

Abstract
We present an improved monomeric form of the red fluorescent protein, mRFP1, as the acceptor in biological fluorescence resonance energy transfer (FRET) experiments using the enhanced green fluorescent protein as donor. We find particular advantage in using this fluorophore pair for quantitative measurements of FRET using multiphoton fluorescence lifetime imaging microscopy (FLIM). The technique was exploited to demonstrate a novel receptor-kinase interaction between the chemokine receptor (CXCR4) and protein kinase C (PKC) α in carcinoma cells for both live- and fixed-cell experiments. The CXCR4-EGFP: PKCα-mRFP1 complex was found to be localized precisely to intracellular vesicles and cell protrusions when imaged by multiphoton fluorescence-FLIM. A comparison of the FRET efficiencies obtained using mRFP1-tagged regulatory domain or full-length PKCα as the acceptor revealed that PKCα, in the closed (inactive) form, is restrained from associating with the cytoplasmic portion of CXCR4. Live-cell FLIM experiments show that the assembly of this receptor:kinase complex is concomitant with the endocytosis process. This is confirmed by experimental evidence suggesting that the recycling of the CXCR4 receptor is increased on stimulation with phorbol ester and blocked on inhibition of PKC by bisindolylmaleimide. The EGFP-mRFP1 couple should be widely applicable, particularly to live-cell quantitative FRET assays.
INTRODUCTION
Measurement of the near-field localization of protein complexes may be achieved by the detection of fluorescence (or Förster) resonance energy transfer (FRET) between protein-conjugated fluorophores. FRET is a nonradiative, dipole-dipole coupling process, whereby energy from an excited donor fluorophore is transferred to an acceptor fluorophore in close proximity (Förster, 1948). The dependence of the coupling efficiency varies with the inverse-sixth power of the distance between acceptor and donor and is typically described in terms of the Förster radius (distance at which the efficiency of energy transfer is 50%), typically of the order 1–10 nm. Excitation of the donor sensitizes emission from the acceptor that ordinarily would not occur. Since the process depletes the excited state population of the donor, FRET will both reduce the fluorescence intensity and the fluorescence lifetime of the donor. The efficiency of energy transfer may be used as a molecular scale ruler to determine the interaction scale of a particular interaction (Stryer, 1978). Expressible fluorophore-coupled protein partners for FRET are necessary to investigate protein-protein interactions in live cultured cells and ultimately in vivo. In the context of imaging the dynamics of intermolecular interactions in cells (when the FRET donor and acceptor are on two separate protein molecules rather than a fused chimera), an accurate assessment of the proportion of donor fluorophore-labeled protein molecules that undergo FRET at each cell pixel is desirable.
Fluorescence lifetime imaging microscopy (FLIM) is a well-established technique for studying spatiotemporal protein-protein interactions in situ through the detection of FRET between protein-bound fluorophores (Anilkumar et al., 2003; Gadella and Jovin, 1995; Herreros et al., 2001; Legg et al., 2002; Ng et al., 2001, 1999a,b; Parsons et al., 2002). For intermolecular FRET a key advantage of donor FLIM is that fluorescence-lifetime measurements of donor emission are essentially independent of acceptor concentration (assuming excess acceptor) and is therefore well suited to studies in intact cells (see, for example, Bastiaens and Pepperkok, 2000; Ng et al., 1999b; Parsons et al., 2004; Peter and Ameer-Beg, 2004; Wouters et al., 2001). Combined with confocal or multiphoton microscopic techniques to examine the localization of effects in cellular compartments, FLIM/FRET techniques allow us to determine populations of interacting protein species on a point-by-point basis at each resolved voxel in the cell (Becker et al., 2001). Time-correlated single-photon counting (TCSPC) is a mature technique that exploits the quantum nature of light to recover the fluorescence lifetime of a fluorophore by statistical analysis of the arrival times of photons with respect to a reference (i.e., the excitation pulse) (O'Connor and Phillips, 1984). FLIM analysis can be enhanced by application of global analysis (i.e., assumption of globally invariant fluorescence lifetime components and calculation over all available data sets, or pixels) to data obtained using either time-domain (Beechem and Brand, 1986; Beechem and Haas, 1989) or frequency domain-based methodology (Verveer and Bastiaens, 2003; Verveer et al., 2000a,b, 2001). The spatially invariant lifetimes are assumed to pertain to the donor molecular species in the presence or absence of FRET, the relative amplitudes of the two components being proportional to the molar fractions of the two species. In the case of protein events that take place in localized regions or specific organelles of the cell, acquisition of fluorescence lifetimes by two-photon excitation, single-photon excitation through a confocal aperture or fluorescence deconvolution (Bastiaens and Pepperkok, 2000; Neil et al., 2000; Schonle et al., 2000; Squire and Bastiaens, 1999) enhances the sensitivity of FRET detection, by removing the contribution of noninteracting species in out-of-plane regions.
There are both advantages and disadvantages to using any of the existing pairs of EGFP mutants in donor FLIM/FRET assays and this has been the subject of a number of review articles (Miyawaki, 2003; Periasamy, 2001; Pollock and Heim, 1999; Selvin, 2000). In brief, for the most commonly used CFP-YFP pair, CFP alone may exhibit bi-exponential decay kinetics when observed in the cellular environment (Tramier et al., 2002), making quantitative analysis of an interacting FRET population impractical. Recent reports of an improved variant of CFP, the Cerulean (Rizzo et al., 2004), show an encouraging improvement in its photophysical parameters (quantum yield and extinction coefficient). Cerulean is reported to have a monoexponential fluorescence decay in vitro, and provided that these kinetics can be reproduced with different biological constructs in cells, this new variant will serve as a good alternative to EGFP when employed in donor FLIM assays. Other fluorescent protein variants suffer from additional problems such as photo-instability for the BFP-EGFP pair (Ellenberg et al., 1998). In addition, there is a substantial emission cross talk due to spectral overlap for the most widely used CFP-YFP pair, complicating quantification of FRET efficiency by intensity-based methods (Gordon et al., 1998). Given the significant Stokes shift between the CFP and YFP emission, acceptor emission cross talk for donor-FLIM can be eliminated with judicious choice of interference filters at the expense of photon-efficient collection. This problem is, however, exacerbated by the 4.9-fold increase in brightness (defined by quantum yield multiplied by extinction coefficient) between CFP and YFP (Patterson et al., 2001).
There has been interest in improving the current techniques through the use of red biofluorescent proteins HcRed and DsRed (Lippincott-Schwartz and Patterson, 2003; Zhang et al., 2002). DsRed has been used successfully with EGFP in limited applications (Erickson et al., 2003; Mizuno et al., 2001). However, HcRed and DsRed exist in oligomeric complexes, which can inhibit both protein distribution and function. For example, when fused to the gap function protein Cx43 (Campbell et al., 2002) and when fused with calmodulin (Mizuno et al., 2001), DsRed has been consistently observed to form perinuclear red fluorescent aggregates. The recently reported (Campbell et al., 2002) monomeric variant of DsRed (mRFP1) has a significant advantage over its progenitor, since it does not form oligomers and matures rapidly with little or no green component in its fluorescence emission. The low quantum efficiency reported for mRFP1 makes it less advantageous as a FRET acceptor when used in intensity-based methodologies. However, since the emissive behavior of the acceptor has little or no effect on the measurement of FRET by donor FLIM, mRFP1 makes a likely candidate as an acceptor for EGFP or YFP donors in lifetime-based assays.
The chemokine receptor CXCR4 has been implicated in promoting the migratory phenotype of a variety of tumors including breast, prostate, and pancreatic cancers, as well as myeloma and several tumors of hematopoietic origin (reviewed by Moore, 2001). CXCR4 is found to be consistently upregulated in human breast cancer cells relative to its level in normal mammary epithelial cells (Muller et al., 2001). Stromal-cell-derived factor 1, the ligand for CXCR4, is expressed at high levels in all target organs for breast cancer metastasis and at low levels in all other organs. CXCR4 undergoes endocytosis after ligand binding or protein kinase C (PKC) activation by phorbol esters (Guinamard et al., 1999; Signoret et al., 1997, 1998). The C-terminal domain of CXCR4, which contains several putative PKCα phosphorylation sites, is required for its endocytosis. We have identified and characterized a phorbol ester-induced CXCR4:PKCα complex in the endosomal structures of MDA-MB-231 breast carcinoma cells, using FRET detection by multiphoton FLIM (between CXCR4-EGFP and PKCα-mRFP1).
MATERIALS AND METHODS
Cell culture
Human breast carcinoma MDA-MB-231 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum at 37°C, in a 10% CO2 atmosphere. MDA-MB-231 cells were transfected with the CXCR4-EGFP plasmid (see below) with Lipofectamine Plus (Gibco Invitrogen, Carlsbad, CA). 48 h after transfection was initiated, cells were replated in complete medium at 1:5 ratio. Selection for neomycin resistance was started the following day by the addition of the antibiotic G-418 (Calbiochem, San Diego, CA) at 1 mg/ml. CXCR4-EGFP expressing cells were enriched by fluorescence-activated cell sorting. After 10–14 days, G418-resistant cells were isolated as a mixed clone and subsequently maintained in complete growth medium containing 1 mg/ml G418. Stable transfectants were cultured for up to 20 passages.
Plasmid constructs
The EGFP-tagged CXCR4 construct was obtained by subcloning the CXCR4 coding sequence into the HindIII and EcoRI sites of the pEGFP-N1 (Clontech, Palo Alto, CA). mRFP1 was obtained from Professor R. Tsien and cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA). Full-length or regulatory domain (RD) (aa 1–337) PKCα-mRFP1 was generated by subcloning the PKCα coding sequence into the NheI and EcoRI sites of pcDNA-mRFP1. PKCα-DsRed was similarly constructed for the purpose of direct comparison.
CXCR4 recycling assay
Live MDA-MB-231 cells stably expressing CXCR4-EGFP and plated on coverslips were either left untreated at 37°C or incubated with media containing 10 μM bisindolylmaleimide (BIM) (Calbiochem) or 1 mM primaquine (Sigma-Aldrich, Poole, Dorset, UK). After 1 h, the cells were incubated with media containing 10 μg/ml of anti-CXCR4 monoclonal antibody (12G5) (R&D Systems, Minneapolis, MN) for 20 min to label both the plasma membrane and internal pools of the receptor, and subsequently washed with PBS to remove the excess antibody. To examine the effect of phorbol ester on the recycling, 1 μM PDBu (Sigma-Aldrich) was added to the media containing the anti-CXCR4 antibody. All cells were then incubated for 2 min in cold culture media adjusted to pH 4.0 to remove cell surface antibody as described (Signoret et al., 1997, 1998). Subsequently this low pH media was washed out, replaced by pH 7.4 media, and the cells were returned to 37°C. At different time points, cells were stained with an anti-mouse Cy-5 conjugated antibody (Jacksons Immunoresearch Laboratories, West Grove, PA) at 4°C for 20 min to label the pool of receptors that had recycled back to the membrane, then fixed in 4% paraformaldehyde/PBS for 30 min. For cells treated with BIM or primaquine, the inhibitor was maintained throughout the experiment.
Cell microinjection, fixation, and preparation
MDA-MB-231 cells stably expressing CXCR4-EGFP were microinjected using a FEMTOJET microinjection system (Eppendorf UK Ltd., Histon, Cambridge, UK). After 16 h, cells were either imaged immediately using a multiphoton fluorescence-FLIM system equipped with a cabinet incubator or fixed in 4% (w/v) paraformaldehyde for 30 min and permeabilized with 0.2% (v/v) Triton X-100/PBS. Coverslips were finally prepared with Mowiol Mounting Medium (ICN, Costa Mesa, CA) containing 2.5% (w/v) 1,4-diazabicyclo (2.2.2) octane (Sigma-Aldrich) as an antifade reagent.
Confocal microscopy
Confocal images were acquired with a confocal laser-scanning microscope (LSM 510, Carl Zeiss, Jena, Germany) equipped with a 63×/1.4 Plan-Apochromat oil immersion objective. Z-series data were acquired by interline scanning for EGFP (488 nm excitation; 510 nm emission) and mRFP1 (514 nm excitation; 600 nm emission), respectively. The confocal aperture was set to one Airy unit for the longest wavelength (typically 0.7 μm). Each image represents a two-dimensional projection of two or three slices in the Z-series, taken across the cell at 0.2-μm intervals.
Absorption and fluorescence emission measurements
Absorption spectra were taken with an 8452A diode array spectrophotometer (Hewlett-Packard, Palo Alto, CA). The fluorescence spectra were measured with an LS 50B (Perkin-Elmer, Boston, MA) at a resolution of 5 nm.
Time-resolved multiphoton microscopy
All FLIM measurements were undertaken with a 40× (1.3 NA) Nikon Plan-Fluor oil objective lens on a modified multiphoton microscopy system which has been described previously (Ameer-Beg et al., 2002, 2003). In brief, the system consists of a MRC1024 MP dual confocal/multiphoton system (BioRad Microscience, Hemel Hempstead, UK) coupled to a TE200 inverted microscope (Nikon, Tokyo, Japan). Time-resolved detection is afforded by the addition of a non-descanned detection channel with a fast photomultiplier and SPC700 time-correlated single-photon counting electronics (Becker and Hickl GmbH, Berlin, Germany). Either a 500 ± 20 nm or 600 ± 20 nm bandpass filter was used in the detector channel (Coherent, Santa Clara, CA). Laser power was adjusted to give average photon counting rates of the order 104–105 photons s−1 (0.0001–0.001 photons/excitation event) and with peak rates approaching 106 photons s−1, below the maximum counting rate afforded by the TCSPC electronics to avoid pulse pile-up. Acquisition times of the order of 300 s at low excitation power were used to achieve sufficient photon statistics for fitting, while avoiding either pulse pile-up or significant photobleaching. Excitation was at 890 nm for EGFP-mRFP1 samples and at 980 nm for mRFP1 lifetime measurements.
Analysis of time-resolved fluorescence data for FRET experiments
In analysis of FRET data (particularly for protein-interaction applications), there are usually two elements that must be considered—interacting fluorophore population and FRET efficiency. Bulk measurements of FRET efficiency (i.e., intensity-based methods) cannot distinguish between an increase in FRET efficiency (i.e., coupling efficiency) and an increase in FRET population (concentration of FRET species) since the two parameters are not resolved. Measurements of FRET based on analysis of the fluorescence lifetime of the donor may resolve this dichotomy when analyzed using multi-exponential decay models. The assumption that noninteracting and interacting fractions are present allows us to determine both the efficiency of interaction and the fractional population of interacting population. Such an approach may be extended from a pixel-by-pixel analysis (where we make no spatial restraints on the fitting parameters) to analysis of all pixels globally under the assumption that the FRET efficiency is constant across all pixels (i.e., global analysis; Verveer et al., 2000a).
In the first instance, the presence/absence of FRET is determined by fitting of the experimental data to a single-exponential decay. Sufficient reduction in the measured lifetime indicates FRET, and since the interactions are intermolecular, we perform additional analysis to determine the source of lifetime reduction. In this instance, we apply a bi-exponential fluorescence decay model to the data to determine the fluorescence lifetime of noninteracting and interacting subpopulations. The data may be fit by iterative re-convolution to
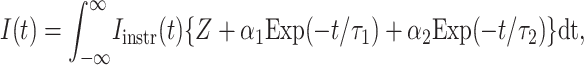 |
(1) |
where Iinstr is the instrumental response, Z is the baseline offset, τ1 and τ2 are lifetimes of the interacting/noninteracting populations, and α1 and α2 are the pre-exponential factors relating to absolute species concentration. The fractional proportions of the two populations, f1 and f2, are related to the absolute concentration via the relationships
 |
(2) |
The reduced goodness-of-fit parameter, 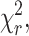 is used as defined by Lakowicz (1999) as
is used as defined by Lakowicz (1999) as
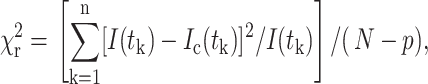 |
(3) |
where I(tk) is the data and Ic(tk) the fit value at the kth time point, tk. N is the number of time points and p the number of variable fit parameters. The value 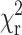 is minimized using a modified Marquardt algorithm and compared alongside plots for the weighted residuals (R = (I(tk)−Ic(tk))/I(tk)) to determine the validity of the decay model.
is minimized using a modified Marquardt algorithm and compared alongside plots for the weighted residuals (R = (I(tk)−Ic(tk))/I(tk)) to determine the validity of the decay model.
Given the level of uncertainty in determining all of the free parameters using this model, we use a minimally restrained system of fitting to determine both the interaction efficiency and population. By fixing the noninteracting species lifetime using data obtained from control experiments (in the absence of FRET), more accurate estimations of the remaining free parameters may be made. Furthermore, by assuming invariance in the efficiency of interaction between pixels throughout the measured cell one can determine the interaction efficiency by summation of data throughout the cell and thereby fix both lifetime parameters for single pixel fitting.
The FRET efficiency is related to the molecular separation of donor and acceptor and the fluorescence lifetime of the interacting fraction by
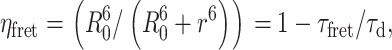 |
(4) |
where R0 is the Förster radius, R the molecular separation, τfret is the lifetime of the interacting fraction, and τd the lifetime of the donor in the absence of acceptor. Clearly in this instance we can also take τfret and τd to be the lifetimes of the interacting fraction and noninteracting fraction, respectively. For measurements of bulk interactions (i.e., where only single-exponential decays are fit to the data), measured efficiencies will appear significantly lower due to the, possibly incorrect, assumption that all donors are associated with one or more acceptors. Where a multi-exponential model is assumed, partially interacting subpopulations may be identified distinctly from the bulk response. The caveat to this is that sufficiently high photon counts are required to resolve these complex dynamics.
RESULTS AND DISCUSSION
Monomeric red fluorescent protein (mRFP1) as a FRET acceptor
We have compared mRFP1 with the more commonly used fluorescent protein pair used for FRET experiments CFP-YFP in Fig. 1 A. Fluorescent proteins (His-tagged) encoded by pRSET vectors were expressed in bacteria and purified using Ni-NTA-agarose (Qiagen, Valencia, CA) as per manufacturer's protocol. The spectra for the CFP-YFP pair were kindly provided by Dr. V. Subramaniam (AstraZeneca, Luton, Bedfordshire, UK) for the purpose of comparison. There is substantially less spectral emission overlap between the EGFP-mRFP1 pair in comparison to CFP-YFP, leading to more relaxed spectral filtering (or indeed more straightforward spectral un-mixing). For measurements of FRET by FLIM we and others (Tramier et al., 2002) have observed multi-exponential decay kinetics for CFP even in the absence of an acceptor (Fig. 1 B and Table 1). In this example, the fluorescent proteins were fused to IκB and p65 (protein components of the NF-κB transcription pathway) which are complexed constitutively (Morton et al., 2003). If a mono-exponential model (giving a single lifetime τ) is used to fit the decay kinetics either in the absence or presence of the YFP acceptor, we find that χ2 is increased and modulation is observed in the residuals: a clear indication that a bi-exponential model is more appropriate in both cases. Although the average fluorescence lifetime may be employed to determine the presence of FRET (and the average FRET efficiency), no quantitative information on the molar concentration of conjugated proteins can be reasonably recovered (in situ) since this would require fitting of at least four exponential components and superlative photon statistics (and by inference unacceptably long acquisition times). We have also observed the CFP moiety to be susceptible to modification in lifetime in the presence of external stimulus (i.e., tumor necrosis factor α (Morton et al., 2003). Unfortunately, the reportedly low quantum yield of ∼0.25 of mRFP1 does not make it an ideal acceptor for use in intensity-based FRET measurements. However, this should not preclude its use for either donor fluorescence quenching or acceptor photobleaching methods.
FIGURE 1.

(A) Emission spectra of cyan (CFP), yellow (YFP), enhanced green (EGFP), and monomeric red (mRFP1) fluorescent-protein variants. (B) Fluorescence transients for CFP in the absence (I) and presence (II) of FRET.
TABLE 1.
Fluorescence decay analysis for cyan and yellow fluorescent proteins (from Fig. 1 B)
| Proteins | τ1 (ns) | f1 | τ2 (ns) | f2 | χ2 |
|---|---|---|---|---|---|
| CFP control | 1.78 ± 0.2 | 30.9 | |||
| 0.36 ± 0.08 | 0.32 ± 0.05 | 2.29 ± 0.05 | 0.68 ± 0.05 | 1.26 | |
| CFP/YFP
|
1.12 ± 0.2 | 35.2 | |||
| 0.33 ± 0.10 | 0.49 ± 0.05 | 1.58 ± 0.15 | 0.51 ± 0.05 | 1.88 |
As a fluorescent probe for functional biology, mRFP1 confers a number of advantages over the DsRed progenitor; it matures rapidly, exhibits negligible green fluorescence, and is less inhibitory to protein function. In illustration, the decay kinetic of the fluorescence at 500 ± 20 nm of PKCα-DsRed is shown to be bi-exponential (data accumulated from a region inside transfected cells) in Fig. 2 A and Table 2. Even in the absence of FRET, the measurement of CXCR4-EGFP lifetime can be contaminated with the 0.4-ns component of the green emission from DsRed (Fig. 2 B and Table 2). Furthermore, the excitation and emission spectral properties of DsRed are complex, fluorescence correlation spectroscopy data suggesting that there might be at least three interconvertible states, apparently fluorescent, with different excitation and emission properties (Malvezzi-Campeggi et al., 2001). In contrast to the DsRed data the mRFP1 probe does not produce significant green fluorescence after the maturation phase and, when used in conjunction with EGFP, no cross talk is observed in situ. The fluorescence lifetime of mRFP1 observed at 600 ± 20 nm is given in Fig. 3. We observe a single-exponential decay (as confirmed by both 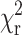 and analysis of residuals) with a lifetime of 2.05 ± 0.1 ns.
and analysis of residuals) with a lifetime of 2.05 ± 0.1 ns.
FIGURE 2.

Individual transient decays for two-photon excitation at 890 nm and emission at 500 nm (±20 nm), indicating contamination of the measurement of CXCR4-EGFP in the presence of full-length PKCα-DsRed in unstimulated cells. (A) PKCα-DsRed, bi-exponential kinetics are indicated. (B) CXCR4-EGFP alone; single-exponential kinetics are observed. (C) PKCα-DsRed and CXCR4-EGFP, Kinetics observed are contaminated with the fast component of PKCα-DsRed.
TABLE 2.
Fluorescence decay analysis for EGFP and DsRed (from Fig. 2)
| Proteins | τ1 (ns) | f1 | τ2 (ns) | f2 | χ2 |
|---|---|---|---|---|---|
| DsRed | 1.88 ± 0.4 | 6.72 | |||
| 2.10 ± 0.05 | 0.65 | 0.43 ± 0.05 | 0.35 | 1.25 | |
| eGFP | 2.08 ± 0.05 | 1.09 | |||
| 2.21 ± 0.05 | 0.8 | 1.63 ± 0.05 | 0.2 | 1.11 | |
| GFP/DsRed
|
2.04 ± 0.2 | 4.27 | |||
| 2.17 ± 0.05 | 0.79 | 0.46 ± 0.05 | 0.21 | 1.13 |
FIGURE 3.

Transient decay for PKCα-mRFP1 by two-photon excitation, and emission at 980 nm and 600 nm (±20 nm), respectively, exhibiting single-exponential kinetics.
Estimation of the Förster radius for EGFP-mRFP1 interaction
To calculate the Förster radius for the interaction between EGFP and mRFP1 we undertook spectroscopic measurements (Fig. 4) of the absorption and emission of mRFP1 and EGFP, respectively. The peak emission of mRFP1 is shifted from 583 nm to 607 nm relative to DsRed, conferring a modest advantage for filtering when co-expressed with either EGFP or YFP. Unfortunately, this may be offset in FRET experiments by the concomitant reduction in the Förster radius. The secondary peak in the absorption spectrum corresponds to a primarily nonradiative state and therefore does not appear on the excitation spectra (data not shown) but is significant in the context of calculation of the spectra overlap and Förster radius. This feature of the mRFP1 photophysics contributes significantly to the sub-optimal conditions for intensity-based FRET assays using this probe. The spectral overlap integral was calculated from Lakowicz (1999) as
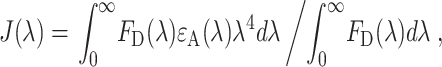 |
(5) |
where FD(λ) is the corrected fluorescence intensity of the donor with the total intensity (area under the curve) normalized to unity. The value ɛA is the extinction coefficient of the acceptor at λ (in units M−1cm−1). The Förster radius (R0) is then given by
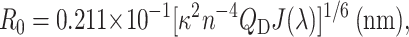 |
(6) |
where n is the refractive index, QD is the fluorescence quantum yield of the donor, and κ is a parameter relating to the orientation of donor and acceptor. Assuming that the dipoles of the acceptor are randomly oriented with respect to the donor we take the κ2 = 2/3. The refractive index is taken to be that of water (n = 1.3). The quantum efficiency of EGFP was taken to be 0.6 (Patterson et al., 1997) and the molar extinction coefficient for mRFP1 to be 44,000 cm−1 M−1 (Campbell et al., 2002). By inserting these values into Eq. 5 we obtain a Förster radius of 4.7 ± 0.5 nm. This is comparable to that calculated for CFP-YFP (4.9–5.2 nm; Tsien, 1998) and EGFP-DsRed (4.71–5.76 nm; Erickson et al., 2003). The estimation of R0 at the lower end of what is expected for DsRed is due to the 25-nm Stokes shift in absorption which leads to slightly less favorable spectral overlap, the more distinct absorption band-shape of mRFP1 (smaller spectral half-width), and the 1.3-fold reduction in molar extinction coefficient (note that there is some two-to-threefold discrepancy in the literature regarding the extinction coefficient of DsRed, almost certainly due to the complex nature of the excited state and the slow maturation of the red species (Erickson et al., 2003 and references therein). There may be some future advantage in substituting the EGFP donor for the red-shifted variant YFP to increase the Förster radius without significant degradation in the spectral filtering between donor and acceptor. However, we foresee the greatest possible advantage from further mutation of mRFP1 to produce variants with higher quantum yields (and/or increased extinction coefficient) to more fully exploit intensity-based methods for FRET assays.
FIGURE 4.

Emission and absorption spectra of EGFP and mRFP1, respectively, from protein extract solutions.
Highly localized FRET between EGFP-CXCR4 and mRFP1-PKCα
Recently, a novel protein-signaling complex involved in the control of breast cancer cell motility was identified biochemically between the chemokine receptor CXCR4, and the N-terminal regulatory domain (RD) of PKCα in breast carcinoma cells (M. Peter and T. Ng, unpublished data). To confirm this interaction in situ, FRET experiments were performed between an EGFP-labeled construct of CXCR4 (CXCR4-EGFP) and an mRFP1-conjugated, full-length PKCα (PKCα-mRFP1). The fluorescence lifetime (assuming a single-exponential decay) in CXCR4-EGFP stably transfected cells was measured in the presence or absence of PKCα-mRFP1 expressed through nuclear microinjection of an expression plasmid (Fig. 5). For both control (PKCα-mRFP1 negative) and co-expressing cells (PKCα-mRFP1 positive) we observe no significant reduction in the fluorescence lifetime of EGFP suggesting that little or no short-range interaction occurs under quiescent conditions. In contrast, after stimulation with phorbol ester (phorbol dibutyrate, PDBu), the fluorescence lifetime is observed to reduce significantly throughout the cell relative to the stimulated, uninjected control cells (Fig. 5 A). Critical analysis of the data (examination of the residuals and 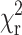 ) indicates that a bi-phasic decay model is more appropriate suggesting the presence of interacting and noninteracting protein populations. Assuming the interaction distance to be constant, we found the invariant lifetimes of the two decay species were 0.95 ± 0.1 ns (EGFP species undergoing FRET) and 2.20 ± 0.05 ns (EGFP species not undergoing FRET). This corresponds to a population-resolved FRET efficiency of 0.57 ± 0.05 or an interaction distance of ∼4.48 nm (between fluorescent proteins at N-termini and within the assumptions made for the determination of the Förster radius). Fixing these two lifetime parameters for each pixel we can resolve the fractional contribution of the FRET species (Fig. 5 A).
) indicates that a bi-phasic decay model is more appropriate suggesting the presence of interacting and noninteracting protein populations. Assuming the interaction distance to be constant, we found the invariant lifetimes of the two decay species were 0.95 ± 0.1 ns (EGFP species undergoing FRET) and 2.20 ± 0.05 ns (EGFP species not undergoing FRET). This corresponds to a population-resolved FRET efficiency of 0.57 ± 0.05 or an interaction distance of ∼4.48 nm (between fluorescent proteins at N-termini and within the assumptions made for the determination of the Förster radius). Fixing these two lifetime parameters for each pixel we can resolve the fractional contribution of the FRET species (Fig. 5 A).
FIGURE 5.

(A) Images of cells co-expressing CXCR4-EGFP and full-length PKCα-mRFP1 with or without phorbol ester treatment. The microinjected PKCα-positive red fluorescent cells are indicated in each panel (red arrows). Increased FRET populations were observed in the vesicular compartment and filopodial cell protrusions (white arrows). The intensity image for the +ve PDBu panel is taken from confocal data and shows regions of co-localization between CXCR4-EGFP (red) and PKCα-mRFP1 (green). (B) Images of cells expressing CXCR4-EGFP with regulatory domain (RD) PKCα-mRFP1 spanning amino acid residues 1–337 (RD (aa 1–337) PKCα-mRFP1) and the associated RD (aa 1–337) PKCα-mRFP1 negative control. The +ve RD (aa 1–337) PKCα-mRFP1 intensity image is taken from confocal data and shows colocalization of CXCR4-EGFP (green) and RD (aa 1–337) PKCα-mRFP1 (red). Donor lifetime images and the corresponding FRET population cell map in the presence of RD (aa 1–337) PKCα-mRFP1, where fitting with a bi-exponential decay model is possible are shown. The field of view was chosen such that the lifetime map of the microinjected +ve RD (aa 1–337) PKCα-mRFP1 cell could be compared to an adjacent −ve RD (aa 1–337) PKCα-mRFP1 cell. (C) Images of cells co-expressing CXCR4-EGFP and full-length PKCα-DsRed with or without phorbol ester treatment. Confocal images show PKCα-DsRed (red) aggregates that do not colocalize with CXCR4-EGFP (green) but indicate a short lifetime component that contaminates each lifetime map. No increase in association/FRET is observed in the presence of PDBu.
The N-terminal regulatory region of PKCα has several functional domains (Parker and Murray-Rust, 2004). The C1 domain contains a Cys-rich motif that forms the phorbol ester binding site. The C2 domain contains the recognition site for acidic lipids and the Ca2+ binding site. An autoinhibitory pseudosubstrate sequence (PS) precedes C1. When PKCα is inactive, the PS domain is bound to the C-terminal kinase region of PKCα. Phorbol esters serve as hydrophobic anchors to recruit PKCα to the membrane. Moreover, phorbol esters induce the removal of the PS sequence from the PKCα kinase core (Nelsestuen and Bazzi, 1991). PKCα is then in an open conformation, unmasking various potential cryptic binding site(s) in the regulatory domain to its binding partners.
We postulate that before the addition of phorbol ester, the closed conformation of the conventional PKC isozyme PKCα is unfavorable for CXCR4 binding. Upon activation by phorbol ester PKCα assumes a more open conformation, allowing its RD to interact with CXCR4. This hypothesis is partly based on our finding of a physical overlap, within PKCα, between the CXCR4 interaction site and the phorbol-binding domain (M. Peter and T. Ng, unpublished data). The phorbol-binding domain has been shown to be rendered inaccessible by at least two inhibitory intramolecular interactions in conventional PKCs before activation (Oancea and Meyer, 1998; Slater et al., 2002). These FLIM data confirm that CXCR4-EGFP can be induced to associate with full-length PKCα-mRFP1 after stimulation with PDBu with good co-localization observed between the two proteins in the membrane and vesicular compartments (Fig. 5 A). The highest concentration of FRET species was localized to filopodial structures (white arrows) and large vesicular clusters. The large clusters containing low lifetime FRET species (marked with a red arrow) may represent receptor-kinase complexes that are associated with clathrin and its associated adaptor proteins in clathrin-coated vesicles (Brown and Petersen, 1998).
These experiments were repeated for comparison with DsRed as acceptor. Full-length PKCα-DsRed was found to display an abnormal aggregation pattern, apparent in unstimulated cells (Fig. 5 C) in contrast to the diffuse cytosolic distribution of full-length PKCα-mRFP1. The fluorescence lifetime observed in the presence of PKCα-DsRed (in the absence of stimulation) is contaminated (as previously discussed) with a fast lifetime component such that the clusters containing a high concentration of DsRed have anomalously low lifetimes. In the presence of PDBu, full-length PKCα-DsRed does not appear to associate with CXCR4-EGFP (no lifetime reduction occurs relative to the control) in contrast to the earlier result. We conclude that PKCα-DsRed is biologically inactive and a poor choice as a FRET acceptor in this instance.
To address the mechanism of the effect of phorbol ester we compared the data obtained using mRFP1-tagged RD (RD (aa 1–337) PKCα-mRFP1) to those of the full-length PKCα as the acceptor. Unlike the full-length PKCα-mRFP1 that was found to be cytosolic and does not co-localize to any significant degree with CXCR4-EGFP in quiescent cells, the RD construct is localized predominantly to membrane and vesicular structures (Fig. 5 B). We find that the fluorescence lifetime is reduced from the control for cells containing the RD (aa 1–337) PKCα-mRFP1 construct. Further analysis of the data revealed the presence of at least two fluorescent species and fitting to a bi-exponential model was performed assuming invariant lifetimes. The lifetimes were found to be 2.1 ± 0.05 ns and 1.2 ± 0.2 ns, respectively, similar to those observed for full-length PKCα upon stimulation. Localization of the highest concentration of FRET species to intracellular vesicular structures is particularly evident in the fractional contribution (FRET population) cell map Fig. 5 B. The population-resolved FRET efficiency (43%) for RD (aa 1–337) PKCα-mRFP1, which does not have an inhibitory domain, indicates that this construct interacts constitutively with CXCR4. These data suggest that, in the closed form, PKCα is conformationally restrained from associating with the cytoplasmic portion of CXCR4. The observed effect of phorbol ester on the receptor-kinase complex formation is probably due to a phorbol ester-induced opening of the inhibitory conformation leading to an unmasking of the CXCR4 binding site in PKCα.
To further analyze the intracellular distribution of the CXCR4-EGFP FRET species, a series of two-photon excited fluorescence lifetime image slices was taken in the z plane at 1-μm intervals toward the dorsal surface of the cell starting from the cell coverslip interface (marked 0 μm in Fig. 6). At this pixel resolution (128 × 128 at 1 μm/pixel) it is hard to resolve individual vesicles but it is clear that areas of locally reduced lifetimes correspond to a subset of the clustered intracellular vesicles. This was not found to be due to concentration-dependent quenching when compared to similar controls (−ve PKCα (RD+V3)−mRFP). We observe that the CXCR4-EGFP species were noticeably more perinuclear in their distribution as we moved toward the dorsal surface of the cell. In an effort to demonstrate the variable degree of GFP-mRFP1 FRET among vesicles of similar EGFP intensity (i.e., concentration-independent), we recorded the fluorescence lifetime for those vesicles that could be individually resolved by thresholding in intensity (to isolate the vesicle) and then summing photon counts over the vesicle. The results of this are shown in Fig. 7 A where we observe a vesicle lifetime histogram which shows a distinct shift from the 2.1- to 2.2-ns lifetime observed for control cells. Plotting the scattergram of fitted peak intensity values (i.e., α in Eq. 1) versus lifetime (Fig. 7 B) we observe no obvious correlation between short lifetime measurement and peak intensity, indicating that the reduction in lifetime is not due to concentration-dependent quenching of EGFP in the vesicles. The distinct localization of the FRET species to these vesicles does not, however, demonstrate whether the EGFP-CXCR4:mRFP1-PKCα complexes are preformed on vesicles and transported to the plasma membrane or assembled in response to endocytosis of the chemokine receptor. A live-cell experiment was therefore performed to address this mechanistic issue.
FIGURE 6.

Series of two-photon excited fluorescence lifetime image slices of 1-μm separation in the z plane toward the dorsal surface of a cell co-expressing CXCR4-EGFP and RD (aa 1–337) PKCα-mRFP1. The perinuclear concentration of FRET species is more apparent in the dorsal sections (arrows).
FIGURE 7.

(A) Vesicle lifetime histogram comprising vesicles from throughout the cell in Fig. 6. (B) Scattergram of fitted vesicle amplitude versus lifetime showing no correlation between vesicle intensity and measured lifetime.
In situ dynamics of CXCR4-EGFP and PKCα-mRFP1 interaction
One of the key advantages in using expressible probes for FRET experiments is the ability to measure dynamic events in live cells. We have performed exemplar multiphoton fluorescence-FLIM measurements using live cells in an incubation chamber attached to a multiphoton microscope to observe the association of CXCR4-EGFP with PKCα-mRFP1 after stimulation with PDBu. Cells, stably expressing CXCR4-EGFP were cultured on glass-bottomed cell culture dishes (MatTek, Ashland, MA) and transiently transfected with PKCα-mRFP1 and allowed to express over a period of 36 h. Growth media was replaced with 37°C phenol red free media and transported to the microscope incubator where they were allowed to equilibrate for ∼30 min. Controls with cells expressing both CXCR4-EGFP and PKCα-mRFP1 were observed for 1 min acquisitions at 0, 5, 10, 15, and 20 min to check for phototoxicity at a laser power of 1–2 mW (data not shown). The control experiment in Fig. 8 A (CXCR4-EGFP alone) was performed using a wider field of view than for the mRFP1 positive cells to allow cell-by-cell quantitation of the fluorescence lifetime (i.e., cell-to-cell variation was minimal). We did not observe any significant photobleaching or morphological changes in the imaged cells to indicate short-term damage due to these exposures. For live cells in media we observe control lifetimes for CXCR4-EGFP of the order of 2.4 ± 0.1-ns slightly longer than observed for fixed cells. In agreement with the experiments performed using fixed cells we observed no significant FRET between CXCR4-EGFP and PKCα-mRFP1 in the cytoplasmic areas of the cell before addition of PDBu (Fig. 8, −1 min). However, some reduction in lifetime was observed in the vesicular structures indicative of basal level activity in the cell. After addition of PDBu, we observed a significant morphological change to the cells within a few minutes as the cell appears to round up while remaining attached to the coverslip. Refocusing after 5 min a significant portion of CXCR4 has internalized to form large vesicular structures (as observed for fixed cells) with a significantly reduced fluorescence lifetime. By 20 min we are almost certainly viewing the base of the cell below the nucleus with the images showing large vesicular aggregates indicating reduced lifetime and, by inference, FRET and protein interaction. It is possible that the vesicle environment modifies the EGFP lifetime (this may be particularly true if the environment is acidic). However, we observe a lifetime of 2.38 ± 0.05 ns for mRFP1 negative cells (Fig. 8 A) after ∼25 min of PDBu treatment compared to 2.17 ± 0.03 ns for mRFP1 positive cells. The effect of addition of PDBu can be seen clearly in the images at each time point and is confirmed by the histograms of FRET efficiency shown in Fig. 8 C for −1 and 20 min (taking into account the effects of both local environment and phorbol treatment on the mRFP1 negative control). We see a distribution of FRET efficiency values peaking ∼0.1–0.15, significantly greater than the observed basal level of <0.05. These results indicate that, as phorbol ester induces endocytosis of the CXCR4 receptor to form large vesicular clusters (as shown in fixed cells, Fig. 5 A), there is a significant increase in the proportion of CXCR4-EGFP that is complexed to PKCα-mRFP1. The assembly of this receptor:kinase complex is therefore demonstrated to be a phorbol ester-induced signaling response which is concomitant with the process of endocytosis.
FIGURE 8.

Time course of protein-protein interaction after stimulation with PDBu in live cells. (A) Control time sequence CXCR4-EGFP alone, showing no reduction in fluorescence lifetime as a function of time. (B) Cells imaged under the same conditions as A, during PDBu stimulation in the presence of both CXCR4-EGFP and PKCα-mRFP1. The fluorescence lifetime is reduced particularly in the vesicular inclusions. (C) Histogram of FRET efficiency for the stimulated cells in B using the control lifetime derived from A as the divisor in Eq. 4.
To further confirm the significance of these results, CXCR4 recycling experiments were performed to examine the effect of both stimulation and inhibition of PKC on the recycling rate. The experiments were performed as described in Materials and Methods and the results are summarized in Fig. 9. Stimulation with PDBu (Fig. 9, D–F) appears to increase the rate of recycling of CXCR4 to the membrane compared to the unstimulated control by confocal imaging (Fig. 9, A–C). This increase in recycling is biologically significant in terms of its effect on the recovery of receptor function after downregulation through endocytosis. To make objective comparison between experimental conditions, images of cells at each time point were thresholded slightly above background and the average pixel value taken corresponding to the cellular membrane and the values normalized to the full scale and plotted as a function of time (Fig. 9 G). Inhibition of PKC by bisindolylmaleimide (BIM) appears to abolish recycling over the timescale of the experiment to a similar extent to that observed with primaquine, a potent inhibitor for membrane transport from the endosomes to the plasma membrane (van Weert et al., 2000.). These results suggest that the relationship between PKCα and CXCR4 is highly significant in the context of endocytic recruitment and recycling.
FIGURE 9.

Time course of endocytic recycling measured by selective immunolabeling of CXCR4 at the plasma membrane, for (A–C) unstimulated cells and (D–F) cells stimulated with PDBu. The pixel intensity of cells under a variety of experimental conditions are plotted versus time (G) to show increased recycling of CXCR4 at the membrane under stimulation with PDBu and retardation of the recycling process after BIM or primaquine treatment.
CONCLUSION
In this article we have demonstrated that the combination of EGFP and mRFP1 offers advantages over the currently available FRET pairs with a specific biological interaction (a novel receptor:kinase complex). Both fluorescent proteins are genetically encodable, their emission spectra are well separated and mRFP1 is monomeric, reducing the possibility of protein function inhibition. We have calculated the Förster radius for the EGFP:mRFP1 pair to be ∼4.7 nm, which is comparable to that of CFP:YFP (4.9 nm). When calculating the Förster radius, we had assumed the refractive index to be that of water, n = 1.3, which is close to that of cytoplasm (Johnsen and Widder, 1999). The higher refractive index of lipids (in the membrane lipid bilayer for instance) of ∼n = 1.48, could account for some of the heterogeneity of CXCR4-EGFP lifetimes in cells that do not express PKCα-mRFP1 (Suhling et al., 2002) and marginally reduce the Förster radius to 4.35 nm. However, one would expect an increase in fluorescence lifetime for increasing refractive index (due to reduction in the nonradiative decay rate). In contrast we observe a minor decrease in the fluorescence lifetime, suggesting that the effect may be due more to local reduction in pH than the increased refractive index (EGFP has been shown to be sensitive to pH; Heikal et al., 2001; Kneen et al., 1998). Despite this, we show that the shortening of EGFP lifetimes due to EGFP:mRFP1 energy transfer is of such extent that it is easily distinguished from the variation of EGFP lifetimes due to local environmental effects.
We were able to confirm the DsRed-induced aggregation using a PKCα fusion construct as has been previously reported for other proteins such as calmodulin (Mizuno et al., 2001). Similarly, the ability of PKCα to translocate to the membrane upon phorbol ester treatment is impaired by tagging with the tetrameric DsRed. The lack of membrane localization can only partially account for the perturbation of protein function since the dimeric mutants of DsRed (tdimer and dimer2) do not affect the membrane localization and yet still perturb the gap junction formation function of Connexin 43 (Campbell et al., 2002).
High spatial resolution imaging in fixed cells combined with the determination of fluorescence lifetime changes in real time in live cells have provided us important additional insights into the mechanisms of phorbol-ester induced CXCR4:PKCα complex formation. In live cells, as phorbol ester induces endocytosis of the CXCR4 receptor to internalize into large vesicular clusters, we observe a significant increase in the proportion of CXCR4-EGFP that is complexed to PKCα-mRFP1. The assembly of this receptor:kinase complex is therefore likely to represent a signaling process that is concomitant with the process of endocytosis, and not the transcytosis of preformed complexes to the plasma membrane. Moreover, we show that when the receptor is endocytosed in the presence of a PKC activator, the rate of receptor recycling to the plasma membrane is significantly increased. This increase in recycling is important biologically in terms of its effect on the recovery of receptor, function after downregulation through endocytosis. PKCs have previously been shown to regulate the endocytosis and recycling of the integrin family of extracellular matrix receptors (Ng et al., 1999a; Roberts et al., 2001). Here we show for the first time that the recycling of a chemotactic receptor is enhanced concomitantly with the formation of a kinase-receptor complex in endosomes. Also, it was shown that dynamic actin-rich comet tails appeared on a subset of cytoplasmic vesicles that were enriched in PKC and the Wiskott-Aldrich syndrome proteins (specifically N-WASP; Taunton, 2001; Taunton et al., 2000). Together these data suggest that the significance of PKC binding/recruitment to the CXCR4 receptor is to stimulate a focal actin polymerization, which generates a physical force that propels the internalized endosome. The concept of a signaling endosome assembly is relatively established with another integral membrane receptor, the epidermal growth factor (EGF) receptor (Seto et al., 2002). After binding of its ligand, EGF, a variety of signaling molecules are recruited to the activated receptor complex. Likewise elements of the clathrin-mediated endocytosis machinery are recruited and serve to promote the formation of clathrin-coated vesicles containing the ligand-bound receptor associated with signaling molecules (Bivona and Philips, 2003). This endomembrane compartment therefore represents a point of convergence of molecular machinery in signaling and endocytosis. Further characterization of the spatiotemporal composition of the CXCR4-mediated signaling endosome using similar biophysical approaches may provide new insights for understanding the poorly understood relationship among receptor trafficking, signaling, and cancer cell behavior (Polo et al., 2004).
When used in combination with two-photon TCSPC imaging, EGFP:mRFP1 offers a quantitative, high-resolution assessment of protein-protein interactions in situ both spatially and temporally. These advantages should make this fluorophore pair widely applicable particularly to live-cell FRET/FLIM assays.
Acknowledgments
We are grateful to Prof. Roger Tsien for the kind gift of the mRFP1 in the bacterial expression vector pRSET. We thank Julian Gilbey, Rosalind Locke, and Paul Barber for their work on the donor-fluorescence data collection and analysis program, which we used to analyze some of the data. We are indebted to Dr. Vinod Subramaniam (AstraZeneca) for providing the spectra for the eCFP-eYFP pair for the purposes of comparison.
Marion Peter is a recipient of a Human Frontier Science Program Long-Term Fellowship. Borivoj Vojnovic and Simon Ameer-Beg acknowledge the support of the Cancer Research UK (under program grant C133/A1812), Research Councils UK, Basic Technology Program, and the Paul Instrument Fund of the Royal Society. This work would not be possible without the technical support of the GCI electronics and mechanical workshops.
Marion Peter and Simon M. Ameer-Beg contributed equally to this work.
References
- Ameer-Beg, S. M., P. R. Barber, R. J. Hodgkiss, R. J. Locke, R. G. Newman, G. M. Tozer, B. Vojnovic, and J. Wilson. 2002. Application of multiphoton steady-state and lifetime imaging to mapping of tumour vascular architecture in vivo. Proc SPIE. 4620:85–95. [Google Scholar]
- Ameer-Beg, S. M., N. Edme, M. Peter, P. R. Barber, and T. B. V. Ng. 2003. Imaging protein-protein interactions by multiphoton FLIM. Proc. SPIE. 5139:180–189. [Google Scholar]
- Anilkumar, N., M. Parsons, R. Monk, T. Ng, and J. C. Adams. 2003. Interaction of fascin and protein kinase Cα: a novel intersection in cell adhesion and motility. EMBO J. 22:5390–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaens, P. I., and R. Pepperkok. 2000. Observing proteins in their natural habitat: the living cell. Trends Biochem. Sci. 25:631–637. [DOI] [PubMed] [Google Scholar]
- Becker, W., K. Benndorf, A. Bergmann, C. Biskup, K. König, U. Tirlapur, and T. Zimmer. 2001. FRET measurements by TCSPC laser scanning microscopy. Proc. SPIE. 4431:414–419. [Google Scholar]
- Beechem, J. M., and L. Brand. 1986. Global analysis of fluorescence decay: applications to some unusual experimental and theoretical studies. Photochem. Photobiol. 44:323–330. [DOI] [PubMed] [Google Scholar]
- Beechem, J. M., and E. Haas. 1989. Simultaneous determination of intramolecular distance distributions and conformational dynamics by global analysis of energy transfer measurements. Biophys. J. 55:1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivona, T. G., and M. R. Philips. 2003. Ras pathway signaling on endomembranes. Curr. Opin. Cell Biol. 15:136–142. [DOI] [PubMed] [Google Scholar]
- Brown, C. M., and N. O. Petersen. 1998. An image correlation analysis of the distribution of clathrin associated adaptor protein (AP-2) at the plasma membrane. J. Cell Sci. 111:271–281. [DOI] [PubMed] [Google Scholar]
- Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg, J., J. Lippincott-Schwartz, and J. F. Presley. 1998. Two-color green fluorescent protein time-lapse imaging. Biotechniques. 25:838–846. [DOI] [PubMed] [Google Scholar]
- Erickson, M. G., D. L. Moon, and D. T. Yue. 2003. DsRed as a potential FRET partner with CFP and GFP. Biophys. J. 85:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster, T. 1948. Intermolecular energy migration and fluorescence. Annals Phys. 6:55–75. [Google Scholar]
- Gadella, T. W., Jr., and T. M. Jovin. 1995. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 129:1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, G. W., G. Berry, X. H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard, R., N. Signoret, M. Ishiai, M. Marsh, T. Kurosaki, J. V. Ravetch, and I. Masamichi. 1999. B cell antigen receptor engagement inhibits stromal cell-derived factor (SDF)-1α chemotaxis and promotes protein kinase C (PKC)-induced internalization of CXCR4. J. Exp. Med. 189:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikal, A. A., S. T. Hess, and W. W. Webb. 2001. Multiphoton molecular spectroscopy and excited state dynamics of enhanced green fluorescent protein (EGFP): acid-base specificity. Chem. Phys. 274:37–55. [Google Scholar]
- Herreros, J., T. Ng, and G. Schiavo. 2001. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol. Biol. Cell. 12:2947–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen, S., and E. A. Widder. 1999. The physical basis of transparency in biological tissue: ultrastructure and the minimization of light scattering. J. Theor. Biol. 199:181–198. [DOI] [PubMed] [Google Scholar]
- Kneen, M., J. Farinas, Y. Li, and A. S. Verkman. 1998. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 74:1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. 1999. Principles of fluorescence spectroscopy. Kluwer Academic/Plenum Publishers, New York.
- Legg, J. W., C. A. Lewis, M. Parsons, T. Ng, and C. M. Isacke. 2002. A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat. Cell Biol. 4:399–407. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., and G. H. Patterson. 2003. Development and use of fluorescent protein markers in living cells. Science. 300:87–91. [DOI] [PubMed] [Google Scholar]
- Malvezzi-Campeggi, F., M. Jahnz, K. G. Heinze, P. Dittrich, and P. Schwille. 2001. Light-induced flickering of DsRed provides evidence for distinct and interconvertible fluorescent states. Biophys. J. 81:1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki, A. 2003. Visualisation of the spatial and temporal dynamics of intracellular signaling. Dev. Cell. 4:295–305. [DOI] [PubMed] [Google Scholar]
- Mizuno, H., A. Sawano, P. Eli, H. Hama, and A. Miyawaki. 2001. Red fluorescent protein from Discosoma as a fusion tag and a partner for fluorescence resonance energy transfer. Biochemistry. 40:2502–2510. [DOI] [PubMed] [Google Scholar]
- Moore, M. A. 2001. The role of chemoattraction in cancer metastases. Bioessays. 23:674–676. [DOI] [PubMed] [Google Scholar]
- Morton, P. E., T. C. Ng, S. A. Roberts, B. Vojnovic, and S. M. Ameer-Beg. 2003. Time-resolved multiphoton imaging of the interaction between the PKC and the NFkB cell signalling pathway. Proc. SPIE. 5139:216–222. [Google Scholar]
- Muller, A., B. Homey, H. Soto, N. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verastegui, and A. Zlotnik. 2001. Involvement of chemokine receptors in breast cancer metastasis. Nature. 410:50–56. [DOI] [PubMed] [Google Scholar]
- Neil, M. A., A. Squire, R. Juskaitis, P. I. Bastiaens, and T. Wilson. 2000. Wide-field optically sectioning fluorescence microscopy with laser illumination. J. Microsc. 197:1–4. [DOI] [PubMed] [Google Scholar]
- Nelsestuen, G. L., and M. D. Bazzi. 1991. Activation and regulation of protein kinase C enzymes. J. Bioenerg. Biomembr. 23:43–61. [DOI] [PubMed] [Google Scholar]
- Ng, T., M. Parsons, W. E. Hughes, J. Monypenny, D. Zicha, A. Gautreau, M. Arpin, S. Gschmeissner, P. J. Verveer, P. I. Bastiaens, and Peter J. Parker. 2001. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J. 20:2723–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T., D. Shima, A. Squire, P. I. Bastiaens, S. Gschmeissner, M. J. Humphries, and P. J. Parker. 1999a. PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 18:3909–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T., A. Squire, G. Hansra, F. Bornancin, C. Prevostel, A. Hanby, W. Harris, D. Barnes, S. Schmidt, H. Mellor, P. I. Bastiaens, and P. J. Parker. 1999b. Imaging protein kinase Cα activation in cells. Science. 283:2085–2089. [DOI] [PubMed] [Google Scholar]
- Oancea, E., and T. Meyer. 1998. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 95:307–318. [DOI] [PubMed] [Google Scholar]
- O'Connor, D. V., and D. Phillips. 1984. Time Correlated Single Photon Counting. Academic Press, London, UK.
- Parker, P. J., and J. Murray-Rust. 2004. PKC at a glance. J. Cell Sci. 117:131–132. [DOI] [PubMed] [Google Scholar]
- Parsons, M., S. M. Ameer-Beg, and B. Vojnovic. 2004. Imaging protein-protein interactions in cell motility using FRET. Biochem. Soc. Trans. 32:431–433. [DOI] [PubMed] [Google Scholar]
- Parsons, M., M. D. Keppler, A. Kline, A. Messent, M. J. Humphries, R. Gilchrist, I. R. Hart, C. Quittau-Prevostel, W. E. Hughes, P. J. Parker, and T. Ng. 2002. Site-directed perturbation of protein kinase C-integrin interaction blocks carcinoma cell chemotaxis. Mol. Cell. Biol. 22:5897–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, G., R. N. Day, and D. Piston. 2001. Fluorescent protein spectra. J. Cell Sci. 114:837–838. [DOI] [PubMed] [Google Scholar]
- Patterson, G. H., S. M. Knobel, W. D. Sharif, S. R. Kain, and D. W. Piston. 1997. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 73:2782–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy, A. 2001. Fluorescence resonance energy transfer microscopy: a mini review. J. Biomed. Opt. 6:287–291. [DOI] [PubMed] [Google Scholar]
- Peter, M., and S. M. Ameer-Beg. 2004. Imaging molecular interactions by multiphoton FLIM. Biol. Cell. 96:231–236. [DOI] [PubMed] [Google Scholar]
- Pollock, B. A., and R. Heim. 1999. Using GFP in FRET-based applications. Trends Cell Biol. 9:57–60. [DOI] [PubMed] [Google Scholar]
- Polo, S., S. Pece, and P. P. Di Fiore. 2004. Endocytosis and cancer. Curr. Opin. Cell Biol. 16:156–161. [DOI] [PubMed] [Google Scholar]
- Rizzo, M. A., G. H. Springer, B. Granada, and D. W. Piston. 2004. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 22:445–449. [DOI] [PubMed] [Google Scholar]
- Roberts, M., S. Barry, A. Woods, P. van der Sluijs, and J. Norman. 2001. PDGF-regulated rab4-dependent recycling of αvβ3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr. Biol. 11:1392–1402. [DOI] [PubMed] [Google Scholar]
- Schonle, A., M. Glatz, and S. W. Hell. 2000. Four-dimensional multiphoton microscopy with time-correlated single-photon counting. Appl. Optics. 39:6306–6311. [DOI] [PubMed] [Google Scholar]
- Selvin, P. R. 2000. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 7:730–734. [DOI] [PubMed] [Google Scholar]
- Seto, E. S., H. J. Bellen, and T. E. Lloyd. 2002. When cell biology meets development: endocytic regulation of signaling pathways. Genes Dev. 16:1314–1336. [DOI] [PubMed] [Google Scholar]
- Signoret, N., J. Oldridge, A. Pelchen-Matthews, P. J. Klasse, T. Tran, L. F. Brass, M. M. Rosenkilde, T. W. Schwartz, W. Holmes, W. Dallas, and M. A. Luther, T. N. Wells, J. A. Hoxie, and M. Marsh. 1997. Phorbol esters and SDF-1 induce rapid endocytosis and down-modulation of the chemokine receptor CXCR4. J. Cell Biol. 139:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoret, N., M. M. Rosenkilde, P. J. Klasse, T. W. Schwartz, M. H. Malim, J. A. Hoxie, and M. Marsh. 1998. Differential regulation of CXCR4 and CCR5 endocytosis. J. Cell Sci. 111:2819–2830. [DOI] [PubMed] [Google Scholar]
- Slater, S. J., J. L. Seiz, A. C. Cook, C. J. Buzas, S. A. Malinowski, J. L. Kershner, B. A. Stagliano, and C. D. Stubbs. 2002. Regulation of PKCα activity by C1–C2 domain interactions. J. Biol. Chem. 277:15277–15285. [DOI] [PubMed] [Google Scholar]
- Squire, A., and P. I. Bastiaens. 1999. Three-dimensional image restoration in fluorescence lifetime imaging microscopy. J. Microsc. 193:36–49. [DOI] [PubMed] [Google Scholar]
- Stryer, L. 1978. Fluorescence energy transfer as a spectroscopic ruler. Ann. Rev. Biochem. 47:819–846. [DOI] [PubMed] [Google Scholar]
- Suhling, K., J. Siegel, D. Phillips, P. M. French, S. Leveque-Fort, S. E. Webb, and D. M. Davis. 2002. Imaging the environment of green fluorescent protein. Biophys. J. 83:3589–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton, J. 2001. Actin filament nucleation by endosomes, lysosomes and secretory vesicles. Curr. Opin. Cell Biol. 13:85–91. [DOI] [PubMed] [Google Scholar]
- Taunton, J., B. A. Rowning, M. L. Coughlin, M. Wu, R. T. Moon, T. J. Mitchison, and C. A. Larabell. 2000. Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J. Cell Biol. 148:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramier, M., I. Gautier, T. Piolot, S. Ravalet, K. Kemnitz, J. Coppey, C. Durieux, V. Mignotte, and M. Coppey-Moisan. 2002. Picosecond-hetero-FRET microscopy to probe protein-protein interactions in live cells. Biophys. J. 83:3570–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- van Weert, A. W., H. J. Geuze, and W. S. B. Groothuis. 2000. Primaquine interferes with membrane recycling from endosomes to the plasma membrane through a direct interaction with endosomes which does not involve neutralisation of endosomal pH nor osmotic swelling of endosomes. Eur. J. Cell Biol. 79:394–399. [DOI] [PubMed] [Google Scholar]
- Verveer, P. J., and P. I. Bastiaens. 2003. Evaluation of global analysis algorithms for single frequency fluorescence lifetime imaging microscopy data. J. Microsc. 209:1–7. [DOI] [PubMed] [Google Scholar]
- Verveer, P. J., A. Squire, and P. I. Bastiaens. 2000a. Global analysis of fluorescence lifetime imaging microscopy data. Biophys. J. 78:2127–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verveer, P. J., A. Squire, and P. I. Bastiaens. 2001. Improved spatial discrimination of protein reaction states in cells by global analysis and deconvolution of fluorescence lifetime imaging microscopy data. J. Microsc. 202:451–456. [DOI] [PubMed] [Google Scholar]
- Verveer, P. J., F. S. Wouters, A. R. Reynolds, and P. I. Bastiaens. 2000b. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 290:1567–1570. [DOI] [PubMed] [Google Scholar]
- Wouters, F. S., P. J. Verveer, and P. I. Bastiaens. 2001. Imaging biochemistry inside cells. Trends Cell Biol. 11:203–211. [DOI] [PubMed] [Google Scholar]
- Zhang, J., R. E. Campbell, A. Y. Ting, and R. Y. Tsien. 2002. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 3:906–918. [DOI] [PubMed] [Google Scholar]