, in the POPE-POPG and POPC bilayers. The errors in the average values are standard error estimates.
The thickness of a bilayer is not a well-defined quantity (Nagle et al., 1996), therefore, the thickness of the POPE-POPG bilayer was evaluated in comparison to the POPC bilayer as a distance between average positions of phosphorus (P) atoms in the two leaflets of a bilayer (P-P distance). The values for POPE-POPG and POPC bilayers are 36.0 ± 0.1 and 35.5 ± 0.1 Å (Róg et al., 2003), respectively (Table 1).
Average positions of P and carbonyl oxygen, O22, atoms (cf. Fig. 1) of POPE and POPG along the bilayer normal were calculated. P and O22 atoms of PG are located deeper in the membrane interface than the corresponding PE atoms by 0.4 ± 0.1 Å and 0.5 ± 0.1 Å, respectively.
Water/membrane interface
In the following analyses we focus on the short-distance interactions among polar groups of POPG and POPE as well as interactions of these groups with water molecules and Na+. In this study we use the same geometrical definitions of H-bonding and water bridging as in our previous articles (Pasenkiewicz-Gierula et al., 1997; Murzyn et al., 2001). Specifically, an H-bond is judged to be formed when the distance between the H donor (D) and H acceptor (A) is ≤3.25 Å and the angle between the D⋯A vector and the D-H bond (the A⋯D-H angle) is ≤35°. A water bridge is made by a water molecule that is simultaneously H-bonded to two lipid oxygen atoms. We distinguish between intermolecular and intramolecular water bridges. As was shown in Pasenkiewicz-Gierula et al. (1997), although a simple TIP3P potential for water was used, simulation reproduced well both the average numbers of water-water and PC-water H-bonds and the H-bond geometry.
Inter- and intralipid H-bonds
Both the ammonium group of POPE (PE-NH3) and the hydroxyl group of POPG (PG-OH) are H-bond donors, whereas the phosphate and carbonyl groups of both lipids as well as PG-OH are H-bond acceptors. These groups participate in numerous interlipid H-bonds and, to a lesser degree, in intralipid H-bonds (Table 2). PE makes interlipid H-bonds both with PE and PG, whereas PG⋯PG H-bonds practically do not form (0.02/POPG) (Table 2). One of the reasons may be a high “dilution” of PG in the bilayer (the PE/PG ratio is 3). PE is the main donor in PE⋯PG H-bonds (in 91% of cases; Table 2), probably because the ammonium group of the neutral POPE molecule bears a positive charge, whereas the hydroxyl group of the negatively charged POPG molecule is neutral. The number of PE⋯PG H-bonds is 0.44/PE and the number of PE⋯PE H-bonds is 0.63/PE (Table 2). Since the PE/PG ratio is 3, one can conclude that PE⋯PG H-bonds form two times more readily than PE⋯PE H-bonds. The most obvious reason is that PG has a greater number of groups that are H-bond acceptors than PE (Table 2). An average PG makes 1.3 H-bonds with PEs (Table 2). An example of a direct H-bond between POPE and POPG is given in Fig. 3 a.
TABLE 2.
Inter- and intralipid H-bonds
| H-donor | H-acceptor | Per PE | Per PG | Intramolecular |
|---|---|---|---|---|
| PE | PE | 0.63 ± 0.01 | — | 0.22 ± 0.01 |
| NH3 | Op | 0.51 ± 0.01 | — | 0.02 ± 0.01 |
| NH3 | O22 | 0.05 ± 0.01 | — | 0.18 ± 0.01 |
| NH3 | O32 | 0.07 ± 0.01 | — | 0.01 ± 0.01 |
| PE | PG | 0.40 ± 0.01 | 1.19 ± 0.01 | — |
| NH3 | Op | 0.23 ± 0.01 | 0.70 ± 0.02 | — |
| NH3 | O22 | 0.06 ± 0.01 | 0.17 ± 0.02 | — |
| NH3 | O32 | 0.02 ± 0.01 | 0.06 ± 0.02 | — |
| NH3 | O15 | 0.02 ± 0.01 | 0.06 ± 0.02 | — |
| NH3 | O16 | 0.07 ± 0.01 | 0.20 ± 0.02 | — |
| PG | PE | 0.04 | 0.11 | — |
| PG | PG | — | 0.02 | 0.29 |
| O15 | Op | — | 0.01 | 0.28 |
| Total: 1.07 ± 0.01 | Total: 1.30 ± 0.01 |
Average numbers of inter- and intramolecular H-bonds per phospholipid in the POPE-POPG and POPC bilayers. H-bond donor and acceptor molecules as well as groups are distinguished. The errors in the average values are standard error estimates.
FIGURE 3.

Examples of lipid-lipid pairs and lipid-water H-bonds in the POPE-POPG bilayer. A direct H-bond between ammonium group of PE (PE-NH3) and hydroxyl group of PG (PG-OH) (a); a water bridge between phosphate oxygen atoms (Op) of PE and PG (b); a water bridge between PE-NH3 and Op of PE (c); and water molecules H-bonded to an arbitrary chosen POPE headgroup (d). The image was produced with MolScript (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997).
On average, PE-NH3 forms ∼0.2 intramolecular H-bonds, mainly with O22; PG-OH forms ∼0.3 such bonds, mainly with nonestrified phosphate oxygen atoms, Op (Table 2).
PE-PG H-bonded pairs
As diagram in Fig. 4 a shows, during the 10-ns analysis time each PG is H-bonded to at least one PE, but most often, consecutively or simultaneously, to two different PEs. A detailed analysis shows that at a given time, on average, 13 PG molecules are H-bonded to 18 PE molecules. Over 45% of all PGs are H-bonded to one PE, 22% to two PEs, ∼3% to three PEs, and <30% are not H-bonded (Fig. 5 a). In effect, a PG remains in the H-bonded state for 71 ± 13% (mean ± SD) of the analysis time (the range is from 50 to 95%), that is, ∼7 ns. In 92% of cases a PE-PG pair is linked via a single H-bond, and in the remaining 8% via two H-bonds. Fig. 6 a illustrates the dynamic character of PE-PG pairing via H-bonds. Each vertical line indicates a transition between bonded and nonbonded states. Due to a large number of transitions and breaks in bonding of various duration, it is difficult to evaluate precisely the bonding time for a given pair; nevertheless, a coarse estimate is color-coded in Fig. 4 a.
FIGURE 4.

Diagrams showing PE-PG interlipid links in the POPE-POPG bilayer due to direct H-bonding (a), and water bridging (b) formed during the whole analysis time of 10 ns. The color roughly codes the linking time as a percentage of the analysis time: black, 80–100%; blue, 60–80%; green, 40–60%; red, 20–40%, and white, 0–20%. The apparent prevalence of interactions along a diagonal results mainly from the way the lipids were numbered. However, lipid molecules undergo translational diffusion and their interacting partners change during the simulation time.
FIGURE 5.

Distribution of the number of lipids bound to a given lipid via H-bonding (HB) (a), water bridging (WB) (b), or both (HB, WB) (c, d, e, and f). All in f indicates links with PE and/or PG.
FIGURE 6.

Time profiles of binding of a chosen PG molecule (PG 1) to PE molecules in the POPE-POPC bilayer. During the analysis time of 10 ns, PG 1 is bound via H-bonds (a) with five PE molecules (PE 2, 4, 10, 19, and 47), and via water bridges (b) with seven PE molecules. 1, binding; 0, nonbinding. Vertical lines indicate transitions between binding and nonbinding states.
Lipid-water H-bonds
Average numbers of H-bonds formed between water molecules and polar groups of POPE and POPG in the POPE-POPG bilayer, as well as numbers of H-bonded water molecules, are given in Table 3 and compared with those for POPC in the POPC bilayer. Because POPE has more groups that can participate in H-bonding than the other two lipids, it binds more water molecules (5.8/PE) than PG (5.1/PG) or PC (4.9/PC). However, if water molecules forming clathrates around the PC choline group (Pasenkiewicz-Gierula et al., 1997) are also considered, then the total hydration of POPC of 11.3 ± 0.1 (Róg et al., 2003) is significantly higher than that of POPE and POPG. Since the area per POPC is larger than the area per POPE or POPG, this result is consistent with the observed correlation between the average surface area available to a headgroup and the headgroup hydration (McIntosh and Simon, 1986; Yeagle et al., 1977; Murzyn et al., 2001). Slightly smaller numbers of H-bonds that water molecules form with Op and O22 of PG than of PE might result from lower location of these PG atoms in the interfacial region. An example of water molecules H-bonded to POPE is shown in Fig. 3 d.
TABLE 3.
Water-lipid H-bonds
| POPE | POPG | POPC | |
|---|---|---|---|
| Group/molecule | No. of H-bonds | No. of H-bonds | No. of H-bonds |
| O13 | 1.96 ± 0.01 | 1.75 ± 0.02 | 2.1 ± 0.1 |
| O14 | 1.96 ± 0.01 | 1.75 ± 0.02 | 2.1 ± 0.1 |
| O22 | 0.50 ± 0.01 | 0.41 ± 0.02 | 0.7 ± 0.1 |
| O32 | 0.58 ± 0.01 | 0.69 ± 0.02 | 0.6 ± 0.1 |
| NH3 | 1.57 ± 0.01 | — | — |
| O15 | — | 0.44 ± 0.02 | — |
| O16 | — | 1.07 ± 0.02 | — |
| Total | 6.57 ± 0.01 | 6.11 ± 0.01 | 5.5 ± 0.1 |
| NCH3 | — | — | 6.4 ± 0.1 |
| No. of H-bonded | 5.78 ± 0.01 | 5.12 ± 0.01 | 4.9 ± 0.1 |
| water (bound) | — | — | (11.3 ± 0.1) |
Numbers of H-bonds between water and polar groups of POPE and POPG per group and per PE and PG molecules (Total) as well as the total number of H-bonded water molecules per PE and PG in the POPE-POPG bilayer. For comparison, numbers of H-bonds per group and per PC (Total) and H-bonded water molecules per POPC together with the number of water molecules in a clathrate around the POPC choline group (NCH3) in the POPC bilayer are also given. For oxygen atom symbols, cf. Fig. 1. The number in parentheses is the number of water molecules bound (H-bonded and clathrating) to a POPC (bound). The errors in the average values are standard error estimates.
Water bridges
The smaller number of water molecules H-bonded to PE and PG than H-bonds formed between phospholipids and water (Table 3) indicates that water molecules can be simultaneously H-bonded to two phospholipid molecules and form water bridges (Pasenkiewicz-Gierula et al., 1997). The number of water bridges per PE and PG in the POPE-POPG bilayer are given in Table 4 and compared to that for the POPC bilayer. Most of the water bridges are intermolecular (1.59/PE, 2.19/PG, and 0.6/PC); intramolecular ones constitute <10% of all bridges in the POPE-POPG bilayer (0.16/PE and 0.14/PG) (Table 4). The number of PE∼PG and PE∼PE water bridges is 0.68/PE and 0.91/PE, respectively. Thus, one can draw a similar conclusion as in the case of PE⋯PG H-bonding, that in the PE-PG bilayer, PE∼PG water bridges are formed more readily than PE∼PE water bridges. In contrast to PG⋯PG H-bonds that practically do not form, PG∼PG water bridging takes place in the bilayer but not often. There are just over three PG molecules in the bilayer mutually linked by one (70% of cases) and two or more (30% of cases) water bridges.
TABLE 4.
Water bridges
| Bridge: | POPE | POPG | POPC |
|---|---|---|---|
| PE-PE | 0.91 ± 0.01 (0.16 ± 0.02) | — | — |
| PE-PG | 0.68 ± 0.01 | 2.04 ± 0.01 | — |
| PG-PG | — | 0.15 ± 0.02 (0.14 ± 0.02) | — |
| PC-PC | — | — | 0.60 ± 0.01 (0.19 ± 0.01) |
| Total | 1.59 ± 0.01 | 2.19 ± 0.02 | 0.60 ± 0.01 |
Average numbers of intermolecular water bridges between phospholipids of the same (PE-PE, PG-PG, and PC-PC) or different (PE-PG) types per phospholipid as well as the total number (Total) of intermolecular water bridges per phospholipid in the POPE-POPG and POPC bilayers. For comparison, average numbers of intramolecular water bridges (not included in Total) are listed in parentheses. The errors in the average values are standard error estimates.
PE∼PG water bridged pairs
As the diagram in Fig. 4 b shows, during the 10-ns analysis time each PG is, consecutively or simultaneously, linked via water bridges to several PEs. A detailed analysis shows that at a given time, on average ∼16 PG molecules are water bridged to ∼25 PE molecules; ∼33% of all PGs are bridged to one PE, ∼35% to two PEs, ∼16% to three PEs, ∼4% to four or more PEs, and <13% are not bridged (Fig. 5 b). In effect, a PG remains in the water bridged state for 88 ± 7% (mean ± SD) of the analysis time (the range is from 69% to 97%), which is ∼9 ns. In 83% of cases a PE∼PG pair is linked via a single water bridge, in ∼15% via a double water bridge, in the remaining 2% via more than two bridges. As Fig. 6 b shows, PE∼PG water bridging is dynamic in a way similar to PE⋯PG H-bonding (cf. Fig. 6 a). Again, due to a large number of transitions and breaks in bridging of various duration, it is difficult to evaluate precisely the time for which a given pair is linked via a water bridge; nevertheless, a coarse estimate is color-coded in Fig. 4 b. Examples of POPE∼POPG and POPE∼POPE water bridges are shown in Fig. 3, b and c, respectively.
Interlipid pairs
In the interfacial region of the POPE-POPG bilayer, polar groups of the lipid molecules interact via H-bonding and water bridging. These interlipid interactions involve a majority of the bilayer lipids. On average, one PE (Fig. 5 f) and two PG (Fig. 5 c) molecules are not linked with other molecules by either H-bonds or water bridges at a given time. Nevertheless, during the whole analysis time of 10 ns, all PEs and PGs were interlinked for 98 ± 4% (mean ± SD; the range is from 77% to 100%) and 88 ± 9% (the range is from 84% to 100%), respectively, of the analysis time. PE interacts with both PE and PG, whereas PG interacts practically only with PE. The number of cases in which PG is linked with zero, one, and more PEs via H-bonding and/or water bridging is shown in Fig. 5 c. On average, each PG is linked with 1.7 PEs. The number of cases when PE is linked with zero, one, and more PEs and PGs via H-bonding and/or water bridging is shown in Fig. 5, d and e, respectively. On average, each PE molecule is linked with 2.2 PE and 0.9 PG molecules. Since the PE/PG ratio is 3, one can conclude that PE-PG links form more readily than PE-PE links. The number of cases when PG is linked with zero, one, and more other lipids (PEs and PGs) via H-bonding and/or water bridging is shown in Fig. 5 f.
Lipid-ion interaction
Radial distribution functions (RDF) of the Na+ relative to Op, Oc (carbonyl oxygen atoms O22 and O32, cf. Fig. 1), and Oe (glycerol oxygen atoms O15 and O16, cf. Fig. 1 c) atoms of POPE and POPG are shown in Fig. 7. Each RDF has a distinct maximum at the distance of 2.5 Å, a minimum at ∼3 Å, and a broader second maximum at 4.5–5 Å. The first maximum indicates binding of ions by Op, Oc, and Oe; the second maximum suggests that ions interact also with more distant oxygen atoms belonging to the same or neighboring lipid molecules. Based on the position of the first minimum in the RDFs in Fig. 7, it was assumed that an ion is bound to the lipid when its distance from a lipid oxygen atom is ≤3 Å. On average, 42% of Na+ ions are bound to either PE or PG, ∼39% to PE, and ∼15% to PG, but in over 10% of cases, Na+ is simultaneously bound to two lipid molecules (ion bridge). Ion bridges are thus not common and they form only between PE and PG; 10% of PGs are linked to PEs via Na+. Remembering that in the PE-PG bilayer the PE/PG ratio is 3, one can conclude that Na+ interacts preferentially with PG compared to PE; however, PG-Na+ interactions are not numerous as they involve only 15% of PGs. Na+ interacts more often with Op (83%) than Oc (17%) of PE but more often with Oc (42%) and Oe (34%) than Op (24%) of PG. This latter result is in qualitative agreement with that of Mukhopadhyay et al. (2004), showing that Na+ binds more readily to carbonyl than to phosphate oxygen atoms of the negatively charged palmitoyloleoylphosphatidylserine.
FIGURE 7.

Radial distribution functions (RDF) of sodium ions (Na+) relative to the phosphate, Op (a), carbonyl, Oc (b), and glycerol, Oe (c) oxygen atoms of POPE (thin line) and POPG (thick line) in the POPE-POPC bilayer. The percentage contribution of the PE and PG oxygen atoms in binding Na+ is given in the text.
Headgroup orientation
The strength of interactions among polar groups of the phospholipids depends on the relative orientations of these groups. To calculate the orientation of a phospholipid headgroup, two vectors were defined. One, representing the lipid α-chain, is the vector linking P and C12 (cf. Fig. 1) atoms (P-C vector); the other, representing the glycerol backbone, is the vector linking C1 and C3 (cf. Fig. 1) atoms (C-C vector). The orientation of both vectors was described as an angle between the vector and the bilayer surface. Distributions of angles for PE and PG are shown in Fig. 8 and average values are given in Table 1. Both the α-chain and glycerol backbone of PE are less inclined relative to the bilayer surface than PG (Table 1). For comparison, the inclination of the POPC α-chain is between that of POPE and POPG, whereas the glycerol backbone of POPC is more inclined than those of POPE and POPG (Table 1).
FIGURE 8.

Distributions of angles between the membrane surface and the P-C vector (a) and C-C vector (b) of POPE (thick line) and POPG (thin line) in the POPE-POPC bilayer. The vector notation is given in the text.
Membrane core
Chain order
The magnitude of interlipid interactions at the bilayer interface affects the order and packing of the hydrocarbon chains of phospholipids. In the following analyses, the correlation between the properties of the bilayer interface and core are investigated. The molecular order parameter, Smol, profiles (for definition, see Hubbell and McConnell, 1971) along the β- and γ-chain of POPE, POPG, and POPC molecules are shown in Fig. 9, and mean values (averaged over appropriate segmental vectors ≥4; for details see Róg and Pasenkiewicz-Gierula, 2001a) of Smol are given in Table 5. Although POPE and POPG comprise one bilayer, the hydrocarbon chains of POPG are more ordered than those of POPE. The higher values of Smol for both the β- and γ-chain of POPG are due to smaller tilts of the chains and longer average lifetimes of the trans conformation (Table 5).
FIGURE 9.

Molecular order parameter (Smol) profiles along the β-chain (a), and the γ-chain (b), of POPE (thick black line) and POPG (thin black line) in the POPE-POPG bilayer, as well as of POPC (shaded line) in the POPC bilayer. The maximal standard error in the derived average values of Smol is indicated as an error bar. For comparison, the values of Smol obtained in a 2H-NMR experiment for the POPC β-chain at 42°C (•; Seelig and Waespe-Šarčević, 1978) are given.
TABLE 5.
Order of hydrocarbon chains
| Membrane | POPE
|
POPG
|
POPC
|
|||
|---|---|---|---|---|---|---|
| Chain | β-chain (Δ; ω) | γ-chain | β-chain (Δ; ω) | γ-chain | β-chain (Δ; ω) | γ-chain |
| Smol | 0.30 ± 0.1 | 0.39 ± 0.1 | 0.35 ± 0.1 | 0.45 ± 0.1 | 0.30 ± 0.1 | 0.37 ± 0.1 |
| Tilt (°) | 23.0 ± 0.5 (28.7; 31.8) | 23.5 ± 0.5 | 19.5 ± 0.5 (28.4; 28.4) | 19.5 ± 0.5 | 20.0 ± 0.5 (25.9; 33.3) | 23.5 ± 0.5 |
| No. of gauche | 3.20 ± 0.1 | 2.94 ± 0.1 | 3.08 ± 0.1 | 2.67 ± 0.1 | 3.38 ± 0.1 | 2.97 ± 0.1 |
| Lifetime (ps) trans/gauche | 195 ±2 | 210 ± 5 | 200 ± 5 | 225 ± 5 | 180 ± 5 | 195 ± 5 |
| 60 ± 2 | 55 ± 2 | 60 ± 2 | 50 ± 2 | 60 ± 2 | 50 ± 2 | |
Average values of the molecular order parameter, Smol; chain tilt angle; number of gauche/chain; and lifetimes of trans and gauche conformations, for the β- and γ-chain of POPE, POPG, and POPC in POPE-POPG and POPC bilayers. Also, tilt angles of Δ (above the double bond) and ω (below the double bond) fragments of POPE, POPG, and POPC β-chains (Δ; ω). The tilt angle was calculated according to the definition in Róg et al. (2004). The errors in the average values are standard error estimates.
Atom packing
A neighbor is a carbon atom of an alkyl chain that is located not further than 7 Å away from a chosen carbon atom in the bilayer core and belonging to a different molecule. The distance of 7 Å is the position of the first minimum in the carbon-carbon RDF (for details, see Róg and Pasenkiewicz-Gierula, 2001b). The number of neighbors (NS) is a suitable parameter to sense the packing of atoms in the bilayer core. Profiles of NS along the β- and γ-chains of POPE and POPG in the POPE-POPG bilayer as well as POPC in the POPC bilayer are shown in Fig. 10. Average NS for POPE, POPG, and POPC are given in Table 1. Fig. 10 demonstrates that packing of atoms in upper fragments of the chains is much denser in the POPE-POPG bilayer than in the POPC bilayer. The effect for the γ-chains extends deeper into the core than for the β-chains. In general, packing of atoms around POPG chains is less dense than around POPE chains, and in the lower parts of the chains, less dense than around POPC chains.
FIGURE 10.

Profiles of the number of neighbors (NS) (cf. text) along the β-chain (a), and the γ-chain (b) of POPE (thick black line) and POPG (thin black line) in the POPE-POPG bilayer, as well as of POPC (shaded line) in the POPC bilayer. The maximal standard error is indicated as an error bar.
Lipid diffusion
The last 10-ns fragment of the 25-ns trajectory generated in MD simulation was analyzed to obtain results discussed in this article. However, as Böckmann et al. (2003) show, this timescale is too short to make derivation of translational self-diffusion coefficients for Brownian motion of the membrane lipids possible. This timescale allows only getting self-diffusion coefficients for faster short-distance, short-timescale unconfined diffusion. Diffusion coefficients for the short-timescale translational self-diffusion of POPE and POPG in the POPE-POPG bilayer plane, as well as that of POPC in the POPC bilayer plane, were calculated from linear parts of the respective mean square displacement curves for the centers of masses of PE, PG, and PC molecules in the bilayers. The coefficients are given in Table 1. Their comparison indicates that the rate of the short-timescale diffusion of POPE is similar to POPG but is slower than POPC (Table 1).
DISCUSSION
A stable computer model of the bacterial membrane, consisting of POPE and POPG molecules in the proportion 3:1, counterions, and water was generated in a 25-ns MD simulation. Direct comparison of this to experimental models is difficult because we have found only three articles devoted to PE-PG bilayers (Garidel and Blume, 2000; Inoue and Nibu, 1999; Tari and Huang, 1989). Using differential scanning calorimetry, Garidel and Blume (2000) detected preferential formation of mixed pairs in PE-PG bilayers at pH 7. This result corresponds well with our observations and will be discussed below, but it is at variance with the results of Inoue and Nibu (1999), who, however, did not strictly control experimental conditions. Other data available in the literature that might seem relevant to our model concern pure PE (e.g., Urbina et al., 1998; Thurmond et al., 1991; Zhou and Schulten, 1995; Hubner and Blume, 1998; Pink et al., 1998; Damodaran and Merz, 1994) and pure PG (e.g., Zhang et al., 1997; Dicko et al., 1998; Kurze et al., 2000; Kaznessis et al., 2002; Lamy-Freund and Riske, 2003) bilayers and monolayers. There is also a limited number of articles on mixed PC-PG bilayers (Kurze et al., 2000; Kaznessis et al., 2002), as well as a series of articles dealing with interactions of sodium ions with polar and charged phospholipids (Pandit et al., 2003a,b; Mukhopadhyay et al., 2004; Pandit and Berkowitz, 2002; Cascales and de la Torre, 1997; Böckmann et al., 2003). Nevertheless, single-molecule properties of a phospholipid in pure-lipid and mixed-lipid bilayers are different as they depend on other membrane components. Therefore, verification of the PE-PG bilayer against available data for pure PE and PG, as well as mixed PC-PG systems, is not fully justified; nevertheless, some trends might be common for all these bilayers. In general, the surface area available to a PE headgroup in the bilayer is smaller and the order of PE hydrocarbon chains is higher than those of a PC (Thurmond et al., 1991; Urbina et al., 1998). The ammonium group of PE forms intermolecular H-bonds with PE phosphate (Boggs, 1987) and carbonyl (Lewis and McElhaney, 1998) groups. The hydroxyl group of PG was postulated to form scarce intermolecular H-bonds with the PG phosphate group (Dicko et al., 1998; Zhang et al., 1997; Kaznessis et al., 2002), and hydrocarbon chains of PG are more ordered than those of PC (Kaznessis et al., 2002). The number of water molecules directly interacting with a headgroup is smaller for PE than PC (McIntosh, 1996). The above characteristics are similar to those displayed by PE and PG in the PE-PG bilayer generated in this study as a model of the bacterial membrane. The model was used to investigate basic interactions between neutral (POPE) and charged (POPG) lipids comprising the membrane. Elucidation of these interactions provides a better understanding of the bacterial membrane organization, but in this study it is the key to explaining why an increase in the concentration of PG relative to PE in the membrane of solvent-tolerant bacteria might be beneficial for cell survival.
The first important result of this study is the identification of specific interactions among lipids in the POPE-POPG bilayer. The prevailing interaction is indirect water bridging, then direct H-bonding and, to a much lesser degree, ion bridging. On average, <1 PE and <2 PG molecules in the bilayer are not mutually bound at any instant via H-bonds and/or water bridges. PE is bound to either PE or PG, whereas PG is bound practically only to PE.
The second important result is a higher preference of PE to interact with PG than with PE. On average each PG is linked with 1.7 PEs. Calculating average numbers of PEs and PGs linked to a PE is not straightforward, because the proportion of PE to PG in the bilayer is 3:1. Assuming that PE interacts with PE and PG with the same preference, then three times more PEs should interact with PEs than PGs. But the number is 2.56, thus <3. Furthermore, PE-PG pairs are linked by a larger number of individual H-bonds and water bridges than PE-PE pairs (Tables 2 and 4). These results correspond well with calorimetric studies of Garidel and Blume (2000), who detected preferential formation of mixed PE-PG pairs in PE-PG bilayers at pH 7. Such a result is not surprising if one remembers that 1), the ammonium group of PE is a stronger H-bond donor than the OH groups of PG; 2), the number of groups accepting H-bonds in PG is larger than in PE; and 3), water readily makes H-bonds with polar groups of both PE and PG. Ion bridges, observed in MD simulations of PC-NaCl (Pandit et al., 2003b), PS-Na+ (Mukhopadhyay et al., 2004), and 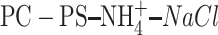 (Pandit et al., 2003a) bilayers, are rare in the PE-PG bilayer. This is most likely due to stronger direct and water-mediated interlipid interactions in the POPE-POPG bilayer.
(Pandit et al., 2003a) bilayers, are rare in the PE-PG bilayer. This is most likely due to stronger direct and water-mediated interlipid interactions in the POPE-POPG bilayer.
The third important result of this study is a much denser packing of chain atoms in the near-the-interface regions of the hydrophobic core of the POPE-POPG as compared with the POPC bilayer (Fig. 10). The denser packing is more strongly sensed by the γ-chains of both PE and PG than by the β-chains, undoubtedly because of the rigid bend in the β-chain at the cis double bond. More compact off-center parts of the hydrophobic core in the POPE-POPG than in the POPC bilayer results from interrelated effects. In particular, specific interactions among lipid headgroups are more numerous, and PE and PG are less hydrated and closer to one another. Moreover, high probability of gauche in the beginning of the POPC β-chain (Róg et al., 2004), not observed for β-chains of POPE and POPG (data not shown), may disturb the packing of atoms in the near-the-interface regions of the POPC bilayer core. Consistent with stronger interlipid interactions in both the bilayer interface and the core is the reduced dynamics of the lipids; indeed, both translational self-diffusion (Table 1) and trans-gauche isomerization (longer lifetime of trans conformations, Table 5) are slower in the PE-PG than in the PC bilayer.
Although POPG and POPE comprise one bilayer, their bilayer properties differ. Most remarkable differences between PG and PE are a higher order of the PG chains and a less dense atom packing along the PG chains. The reasons for such differences are not that apparent. They may stem from relative geometries of PG and PE headgroups that facilitate interlipid H-bonding and water bridging. These interactions, either in a PE-PE or PE-PG pair, involve predominantly the NH3 group as an H-bond donor and the PO4 group as an H-bond acceptor. To interact effectively, these groups should be close to each other. Minimization of their vertical distance can be achieved by orientation of the P-C and C-C vectors of the PE headgroup more parallel to the bilayer surface to level up the position of NH3 with that of PO4. The P-C and C-C vectors of the PG headgroup have to be more inclined to accommodate the glycerol group that is much larger than the NH3 group. These headgroup-orienting preferences are observed in the POPE-POPG bilayer. A more vertical orientation of the C-C vector of POPG is the likely reason for a smaller tilt of PG chains and consequently higher values of Smol along PG chains than PE chains (Table 5). A smaller tilt is particularly apparent for the ω (below the double bond) fragment of the PG β-chain. Thus, lower fragments of less numerous and less tilted POPG chains are surrounded by more tilted fragments of POPE chains; in effect their neighborhood is less densely packed than that of POPE chains.
In general, organic solvents destabilize the lamellar structure of a PE bilayer by decreasing the lamellar-to-inverted-hexagonal-phase transition temperature and/or increasing the bilayer permeability (for recent reviews, see Weber and de Bont, 1996; Isken and de Bont, 1998). To counteract these effects, solvent-tolerant bacteria, whose lipid matrix consists mainly of PE and PG in the proportion 3:1 (Dowhan, 1997), developed several adaptation mechanisms. One of them is cis-trans isomerization, discussed in our previous article (Róg et al., 2004); another is changes in the headgroup composition of the membrane. Stability of the membrane lamellar phase can be achieved by increasing an average surface area available to the phospholipid headgroup. Indeed, in the POPE-POPG bilayer the area is larger than that of a PE headgroup and will increase with the increasing PG/PE ratio. A stabilizing effect of PG on a PE bilayer was shown in model studies (Tari and Huang, 1989). However, our results do not support the hypothesis that the decreased permeability of the membrane is due to higher ordering of the alkyl chains; the values of Smol for upper segments of the PE, PG, and PC chains are similar, although the packing of upper-segment atoms is much higher in the PE-PG than the PC bilayer. In light of our results, the more compact packing of atoms is due to strong short-distance PE-PG interactions in the membrane interfacial region. And because PE-PG interactions are stronger than PE-PE interactions, with the increasing PG/PE ratio the membrane becomes less permeable for lipophilic and polar molecules and, at the same time, more stable. In effect, the bacteria can tolerate toxic effects of organic solvents better.
CONCLUSIONS
A stable computer model of the fully hydrated mixed-lipid charged bilayer made of POPE and POPG in the proportion 3:1 and counterions was generated using MD simulation. This bilayer constitutes a suitable model of the inner bacterial membrane.
The main interlipid interactions in the bilayer/water interfacial region are water bridging and direct H-bonding, and, to a much lesser degree, ion bridging. POPE interacts with both POPE and POPG, whereas POPG interacts practically only with POPE.
POPE has a higher preference to interact with POPG than with POPE.
In the near-the-interface regions of the hydrophobic core of the POPE-POPG bilayer the packing of chain atoms is much denser than in the bilayer made of POPC.
The translational self-diffusion of lipids and trans-gauche isomerization are slower in the POPE-POPG than the POPC bilayer.
Bilayer properties of POPG differ from those of POPE. Due to relative geometries of PG and PE headgroups POPG chains are less tilted (more ordered) and less densely packed than those of POPE.
Acknowledgments
All calculations were performed in the Academic Computer Centre Cyfronet, Kraków, Poland, computational grant numbers KBN/SGI_ORIGIN_2000/UJ/048/1999 and KBN/SGI_ORIGIN_200/UJ/004/2000. T.R. and K.M. held fellowship awards from the Polish Foundation for Science in 2003.
References
- Aqvist, J. 1990. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 94:8021–8024. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Böckmann, R. A., A. Hac, T. Heimburg, and H. Grubmüller. 2003. Effect of sodium chloride on a lipid bilayer. Biophys. J. 85:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs, J. M. 1987. Lipid intermolecular hydrogen bonding: influence on structural organization and membrane function. Biochim. Biophys. Acta. 906:353–404. [DOI] [PubMed] [Google Scholar]
- Boggs, J. M., and B. Tümmler. 1993. Interdigitated gel phase bilayers formed by unsaturated synthetic and bacterial glycerolipids in the presence of polymyxin B and glycerol. Biochim. Biophys. Acta. 1145:42–50. [DOI] [PubMed] [Google Scholar]
- Cascales, J. J. L., and J. G. de la Torre. 1997. Effect of lithium and sodium ions on a charged membrane of dipalmitoylphosphatidylserine: a study by molecular dynamics simulation. Biochim. Biophys. Acta. 1330:145–156. [DOI] [PubMed] [Google Scholar]
- Cascales, J. J. L., J. G. de la Torre, S. J. Marrink, and H. J. C. Berendsen. 1996. Molecular dynamics simulation of a charged biological membrane. J. Chem. Phys. 104:2713–2720. [Google Scholar]
- Case, D. A., D. A. Pearlman, J. W. Caldwell, T. E. Cheatham III, W. S. Ross, C. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, D. M. Ferguson, R. J. Radmer, G. L. Seibel, U. C. Singh, P. K. Weiner, and P. A. Kollman. 1997. AMBER 5.0. University of California, San Francisco.
- Charifson, P. S., R. G. Hiskey, and L. G. Pedersen. 1990. Construction and molecular modeling of phospholipid surfaces. J. Comput. Chem. 11:1181–1186. [Google Scholar]
- Damodaran, K. V., and K. M. Merz. 1994. A comparison of DMPC- and DLPE-based lipid bilayers. Biophys. J. 66:1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damodaran, K. V., K. M. Merz, and B. P. Gaber. 1992. Structure and dynamics of the dilauroylphosphatidylethanolamine lipid bilayer. Biochemistry. 31:7656–7664. [DOI] [PubMed] [Google Scholar]
- Dicko, A., H. Bourque, and M. Pezolet. 1998. Study by infrared spectroscopy of the conformation of dipalmitoylphosphatidylglycerol monolayers at the air-water interface and transferred on solid substrates. Chem. Phys. Lipids. 96:125–139. [Google Scholar]
- Dowhan, W. 1997. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 66:199–232. [DOI] [PubMed] [Google Scholar]
- Egberts, E., S.-J. Marrik, and H. J. C. Berendsen. 1994. Molecular dynamics simulation of phospholipid membrane. Eur. Biophys. J. 22:423–436. [DOI] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Falck, E., M. Patra, M. Karttunen, M. T. Hyvönen, and I. Vattulainen. 2004. Lessons of slicing membranes: interplay of packing, free area, and lateral diffusion in phospholipid/cholesterol bilayers. Biophys. J. 87:1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garidel, P., and A. Blume. 2000. Miscibility of phosphatidylethanolamine-phosphatidylglycerol mixtures as a function of pH and acyl chain length. Eur. Biophys. J. 28:629–638. [DOI] [PubMed] [Google Scholar]
- Gurtovenko, A. A., M. Patra, M. Karttunen, and I. Vattulainen. 2004. Cationic DMPC/DMTAP lipid bilayers: molecular dynamics study. Biophys. J. 86:3461–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. H., and S. S. Li. 1999. Calorimetric and molecular mechanics studies of the thermotropic phase behavior of membrane phospholipids. Biochim. Biophys. Acta. 1422:273–307. [DOI] [PubMed] [Google Scholar]
- Hubbell, W. L., and H. M. McConnell. 1971. Molecular motion in spin-labeled phospholipids and membranes. J. Am. Chem. Soc. 93:314–326. [DOI] [PubMed] [Google Scholar]
- Hubner, W., and A. Blume. 1998. Interactions at the lipid-water interface. Chem. Phys. Lipids. 96:99–123. [Google Scholar]
- Hyslop, P. A., B. Morel, and R. D. Sauerheber. 1990. Organization and interaction of cholesterol and phosphatidylcholine in model bilayer membranes. Biochemistry. 29:1025–1038. [DOI] [PubMed] [Google Scholar]
- Inoue, T., and Y. Nibu. 1999. Phase behavior of hydrated lipid bilayer composed of binary mixture of phospholipids with different head groups. Chem. Phys. Lipids. 100:139–150. [Google Scholar]
- Isken, S., and J. A. M. de Bont. 1998. Bacteria tolerant to organic solvents. Extremophiles. 2:229–238. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Jorgensen, W. L., and J. Tirado-Rives. 1988. The OPLS potential functions for proteins: energy minimization for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- Kaznessis, Y. N., S. T. Kim, and R. G. Larson. 2002. Simulations of zwitterionic and anionic phospholipid monolayers. Biophys. J. 82:1731–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P. 1991. MolScript: a program to produce both detailed and schematic plots of proteins. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- Kurze, V., B. Steinbauer, T. Huber, and K. Beyer. 2000. A H-2 NMR study of macroscopically aligned bilayer membranes containing interfacial hydroxyl residues. Biophys. J. 78:2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy-Freund, M. T., and K. A. Riske. 2003. The peculiar thermo-structural behavior of the anionic lipid DMPG. Chem. Phys. Lipids. 122:19–32. [DOI] [PubMed] [Google Scholar]
- Lewis, R. N. A. H., and R. N. McElhaney. 1998. The structure and organization of phospholipid bilayers as revealed by infrared spectroscopy. Chem. Phys. Lipids. 96:9–21. [Google Scholar]
- McIntosh, T. J. 1996. Hydration properties of lamellar and non-lamellar phases of phosphatidylcholine and phosphatidylethanolamine. Chem. Phys. Lipids. 81:117–131. [DOI] [PubMed] [Google Scholar]
- McIntosh, T. J., and S. A. Simon. 1986. Area per molecule and distribution of water in fully hydrated dilauroylphosphatidylethanolamine bilayer. Biochemistry. 25:4948–4952. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505–524. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, P., L. Monticelli, and D. P. Tieleman. 2004. Molecular dynamics simulation of a palmitoyl-oleoyl-phosphatidylserine bilayer with Na+ counterions and NaCl. Biophys. J. 86:1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzyn, K., and M. Pasenkiewicz-Gierula. 1999. Construction and optimization of a computer model for a bacterial membrane. Acta Biochim. Pol. 46:631–639. [PubMed] [Google Scholar]
- Murzyn, K., T. Róg, G. Jezierski, Y. Takaoka, and M. Pasenkiewicz-Gierula. 2001. Effects of phospholipid unsaturation on the membrane/water interface: a molecular simulation study. Biophys. J. 81:170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F., R. Zhang, S. Tristram-Nagle, W. Sun, H. I. Petrache, and R. M. Suter. 1996. X-ray structure determination of fully hydrated L alpha phase dipalmitoylphosphatidylcholine bilayers. Biophys. J. 70:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S. A., and M. L. Berkowitz. 2002. Molecular dynamics simulation of dipalmitoylphosphatidylserine bilayer with Na+ counterions. Biophys. J. 82:1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003a. Mixed bilayer containing dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylserine: lipid complexation, ion binding, and electrostatics. Biophys. J. 85:3120–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003b. Molecular dynamics simulation of dipalmitoylphosphatidylcholine bilayer with NaCl. Biophys. J. 85:3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula, M., Y. Takaoka, H. Miyagawa, K. Kitamura, and A. Kusumi. 1997. Hydrogen bonding of water to phosphatidylcholine in the membrane as studied by a molecular dynamics simulation: location, geometry, and lipid-lipid bridging via hydrogen-bonded water. J. Phys. Chem. 101:3677–3691. [Google Scholar]
- Pasenkiewicz-Gierula, M., Y. Takaoka, H. Miyagawa, K. Kitamura, and A. Kusumi. 1999. Charge pairing of headgroups in phosphatidylcholine membranes: a molecular dynamics simulation study. Biophys. J. 76:1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman, D. A., D. A. Case, J. C. Caldwell, G. L. Seibel, U. C. Singh, P. K. Weiner, and P. A. Kollman. 1991. Amber 4.0. University of California, San Francisco.
- Pink, D. A., S. McNeil, B. Quinn, and M. J. Zuckermann. 1998. A model of hydrogen bond formation in phosphatidylethanolamine bilayers. Biochim. Biophys. Acta. 1368:289–305. [DOI] [PubMed] [Google Scholar]
- Raghavan, K., M. R. Reddy, and M. L. Berkowitz. 1992. A molecular dynamics study of the structure and dynamics of water between dilauroylphosphatidylethanolamine bilayers. Langmuir. 8:233–240. [Google Scholar]
- Róg, T., and M. Pasenkiewicz-Gierula. 2001a. Cholesterol effects on the phosphatidylcholine bilayer nonpolar region: A molecular simulation study. Biophys. J. 81:2190–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Róg, T., and M. Pasenkiewicz-Gierula. 2001b. Cholesterol effects on the phospholipid condensation and packing in the bilayer: a molecular simulation study. FEBS Lett. 502:68–71. [DOI] [PubMed] [Google Scholar]
- Róg, T., K. Murzyn, and M. Pasenkiewicz-Gierula. 2003. Molecular dynamics simulations of charged and neutral lipid bilayers: treatment of electrostatic interactions. Acta Biochim. Polon. 50:789–798. [PubMed] [Google Scholar]
- Róg, T., K. Murzyn, R. Gurbiel, Y. Takaoka, A. Kusumi, and M. Pasenkiewicz-Gierula. 2004. Effects of phospholipid unsaturation on the bilayer nonpolar region: a molecular simulation study. J. Lipid Res. 45:326–336. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Cicotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 22:327–341. [Google Scholar]
- Scott, H. L. 2002. Modeling the lipid component of membranes. Curr. Opin. Struct. Biol. 12:495–502. [DOI] [PubMed] [Google Scholar]
- Seelig, J., and N. Waespe-Šarčević. 1978. Molecular order in cis and trans unsaturated phospholipid bilayers. Biochemistry. 17:3310–3315. [DOI] [PubMed] [Google Scholar]
- Shinoda, W., and S. Okazaki. 1998. A Voronoi analysis of lipid area fluctuation in a bilayer. J. Chem. Phys. 109:1517–1521. [Google Scholar]
- Smaby, J. M., M. Momsen, H. L. Brockman, and R. E. Brown. 1997. Phosphatidylcholine acyl unsaturation modulates the decrease in interfacial elasticity induced by cholesterol. Biophys. J. 73:1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tari, A., and L. Huang. 1989. Structure and function relationship of phosphatidylglycerol in the stabilization of the phosphatidylethanolamine bilayer. Biochemistry. 28:7708–7712. [DOI] [PubMed] [Google Scholar]
- Thurmond, R. L., S. W. Dodd, and M. F. Brown. 1991. Molecular areas of phospholipids as determined by 2H NMR spectroscopy. Biophys. J. 59:108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uran, S., A. Larsen, P. B. Jacobsen, and T. Skotland. 2001. Analysis of phospholipid species in human blood using normal-phase liquid chromatography coupled with electrospray ionization ion-trap tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 758:265–275. [DOI] [PubMed] [Google Scholar]
- Urbina, J. A., B. Moreno, W. Arnold, C. H. Taron, P. Orlean, and E. Oldfield. 1998. A carbon-13 nuclear magnetic resonance spectroscopic study of inter-proton pair order parameters: a new approach to study order and dynamics in phospholipid membrane systems. Biophys. J. 75:1372–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, F. J., and J. A. M. de Bont. 1996. Adaptation mechanisms of microorganisms to the toxic effects of organic solvents on membranes. Biochim. Biophys. Acta. 1286:225–245. [DOI] [PubMed] [Google Scholar]
- Yeagle, P. L., W. C. Hutton, G. Huang, and R. B. Martin. 1977. Phospholipid head-group conformations: intermolecular interactions and cholesterol effects. Biochemistry. 16:4344–4349. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. P., R. N. A. H. Lewis, and R. N. McElhaney. 1997. Calorimetric and spectroscopic studies of the thermotropic phase behavior of the n-saturated 1,2-diacylphosphatidylglycerols. Biophys. J. 72:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, F., and K. Schulten. 1995. Molecular dynamics study of a membrane-water interface. J. Phys. Chem. 99:2194–2207. [Google Scholar]