

Abstract
Elastic ellipsoidal functions defined by the observed hydration patterns around the DNA bases provide a new basis for measuring the recognition of ligands in the grooves of double-helical structures. Here a set of knowledge-based potentials suitable for quantitative description of such behavior is extracted from the observed positions of water molecules and amino acid atoms that form hydrogen bonds with the nitrogenous bases in high resolution crystal structures. Energies based on the displacement of hydrogen-bonding sites on drugs in DNA-crystal complexes relative to the preferred locations of water binding around the heterocyclic bases are low, pointing to the reliability of the potentials and the apparent displacement of water molecules by drug atoms in these structures. The validity of the energy functions has been further examined in a series of sequence substitution studies based on the structures of DNA bound to polyamides that have been designed to recognize the minor-groove edges of Watson-Crick basepairs. The higher energies of binding to incorrect sequences superimposed (without conformational adjustment or displacement of polyamide ligands) on observed high resolution structures confirm the hypothesis that the drug subunits associate with specific DNA bases. The knowledge-based functions also account satisfactorily for the measured free energies of DNA-polyamide association in solution and the observed sites of polyamide binding on nucleosomal DNA. The computations are generally consistent with mechanisms by which minor-groove binding ligands are thought to recognize DNA basepairs. The calculations suggest that the asymmetric distributions of hydrogen-bond-forming atoms on the minor-groove edge of the basepairs may underlie ligand discrimination of G·C from C·G pairs, in addition to the commonly believed role of steric hindrance. The analysis of polyamide-bound nucleosomal structures reveals other discrepancies in the expected chemical design, including unexpected contacts to DNA and modified basepair targets of some ligands. The ellipsoidal potentials thus appear promising as a mathematical tool for the study of drug- and protein-DNA interactions and for gaining new insights into DNA-binding mechanisms.
INTRODUCTION
Comprehension and prediction of nucleic acid-ligand interactions are key to the rational design of drugs that are targeted to the nucleic acid components of living cells. Although many computational approaches have been developed to study protein-ligand interactions (Kuntz et al., 1982; Bohm, 1992; Gillet et al., 1993; Rotstein and Murcko, 1993; Eisen et al., 1994; Klebe and Abraham, 1994; Miller et al., 1994; Oshiro et al., 1995; Jones et al., 1997), the unique aspects of nucleic acid interactions are not necessarily considered in their design.
The hydrogen-bond donor and acceptor atoms that line the grooves of the DNA double helix serve as recognition elements for interactions with proteins, drugs, and solvent. Water molecules form a distinctive spine of hydration in the minor grooves of numerous B-DNA structures (Kopka et al., 1983) and ordered networks of fused polygons in the major grooves of many A-DNA structures (Shakked et al., 1981). Moreover, the minor-groove spine of associated water molecules in AT-rich duplexes can be displaced by small, positively charged, crescent-shaped molecules, such as the antibiotic netropsin (Kopka et al., 1985a,b), with proton donor and acceptor atoms arranged to mimic the crystallographically observed configurations of bound waters. The positive charges on the drug molecules and the cationic amino-acid side groups on the proteins are thought to facilitate ligand access past the negatively charged sugar-phosphate backbone.
Some small molecules have capabilities of recognizing short DNA sequences via a code that complements the chemical information on the minor-groove edges of the basepairs (Trauger et al., 1996; White et al., 1998; Dervan and Burli, 1999; Wemmer, 2001). In contrast to other binding ligands, which form 1:1 complexes in the minor groove and possess only partial sequence-reading capabilities, the polyamide molecules designed to recognize specific basepair sequences form 2:1 complexes with DNA. The DNA sequence-reading abilities of these so-called lexitropsins (Goodsell, 2001; Wemmer, 2001) are realized by the combination of three ring subunits—imidazole (Im), pyrrole (Py), and hydroxypyrrole (Hp). A pair of pyrrole residues is used to discriminate A·T or T·A from G·C and C·G (Pelton and Wemmer, 1989; White et al., 1996), an Im·Py pair to differentiate G·C from C·G, and both G·C and C·G from A·T and T·A (Trauger et al., 1996; White et al., 1997), and a pair of Hp and Py units to distinguish T·A from A·T, and both T·A and A·T from G·C and C·G (White et al., 1998). The discrimination mechanisms are thought to reflect both steric hindrance and the asymmetric distributions of hydrogen-bond donor and acceptor groups on the minor-groove edges of the basepairs (Goodsell, 2001; Wemmer, 2001). Specifically, the Py·Py pair is expected to clash with the exocyclic amino group of guanine when brought into the vicinity of a G·C or C·G basepair, thereby favoring its association with A·T or T·A. The Hp·Py pair is believed to use a similar steric mechanism to discriminate against G·C and C·G and to interact preferentially with A·T over T·A via hydrogen-bond formation (involving the exocyclic OH of Hp and the N3 atom of adenine). The Im·Py pair has the capacity to exclude C·G basepairs on the basis of steric hindrance and to associate preferentially with G·C over A·T and T·A via hydrogen bonding (between the imidazole ring nitrogen and the guanine exocyclic amino group).
One of the best sources of information about nucleic acid-ligand interactions is the database of experimental nucleic acid structures (Berman et al., 1992), which is now at the point where there are enough data to extract the preferred positions of different ligands in close contact with the constituent bases, sugars, and phosphates. For example, water molecules cluster in distinct hydrogen-bonding sites around the bases as opposed to being evenly spread over the molecular surface (Schneider et al., 1993, 1998; Schneider and Berman, 1995). Moreover, the bound solvent clusters serve as recognition motifs for specific interactions of DNA with proteins, drugs, and other ligands (Woda et al., 1998; Howerton et al., 2001; Moravek et al., 2002).
These findings have stimulated our interest in developing a concise, more quantitative description of the ligand-binding sites around DNA and RNA. To make use of the observed hydration sites in ligand-docking calculations, an effective mathematical framework must be constructed for precise description of the sites of intermolecular association. The approach taken here follows that of Olson et al. (1998), who derived a set of elastic functions that reflect the sequence-dependent bending, twisting, and stretching of nucleic acid basepair steps. This class of ellipsoidal expressions can also be used to characterize the distributions of water and other ligands around the chemical components of DNA or RNA. Once the binding functions are defined, the interactions of drugs and proteins with DNA can be converted to knowledge-based energies, i.e., statistical scores, for molecular docking applications.
In this article, we first determine a set of elastic functions at the hydration and protein binding sites of the Watson-Crick basepairs with three different approaches: a previously described Fourier averaging of the binding patterns of ligands around individual bases (Schneider et al., 1993), here termed local densities; a similar analysis of ligands in longer stretches of DNA yielding global densities (Schneider et al., 1993); and a statistical clustering algorithm combined with principal component analysis. The resulting ligand-scoring functions are then used to compare the binding of various small molecules in the B-DNA minor groove with the known sites of bound water in well-resolved crystal structures. We consider a series of 2:1 drug-DNA complexes with sequence-recognition capabilities as well as minor-groove binders that form 1:1 complexes with DNA. The ligands are assigned energy scores based on the positioning of the hydrogen-bonding sites on the drugs, relative to the preferred locations of water around the DNA bases. The knowledge-based functions are also used in a series of sequence substitution studies to test the hypotheses that underlie the drug design, e.g., the relative contribution of steric or hydrogen-bonding factors to ligand-binding preferences. The energies of incorrect sequences are obtained by superimposing different bases (without conformational adjustment) on the observed side groups in high resolution DNA-polyamide structures and measuring the relative positions of the hydration sites of the modified duplexes with respect to the unmodified ligand positions. The binding scores of computationally “synthesized” drug-DNA complexes are also compared with the known DNA-binding affinities of polyamide hairpin molecules in solution. The structures of the computer-generated species are checked against those of related hairpin molecules bound to nucleosomal DNA. The observed spatial positioning of the polyamide ligands with respect to sequence-specific DNA targets on the surface of the nucleosome core particle is assessed with the hydration density functions.
METHODS
Data collection
We started by collecting the coordinates of all nucleic acid bases from well-resolved crystal structures of A- and B-DNA double helices and protein-bound DNA complexes in the Nucleic Acid Database (NDB) (Berman et al., 1992) at a resolution cutoff of 2.0 Å (Table S1 in Supplementary Material). The selected structures were filtered to exclude identical DNA sequences and over-represented protein structures to obtain a balanced sample of different spatial forms. In total, 30 A-DNA, 27 B-DNA, and 27 protein-DNA structures were examined. All bases at the ends of strands were excluded, as were those that form non-Watson-Crick basepairs or are unpaired, chemically modified, or located in the vicinity of metal ions, spermine, and other non-water molecules. We next extracted the coordinates of all water molecules and amino acid atoms that lie within 3.4 Å of any of the heavy (non-hydrogen) atoms on the selected bases. We then superimposed an ideal standard planar base (Clowney et al., 1996) on each crystallographically determined base using a least-squares fitting procedure (Horn, 1987). The latter step makes it possible to express the coordinates of bound waters and protein donor and acceptor atoms in different structures in a common reference frame (Olson et al., 2001) on the ideal base (Fig. 1). Because the number of water positions associated with the cytosines and guanines in the A-DNA structures determined to-date is much larger than the number around adenine and thymine, a random subset of the waters around cytidine and guanine (30% of the original positions) was selected so that the distributions used in the determination of elastic functions are closer in size to those available for adenine and thymine.
FIGURE 1.

Scatter maps (a) and ellipsoidal functions (b–d) representing the distributions of water oxygens and amino acid atoms in contact with adenine and thymine in B-DNA (b and c) and protein-DNA structures (d). Ligand-binding ellipsoids around the ideal A·T basepairs in b and d are generated by local Fourier averaging and those in c by local clustering. The contour surfaces correspond to an energy level of 2, where the lengths along the principal axes are equal to twice the variance in these directions. Donor ligands in the vicinity of proton acceptor atoms on the bases are illustrated in blue and acceptor ligands in contact with proton donor atoms on the bases are shown in red.
Knowledge-based potentials
Local pseudoelectron density functions
The Fourier averaging of DNA hydration sites, originated by Schneider and Berman (1995) and Schneider et al. (1993, 1998), converts a set of observed points into a pseudoelectron density map using standard crystallographic procedures. Here, the CCP4 program suite (Collaborative Computational Project No. 4, 1994) is used to convert distributions of ligand donor and acceptor atoms in contact with the DNA bases to pseudoelectron density representations, and the Shelxl program suite (Sheldrick and Schneider, 1997) to generate thermal ellipsoids. (Complete details of the procedure are described at http://rutchem.rutgers.edu/∼olson/ligands.html.)
Global pseudoelectron density functions
Whereas the above local densities are based on the distribution patterns of bound ligand atoms around one of the four bases, the global ligand-binding functions are generated at the level of overall structure (Schneider and Berman, 1995; Schneider et al., 1993). The discrete water or protein contact sites compiled for the individual bases are superimposed on the global structure, e.g., a basepair or a set of bases, and Fourier averaging of the superimposed points is performed in the global reference frame.
Quantitative description of interactions in the major or minor groove is based on the superposition of ligand-binding sites in the groove of interest. Because the global density refinement typically fails to generate satisfactory ellipsoids in the major groove (see Discussion), this approach has only been used to generate minor-groove ellipsoids, and to study minor-groove ligand-DNA interactions.
Local clustering
The ligand-binding data have also been analyzed with a hierarchical, agglomerative clustering algorithm (Auf der Heyde, 1990). Each observation is initially treated as an individual cluster, and the clusters are merged one-by-one according to their distances of separation. Four ways of calculating the cluster distances are considered: single linkage; complete linkage; average linkage; and centroid linkage. The complete and centroid linkage distances are used to cluster ligand positions in the minor groove. These choices are empirical, based on the quality of the clusters generated with the different methods.
To reduce noise, only clusters with >5% of the total number of positions are characterized by elastic functions. The formulation of the energy expression is based on principal component and factor analysis of the clustered data (Auf der Heyde, 1990). The three orthogonal axes used to specify the positions of associated waters or amino acid atoms with respect to a given base are transformed into three new axes, so that these new axes coincide with the directions of maximum variance. Thus, each cluster is represented by an ellipsoid using the new axes (eigenvectors) as the ellipsoidal axes, and the variance along these axes (square-roots of the eigenvalues) as the axis lengths. The force constant matrix F used to calculate the energy of a given ligand-binding site (see Eq. 1 below) is the inverse of the covariance matrix. The mathematical protocol for obtaining this set of potentials is described in full at http://rutchem.rutgers.edu/∼olson/ligands.html.
Global clustering
The global clustering of observed DNA contacts is similar to the global density refinement in using the water molecules around all the bases in a fragment of double-helical structure to predict ligand-binding sites. Standard bases are overlapped on the real bases in a given structure, and the water distributions around the standard bases are automatically converted to the global reference frame. Ellipsoidal functions are then generated in the global reference frame with the same clustering procedure used at the local level.
Global clustering, however, is inferior to the preceding three methods of generating ligand-binding ellipsoids in several respects:
The calculations require excessive human intervention.
The ellipsoidal distributions are highly exaggerated, too long, and/or too flat.
The donor/acceptor properties of ligand and base atoms must be assigned manually.
The positions of the centers of derived ellipsoids are irregular.
Despite these disadvantages, initial sensitivity tests have been carried out for sets of ligand-binding ellipsoids generated by global clustering. The calculated results further confirm the inferiority of this method (see below).
Selection of drug-DNA structures
The knowledge-based potentials have been tested against 18 well-resolved (⩽2.4 Å resolution) oligonucleotide duplex structures with one or more drug molecules positioned in the minor groove (Table 1). Among the test structures are seven drug-DNA complexes (Kopka et al., 1997; Kielkopf et al., 1998a,b, 2000; Mitra et al., 1999) with 2:1 binding stoichiometry, including five structures with polyamide ligands designed to bind the minor-groove edges of basepairs (bdd002, bdd003, gdj057, dd0020, and dd0021) (Kielkopf et al., 1998a,b, 2000). These five drugs, which incorporate DNA sequence-reading capabilities, are analyzed in more detail than the two remaining structures, gdh060 (Mitra et al., 1999) and gdj054 (Kopka et al., 1997), with 2:1 binding stoichiometry and similar chemical makeup but with limited DNA sequence-reading capabilities. The distamycin ligand in the former complex is made up of a string of pyrrole (Py) rings with the capacity only to differentiate A·T and T·A from G·C and C·G basepairs. The pairs of imidazole (Im) rings that constitute the diimidazole lexitropsin bound to DNA in gdj054 cannot distinguish any differences among basepairs, since the Im·Im pair has equal affinity for all four Watson-Crick interactions (Wemmer, 2001). Four other drug-DNA complexes with 2:1 binding stoichiometry—gdhb25, gdlb49, gdlb50, gdlb51 (Chen et al., 1994, 1997)—are excluded from the test set because they contain modified (hypoxanthine) bases. The remaining 11 ligands (Coll et al., 1987, 1989; Larson et al., 1989; Sriram et al., 1992; Balendiran et al., 1995; Goodsell et al., 1995; Wood et al., 1995; Clark et al., 1996a, 1996b, 1997; Aymami et al., 1999) form 1:1 drug-oligonucleotide complexes. These complexes are selected from all 1:1 drug-DNA complexes on the basis of the relatively large number of drug atoms in each structure (⩾3) potentially involved in intermolecular hydrogen bonding with DNA. Such interactions are expected to play an important role in sequence-specific DNA-binding interactions. The (2.3–2.65 Å resolution) structures of three polyamide-DNA complexes with ligands designed to target specific sequences on the surface of the nucleosome core particle (pd0328, pd0329, and pd0330) (Suto et al., 2003) are also examined. The drugs in the latter complexes are covalently connected by a peptide linker, whereas those associated with the oligonucleotide duplexes are chemically independent species.
TABLE 1.
Drug-DNA structures examined with knowledge-based energy functions
| NBD_ID* | DNA sequence | Drug composition† |
|---|---|---|
| 2:1 Drug-DNA complexes | ||
| bdd002 | CCAGTACTGG | ImHpPyPy-β-Dp |
| bdd003 | CCAGTACTGG | ImPyPyPy-β-Dp |
| gdj057 | CCAGGCCTGG | ImImPyPy-β-Dp |
| dd0020 | CCAGATCTGG | ImPyHpPy-β-Dp |
| dd0021 | CCAGATCTGG | ImPyPyPy-β-Dp |
| gdh060 | GTATATAC | PyPyPy (distamycin) |
| gdj054 | CATGGCCATG | ImIm (di-imidazole lexitropsin) |
| 1:1 Drug-DNA complexes | ||
| dd0014 | CGCATATTTGCG | PlBiBiBiBz (tri-benzimidazole) |
| gd1003 | CGCAAATTTGCG | PyPyPy (distamycin) |
| gd1004 | CGCGATATCGCG | PyPy (netropsin) |
| gd1008 | CGCGAATTCGCG | IdBz (DAPI) |
| gd1018 | CGCGAATTCGCG | PyPy (netropsin) |
| gd1030 | CGCGTTAACGCG | PyPy (netropsin) |
| gd1033 | CGCGAATTCGCG | PiBiBiBz (benzimidazole derivative) |
| gd1038 | CGCGAATTCGCG | ImPy (imidazole-pyrrole lexitropsin) |
| gd1039 | CGCAAATTTGCG | PlBiBiBiBz (tri-benzimidazole) |
| gd1047 | CGCGAATTCGCG | PrBiBiBz (Hoescht 33258) |
| gd1052 | CGCGAATTCGCG | BiBiBz (Hoescht 33258 analog) |
| Drug-nucleosomal DNA complexes | ||
| pd0328 | …AGTGTA… | ImPyImPy-γ-PyPyPyPy-β-Dp |
| pd0329 | …CGTGTT…, …GTGTAT… | ImPyPyPy-γ-PyPyPyPy-β-Dp |
| …AGTTTC…, …GGAATT… | ||
| pd0330 | …AGGATA… | ImImPyPy-γ-PyPyPyPy-β-Dp |
NDB_ID refers to the identification code of the complex in the Nucleic Acid Database (Berman et al., 1992).
The following abbreviations are used for drug subunits: Im, imidazole; Py, pyrrole; Hp, hydroxypyrrole; Pr, piperazine; Bz, benzene; Pl, pyrrolidine; Bi, benzimidazole; Id, indole. The β in the chemical formulae refers to a β-alanine that follows the sequence of peptide-linked subunits in the polyamide ligands and the Dp to the 3-amino-(dimethylpropylamine) group at the tail of these molecules. The γ refers to a γ-aminobutyric acid hairpin turn used to link pairs of polyamide chains bound to nucleosomal DNA.
Drug-DNA interaction energies
Drug-DNA interaction pairs
Calculation of the energy of a drug-DNA system entails the enumeration of a set of critical atoms on the drug that may interact with the ligand-binding sites around the DNA bases. Each of the potential hydrogen-bond donor or acceptor atoms on the drug is assigned a DNA-binding ellipsoid with complementary acceptor or donor properties. The partner ellipsoid is selected on the basis of the magnitude of interaction with the drug atom, i.e., the interaction score of lowest value. The number of interactions with DNA is limited by the hydrogen-bonding quotas of the unfulfilled proton donor and acceptor sites on the edges of the Watson-Crick basepairs. Except for the O2 atom of thymine, each of the minor-groove atoms can form only a single hydrogen bond with drug. Thus, only one of the two ligand-binding sites generated near the exocyclic N2 of guanine can be filled in a given complex. Although the presence of two free electron pairs on thymine O2 allows for two hydrogen-bonding interactions with drugs, the geometry of the A·T basepair naturally incorporates one of the two sites in a weak C2(A)–H⋯O2(T) hydrogen bond (Leonard et al., 1995). Interactions of ligands with the unfulfilled hydrogen-bonding sites on melted Watson-Crick basepairs, in which one or more hydrogen donor-acceptor interactions between complementary residues are broken, are not considered.
Interaction score
The total energy, ETot, of the drug-DNA complex is calculated as the sum of interaction energies E for all critical drug atom-DNA ellipsoid pairs. The interaction energy of a given hydrogen-bond donor or acceptor atom on a drug at position Xa = (xa, ya, za), with respect to a preferred DNA ligand-binding site centered at Xe = (xe, ye, ze), is approximated by the harmonic energy expression
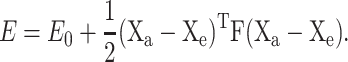 |
(1) |
Here E0 is the minimum energy and F is the force matrix based on the size and shape of the binding ellipsoid and expressed in the global frame of the DNA-drug assembly. (See Table S2 in Supplementary Material for the components of the force matrices at selected ligand-binding sites around the standard bases.) Although the value of E0 can be adjusted to represent the relative strength of different ligand-binding sites, e.g., a value of E0 > 0 can be assigned to a weak hydrogen-bonding site, E0 is set to zero for all interactions in the present work.
The force constants and ligand-binding sites used in the analysis of minor-groove binding are based on the positions of bound water molecules in well-resolved B-DNA structures. The knowledge-based binding potentials derived from the waters in A-DNA structures are not considered since all known DNA complexes with minor-groove bound ligands retain the B-form. Energies derived on the basis of the hydration patterns in protein-DNA complexes are expected, because of similarities in solvent binding patterns (see Results), to resemble the reported B-DNA-based values. Here the elastic potentials, i.e., probable water binding positions, associated with each of the four bases are superimposed by least-squares fitting (Horn, 1987) of a standard base with known hydration sites on the bases in a given drug-DNA complex (Fig. 2). Because of the symmetric properties of the ideal base reference frame (Olson et al., 2001), it is easy to substitute one base for another in the calculations. Modifications of DNA-binding ligands are performed by analogous substitutions of standard drug fragments (see below).
FIGURE 2.

Schematic illustration of the construction of base-substituted DNA duplex structures with associated ligand-binding sites (ellipsoids).
Steric contributions
Configurations with severe nonbonded clashes between drug and DNA atoms are excluded from the calculations. The selection of allowed states is based on the extreme Ramachandran distance thresholds (Ramachandran et al., 1963; Sasisekharan et al., 1967). Although a steric term is not generally needed in the assessment of interactions in known (typically contact-free) drug-DNA structures, the check of disallowed contacts is important in new situations generated by sequence substitutions. Because steric factors are thought to underlie the minor-groove discrimination of C·G and G·C basepairs by polyamide drugs (Trauger et al., 1996; White et al., 1997), the contacts of drug atoms to the free hydrogens attached to the guanine exocyclic amino nitrogen are explicitly enumerated. The positions of other hydrogen atoms are not considered.
Interaction energy ceiling
A critical drug atom may not necessarily be close to one of the DNA-binding ellipsoids. Because of the quadratic nature of the knowledge-based potentials, the energy assigned to a drug atom and its (possibly distant) DNA ellipsoidal partner could be quite large. Such behavior is inconsistent with physical modeling, where the formation of a hydrogen bond can significantly decrease the total interaction energy, but the loss of a hydrogen bond introduces no energetic penalty. An energy ceiling has therefore been introduced to limit the quadratic growth of the calculated hydrogen-bonding energies. That is, if the energy assigned to a critical drug atom is greater than some upper limit, the potential energy of the critical atom is equated to that limit.
The value of the energy ceiling is based on the interaction energies of water dimers estimated from ab initio molecular orbital calculations (Singh and Kollman, 1985). The computed hydrogen-bonding energy in such structures is lowest (∼−6 kcal/mol) when the waters are separated by a distance of 2.9 Å, and approaches a value of zero when the molecules are at a distance of 7–8 Å, i.e., 4–5 Å beyond the ideal distance of separation. The principal axes of the current set of knowledge-based ellipsoids range from 0.5 Å to 2.2 Å in length (with an average value of 1.4 Å and an average variance σ = 0.7 Å). An atom located 5.7σ −7.1σ from the center of a binding ellipsoid is thus as far from a potential binding site as a pair of non-interacting waters are from their ideal hydrogen-bonding positions, i.e., 4–5 Å ÷ 0.7 Å/σ = 5.7–7.1σ. According to Eq. 1, the energy of a drug atom displaced from the center of a DNA-binding ellipsoid by nσ along one of the three principal axes is raised to a level of n2/2. Separation distances of 5.7–7.1σ therefore correspond to an energy ceiling of 16–25. Preliminary calculations testing these two energy limits yield similar results. The data reported below are based on the higher energy ceiling.
Nomenclature of drug and base atoms in polyamide-DNA complexes
For purposes of analysis, a local numbering scheme is introduced to account, at the level of drug subunits, for the atoms comprising the polyamides complexed to DNA rather than the standard chemical nomenclature based on the structure of the ligands as a whole (Fig. 3, a and b). The drug subunit on which an atom is located is further distinguished by a residue name and number and different drugs are assigned a numerical identifier. Thus, one can easily relate a particular atom, residue, or drug in a bound polyamide-DNA complex to an atom or base on DNA. The drug residues are numbered in the same sense as the base sequence, with subunits of lower numerical value associated with the coding strand and residues with higher values bound to the complementary strand (Fig. 3 c).
FIGURE 3.

Nomenclature of drug atoms and residues, DNA bases, and individual molecules in 2:1 polyamide-DNA crystal complexes. Drug atoms on pyrrole (Py), imidazole (Im), and hydroxypyrrole (Hp) rings are numbered in a according to the positions on the ring, and can be compared in b to the conventional chemical nomenclature for hetero atoms of ImHpPyPy-β-Dp based on the structure of the molecule as a whole. The five 2:1 crystal complexes (Kopka et al., 1997; Kielkopf et al., 1998a,b, 2000; Mitra et al., 1999) are made up of two crescent-shaped polyamide strands bound at the centers of 10-bp DNA duplexes. The bases on the coding strand are numbered in c from 1 to 10 and those on the complementary strand from 11 to 20. Drug I binds to the minor-groove edge of the coding strand, and drug II to the minor-groove edge of the complementary strand. Ring subunits of drug I and drug II are represented by circles and numbered 1–4 and 5–8, respectively. The alanyl-3-amino-(dimethylpropylamine) group (Dp) at the tail of each drug molecule is denoted by the linked diamonds and semicircles.
Each drug atom is denoted, like each base atom, by a subunit name, subunit_ID, atom name, and atom number. The bases are represented by standard symbols (A, C, G, T) and the polyamide subunits by abbreviations (Hp, Im, Py, Dp). The subunit_ID refers to the sequential location of the drug subunit in the polyamide chain or the position of the base in the DNA strand. The atom name is based on chemical identity and the atom number is assigned according to its position on the drug subunit or base. For instance, the atom of ImHpPyPy-β-Dp which docks to the coding strand of DNA, designated N8 by the standard naming convention for the molecule as a whole (Fig. 3 b), is termed Hp2(N4) (Fig. 3, b and c), and the DNA atom with which it is in contact, the thymine O2 at base 5, is denoted T5(O2) (Fig. 3 c). (The β in the preceding chemical formula refers to a β-alanine that follows the sequence of peptide-linked subunits and the Dp to the 3-amino-(dimethylpropylamine) group at the tail of the drug molecule.)
Modification of polyamide ligands
Polyamide drug models are constructed by overlapping standard drug subunits with the polyamide ligand templates in known 2:1 crystal complexes. The Cartesian coordinates of the ring atoms of the standard subunits are determined with a downhill simplex procedure (Clowney et al., 1996), which minimizes the difference between the internal chemical parameters (bond lengths and valence angles) of the derived ring structures and the corresponding mean values in the five polyamide complexes. The computational “synthesis” of new ligands is effected by fitting standard ring subunits on a selected polyamide drug template with a least-squares procedure (Horn, 1987) and then connecting the exocyclic atoms on successively positioned subunits, i.e., the C2′ carbonyl carbon of unit i and the N4 amide nitrogen of unit i+1. The conformation of the intervening peptide linker—the C2–C2′–N4–C4 torsion angle, the C2–C2′–N4 and C2′–N4–C4 valence angles, and the C2′–N4 bond length—is automatically determined by the locations of the drug subunits.
RESULTS
Distributions of water molecules and amino acid atoms around the DNA bases
Fig. 1 a illustrates the distributions of the water oxygens in contact with adenine and thymine in B-DNA crystal structures. The collective positions show the same patterns reported previously (Schneider et al., 1993; Schneider and Berman, 1995). Except for the water clusters near the C8 atoms of purines (R) and the C6 atoms of pyrimidines (Y), the hydration patterns are similar in all structural categories (A-DNA, B-DNA, and protein-DNA). In general, water accumulates near the base carbon atoms only in B-type conformers.
Careful examination of A-DNA and B-DNA structures reveals the reason for the difference in water positioning in the two helical forms. In A-DNA, the 5′-phosphate group of the chain backbone lies very close to the R(C8) and Y(C6) atoms. Moreover, the 5′-phosphorus atom lies roughly in the same plane as the base (Lu et al., 2000), leaving almost no space near R(C8) or Y(C6) for a water molecule. Indeed, the O5′ atom is generally in close contact with R(C8) and Y(C6) atoms in A-DNA structures, and the stabilizing contribution of C–H⋯O5′ hydrogen bonding to RNA (A-type) structure has long been appreciated (Shefter and Trueblood, 1965; Sussman et al., 1972; Rosenberg et al., 1973; Wahl and Sundaralingam, 1997). In B-DNA structures, by contrast, there are no atoms on the sugar-phosphate backbone close to R(C8) or Y(C6), and the 5′-phosphorus atom lies in a different plane from the base (Lu et al., 2000). Thus, there is sufficient room around the R(C8) or Y(C6) atoms in B-DNA for water molecules to associate with the bases at these sites. The DNA in protein-DNA complexes is known from other analyses (A. Colasanti, X.-J. Lu, and W. K. Olson, unpublished data) to be predominantly B-form DNA. There is accordingly enough space near R(C8) or Y(C6) to hold water molecules in most protein-bound structures. The number of such contacts, however, is much smaller than the number of waters associated with other base atoms in B-DNA and protein-DNA structures. There are additional examples of close C–H⋯O interactions in protein-DNA structures, particularly major-groove contacts to the thymine methyl groups and the C5 atoms of cytosines (Mandel-Gutfreund et al., 1998), if the current restriction on 2.0 Å or better structural resolution is relaxed. The analysis of DNA physical characteristics with lower quality data is, however, questionable.
Whereas the distribution of water around the bases is independent of the crystal structures from which the binding sites are collected, the amino acid distribution patterns (not shown) are sensitive to the choice of protein-DNA complexes. Only a few amino acids contact DNA in each protein-DNA complex, and the closely associated atoms are usually concentrated in a small region of the structure, often in only one of the two grooves. For example, eight of the 35 close contacts of amino acid atoms to the N3 of adenine occur in the DNA bound to the yeast TATA-box protein (pdt012; see Table S3 in Supplementary Material for a list of the specific contacts to DNA in the structures considered here).
Ligand-binding potentials based on pseudoelectron densities
Contour maps of the elastic energy functions obtained by pseudoelectron density analysis of the water molecules and amino acid atoms in contact with the bases of B-DNA and protein-DNA structures are illustrated in Fig. 1, b and d. The ellipsoidal contours (depicted at a level corresponding to twice the variances along the principal axes) are determined separately for the individual bases, but pictured for the composite A·T pair. As is clear from this example, the water ellipsoids are very similar for a given base in different types of DNA structures (also see Table S4 in Supplementary Material). The centers of corresponding ellipsoids are close, in agreement with well-known restrictions on the hydrogen bonding of electronegative atoms (Llamas-Saiz and Foces-Foces, 1990; Gavezzotti and Filippini, 1994; Pirard et al., 1995). The centroid positions and axes also agree well with previously reported values (Schneider et al., 1993; Schneider and Berman, 1995).
Ligand-binding potentials obtained by clustering
Contour surfaces of elastic potentials obtained by applying clustering techniques to the distributions of water around adenine and thymine are reported in Fig. 1 c. The images are qualitatively similar to those obtained with Fourier averaging. The distances between the centers of corresponding ellipsoids obtained by the two approaches are small (usually <0.7 Å). Many of the ellipsoids generated by the clustering of ligand coordinates, however, are thinner and more elongated than the more nearly spherical shapes obtained by the density calculations. The predicted binding of ligands to the DNA bases on the basis of the clustering of Cartesian coordinates is thus more directional than that expected from the ellipsoids derived from Fourier averaging.
Numerical analysis of the water and amino acid ellipsoids around the DNA bases (Table 2 and, for full description, Table S5 in Supplementary Material) reveals several factors responsible for the shapes of the clustering potentials. First of all, the sizes of the clustering ellipsoids are sensitive to the number and locations of ligand-binding sites in the datasets. The more widely scattered the atomic positions are in a cluster, the larger the ellipsoid is. The ellipsoids characterizing the weak hydrogen-bonding interactions of water with the carbon-base atoms show greater variability in size and center location than the ellipsoids associated with the base nitrogen and oxygen atoms. Second, a few extreme points can influence the shapes, i.e., principal axis lengths and directions, of ellipsoids derived from sparsely populated ligand clusters. Restriction of the current analysis to clusters with 5% or more of the total sites of ligand-base contact helps to minimize the variation in ellipsoidal shape. The ellipsoids obtained by Fourier averaging (Table S4 in Supplementary Material) are less sensitive to small changes in the compiled ensemble than are the ellipsoids derived by direct clustering techniques.
TABLE 2.
Geometric parameters, in Å, of ligand-binding ellipsoids around the DNA bases produced by local clustering of waters in B-DNA structures
| Adenine | Thymine | Guanine | Cytosine | |||||
|---|---|---|---|---|---|---|---|---|
| Minor-groove N, O ellipsoids | ||||||||
| Atom | N3 | O2 | N2 | N3 | O2 | |||
| #Ligand contacts | 82 | 77 | 51 | 67 | 68 | |||
| 〈x〉 | −5.06 | −5.07 | −5.73 | −4.97 | −5.19 | |||
| 〈y〉 | 2.31 | 2.03 | 0.24 | 2.79 | 2.46 | |||
| 〈z〉 | −0.41 | −0.71 | 0.87 | −0.96 | −0.80 | |||
| λ1 | 0.63 | 0.87 | 0.42 | 0.61 | 0.68 | |||
| λ2 | 1.15 | 1.66 | 2.05 | 1.20 | 1.44 | |||
| λ3 | 1.81 | 2.41 | 2.63 | 1.99 | 2.07 | |||
| λ3,1 | −0.02 | 0.05 | 0.31 | −0.26 | −0.14 | |||
| λ3,2 | −0.36 | 0.28 | 0.23 | −0.18 | −0.19 | |||
| λ3,3 | 0.93 | 0.96 | 0.92 | 0.95 | 0.97 | |||
| Major-groove N, O ellipsoids | ||||||||
| Atom | N6 | N7 | O4 | O6 | N7 | N4 | ||
| #Ligand contacts | 56 | 62 | 77 | 58 | 57 | 61 | ||
| 〈x〉 | 4.31 | 3.48 | 4.58 | 4.03 | 3.53 | 4.42 | ||
| 〈y〉 | 1.85 | 4.21 | 2.03 | 1.73 | 4.45 | 3.36 | ||
| 〈z〉 | −0.04 | 0.50 | 0.35 | −0.25 | 0.67 | 0.17 | ||
| λ1 | 1.06 | 0.55 | 0.92 | 0.54 | 0.64 | 0.61 | ||
| λ2 | 1.44 | 1.67 | 1.56 | 1.29 | 0.89 | 0.96 | ||
| λ3 | 1.97 | 2.56 | 2.37 | 1.82 | 2.07 | 1.95 | ||
| λ3,1 | 0.26 | −0.32 | −0.21 | 0.96 | −0.24 | −0.63 | ||
| λ3,2 | 0.68 | 0.89 | −0.13 | −0.04 | −0.27 | 0.18 | ||
| λ3,3 | 0.69 | 0.34 | 0.97 | 1.00 | 0.93 | 0.76 | ||
| Major-groove C ellipsoids | ||||||||
| Atom | C8 | C5M | C6 | C8 | C5 | C6 | ||
| #Ligand contacts | 21 | 10 | 22 | 19 | 13 | 36 | ||
| 〈x〉 | 1.15 | 4.59 | 0.64 | 1.26 | 3.47 | 1.20 | ||
| 〈y〉 | 7.66 | 6.62 | 7.93 | 7.81 | 5.08 | 7.67 | ||
| 〈z〉 | 0.97 | −1.36 | 0.73 | 0.43 | −0.01 | 0.59 | ||
| λ1 | 0.43 | 0.48 | 0.69 | 0.11 | 0.52 | 0.66 | ||
| λ2 | 1.08 | 1.63 | 0.87 | 0.94 | 1.57 | 2.11 | ||
| λ3 | 1.61 | 1.92 | 3.23 | 2.28 | 3.04 | 2.87 | ||
| λ3,1 | 0.71 | −0.31 | 0.15 | −0.65 | −0.33 | −0.36 | ||
| λ3,2 | −0.08 | 0.82 | −0.47 | 0.21 | −0.23 | −0.04 | ||
| λ3,3 | 0.79 | 0.48 | 0.87 | 0.73 | 0.92 | 0.93 | ||
Data based on the positions of waters around 110 bases in 27 B-DNA structures. Rows labeled 〈x〉, 〈y〉, and 〈z〉 are coordinates of ellipsoidal centers, rows labeled λ1, λ2, and λ3 are lengths (twice the variances) of principal axes, and rows labeled λ3,i (i = 1,3) are direction cosines of longest axis.
Despite the issues of computational sensitivity noted above, the clustering of ligand-binding sites accounts satisfactorily for the major features of water and protein interaction with the nucleic acid bases. Moreover, the long, thin ellipsoids reproduce certain subtle attributes of molecular association particularly well, e.g., the bifurcated hydrogen bonding of some water molecules to adenine N6 and N7. The boundary between the clusters near N6 and N7 on adenine in Fig. 1 a is not clear because such shared positions can be represented by two closely spaced ellipsoids.
Some of the widely scattered waters near the N2 atom of guanine (not shown) form hydrogen bonds with both the N2 atom of G and the O2 atom of C. This feature is well represented by the ellipsoids computed with both Fourier averaging and clustering, even though the former calculations result in two distinct hydration sites and the latter yields a single elongated binding volume. To assess the influence of shared water molecules on ellipsoidal location, size, and direction, the water molecules, which are near guanine in B-DNA structures and also in contact with the complementary cytidine base, were removed from the set of observed binding sites. The regenerated N2 ellipsoid is only slightly smaller than the original ellipsoid and still overlaps the O2 ellipsoid of C.
Interestingly, the minor-groove N3 and O2 atoms are contacted preferentially via their lower faces in most B-DNA and protein-bound duplexes. Specifically, the centers of the N3 and O2 binding ellipsoids are displaced −0.5 to −1.0 Å below the planes of the heterocyclic rings, and the major (longest) axis of each ellipsoid lies roughly parallel to the base normal in most cases considered here; see direction cosines λ3,i (i = 1,3) in Table 2, and Table S6 in Supplementary Material.
By contrast, the approach of ligands in the major groove depends on sequence. The long axes of the guanine O6 and N7 binding ellipsoids are consistently parallel to the normal of G, as opposed to the many examples where the approach to the corresponding N6 and N7 sites on adenine is more lateral. The directionality of interactions of the pyrimidines tends to be opposite to that of the complementary purines. That is, the cytidine N4 is contacted more laterally and the thymine O4 is approached from above or below, i.e., parallel to the base normal.
Comparison of density and clustering potentials
The elastic energy functions generated by clustering and Fourier averaging of B-DNA water sites are compared in Table 3 in terms of the relative “chemical” placement of the binding-site ellipsoids with respect to each of the contacted base atoms. The comparable lengths and angles of hydrogen bonds between the ellipsoidal centers and associated base atoms confirm the similar placement of water ellipsoids seen in Fig. 1. Corresponding hydrogen-bonding distances differ in most cases by 0.10 Å or less, and virtual valence and torsion angles generally agree within 5° and 10°, respectively. The differences are greatest for the least well-determined ellipsoids—N2 of G, C5 of C, C5M of T, C6 of pyrimidines—associated with the base atoms that bind the fewest water molecules. The ligand-binding potentials computed at these sites are thus less accurate than the potentials at other binding locations. Despite these uncertainties, it is noteworthy that the hydrogen bonds involving the cytidine C5 atom are appreciably shorter than those of other C–H⋯O interactions and, in fact, are shorter than most N–H⋯O contacts. The C5 atom of cytidine bears a substantially larger negative charge than many nitrogen atoms in popular nucleic acid force fields (see below).
TABLE 3.
“Chemical” comparison of B-DNA ligand-binding functions generated by local clustering and pseudoelectron density refinements
| Atom*
|
Distance† (C⋯E, Å)
|
Valence‡ (B–C⋯E, °)
|
Torsion§ (A–B–C⋯E, °)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | Clust. | Local | Global | Clust. | Local | Global | Clust. | Local | Global |
| Adenine | |||||||||||
| N1 | C2 | N3 | 2.77 | 2.80 | 2.80 | 108 | 108 | 108 | 171 | 169 | 169 |
| C5 | C6 | N6 | 2.86 | 2.94 | 2.96 | 139 | 135 | 135 | −1 | −1 | 0 |
| N9 | C8 | N7 | 2.67 | 2.68 | 2.71 | 123 | 123 | 123 | −167 | −169 | −168 |
| C4 | N9 | C8 | 3.14 | 3.18 | 2.60 | 127 | 128 | 116 | −157 | −156 | 170 |
| Thymine | |||||||||||
| N1 | C2 | O2 | 2.67 | 2.78 | 2.78 | 160 | 159 | 159 | 52 | 62 | 61 |
| N3 | C4 | O4 | 2.66 | 2.74 | 2.74 | 142 | 141 | 140 | −168 | −174 | −173 |
| C4 | C5 | C5M | 3.02 | 3.29 | 3.15 | 150 | 147 | 147 | 115 | 99 | 100 |
| C2 | N1 | C6 | 3.04 | 3.17 | 2.83 | 126 | 124 | 137 | −163 | −172 | −146 |
| Guanine | |||||||||||
| N1 | C2 | N2 | 2.92 | 3.02 | 3.34 | 131 | 129 | 136 | 157 | 178 | 175 |
| N1 | C2 | N2 | — | 3.26 | — | — | 142 | — | — | 140 | — |
| N1 | C2 | N3 | 2.83 | 2.87 | 2.91 | 113 | 114 | 115 | 159 | 157 | 158 |
| C5 | C6 | O6 | 2.60 | 2.64 | 2.63 | 138 | 136 | 137 | −8 | −8 | −14 |
| N9 | C8 | N7 | 2.78 | 2.81 | 2.82 | 119 | 119 | 119 | −164 | −168 | −165 |
| C4 | N9 | C8 | 3.13 | 3.11 | 3.06 | 130 | 130 | 119 | −170 | −171 | −149 |
| Cytosine | |||||||||||
| N1 | C2 | O2 | 2.70 | 2.75 | 2.66 | 157 | 155 | 159 | 49 | 49 | 42 |
| N3 | C4 | N4 | 2.88 | 2.93 | 2.95 | 113 | 114 | 114 | −176 | −178 | −174 |
| N3 | C4 | C5 | 2.55 | 2.78 | 2.69 | 117 | 116 | 119 | −180 | −169 | 178 |
| C2 | N1 | C6 | 2.93 | 2.99 | 3.20 | 137 | 125 | 123 | −163 | −146 | −168 |
Comparison criteria proposed by Schneider et al. (1993) for ellipsoids generated by three methods: Clust, local clustering; Local, local density refinement; and Global, global density refinement.
The columns labeled Atom refer to atoms on the base. C is the base atom closest to the ellipsoid. B and A are used to measure the virtual valence and torsion angles reported in the following columns, with B covalently linked to C and A covalently linked to B.
Hydrogen-bonding distances between the centers E of corresponding ellipsoids generated by the three methods and the base atoms marked C.
Virtual valence angles formed by the centers E of the ellipsoids generated by the three methods and the base atoms marked C and B.
The columns under Torsion (A–B–C–E, °) list the virtual torsion angles formed by the centers E of the ellipsoids generated by the three methods and the base atoms marked C, B, and A.
Although the positions of the ellipsoidal centers are similar, their shapes are very different. As noted above, the ellipsoids constructed from pseudoelectron density maps tend to be spherical or egg-like due to the periodicity of the Fourier transformation used to calculate the pseudoelectron densities. On the other hand, many of the ellipsoids generated with the clustering algorithm are thin and/or flat. The differences in the shapes of these ellipsoids affect the computed energies, i.e., binding scores, of minor-groove binding species in DNA·drug complexes.
Occupancy values, reported in previous work (Schneider et al., 1993, 1998; Schneider and Berman, 1995), are not considered for two reasons. First, there is no consistent way to define occupancy for the two ellipsoid-generating methods considered here. Second, the calculated occupancy values obtained by either method are highly volatile.
Dependence of derived hydration potentials on base charges
The strength of hydrogen bonding is often attributed to the magnitude of charges on associated proton donor and acceptor atoms (Jeffrey, 1997). The ellipsoidal features of the present set of DNA-binding potentials may thus reflect the partial charges on the contacted atoms of the interacting bases and water molecules.
To test this hypothesis, we have compared the ellipsoidal parameters of the derived water-binding potentials with the partial atomic charges on the bases given in three popular nucleic acid force fields—AMBER (Weiner et al., 1984; Cornell et al., 1995; Cieplak et al., 2001), CHARMM (Brooks et al., 1983), and the Poltev atomic potentials (Zhurkin et al., 1981).
Three different forms of the partial charges were considered:
The charges of the contacted heavy base atoms.
The sum of the charges of the contacted base atoms and all attached hydrogens.
The sum of the charges of the heavy base atoms and the one hydrogen atom involved in the interaction of interest.
Both the signs and the magnitudes of the charges were considered in testing for statistical relationships between the partial base charges and derived potential functions.
The hydration potentials were also expressed in terms of several different variables:
The mean hydrogen-bonding distances between the centers of each ellipsoid and the contacted base atom.
The number of hydrogen-bonding atoms per hydration site.
The volume of each binding site, i.e., the product of the eigenvalues of the ellipsoid of interest.
The direction of binding, as measured by the scalar product of a unit vector directed from the contacted base atom to the center of the associated binding site and a unit vector along one of the three axes of the ellipsoid.
Four forms of each selected parameter (original, square, inverse, and inverse square) were tested.
Pairwise linear regression analysis was then performed on all combinations of the partial atomic charges and ellipsoidal parameters of selected ligand-binding potentials, and the resulting correlation coefficients were determined. By restricting the analysis to the bound waters, there is no need to consider the effects of ligand partial charge on ellipsoidal properties. A total of 240 or 288 partial charge-parameter combinations (four force fields × three charge measures/force field × four forms of each charge measure × five or six variables per ellipsoid) were thus considered for each of the derived ellipsoidal potentials. The different numbers reflect the fact that one of the ellipsoidal variables, the number of atoms per hydration site, is not determined in Fourier averaging.
The AMBER2 charges, if expressed as the sum of the partial charges on the base atoms and one or all attached hydrogens, are found to be strongly correlated with the derived hydration potentials. For example, the mean hydrogen-bonding distances between the centers of the density-refined ellipsoids and the contacted base atoms are coupled, with a linear correlation coefficient of 0.73, to the net base charges (Fig. 4). The AMBER1 and Poltev charge sets show the same dependence on ellipsoidal geometry but the correlation coefficients are lower. The 1983 CHARMM charge set, however, is not correlated with the energy ellipsoids. The magnitudes of the partial atomic charges on the DNA bases are also strongly linked to the number of waters per binding site determined as part of the clustering treatment (correlation coefficient of 0.77). These findings support the simple notion that more highly polarized base atoms with partial charges of greater magnitude will attract their hydrogen-bonding water partners more strongly and thereby draw them closer and in greater number to the DNA. Conversely, the C–H and N–H proton donor groups on the bases with generally smaller partial charges (of magnitude 0.4 esu or less in the various force fields) are more distant from the surrounding water molecules than the acceptor oxygens or nitrogens of larger partial charges. The poor correlation (not shown) of the derived hydrogen-bonding distances with the surface electrostatic potentials of the basepairs (electrostatic potentials of specific basepair sites were obtained by averaging the potential determined by solution of the Poisson equation at accessible sites 1 Å beyond the van der Waals' surface (A. R. Srinivasan, R. R. Sauers, M. O. Fenley, A. H. Boschitsch, A. Matsumoto, A. V. Colasanti, and W. K. Olson, unpublished data)), confirms the apparent dominance of short-range effects on the water-base contacts. A compilation of charge sets is included in Table S7 in Supplementary Material.
FIGURE 4.

Dependence of ligand-binding potentials on partial charges of the DNA bases. Scatter maps of atomic charges (original AMBER2 (Cornell et al., 1995) values expressed as the sum of the charges, in esu, on the base atom and all attached hydrogens) versus the hydrogen-bonding distances, in Å, between the base atoms and derived ellipsoidal centers, and the fitted linear function. Ellipsoids generated with local density refinement (correlations for ellipsoids generated by clustering are similar). All C–H groups bear positive charges except that at C5 of C, which is assigned a net charge of −0.34 esu in the force field. Red denotes proton donor atoms, and blue proton acceptors. The solid circles indicate atoms on A, the open triangles atoms on C, the solid squares atoms on G, and the open diamonds atoms on T.
Ligand-binding potentials based on global modeling
Comparison of potentials determined by local and global density refinement
Compared with the local density refinement, the global refinement has the advantage of identifying water and amino acid positions shared by one base and its complement or neighbor. Ellipsoids generated locally for individual bases may overlap when converted to the global reference frames, and may compromise their accuracy in the calculation of energies of critical atoms. On the other hand, the water molecules or amino acid atoms counted twice in the local refinement will overlap in the global refinement, and be combined, in principle, into one specific shared binding site. Thus, ellipsoids generated in the global frame should be more accurate in assessing the interactions of critical atoms.
Examination of Table 3, which includes a “chemical” comparison of the relative positions of global versus local B-DNA-binding ellipsoids, reveals the general similarity of the two sets of potentials. The centers of the global and local ellipsoids near nitrogen and oxygen are very close, with mean hydrogen-bonding distances within 0.1 Å and virtual valence and torsion angle differences of <10°. The ellipsoids associated with the weaker hydration sites near carbon, however, show much greater differences in their center coordinates, with some distances differing by >0.3 Å and certain virtual valence or torsion angles showing discrepancies of 20° or more. These differences illustrate the limitations of the density calculations in generating ellipsoidal energy functions associated with weak C–H hydrogen bonds.
The global treatment faces the same problems as the local refinement of ligand pseudoelectron densities and the generation of the corresponding ellipsoids, namely determination of the number of peaks to be retained and treatment of negative eigenvalues of the anisotropic thermal factors. The correction of negative eigenvalues by isotropic refinement of the relevant hydration sites usually resolves the problem. The resulting spherical energy functions are expected to be of lesser accuracy only if many hydration sites must be refined isotropically.
Computational limitations
Although the minor-groove ellipsoids associated with purine N3 or pyrimidine O2 atoms are readily determined, human intervention is needed to generate the ellipsoid near the N2 atom of G, because of the small number of associated ligands. To facilitate automatic global refinement of the binding sites, ligand positions around N2 of G are treated separately from those near other atoms on the molecule. The N2 ellipsoid is constructed by global refinement of ligand-binding positions around an ideal G·C pair, and then superimposed on each G in the global frame of DNA.
The global methodology fails in the treatment of the major groove in that too many ellipsoids are generated in the refinement. A number of factors contribute to the problem. First, the hydrogen-bonding patterns are more complicated in the major groove than in the minor groove. Second, the strong hydration sites at adenine N6 and N7 partially overlap, and third, the hydration sites associated with carbon atoms on the bases—C5 of C, C5M of T, R(C6), R(C8)—contain few scattered waters. Because major-groove ellipsoids cannot usually be generated for these weak hydration sites without human intervention, global refinement can only be applied to the study of ligand-DNA interactions in the minor groove.
Minor-groove ellipsoids
Fig. 5 illustrates, in stereo, the minor-groove binding ellipsoids obtained by global refinement of the water binding sites from high resolution B-DNA structures on a 6-bp double-helical fragment of one of the lexitropic drug-DNA complexes, bdd002 (Kielkopf et al., 1998b), with a polyamide ligand designed to recognize the basepair edges. For simplicity, neither the ligand framework nor the predicted binding sites of the two basepairs at either end of the structure are shown. As seen from the figure, the observed positions of the hydrogen-bond donor and acceptor atoms of the designed (Im-Py-Hp) ligand closely overlap the centers of the predicted binding sites. The positions of drug atoms are denoted by small magenta spheres and the predicted binding sites by colored ellipsoids (red near hydrogen donor atoms and blue near hydrogen acceptor atoms on the bases). Quantitative evaluation of the ligand-binding sites in this and other drug-bound DNA molecules is described below.
FIGURE 5.

Stereo representation of the minor-groove binding ellipsoids obtained by global density refinement of the water-binding sites from high resolution B-DNA structures on a 6-bp fragment of a polyamide drug-DNA complex, bdd002 (Kielkopf et al., 1998b). The proton donor and acceptor atoms on the drug are denoted by small magenta spheres. The ellipsoids of donor ligands in the vicinity of proton acceptor atoms on the bases are illustrated in blue and those of acceptor ligands in contact with proton donors on the base are shown in red.
Identification of intermolecular interactions in drug-DNA complexes
Correspondence with water-binding sites
The energies of critical donor and acceptor atoms from drugs in a representative lexitropic drug-DNA crystal structures are reported in Table 4 (the interaction energies of four other complexes are listed in Table S8 in Supplementary Material). The scores are based on the positions of the atoms with respect to the minor-groove binding sites of water derived by three different refinements of the hydration patterns in drug-free B-DNA structures. A low score confirms the similarity of drug and water binding. The tabulated energies demonstrate the utility of the knowledge-based potentials in identifying intermolecular hydrogen bonds in drug-DNA complexes.
TABLE 4.
Energies of critical drug atoms and DNA-binding ellipsoids in a representative lexitropic polyamide-DNA oligonucleotide complex
| Energies
|
||||||
|---|---|---|---|---|---|---|
| Drug | Drug atom* | A/D† | Minor-groove binding site | Clustering | Local density | Global density |
| bdd002: 2 ImHpPyPy-β-Dp + d(CCAGTACTGG)2 (Kielkopf et al., 1998b) | ||||||
| Drug 1 | Im1(N3) | A | G4(N2) | 2.5 | 2.6 | 2.5 |
| Hp2(N4) | D | T5(O2) | 1.1 | 0.9 | 1.1 | |
| Hp2(O3) | D | T5(O2) | 7.7 | 8.4 | 10.3 | |
| Py3(N4) | D | A6(N3) | 13.8 | 2.0 | 1.6 | |
| Py4(N4) | D | C7(O2) | 7.5 | 3.1 | 3.5 | |
| DpI(N11) | D | T8(O2) | 1.1 | 0.9 | 0.8 | |
| Drug II | Im5(N3) | A | G14(N2) | 1.4 | 1.7 | 1.8 |
| Hp6(N4) | D | T15(O2) | 1.3 | 0.7 | 1.1 | |
| Hp6(O3) | D | T15(O2) | 11.0 | 10.2 | 8.4 | |
| Py7(N4) | D | A16(N3) | 2.2 | 0.3 | 0.3 | |
| Py8(N4) | D | C17(O2) | 9.5 | 4.7 | 1.8 | |
| DpII(N11) | D | T18(O2) | 3.1 | 2.8 | 2.6 | |
| Average‡ | 5.2 (4.5) | 3.2 (3.1) | 3.0 (3.1) | |||
Drug atoms are named according to the nomenclature in Fig. 3 a. Also see Fig. 6 and the legend to Table 1.
A/D refers to the proton acceptor or donor property of the drug atom.
Average refers to the average binding score per critical drug atom in the given complex. The standard deviations of the binding scores are reported in parentheses.
The schematic in Fig. 6 illustrates the interactions considered in the computations. In this example, the contacts of ImHpPyPy-β-Dp with the d(CCAGTACTGG)2 duplex observed in the 2:1 crystal complex (NDB code: bdd002) (Kielkopf et al., 1998b) are reduced to the 12 entries included in Table 4. The identities of the minor-groove binding ellipsoids are defined in terms of the base atoms with which they are associated. Bases without drug contacts are excluded from the figure.
FIGURE 6.

Schematic illustration of the observed interactions between ImHpPyPy-β-Dp and DNA base atoms in the 2:1 drug-DNA complex bdd002 (Kielkopf et al., 1998b). Circles denote binding (hydration) sites in the minor groove with labels corresponding to the atoms on DNA closest to the site. Open circles correspond to sites that interact with donor atoms on the drug, and light-shaded circles to sites that interact with acceptor atoms on the drug. Dashed arrows indicate the critical drug atoms found at the binding sites in the crystal complex. The arrows point from the proton donors on the drug to the proton acceptors on DNA, and vice versa. Drug atoms are numbered according to the nomenclature in Fig. 3. Only atoms in direct contact are labeled.
As shown in Table 5, the energies of most drug atoms in the five 2:1 polyamide-DNA complexes are low in value, i.e., average scores of 5 or less, corresponding to the principal-axis displacement of a drug atom by 3.2σ or less from the center of an expected water binding site. The energies of drug atoms with other binding sites are typically much larger. The energies derived from the ellipsoids obtained by Fourier averaging of water contacts are generally lower in value and thus more consistent with ligand positioning in the five complexes.
TABLE 5.
Summary of computed energies of critical atoms on drug molecules in drug-DNA complex structures
| Energies
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Clustering
|
Local density
|
Global density
|
||||||
| NBD_ID | Drug_ID* | Critical atoms | <5 | <10 | <5 | <10 | <5 | <10 |
| 2:1 Drug-DNA complexes | ||||||||
| bdd002 | ImHpPyPy I | 6 | 3 | 5 | 5 | 6 | 5 | 5 |
| II | 6 | 4 | 5 | 5 | 5 | 5 | 6 | |
| bdd003 | ImPyPyPy I | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| II | 5 | 5 | 5 | 5 | 5 | 5 | 5 | |
| gdj057 | ImImPyPy I | 6 | 5 | 6 | 6 | 6 | 6 | 6 |
| II | 6 | 6 | 6 | 6 | 6 | 6 | 6 | |
| dd0020 | ImPyHpPy I | 6 | 5 | 6 | 5 | 6 | 6 | 6 |
| II | 6 | 3 | 6 | 4 | 6 | 6 | 6 | |
| dd0021 | ImPyPyPy I | 5 | 2 | 5 | 5 | 5 | 4 | 5 |
| II | 5 | 4 | 5 | 5 | 5 | 5 | 5 | |
| gdj054 | ImIm I | 6 | 5 | 6 | 6 | 6 | 5 | 6 |
| II | 6 | 5 | 6 | 4 | 6 | 4 | 6 | |
| gdh060 | PyPyPy I | 5 | 4 | 5 | 5 | 5 | 4 | 5 |
| (unit 1) | II | 5 | 5 | 5 | 5 | 5 | 4 | 5 |
| gdh060 | PyPyPy I | 5 | 4 | 5 | 5 | 5 | 5 | 5 |
| (unit 2) | II | 5 | 5 | 5 | 5 | 5 | 3 | 5 |
| 1:1 Drug-DNA complexes | ||||||||
| dd0014 | PlBiBiBiBz | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| gd1003 | PyPyPy | 5 | 4 | 4 | 4 | 5 | 3 | 4 |
| gd1004 | PyPy | 6 | 4 | 5 | 5 | 6 | 5 | 5 |
| gd1008 | IdBz | 3 | 2 | 3 | 3 | 3 | 3 | 3 |
| gd1018 | PyPy | 6 | 5 | 5 | 6 | 6 | 5 | 6 |
| gd1030 | PyPy | 6 | 5 | 6 | 6 | 6 | 6 | 6 |
| gd1033 | PlBiBiBz | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| gd1038 | ImPy | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| gd1039 | PlBiBiBiBz | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| gd1047 | PrIpIpBz | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| gd1052 | IpIpBz | 4 | 3 | 3 | 4 | 4 | 3 | 3 |
| Total | 136 | 111 | 130 | 127 | 135 | 121 | 132 | |
Drug_ID refers to the chemical name and number of a polyamide ligand according to the nomenclature in Fig. 3; critical atoms to the number of hydrogen-bond forming atoms on the drug; <5 and <10 to the number of critical atoms with calculated average binding scores <5 or 10, i.e., within 3.2 or 4.5 standard deviations of the preferred positions of water molecules on the minor-groove edges of the DNA bases. See legend to Table 1.
Effectiveness of potentials
A comparison of the intermolecular hydrogen-bonding distances in selected drug-DNA complexes with the distances of the binding ellipsoids from the base atoms reveals the underlying cause of the computed differences in energy. The average distance between proton donor and acceptor atoms in the five polyamide-DNA complexes (3.02 Å) is greater than the ideal separation distances derived from the observed binding sites of water molecules (2.84 Å for the energy functions based on Fourier averaging and 2.76 Å for energy functions obtained with clustering). The drug atoms are apparently constrained by their size and chemical framework, and thus unable to fit the hydration sites perfectly. The closer distances of water to base atoms may reflect bias in the 3.4 Å cutoff limit used in their identification. It is also possible that binding sites identified as water in some of the crystal structures are sodium counterions positioned more closely than water to (partially negatively charged) proton acceptor sites on the bases.
The assessment of ligand-binding interactions on the basis of potentials obtained by the global clustering of hydration sites is unsatisfactory. The computed mean binding scores in the five polyamide structures—bdd002: 5.9; bdd003: 3.6; gdj057: 5.5; dd0020: 5.0; and dd0021: 6.1—are significantly higher than the corresponding values for the other knowledge-based potentials (Table 4, and Table S8 in Supplementary Material). Because of the poor performance of the functions based on global clustering, we omit their further consideration.
The computed energies of ligand atoms in 1:1 drug-DNA structures, presented in Table 5, confirm the effectiveness of the elastic potentials in identifying critical atoms on minor-groove binding ligands. If we set an acceptance limit on the calculated score of a critical atom at 10 or less (4.5σ), 130 of the 136 critical drug atoms (95.6%) in 1:1 and 2:2 drug-DNA complexes are identified with the potentials derived by clustering, 135 atoms (99.3%) with the energies based on local pseudoelectron density refinement, and 132 atoms (97.1%) with the functions obtained by global pseudoelectron analysis. If the acceptance criterion is lowered to 5 (3.2σ), the respective potentials identify 111, 127, and 121 critical atoms (81.6%, 93.4%, and 89.0%) successfully.
All three methods of generating knowledge-based potentials thus yield energies sensitive enough to characterize the drug-DNA interactions in known structures as low in energy. There are relatively few negative predictions, i.e., critical atoms with high energy values. On the other hand, it is possible that a mathematical model that generates a low number of negatives predictions might be overfitted, and the number of false positives with noncritical atoms in low energy positions might be high. To test the model further and to determine whether the knowledge-based potentials can account for sequence-specific binding of drugs in the DNA minor groove, we next examine the binding energies of various ligands with modified DNA sequences.
Energies of minor-groove ligands with modified DNA sequences
Sequence changes in conformationally locked DNA duplexes
As a first approximation, we assume that the substitution of one of the four common basepairs by another has no effect on the overall structure of a DNA complexed with a minor-groove binding ligand. This simplification ignores the well-known, albeit small, sequence-specific differences in the intrinsic structure of DNA basepair steps (Gorin et al., 1995; Olson et al., 1998) and any spatial adjustments of the drug against the surface of DNA, but is consistent with the limited effects of small, groove-binding molecules on ordinary B-DNA structure (see Discussion). We further assume that the intermolecular interactions in the crystal structure of a drug-DNA complex are lower in energy than those of other small molecules or DNA sequences in the same configuration. A change of one or two basepairs in the known drug-DNA crystal complexes is thus expected either to increase the overall hydrogen-bonding score or to introduce steric hindrance into the system.
Table 6 summarizes the interactions of various DNA sequences in contact with one of the five lexitropic polyamides from known 2:1 drug-oligonucleotide complexes (the remaining four are described in Table S9 in Supplementary Material). The binding scores are calculated with the same sets of knowledge-based potentials shown above. The energies of the modified sequences, each with a substitution of one of the four key basepairs in the center of the DNA (12 possible basepair substitutions), are expressed relative to the total score ETot of the sequence found in the crystal structure. The key basepairs contain the critical atoms which are thought to determine the specificity of drug recognition (Fig. 6). Because of the local nature of the knowledge-based functions, the scores associated with multiple basepair changes can be estimated from the sum of energies determined for single basepair substitutions. (This is also true for the energies based on global pseudoelectron density refinement, since the minor-groove binding ligands do not significantly distort the complexed DNA from the B-form.) Unfavorable steric interactions brought about by the substitutions are also tallied.
TABLE 6.
Knowledge-based energies of a representative lexitropic polyamide bound to sequence-modified DNA
| Energies
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Clustering
|
Local density
|
Global density
|
||||||
| Sequence* single-base substitution | Comp_ID* | Steric hindrance | ETot | ΔETot | ETot | ΔETot | ETot | ΔETot |
| bdd002 | ||||||||
| CCAGTACTGG | 62.3 | 38.2 | 35.8 | |||||
| ---C------ | (S1a) | † | 5.0 | −0.3‡ | 0.9 | |||
| ----A----- | (S1b) | 10.2 | 10.2 | 8.5 | ||||
| -----T---- | (S1c) | 8.0 | 5.6 | 7.9 | ||||
| ------G--- | (S1d) | § | 3.2 | 2.8 | 3.6 | |||
| ---A------ | (S1m) | 21.8 | 25.4 | 22.2 | ||||
| ----C----- | (S1n) | § | 7.7 | −0.7 | 1.1 | |||
| -----C---- | (S1o) | § | 0.4 | 0.5 | 1.6 | |||
| ------A--- | (S1p) | 28.4 | 22.7 | 25.0 | ||||
| ---T------ | (S1w) | 25.7 | 21.4 | 23.1 | ||||
| ----G----- | (S1x) | § | 8.5 | 2.5 | 4.8 | |||
| -----G---- | (S1y) | § | 4.7 | −3.1 | −0.2 | |||
| ------T--- | (S1z) | 22.6 | 25.3 | 23.2 | ||||
Only one of the two DNA strands is listed for each drug-DNA structure. Comp_ID is the identification code of the computer substitution experiment referenced in the text.
Steric hindrance that is predicted to occur in the drug design (Kielkopf et al., 1998a,b, 2000; White et al., 1998), is not detected. Such situations occur only when Im·Py drug pairs are used to recognize G·C basepairs.
Basepair substitution results are inconsistent with predictions, i.e., the basepair change neither increases the total energy nor introduces steric hindrance.
Steric hindrance exists between the ligand and modified DNA.
As noted above, a modified sequence is expected to introduce a higher interaction energy score and/or steric clashes at the binding site. A modified sequence of lower total energy and free of steric hindrance, reflected by a negative ΔETot and a null entry in the third column of Table 6, is inconsistent with this assumption. The ligands of two drug-DNA complexes, bdd003 and dd0021, are thought to be incapable of recognizing the difference between A·T and T·A pairs in the middle of the DNA sequences to which they are respectively bound. It is therefore possible that the change of an A·T pair in the middle of these sequences to a T·A pair or vice versa may decrease the total energy. This is the reason why some of the calculations involving bdd003 and dd0021 are considered to be consistent with expectations, even though the basepair substitutions introduce a negative value of ΔETot and the complexes are free of steric hindrance (examples highlighted by symbols (‡) in Table S9 in Supplementary Material). With this proviso, we find that only one of the 60 base-substituted structures is inconsistent with expectations. Specifically, the substitution of G4 by cytosine in bdd002 introduces a small (−0.3) decrease in the interaction score based on the local pseudoelectron density potential and is free of the steric effects with imidazole anticipated by the drug design (see Discussion).
Base substitution scores
The top half of Table 7 summarizes the base substitution scores of the five lexitropic drug-DNA complexes plus the corresponding values obtained for the 2:1 complexes of distamycin and diimidazole lexitropsin with DNA (gdj054 and gdh060, respectively). As noted above, the latter molecules have limited base-recognition capabilities. The binding scores associated with the interactions of theses two ligands are accordingly insensitive to A·T→T·A substitutions and vice versa. The complete exchange of purine and pyrimidine bases, i.e., A·T→G·C or T·A→C·G, however, introduces steric clashes at the ligand-DNA interface and contributes to the A·T binding preferences of these drugs. Most of the basepair changes in the distamycin-DNA and diimidazole lexitropsin-DNA complexes therefore increase the intermolecular energy, either through the drug-DNA interaction score or unpermitted steric interactions. The interchange of certain A·T and T·A pairs in these complexes fails to increase the ligand-binding energy scores or to introduce steric hindrance. Such results are limited to the sites on DNA which the drugs cannot distinguish.
TABLE 7.
Summary of basepair substitution scores for drug-DNA complexes
| Consistency*
|
||||||||
|---|---|---|---|---|---|---|---|---|
| NBD_ID | Substitutions† | Clustering | Local density | Global density | ||||
| 2:1 Drug-DNA complexes | ||||||||
| bdd002 | 12 | 12 (0) | 11 (0) | 12 (0) | ||||
| bdd003 | 12 | 12 (1) | 12 (0) | 12 (0) | ||||
| gdj057 | 12 | 12 (0) | 12 (0) | 12 (0) | ||||
| dd0020 | 12 | 12 (0) | 12 (0) | 12 (0) | ||||
| dd0021 | 12 | 12 (2) | 12 (2) | 12 (2) | ||||
| gdj054 | 15 | 15 (3) | 15 (4) | 15 (4) | ||||
| gdh060 (unit 1) | 18 | 18 (2) | 18 (1) | 18 (1) | ||||
| gdh060 (unit 2) | 18 | 18 (1) | 18 (2) | 18 (2) | ||||
| Totals | 111 | 111 (9) | 110 (9) | 111 (9) | ||||
| 1:1 Drug-DNA complexes | ||||||||
| A·T | A·T | A·T | A·T | A·T | A·T | A·T | A·T | |
| Basepair change | ↕ | ↕ | ↕ | ↕ | ↕ | ↕ | ↕ | ↕ |
| T·A | G·C | T·A | G·C | T·A | G·C | T·A | G·C | |
| dd0014 | 5 | 10 | 3 | 9 | 3 | 10 | 3 | 9 |
| gd1003 | 5 | 10 | 2 | 9 | 3 | 8 | 3 | 10 |
| gd1004 | 4 | 8 | 3 | 6 | 3 | 6 | 2 | 5 |
| gd1008 | 4 | 8 | 2 | 7 | 3 | 6 | 3 | 7 |
| gd1018 | 4 | 8 | 2 | 8 | 2 | 8 | 1 | 8 |
| gd1030 | 4 | 8 | 2 | 7 | 4 | 8 | 2 | 8 |
| gd1033 | 4 | 8 | 3 | 5 | 2 | 5 | 3 | 6 |
| gd1038 | 4 | 8 | 2 | 7 | 3 | 6 | 2 | 6 |
| gd1039 | 5 | 10 | 4 | 8 | 2 | 10 | 4 | 8 |
| gd1047 | 4 | 8 | 3 | 6 | 1 | 7 | 1 | 4 |
| gd1052 | 4 | 8 | 3 | 5 | 2 | 6 | 2 | 6 |
| Totals | 47 | 94 | 29 | 77 | 28 | 80 | 26 | 77 |
Consistency levels correspond to the number of single-base substitutions that either increase the total interaction energy, or introduce steric hindrance into the complex. The values in parentheses for the 2:1 complexes correspond to cases where the basepair change neither increases the total energy nor introduces steric hindrance but which is consistent with the limited recognition capabilities of the drug. Such cases do not exist within the 1:1 drug-DNA complexes.
Substitutions denote the number of single basepair substitutions considered for a particular complex.
The interactions of the 1:1 drug-DNA complexes are even less specific than those of the 2:1 complexes. These ligands typically form a single hydrogen-bond contact with each basepair and associate preferentially with A·T or T·A. The recognition of sequence is again a response of drug to steric clashes with the exocyclic amino group of guanine, rather than to hydrogen-bond specificity. A single hydrogen bond (to the N3 acceptor atom of adenine or the symmetrically placed O2 acceptor atom of thymine) cannot discriminate an A·T from a T·A basepair. The basepair substitutions of 1:1 drug-DNA complexes are thus divided into two categories in Table 7: (1) base interchanges which preserve the chemical composition, e.g., A·T→T·A; and (2) complete base exchange that alters the AT content, e.g., A·T→G·C. Roughly 80% of the latter substitutions either increase the knowledge-based energies or introduce steric hindrance between drug and DNA, but only 60% of the changes in the former category have such effects. The lower number reflects the nonspecific interactions of the ligands with AT-rich DNA. The computed inability of the drugs to bind GC-rich DNA is consistent with the literature (Goodsell, 2001; Wemmer, 2001).
Comparison with in vitro experiments
Experimental systems
As a further test of the ellipsoidal potentials, we compare in Table 8 the computed energies of various drug-DNA complexes with the experimentally measured binding affinities of two polyamide drugs with three different DNA 11-mers (Pilch et al., 1999). The drugs, ImImPy-γ-PyPyPy-β-Dp and ImPyPy-γ-PyPyPy-β-Dp (designed respectively to bind GGT·ACC and GTT·AAC duplex sequences), are single molecules with six sequentially linked rings connected by amide links in either half and at their centers by a γ-aminobutyric acid hairpin turn (denoted by γ in the preceding formulae) rather than the pairs of associated ligands found in the oligonucleotide crystal complexes considered above (see Fig. 7). The targeted DNA-binding sites are located in the middle of two of the three duplexes, 5′-CATTGGTAGAC-3′ and 5′-CATTGTTAGAC-3′ (here denoted by the sequence strands). The third duplex, 5′-CATTATTAGAC-3′, is not expected to bind either ligand tightly.
TABLE 8.
Comparison of experimentally measured DNA-binding affinities of polyamide hairpin ligands to different DNA sequences with knowledge-based energies
| DNA-binding sequence | 5′-TGGTA-3′ | 5′-TGTTA-3′ | 5′-TATTA-3′ |
|---|---|---|---|
| ImImPy-γ-PyPyPy-β-Dp | |||
| ΔGb (kcal/mol)* | −10.0 | −9.4 | −7.1 |
| Energy† | |||
| Local density | 16.8(3.0) | 25.0(5.8) | 64.0(5.0) |
| Clustering | 25.0(6.4) | 48.5(9.0) | 69.4(7.0) |
| Global density | 19.7(3.8) | 27.1(4.5) | 65.0(6.8) |
| ImPyPy-γ-PyPyPy-β-Dp | |||
| ΔGb (kcal/mol)* | −8.0 | −10.7 | −8.8 |
| Energy† | |||
| Local density | 17.9(2.8) | 15.1(4.5) | 39.0(3.3) |
| Clustering | 38.3(5.9) | 24.6(5.7) | 44.6(7.1) |
| Global density | 27.4(4.2) | 17.0(2.5) | 34.9(4.6) |
Data of Pilch et al. (1999) obtained in solution studies at 293 K. ΔGb is the binding free energy, determined from the UV absorption curves.
Energies are the average ligand-binding scores and standard deviations (in parentheses) obtained for 10 drug-DNA models based on the known structures of 2:1 polyamide-DNA complexes and assessed with the designated knowledge-based potential.
FIGURE 7.

Stereo views of computationally “synthesized” structures of the ImPyPy-γ-PyPyPy polyamide hairpin molecule (magenta stick figures) superimposed on the observed structure (space-filled atomic model) of the related ImImPyPy-γ-PyPyPyPy-β-Dp molecule bound to nucleosomal DNA (pd0330) (Suto et al., 2003). The 10 model structures are constructed from the observed positions of pairs of polyamide ligands complexed to DNA in known 2:1 oligonucleotide-ligand complexes (bdd002, bdd003, gdj057, dd0020, and dd0021) (Kielkopf et al., 1998a,b, 2000). The root-mean-square fit of the ring atoms in the predicted models with the corresponding positions in the crystallographically observed structure ranges from 0.4 to 1.0 Å. Atoms of the observed ligand are color-coded such that oxygens are red, nitrogens are blue, and carbons are gray. (a) An “inside” view of the drug surface that binds to the DNA minor groove; (b) an “outside” view showing the overall curvature of the hairpin molecules.
The binding affinities obtained from UV absorption measurements for all six drug-DNA combinations and from circular dichroism titration measurements for complexes with the ligand, ImImPy-γ-PyPyPy-β-Dp, which is expected to bind the GGT-containing 11-mer, support the molecular design. The latter drug binds the predicted sequence with 2.7-fold greater affinity than the GTT-containing molecule, and 158-fold greater affinity than the ATT-containing DNA (Pilch et al., 1999). The three drug pairs (Im·Py, Im·Py, Py·Py) in the ligand match all three basepairs (G·C, G·C, T·A) of GGT, two basepairs of GTT, and one basepair of ATT. The second drug, ImPyPy-γ-PyPyPy-β-Dp, which is designed to associate with the GTT-containing duplex, binds the expected sequence with 24-fold greater affinity than the ATT-containing chain, and with 89-fold greater affinity than the GGT-containing duplex (Pilch et al., 1999). The three drug pairs (Im·Py, Py·Py, Py·Py) match all three basepairs (G·C, T·A, T·A) of GTT, but only two basepairs of GGT or ATT.
Ligand model
For simplicity, we construct hairpin drug structures from the pairs of polyamides bound to DNA in known 2:1 drug-DNA complexes (Fig. 7). The ligands in these complexes share common structural features, which are assumed to be adopted by other polyamide-DNA complexes. The mean internal chemical parameters of the peptide linkers and the imidazole (Im), hydroxypyrrole (Hp), and pyrrole (Py) subunits of the 2:1 complexes (Table S10 in Supplementary Material) are similar to the corresponding values of the hairpin-linked drugs bound to nucleosomal DNA (Suto et al., 2003). The mean absolute differences in the chemical bond lengths and valence angles of ligands bound to the oligonucleotides versus those bound to nucleosomal DNA are 0.02 Å and 1.1°, respectively. A total of 10 hairpin models are considered for each drug-DNA system (by superposition of the appropriate chemical subunits in two orientations on each of the five polyamide-DNA crystal templates). The peptide linkers between drug subunits are automatically generated, but the γ-aminobutyric acid linkers that join the two polyamide strings are not built.
Interaction scores
The energy score assigned to each of the six drug-DNA complexes is the average of the binding energies computed for the 10 complexes surveyed and thus includes the minor entropic effects associated with the variation of structure. The computed scores are compared with the measured thermodynamic properties (Pilch et al., 1999) in Table 8. The predictions based on the knowledge-based functions are consistent with the experimental data in that the ligand-binding scores are lowest for the sequences with highest affinity to a particular drug. Notably, the magnitude of the computed energies of the three ImImPy-γ-PyPyPy-β-Dp complexes varies roughly linearly with the Gibbs free energies (ΔGb values in Table 8), particularly the energies computed with the pseudoelectron density potentials. On the other hand, since the computations incorporate neither hydrogen-bond donor-donor repulsions nor steric hindrance into the energy of the complex, the sterically hindered configuration of ImPyPy-γ-PyPyPy-β-Dp against the 5′-CATTGGTAGAC-3′ duplex cannot be directly compared with the measured free energies. (The exocyclic amino group on the underlined guanine comes too close to the C3 atom on the underlined pyrrole unit in all polyamide-DNA templates.)
Polyamide-nucleosome binding
The binding scores of hairpin polyamides which are complexed to exposed segments of DNA on the nucleosome core particle structure tend to be of greater magnitude than the corresponding interactions of polyamides found in 2:1 drug-oligonucleotide crystal complexes (see Tables 4 and 9, and Table S8 in Supplementary Material). The energies derived from local Fourier averaging and clustering calculations span the respective ranges 1.9–4.9 and 3.1–5.6 for the binding of ImPyPy-γ-PyPyPy-β-Dp to the model oligonucleotide-ligand templates versus 2.6 ± 2.6 and 3.3 ± 2.2 for the interaction of the closely related ImImPyPy-γ-PyPyPyPy-β-Dp molecule with nucleosomal DNA. (The cited values for the oligonucleotide templates are the quotient of the total interaction scores listed in Table 8 and the eight close donor-acceptor contacts in the modeled DNA-drug complex.)
TABLE 9.
Energy scores of polyamide-DNA interactions in lexitropic polyamide-nucleosomal DNA complexes
| DNA-binding site
|
Energies*
|
|||||
|---|---|---|---|---|---|---|
| Ligand | Designed contacts† | Observed contacts on DNA‡ | Ligand direction§ | SH‖ | Clustering | Local density |
| pd0328: ImPyImPy-γ-PyPyPyPy-β-Dp (Suto et al., 2003) | ||||||
| I |  |
A030G031T032G033T034A035T263C262A261C260A259T258 | → | −4 | 6.9(6.4) | 3.2(2.7) |
| II |  |
A176G177T178G179T180A181T117C116A115C114A113T112 | ← | +4 | 5.3(3.9) | 3.0(2.4) |
| pd0329: ImPyPyPy-γ-PyPyPyPy-β-Dp (Suto et al., 2003) | ||||||
| I |  |
G031T032G033T034A035T036C262A261C260A259T258A257 | → | −4 | 6.4(5.3) | 3.9(3.5) |
| II |  |
A248G249T250T251T252C253T045C044A043A042A041G040 | ← | −3 | 6.8(5.5) | 3.2(1.6) |
| III |  |
G070G071A072A073T074T075C223C222T221T220A219A218 | → | 0 | 4.0(5.0) | 1.6(2.0) |
| IV |  |
A102G103T104T105T106C107T191C190A189A188A187G186 | ← | +3 | 5.3(3.5) | 3.3(1.8) |
| V |  |
G177T178G179T180A181T182C116A115C114A113T112A111 | → | +4 | 7.6(7.2) | 4.9(5.8) |
| pd0330: ImImPyPy-γ-PyPyPyPy-β-Dp (Suto et al., 2003) | ||||||
| I | 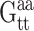 |
T282G283G284A285T286A287A011C010C009T008A007T006 | ← | −6 | 3.3(2.2) | 2.6(2.6) |
Because of the more exhaustive calculations needed to assess DNA-ligand interactions on the basis of global pseudoelectron densities, only scores based on clustering and local density potentials are reported.
The symbol 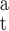 denotes a ligand design which allows for adenine or thymine at the given site on DNA.
denotes a ligand design which allows for adenine or thymine at the given site on DNA.
Bases in direct contact with proton donor and acceptor atoms on polyamide ligands are underlined. The complementary strand is shown below the sequence strand.
The arrows indicate the direction of the polyamide with respect to the sequence strand of nucleosomal DNA, i.e., nucleotides 1–146.
SH refers to the superhelical location of the binding site relative to the central dyad of nucleosomal DNA, i.e., number of helical turns from the dyad.
As expected from the global asymmetry of the nucleosome core particle, i.e., the two halves of the 146-bp structure are 1-bp out of register (Luger et al., 1997; Suto et al., 2003), the designed polyamides do not take the same advantage of hydrogen-bonding sites at sequentially symmetric sites in the complex. The contacts of ImPyImPy-γ-PyPyPyPy-β-Dp with its AGTGTA·TACACT duplex target (pd0328) are scored more favorably in the longer half of the structure (at superhelical position SH +4, i.e., four helical turns past the dyad on basepair 73) than in the shorter half (at SH −4). Moreover, the locations of bound ligands do not necessarily match the expected chemical design. The subunits of ImPyPyPy-γ-PyPyPyPy-β-Dp in pd0329 make unexpected direct contacts with a GTGT·ACAC target at SH −4 and recognize a shortened GTA·ATC target at SH +4. The scores of the latter interactions are generally higher than those of the same ligand in the vicinity of three expected DNA targets—GTTT·AAAC (SH ±3), GAAT·ATTC (SH 0). Furthermore, the lowest scoring contacts of ImPyPyPy-γ-PyPyPyPy-β-Dp (at SH 0) and ImImPyPy-γ-PyPyPyPy-β-Dp (at SH −6) with nucleosomal DNA differ from the preferred binding sites determined in solution. The former ligand binds preferentially at the SH ±4 sites, which make unexpected intermolecular contacts in the crystal form (see above), and the latter ligand contacts DNA at SH ±3 as well as SH ±6 (Gottesfeld et al., 2001).
DISCUSSION
Knowledge-based ligand-binding potentials
Overview
The sets of density ellipsoids reported in this article provide a quantitative framework for characterizing the association of ligands with the DNA bases and a new way to assess the binding of arbitrary molecular species to specific genetic sequences. The composite findings from complementary (Fourier averaging and clustering) analyses of the positions of water molecules and amino acid atoms in contact with the DNA bases in well-resolved crystal structures make it possible to assign numerical descriptors to different base-recognition motifs and to deduce the likely arrangements of other molecules, e.g., drugs, in the grooves of the double helix. The features of molecular association reproduced by the different methods of analysis enhance the reliability of the numerical findings. The similar positioning and direction of approach of ligands to corresponding atoms in different structures help to decipher chemical trends in hydrogen-bonding patterns and binding geometries. The quantified differences in ligand binding to A- and B-DNA helices provide new perspectives on the solvent-induced B→A transformation.
Symmetric recognition and patterns of minor-groove binding
The mean positions of closely associated waters and amino acid hydrogen-bond donors in the vicinity of the purine N3 and pyrimidine O2 atoms offer new insight into the minor-groove recognition of Watson-Crick basepairs. The rough equivalence of purine and pyrimidine binding positions, i.e., mean (〈x〉, 〈y〉, 〈z〉) coordinates and direction cosines (λ3,1, λ3,2, λ3,3) in Table 2, and Tables S4–S6 in Supplementary Material, confirms the well-known pseudosymmetry of Watson-Crick basepairing and the regularity of minor-groove recognition of normal duplex DNA by other molecules. Here we additionally see that the preferential approach of ligands to a B-DNA basepair is pincer-like, with contacts to the base in the leading strand coming from below the common basepair plane and those to the complementary base located above the plane.
The offset of recognition sites, toward the 5′-phosphate groups of the antiparallel strands, promotes an economy of interaction between the bases and the sugar oxygens of the preceding residues on each strand, i.e., the bridging of N3 or O2 and the neighboring sugar by a common water molecule or amino acid residue (Moravek et al., 2002). The sets of vectors connecting the centers of N3 and O2 ellipsoids of sequential bases further describe two right-handed strands of ligand-binding clusters (Fig. 5), corresponding to the well-known spine of hydration in the B-DNA minor groove (Drew and Dickerson, 1981). By contrast, the approach of ligands to the exposed minor-groove edges of A-DNA basepairs shows more lateral, in-plane character. The 〈z〉 component of the N3 and O2 binding centers of the four bases in A-DNA structures is closer to zero (Tables S4 and S5 in Supplementary Material) and the major axes of the binding ellipsoids span a broad range of orientations (Table S6 in Supplementary Material).
Intrinsic asymmetry of major-groove recognition
DNA-binding ligands also appear to approach A·T pairs differently from G·C pairs in the B-DNA major groove (Table S6). That is, the adenine N6 and cytidine N4 are approached laterally and the guanine O6 and thymine O4 are contacted from above or below, i.e., parallel to the base normal. These tendencies may be related to intrinsic chemical features of the exocyclic carbonyl and amino groups (since the approach to the G and T oxygens and the A and C nitrogens are comparable) or to well-known sequence-dependent differences in DNA major-groove width and accessibility, i.e., the major groove of G·C-rich helical stretches in B-DNA structures is typically wider than that of A·T stretches (Heinemann et al., 1992).
Chemical basis of recognition
The present analysis of binding clusters further reveals a clear connection between the hydrogen-bonding distances to different atoms, the number of associated waters per binding site, and the partial atomic charges of the contacted base atoms. The results support the idea that the strength of hydrogen bonding reflects the magnitude of the charges on the associated proton donor and acceptor atoms (Jeffrey, 1997). The base atoms with more negative charges bring ligands closer (Fig. 4) and those with charge of greater absolute magnitude attract a greater number of bound ligands. Although secondary interactions may contribute to the stabilities of multi-hydrogen-bonded complexes of the DNA bases (Pranata et al., 1991), the primary contacts seemingly govern the observed hydrogen-bonded geometry.
The closest, and presumably tightest, hydrogen-bonding interactions involve nitrogen and oxygen acceptor atoms on the bases. The mean (2.6–2.8 Å) displacement of ligand atoms from such sites is consistently smaller than the corresponding (2.9–3.0 Å) distances to nitrogen donor atoms (Table 3). Some of the binding sites identified as waters in the vicinity of the proton acceptor atoms, however, may be localized sodium ions which approach these partially negatively charged sites more closely than water and thereby contribute to the smaller distances. For example, ∼15% of the observed close contacts of water with the N7 atoms of adenine and the O6 atoms of guanine in B-DNA structures are 2.5 Å or less in value, a limit corresponding to the mean distance of direct sodium-DNA contacts in the very best resolved DNA and RNA crystal structures (Tereshko et al., 2001). On the other hand, the rates of water displacement determined in molecular dynamics simulations (Auffinger and Westhof, 2001) tend to be lower for atoms bearing greater partial charge, i.e., electrostatic terms in the force field seemingly contribute to the occupancy times. With the exception of the (2.6–2.8 Å) water and amino acid contacts to the C5 atom of cytidine, the average distances between ligand and base carbons exceed 3 Å. The C5 atom stands out from all other carbons in being assigned a significant negative charge in the AMBER force field (Table S7 in Supplementary Material) and in retaining this feature in quantum mechanical studies, which are more accurate than those used in the parameterization of AMBER (A. R. Srinivasan, R. R. Sauers, M. O. Fenley, A. H. Boschitsch, A. Matsumoto, A. V. Colasanti, and W. K. Olson, unpublished data).
The interactions of ligands with the purine C8 and pyrimidine C6 atoms on the outer edges of the Watson-Crick basepairs depend upon helical context. There are no clusters of waters near such sites in A-DNA helices, nor any closely associated amino acid atoms in the vicinity of R(C8) and Y(C6) in protein-bound DNA structures. The 5′-phosphorus atoms of A-DNA lie roughly in the same plane as the bases attached to the same sugar (Lu et al., 2000), leaving little space for water near the C6 or C8 atoms. The A-DNA phosphate oxygens seemingly displace the C6/C8 water clusters of the B-form structure, allowing the DNA to act as its own solvent in the dehydrated A-form. These intramolecular C–H⋯O interactions contribute a previously unrecognized component to the well-known economy of hydration around the A-DNA phosphates (Saenger et al., 1986).
Knowledge-based assessment of DNA-ligand interactions
Binding scores
The knowledge-based elastic functions that describe the hydration patterns around the DNA bases provide a new means to assess the sequence-specific recognition of proteins and drugs in the grooves of the double helix. The low scores of hydrogen-bonding sites on minor-groove binding ligands relative to the expected locations of water molecules around the DNA bases in well-resolved oligonucleotide crystal complexes confirm the utility of the elastic expressions (Tables 4 and 5). The unfavorable high scores of incorrect sequences superimposed on DNA structures, which are co-crystallized with polyamide molecules designed to recognize the minor-groove edges of specific Watson-Crick basepairs (Kielkopf et al., 1998a, 1998b, 2000), show how the potentials capture aspects of sequence-selective binding (Tables 6 and 7). The capabilities of the knowledge-based functions are also evident in the satisfactory computational accounting of the measured free energies of DNA-polyamide association in solution (Pilch et al., 1999) (Table 8) and the correspondence of low energy scores with the observed sites of polyamide binding on nucleosomal DNA (Suto et al., 2003) (Table 9). With a notable exception discussed below, the numerical findings support the mechanisms by which minor-groove binding ligands are thought to discriminate among DNA sequences. The elastic displacement of key ligand atoms from probable sites of base contact thus provides a convenient mathematical framework for studying the interactions between drug or protein molecules with DNA and shows promise for gaining insight into binding mechanisms. In addition to the analysis of known high resolution DNA-ligand complexes, the structure-based potentials can be used for rapid docking of arbitrary molecules on the surface of DNA and for assessment of the predictions of all-atom, physics-based force fields, e.g., AMBER (Weiner et al., 1984; Cornell et al., 1995; Cieplak et al., 2001) or CHARMM (Brooks et al., 1983; Foloppe et al., 2000). For example, the knowledge-based potentials incorporate new details of the space and directionality of ligand binding not considered in earlier analyses of computer-simulated DNA hydration (Feig and Pettitt, 1999). Versatile DNA docking, however, requires the knowledge-based representation of other DNA-ligand interactions, e.g., metals, anions, etc., with the DNA sugars, phosphates, and bases.
Mechanistic insights
The current findings in many ways confirm published experimental work and earlier ideas about the mechanism of DNA sequence recognition. For instance, the ImPyPyPy-β-Dp polyamide is known to have a higher binding affinity for its target sequence than ImHpPyPy-β-Dp (with dissociation constants of 48 nM and 344 nM, respectively) (Kielkopf et al., 1998b), and this is reflected by the higher energy of ImHpPyPy-β-Dp interaction compared to ImPyPyPy-β-Dp association with the same 5′-GTAC-3′ target sequence (bdd002 versus bdd003 in Table S8 in Supplementary Material). The computational results also confirm the expected discriminatory mechanisms of Py·Py and Hp·Py binding (Goodsell, 2001). That is, the interactions of Py·Py drug pairs with A·T or T·A basepairs are believed to be free of close intermolecular contacts, but those with G·C or C·G basepairs are thought to introduce unfavorable nonbonded contacts (Pelton and Wemmer, 1989; White et al., 1996). The computer substitution studies of the bases at the central TA and AT dimer steps in structures bdd003 and dd0021 support these expectations (calculations labeled S1b, S1c, S1n, S1o, S1x, and S1y in Table S9 in Supplementary Material). The corresponding substitutions of the basepairs in structures bdd002 and dd0020 (Table 6) similarly confirm the notion that steric conflicts between the NH2 group on G and the OH group on the Hp ring impede the binding of Hp·Py drug pairs to either G·C or C·G (Kielkopf et al., 1998a; White et al., 1998), as well as the mechanism by which the Hp·Py pair discriminates T·A from A·T (Kielkopf et al., 1998b; Goodsell, 2001). The calculations coded S1b and S1c show that the interaction of the O3–H group on Hp and the O2 atom on T plays a crucial role in basepair recognition, as anticipated in the molecular design.
Interestingly, the calculations do not support the mechanism by which the Im·Py drug pair is thought to recognize a G·C basepair. The discriminating power—in 8 of 10 interchanges of the G·C and C·G basepairs at the edges of the recognition sites in the five polyamide complexes (calculations coded S1a and S1d in Table 6, and Table S9 in Supplementary Material) and in the two exchanges of G and C in the center of the gdj057 complex (calculations coded S1b and S1c)—arises from the asymmetric distribution of hydrogen-bond forming atoms, i.e., G(N2), on the minor-groove edges of the basepairs rather than steric effects between the Py group and the NH2 group of G (Goodsell, 2001; Wemmer, 2001). The increased energy of the modified sequences reflects the repositioning of proton donor and acceptor atoms between drug and DNA. That is, the imidazole ring is capable of changing its association from the G in the original G·C pair to the G on the complementary strand of the substituted C·G basepair. Only in two cases (calculations labeled S1d for bdd002 and S1a for bdd003) is the steric discrimination of the NH2 on G by a CH group on the pyrrole ring, which is predicted by the recognition code (Trauger et al., 1996; White et al., 1997), also found. These inconsistencies merit further study to determine if the results are correct or reflect inaccuracies in the present work, such as the failure to optimize DNA and drug structures upon base modifications (see below).
The hydrogen-bonding mechanism believed to account for the discriminating power of the Im·Py pair for G·C and C·G over A·T and T·A binding, however, is substantiated by the calculations. The increase in interaction energy found upon substitution of the G·C and C·G basepairs at the ends of the recognition sites in all five polyamide complexes (Table 6, and Table S9 in Supplementary Material, cases coded S1m, S1p, S1w, and S1z) and upon replacement of the central G and C in gdj057 (calculations labeled S1n, S1o, S1x, and S1y), confirms the stabilizing role of the hydrogen bond between the NH2 group of G and the N3 atom on the imidazole ring in the minor-groove complexes, but not the specificity of Im for the guanine on the same side of the complex (Goodsell, 2001; Wemmer, 2001).
DNA sequence-dependent structure
The present calculations ignore the well-known sequence-dependent fine structure of DNA (Gorin et al., 1995; Olson et al., 1998) and may therefore compromise the estimates of binding specificity. The general similarity of DNA local helical structure in the known 2:1 drug-DNA-binding complexes (Fig. 8), however, suggests that omission of sequence-structure contributions may have limited effects on the accuracy of the calculations. The basepair step parameters of different DNA sequences bound to the same ligand are fairly similar, e.g., d(CCAGTACTGG)2 and d(CCAGATCTGG)2 duplexes complexed with ImPyPyPy-β-Dp in structures bdd003 and dd0021. It is not clear whether the subtle differences in key variables in such structures (e.g., RollTA in bdd003 is more positive than RollAT in dd0021) reflect the sequence substitutions or the different space groups in which the DNA complexes are crystallized. Although the observed differences in Roll at the central basepair step of polyamide-bound DNA structures follow trends seen in B-DNA and protein-DNA complexes, i.e., RollGC < RollAT < RollTA, the step parameters and groove widths of the DNA from structures crystallized in the same space group—e.g., C2: bdd002 and bdd003, and P 21 21 21: gdj057 and dd0021—are remarkably similar despite differences in associated ligands and/or bound DNA. In this sense, the present template model of interactions may generate better results than a flexible docking algorithm, which adjusts the drug structure as well as the DNA basepair step parameters in the process of intermolecular fitting.
FIGURE 8.

Variation of key basepair step parameters (Roll, Twist, Slide) and DNA major and minor-groove widths (P–P distances) versus sequence in the five 2:1 polyamide-DNA structures. Data calculated with the 3DNA software package (Lu and Olson, 2003). The range of parameters observed in B-DNA structures, i.e., values within ±1σ of the mean values for a generic dimer step (Olson et al., 1998; Lu et al., 2000), are denoted by dashed lines.
The series of polyamide ligands bound to decamer duplexes consistently widen the minor groove and narrow the major groove over that of ligand-free B-DNA (Fig. 8). Such changes, which are also seen in the nucleosomal polyamide complexes, resemble the deformations in helical structure known to accompany the transition of B-DNA to the A form (Zhurkin et al., 1979; Lu et al., 2000). The computed interstrand P⋯P distances in the figure confirm earlier observations that the side-by-side binding of polyamide ligands widens the minor groove of DNA (Wemmer and Dervan, 1997; Goodsell, 2001). In some cases, the drugs that associate with DNA induce conformational changes in basepair step parameters outside the ligand-binding site (note the uniformly positive Slide values and concomitant overtwisting of successive basepairs in Fig. 8). The latter trends are expected to persist with related drugs and other DNA hosts, and indeed, are found in the polyamide complexes with nucleosomal DNA.
Most of the basepair step parameters in the known 2:1 polyamide-duplex structures are consistent with the sequence-dependent mean values observed in ligand-free B-DNA structures, in the sense that the step parameters lie within a few standard deviations of the reported mean values for individual dimers (Gorin et al., 1995; Berman and Olson, 2003). Specifically, 49.3% of the step parameters in the central drug binding domain of the five lexitropic sequences lie within ±1.0 standard deviation (±1σ) of the mean values, 80.7% within ±2.0σ, and 96.7% within ±3.0σ, where the sequence-dependent standard deviations σ are based on the observed values in protein-DNA crystal complexes (Olson et al., 1998). (The protein-DNA values, which are typically greater than those derived from pure B-DNA structures, are consistent with the persistence length of DNA (Matsumoto and Olson, 2002) and are expected to be more relevant to DNA deformed by groove-binding agents.)
Limitations of knowledge-based studies
The binding scores of DNA-ligand complexes reported here are based on trends in known high resolution structures rather than on physical principles. The combination of the knowledge-based potentials with Ramachandran distance cutoffs excludes the balance of favorable and unfavorable interactions incorporated in traditional all-atom computational treatments (Brooks et al., 1983; Weiner et al., 1984; Cornell et al., 1995; Foloppe et al., 2000; Cieplak et al., 2001). It is therefore not possible to extract the underlying forces behind particular binding motifs with the present approach. Although the functions do not consider either water-mediated hydrogen-bond contacts or the strength of hydrogen bonds, other measures, such as a scale of hydrogen-bond strengths based on the frequency of water-mediated contacts at different hydration sites (Bohm, 1992), can be introduced to mimic these effects. The knowledge-based model implicitly incorporates aspects of water and amino acid binding, such as the orientation and preferred approach of ligands to DNA, which may influence the strength of hydrogen bonding interactions (Jeffrey, 1997). These subtle effects are contained in the shapes and directions of the ellipsoidal energy functions. The interactions between multiple drugs bound simultaneously in the DNA grooves and the intramolecular interactions within specific drug molecules are not considered.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
This work was taken in part from the dissertation of Wei Ge written in partial fulfillment of the requirements for the degree of Doctor of Philosophy, Rutgers University, 2003.
We are grateful to Dr. A. R. Srinivasan for assistance with molecular graphics and Professor Helen M. Berman for discussions and encouragement.
This work has been generously supported by the U.S. Public Health Service (research grant GM20861 to W.K.O.) and the MSMT of the Czech Republic (grant LN00A032 to B.S.). Computations were carried out at the Rutgers University Center for Computational Chemistry and through the facilities of the Nucleic Acid Database project (National Science Foundation grant DBI-0110076).
References
- Auf der Heyde, T. P. E. 1990. Analyzing chemical data in more than two dimensions. J. Chem. Ed. 67: 461–469. [Google Scholar]
- Auffinger, P., and E. Westhof. 2001. Water and ion binding around r(UA)12 and d(TA)12 oligomers—comparison with RNA and DNA (CG)12 duplexes. J. Mol. Biol. 305:1057–1072. [DOI] [PubMed] [Google Scholar]
- Aymami, J., C. M. Nunn, and S. Neidle. 1999. DNA minor groove recognition of a non-self-complementary AT-rich sequence by a tris-benzimidazole ligand. Nucleic Acids Res. 27:2691–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendiran, K., S. T. Rao, C. Y. Sekharudu, G. Zon, and M. Sundaralingam. 1995. X-ray structures of the B-DNA dodecamer d(CGCGTTAACGCG) with an inverted central tetranucleotide and its netropsin complex. Acta Crystallogr. D51:190–198. [DOI] [PubMed] [Google Scholar]
- Berman, H. M., and W. K. Olson. 2003. The many twists of DNA. In DNA50: The Secret of Life. M. Balaban, editor. Faircount, London, UK. 104–124.
- Berman, H. M., W. K. Olson, D. L. Beveridge, J. Westbrook, A. Gelbin, T. Demeny, S.-H. Hsieh, A. R. Srinivasan, and B. Schneider. 1992. The nucleic acid database: a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 63:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm, H. J. 1992. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J. Comp. Aided Mol. Design. 6:61–78. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4:187–217. [Google Scholar]
- Chen, X., B. Ramadrishnan, and M. Sundaralingam. 1997. Crystal structures of the side-by-side binding of distamycin to AT-containing DNA octamers d(ICITACIC) and d(ICATATIC). J. Mol. Biol. 267:1157–1170. [DOI] [PubMed] [Google Scholar]
- Chen, X., B. Ramakrishnan, S. T. Rao, and M. Sundaralingam. 1994. Side-by-side binding of two distamycin A drugs in the minor groove of an alternating B-DNA duplex. Nat. Struct. Biol. 1:169–175. [DOI] [PubMed] [Google Scholar]
- Cieplak, P., J. Caldwell, and P. Kollman. 2001. Molecular mechanical models for organic and biological systems going beyond the atom-centered two-body additive approximation: aqueous solution free energies of methanol and N-methyl acetamide, nucleic acid base, and amide hydrogen bonding and chloroform/water partition coefficients of the nucleic acid bases. J. Comp. Chem. 22:1048–1057. [Google Scholar]
- Clark, G. R., D. W. Boykin, A. Dzarny, and S. Neidle. 1997. Structure of a bis-amidinium derivative of Hoechst 33258 complexed to dodecanucleotide d(CGCGAATTCGCG)2: the role of hydrogen bonding in minor groove drug-DNA recognition. Nucleic Acids Res. 25:1510–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, G. R., E. J. Gray, S. Neidle, Y.-H. Li, and W. Leupin. 1996a. Isohelicity and phasing in drug-DNA sequence recognition: crystal structure of a tris (benzimidazole)-oligonucleotide complex. Biochemistry. 35:13745–13752. [DOI] [PubMed] [Google Scholar]
- Clark, G. R., C. J. Squire, E. J. Gray, W. Leupin, and S. Neidle. 1996b. Designer DNA-binding drugs: the crystal structure of a meta-hydroxy analogue of Hoechst 33258 bound to d(CGCGAATTCGCG)2. Nucleic Acids Res. 24:4882–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowney, L., S. C. Jain, A. R. Srinivasan, J. Westbrook, W. K. Olson, and H. M. Berman. 1996. Geometric parameters in nucleic acids: nitrogenous bases. J. Am. Chem. Soc. 118:509–518. [Google Scholar]
- Coll, M., J. Aymami, G. A. van der Marel, J. H. van Boom, A. Rich, and A. H.-J. Wang. 1989. Molecular structure of the netropsin-d(CGCGATATCGCG) complex: DNA conformation in an alternating AT segment. Biochemistry. 28:310–320. [DOI] [PubMed] [Google Scholar]
- Coll, M., C. A. Frederick, A. H.-J. Wang, and A. Rich. 1987. A bifurcated hydrogen-bonded conformation in the d(A·T) base pairs of the DNA dodecamer d(CGCAAATTTGCG) and its complex with distamycin. Proc. Natl. Acad. Sci. USA. 84:8385–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 50:760–763. [DOI] [PubMed] [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second-generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Dervan, P. B., and R. W. Burli. 1999. Sequence-specific DNA recognition by polyamides. Curr. Opin. Chem. Biol. 3:688–693. [DOI] [PubMed] [Google Scholar]
- Drew, H. R., and R. E. Dickerson. 1981. Structure of a B-DNA dodecamer. III. Geometry of hydration. J. Mol. Biol. 151:535–556. [DOI] [PubMed] [Google Scholar]
- Eisen, M. B., D. C. Wiley, M. Karplus, and R. E. Hubbard. 1994. HOOK: a program for finding novel molecular architectures that satisfy the chemical and steric requirements of a macromolecule binding site. Proteins. 19:199–221. [DOI] [PubMed] [Google Scholar]
- Feig, M., and B. M. Pettitt. 1999. Modeling high-resolution hydration patterns in correlation with DNA sequence and conformation. J. Mol. Biol. 286:1075–1095. [DOI] [PubMed] [Google Scholar]
- Foloppe, N., J. MacKerell, and D. Alexander. 2000. All-atom empirical force field for nucleic acids. I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comp. Chem. 21:86–104. [Google Scholar]
- Gavezzotti, A., and G. Filippini. 1994. Geometry of the intermolecular X–H⋯Y (X, Y = N,O) hydrogen bond and the calibration of empirical hydrogen-bond potentials. J. Phys. Chem. 98:4831–4837. [Google Scholar]
- Gillet, V., A. P. Johnson, P. Mata, S. Sike, and P. Williams. 1993. SPROUT: a program for structure generation. J. Comp. Aided Mol. Design. 7:127–153. [DOI] [PubMed] [Google Scholar]
- Goodsell, D. S. 2001. Sequence recognition of DNA by lexitropsins. Curr. Med. Chem. 8:509–516. [DOI] [PubMed] [Google Scholar]
- Goodsell, D. S., M. Kaczor-Grzeskowiak, and R. E. Dickerson. 1995. Crystal structure of C-T-C-T-C-G-A-G-A-G. Implications for the structure of the Holliday junction. Biochemistry. 34:1022–1029. [DOI] [PubMed] [Google Scholar]
- Gorin, A. A., V. B. Zhurkin, and W. K. Olson. 1995. B-DNA twisting correlates with base pair morphology. J. Mol. Biol. 247:34–48. [DOI] [PubMed] [Google Scholar]
- Gottesfeld, J. M., C. Melander, R. K. Suto, H. Raviol, K. Luger, and P. B. Dervan. 2001. Sequence-specific recognition of DNA in the nucleosome by pyrrole-imidazole polyamides. J. Mol. Biol. 309:615–629. [DOI] [PubMed] [Google Scholar]
- Heinemann, U., C. Alings, and M. Bansal. 1992. Double helix conformation, groove dimensions and ligand binding potential of a G·C stretch in B-DNA. EMBO J. 11:1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, B. K. P. 1987. Closed-form solution of absolute orientation using unit quaternions. J. Opt. Soc. Am. A. 4:629–642. [Google Scholar]
- Howerton, S. B., C. C. Sines, D. VanDerveer, and L. D. Williams. 2001. Locating monovalent cations in the grooves of B-DNA. Biochemistry. 40:10023–10031. [DOI] [PubMed] [Google Scholar]
- Jeffrey, G. A. 1997. Chapt. 10. In Introduction to Hydrogen Bonding. Oxford University Press, New York.
- Jones, G., P. Willett, R. C. Glen, A. R. Leach, and R. Taylor. 1997. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267:727–748. [DOI] [PubMed] [Google Scholar]
- Kielkopf, C. L., E. E. Baird, P. B. Dervan, and D. C. Rees. 1998a. Structural basis for G·C recognition in the DNA minor groove. Nat. Struct. Biol. 5:104–109. [DOI] [PubMed] [Google Scholar]
- Kielkopf, C. L., R. E. Bremer, S. White, J. W. Szewczyk, J. M. Turner, E. E. Baird, P. B. Dervan, and D. C. Rees. 2000. Structural effects of DNA sequence on T·A recognition by hydroxypyrrole/pyrrole pairs in the minor groove. J. Mol. Biol. 295:557–567. [DOI] [PubMed] [Google Scholar]
- Kielkopf, C. L., S. White, J. W. Szewszyk, J. M. Turner, E. E. Baird, P. B. Dervan, and D. C. Rees. 1998b. A structural basis for recognition of A·T and T·A base pairs in the minor groove of B-DNA. Science. 282:111–115. [DOI] [PubMed] [Google Scholar]
- Klebe, G., and U. Abraham. 1994. On the prediction of binding properties of drug molecules by comparative molecular field analysis. J. Med. Chem. 36:70–80. [DOI] [PubMed] [Google Scholar]
- Kopka, M. L., A. V. Fratini, H. R. Drew, and R. E. Dickerson. 1983. A quantitative study of ordered water structure around a B-DNA dodecamer. J. Mol. Biol. 163:129–146. [DOI] [PubMed] [Google Scholar]
- Kopka, M. L., D. S. Goodsell, G. W. Han, T. K. Chiu, J. W. Lown, and R. E. Dickerson. 1997. Defining GC-specificity in the minor groove: side-by-side binding of the di-imidazole lexitropsin to C-A-T-G-G-C-C-A-A-T-G. Structure. 15:1033–1046. [DOI] [PubMed] [Google Scholar]
- Kopka, M. L., C. Yoon, D. Goodsell, P. Pjura, and R. E. Dickerson. 1985a. Binding of an antitumor drug to DNA. Netropsin and C-G-C-G-A-A-T-T-BrC-G-C-G. J. Mol. Biol. 183:553–563. [DOI] [PubMed] [Google Scholar]
- Kopka, M. L., C. Yoon, D. Goodsell, P. Pjura, and R. E. Dickerson. 1985b. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc. Natl. Acad. Sci. USA. 82:1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz, I. D., J. M. Blaney, S. J. Oatley, R. Langridge, and T. E. Ferrin. 1982. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 161:269–288. [DOI] [PubMed] [Google Scholar]
- Larson, T. A., D. S. Goodsell, D. Cascio, K. Grzeskowiak, and R. E. Dickerson. 1989. The structure of DAPI bound to DNA. J. Biomol. Struct. Dyn. 7:477–491. [DOI] [PubMed] [Google Scholar]
- Leonard, G. A., K. McAuley-Hecht, T. Brown, and W. N. Hunter. 1995. Do C–H⋯O hydrogen bonds contribute to the stability of nucleic acid basepairs? Acta Crystallogr. D51:136–139. [DOI] [PubMed] [Google Scholar]
- Llamas-Saiz, A. L., and C. Foces-Foces. 1990. N–H⋯N sp2 hydrogen interactions in organic crystals. J. Mol. Struct. 238:367–382. [Google Scholar]
- Lu, X.-J., and W. K. Olson. 2003. 3DNA: a software package for the analysis, rebuilding, and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 31:5108–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X.-J., Z. Shakked, and W. K. Olson. 2000. A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol. 300:819–840. [DOI] [PubMed] [Google Scholar]
- Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 389:251–260. [DOI] [PubMed] [Google Scholar]
- Mandel-Gutfreund, Y., H. Margalit, R. L. Jernigan, and V. B. Zhurkin. 1998. A role for CH⋯O interactions in protein-DNA recognition. J. Mol. Biol. 277:1129–1140. [DOI] [PubMed] [Google Scholar]
- Matsumoto, A., and W. K. Olson. 2002. Sequence-dependent motions of DNA: a normal mode analysis at the base-pair level. Biophys. J. 83:22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. D., S. K. Kearsley, D. J. Underwood, and R. P. Sheridan. 1994. FLOG: a system to select “quasi-flexible” ligands complementary to a receptor of known three-dimensional structure. J. Comp. Aided Mol. Design. 8:153–174. [DOI] [PubMed] [Google Scholar]
- Mitra, S. N., M. C. Wahl, and M. Sundaralingam. 1999. Structure of the side-by-side binding of distamycin to d(GTATATAC)2. Acta Crystallogr. D55:602–609. [DOI] [PubMed] [Google Scholar]
- Moravek, Z., S. Neidle, and B. Schneider. 2002. Protein and drug interactions in the minor groove of DNA. Nucleic Acids Res. 30:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, W. K., M. Bansal, S. K. Burley, R. E. Dickerson, M. Gerstein, S. C. Harvey, U. Heinemann, X.-J. Lu, S. Neidle, Z. Shakked, H. Sklenar, M. Suzuki, C.-S. Tung, E. Westhof, C. Wolberger, and H. M. Berman. 2001. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 313:229–237. [DOI] [PubMed] [Google Scholar]
- Olson, W. K., A. A. Gorin, X.-J. Lu, L. M. Hock, and V. B. Zhurkin. 1998. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA. 95:11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro, C. M., I. D. Kuntz, and J. S. Dixon. 1995. Flexible ligand docking using a genetic algorithm. J. Comp. Aided Mol. Design. 9:113–130. [DOI] [PubMed] [Google Scholar]
- Pelton, J. G., and D. E. Wemmer. 1989. Structural characterization of a 2:1 distamycin A·d(CGCAAATTGGC) complex by two-dimensional NMR. Proc. Natl. Acad. Sci. USA. 86:5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch, D. S., N. Poklar, E. E. Baird, P. B. Dervan, and K. J. Breslauer. 1999. The thermodynamics of polyamide-DNA recognition: hairpin polyamide binding in the minor groove of duplex DNA. Biochemistry. 38:2143–2151. [DOI] [PubMed] [Google Scholar]
- Pirard, B., G. Baudoux, and F. Durant. 1995. A database study of intermolecular NH⋯O hydrogen bonds for carboxylates, sulfonates, and monohydrogen phosphonates. Acta Crystallogr. B. 51:103–107. [Google Scholar]
- Pranata, J., S. G. Wierschke, and W. L. Jorgensen. 1991. OPLS potential functions for nucleotide bases. Relative association constants of hydrogen-bonded base pairs in chloroform. J. Am. Chem. Soc. 113:2810–2819. [Google Scholar]
- Ramachandran, G. N., C. R. Ramakrishnan, and V. Sasisekharan. 1963. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7:95–99. [DOI] [PubMed] [Google Scholar]
- Rosenberg, J. M., N. C. Seeman, J. J. P. Kim, F. L. Suddath, H. B. Nicholas, and A. Rich. 1973. Double helix at atomic resolution. Nature. 243:150–154. [DOI] [PubMed] [Google Scholar]
- Rotstein, S. H., and M. A. Murcko. 1993. GroupBuild: a fragment-based method for de novo drug design. J. Med. Chem. 36:1700–1710. [DOI] [PubMed] [Google Scholar]
- Saenger, W., W. N. Hunter, and O. Kennard. 1986. DNA conformation is determined by economics in the hydration of phosphate groups. Nature. 324:385–388. [DOI] [PubMed] [Google Scholar]
- Sasisekharan, V., A. V. Lakshminarayanan, and G. N. Ramachandran. 1967. Stereochemistry of nucleic acids and polynucleotides. I. Theoretical determination of the allowed conformations of the monomer unit. In Conformation of Biopolymers. G. N. Ramachandran, editor. Academic Press, New York. 641–654.
- Schneider, B., and H. M. Berman. 1995. Hydration of the DNA bases is local. Biophys. J. 69:2661–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, B., D. M. Cohen, L. Schleifer, A. R. Srinivasan, W. K. Olson, and H. M. Berman. 1993. A systematic method for studying the spatial distribution of water molecules around nucleic acid bases. Biophys. J. 65:2291–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, B., K. Patel, and H. M. Berman. 1998. Hydration of the phosphate group in double-helical DNA. Biophys. J. 75:2422–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakked, Z., D. Rabinovich, W. B. T. Cruse, E. Egert, O. Kennard, G. Sala, G. Salisbury, and M. A. Viswamitra. 1981. Crystalline A-DNA—the X-ray analysis of the fragment d(G-G-T-A-T-A-C-C). Proc. R. Soc. Lond. B213:479–487. [DOI] [PubMed] [Google Scholar]
- Shefter, E., and K. N. Trueblood. 1965. The crystal and molecular structure of D+-barium uridine-5′-phosphate. Acta Crystallogr. 18:1067–1077. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G. M., and T. R. Schneider. 1997. Shelxl: high-resolution refinement. Methods Enzymol. 277:319–343. [PubMed] [Google Scholar]
- Singh, U. C., and P. A. Kollman. 1985. A water dimer potential based on ab-initio calculations using Morokuma component analysis. J. Chem. Phys. 83:4033–4040. [Google Scholar]
- Sriram, M., G. A. van der Marel, H. L. P. F. Roelen, J. H. van Boom, and A. H.-J. Wang. 1992. Structural consequences of a carcinogenic alkylation lesion on DNA: effect of O6-ethylguanine on the molecular structure of the d(CGC[e6G]AATTCGCG)-netropsin complex. Biochemistry. 31:11823–11834. [DOI] [PubMed] [Google Scholar]
- Sussman, J. L., N. C. Seeman, S. H. Kim, and H. M. Berman. 1972. Crystal structure of a naturally occurring dinucleoside phosphate: uridyly3′,5′-adenosine phosphate. J. Mol. Biol. 66:403–421. [DOI] [PubMed] [Google Scholar]
- Suto, R. K., R. S. Edayathumangalam, C. L. White, C. Melander, J. M. Gottesfeld, P. B. Dervan, and K. Luger. 2003. Crystal structures of nucleosome core particles in complex with minor groove DNA-binding ligands. J. Mol. Biol. 326:371–380. [DOI] [PubMed] [Google Scholar]
- Tereshko, V., C. J. Wilds, G. Minasov, T. P. Prakash, M. A. Maier, A. Howard, Z. Wawrzak, M. Manoharan, and M. Egli. 2001. Detection of alkali metal ions in DNA crystals using state-of-the-art x-ray diffraction experiments. Nucleic Acid Res. 29:1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauger, J. W., E. E. Baird, and P. B. Dervan. 1996. Recognition of DNA by designed ligands of subnanomolar concentrations. Nature. 382:559–561. [DOI] [PubMed] [Google Scholar]
- Wahl, M. C., and M. Sundaralingam. 1997. C–H⋯O hydrogen bonding in biology. Trends Biochem. Sci. 22:97–102. [DOI] [PubMed] [Google Scholar]
- Weiner, S. J., P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, J. S. Profeta, and P. Weiner. 1984. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106:765–784. [Google Scholar]
- Wemmer, D., and P. B. Dervan. 1997. Targeting the minor groove of DNA. Curr. Opin. Struct. Biol. 7:355–361. [DOI] [PubMed] [Google Scholar]
- Wemmer, D. E. 2001. Ligands recognizing the minor groove of DNA: development and applications. Biopolymers. 52:197–211. [DOI] [PubMed] [Google Scholar]
- White, S., E. E. Baird, and P. B. Dervan. 1996. Effects of the A·T/T·A degeneracy of pyrrole-imidazole polyamide recognition in the minor groove of DNA. Biochemistry. 35:12532–12537. [DOI] [PubMed] [Google Scholar]
- White, S., E. E. Baird, and P. B. Dervan. 1997. On the pairing rules for recognition in the minor groove of DNA by pyrrole-imidazole polyamides. Chem. Biol. 4:569–578. [DOI] [PubMed] [Google Scholar]
- White, S., J. W. Szewczyk, J. M. Turner, E. E. Baird, and P. B. Dervan. 1998. Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands. Nature. 391:468–471. [DOI] [PubMed] [Google Scholar]
- Woda, J., B. Schneider, K. Patel, K. Mistry, and H. M. Berman. 1998. An analysis of the relationship between hydration and protein-DNA interactions. Biophys. J. 75:2170–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, A. A., C. M. Nunn, A. Czarny, D. W. Boykin, and S. Neidle. 1995. Variability in DNA minor groove width recognised by ligand binding: the crystal structure of a bis-benzimidazole compound bound to the DNA duplex of d(CGCGAATTCGCG)2. Nucleic Acids Res. 23:3678–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurkin, V. B., Y. P. Lysov, and V. I. Ivanov. 1979. Anisotropic flexibility of DNA and the nucleosomal structure. Nucleic Acids Res. 6:1081–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurkin, V. B., V. I. Poltev, and V. L. Florentiev. 1981. Atom-atom potential functions for conformational calculations of nucleic acids. Mol. Biol. USSR (Engl. Ed.) 14:882–895. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.