

Abstract
Point mutations were introduced near the primary electron acceptor sites assigned to A0 in both the PsaA and PsaB branches of Photosystem I in the cyanobacterium Synechocystis sp. PCC 6803. The residues Met688PsaA and Met668PsaB, which provide the axial ligands to the Mg2+ of the eC-A3 and eC-B3 chlorophylls, were changed to leucine and asparagine (chlorophyll notation follows Jordan et al., 2001). The removal of the ligand is expected to alter the midpoint potential of the 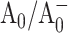 redox pair and result in a change in the intrinsic charge separation rate and secondary electron transfer kinetics from
redox pair and result in a change in the intrinsic charge separation rate and secondary electron transfer kinetics from 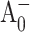 to A1. The dynamics of primary charge separation and secondary electron transfer were studied at 690 nm and 390 nm in these mutants by ultrafast optical pump-probe spectroscopy. The data reveal that mutations in the PsaB branch do not alter electron transfer dynamics, whereas mutations in the PsaA branch have a distinct effect on electron transfer, slowing down both the primary charge separation and the secondary electron transfer step (the latter by a factor of 3–10). These results suggest that electron transfer in cyanobacterial Photosystem I is asymmetric and occurs primarily along the PsaA branch of cofactors.
to A1. The dynamics of primary charge separation and secondary electron transfer were studied at 690 nm and 390 nm in these mutants by ultrafast optical pump-probe spectroscopy. The data reveal that mutations in the PsaB branch do not alter electron transfer dynamics, whereas mutations in the PsaA branch have a distinct effect on electron transfer, slowing down both the primary charge separation and the secondary electron transfer step (the latter by a factor of 3–10). These results suggest that electron transfer in cyanobacterial Photosystem I is asymmetric and occurs primarily along the PsaA branch of cofactors.
INTRODUCTION
Photosystem I (PS I) is a membrane-bound, chlorophyll-protein complex that uses the energy of light to reduce ferredoxin or flavodoxin in cyanobacteria, algae, and higher plants (Brettel, 1997). The discovery and subsequent refinements of the x-ray crystal structure of the PS I reaction center from the cyanobacterium Synechococcus elongatus (Jordan et al., 2001; Klukas et al., 1999; Krauss et al., 1993, 1996) have stimulated continuing interest in the relationship between structure and function. The PS I reaction center (RC) consists of three pairs of chlorophyll (Chl) a molecules and a pair of phylloquinones that are coordinated by a heterodimer of the PsaA and PsaB polypeptides: a Chl a/a′ heterodimer (eC-A1 and eC-A2 in Jordan et al., 2001) spectroscopically identified as the electron donor P700, two Chl a molecules (eC-A2 and eC-B2) identified as accessory pigments, two Chl a molecules (eC-A3 and eC-B3) identified as A0, and two phylloquinones (QK-A and QK-B) identified as A1. These cofactors are arranged into two pseudosymmetric branches that diverge at P700 and converge at the iron-sulfur cluster Fx. According to the conventional electron transfer (ET) model, primary charge separation leads to the reduction of the primary electron acceptor A0, creating the radical pair 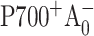 . The unpaired electron migrates to the phylloquinone secondary acceptor A1, then to the [4Fe-4S] center FX, and finally to the terminal iron-sulfur clusters FA and FB before being transferred to soluble ferredoxin (Brettel, 1997).
. The unpaired electron migrates to the phylloquinone secondary acceptor A1, then to the [4Fe-4S] center FX, and finally to the terminal iron-sulfur clusters FA and FB before being transferred to soluble ferredoxin (Brettel, 1997).
The presence of two highly symmetrical branches of cofactors in the RC raises the question of whether one or both are active in ET under physiologically relevant conditions. There is widespread agreement in the field that the PsaA branch of cofactors is active in both prokaryotes and eukaryotes. Evidence that the PsaB branch of cofactors is equally active has mainly been obtained in eukaryotes such as in the algae Chlorella sorokiniana (Joliot and Joliot, 1999) and Chlamydomonas reinhardtii (Boudreaux et al., 2001; Fairclough et al., 2001, 2003; Guergova-Kuras et al., 2001; Muhiuddin et al., 2001), whereas evidence indicating significantly less PsaB-branch electron transfer has been reported in prokaryotes such as the cyanobacteria Synechococcus sp. PCC 7002 and Synechocystis sp. PCC 6803 (Golbeck et al., 2001; Xu et al., 2003a,b). These conclusions have been based primarily on studies involving specific mutations around the respective phylloquinone (A1) binding sites on the PsaA and PsaB polypeptides. The advantage to these mutations is that the spectral and functional changes they induce are so subtle that they are not expected to influence the initial charge separation. The disadvantage is that the subtle nature of the changes induced by these mutations leaves some ambiguity in the interpretation of the data. Moreover, these experiments have focused primarily on measurements of changes in the ET kinetics (optical and EPR), which reflect the relatively slow electron transfer step from the A1 to FX. In this case, the FX electron acceptor is common to both branches, and the changes around A1 in one branch can, in principle, influence the properties of FX and thereby affect the ET kinetics through the other branch. Ramesh et al. (2004), employing optical ultrafast spectroscopy, reported that the replacement of the Met axial ligand to either eC-A3 or eC-B3 by His in the PS I from C. reinhardtii partially blocked electron transfer, and concluded that both branches are active in ET. However, these data were later challenged by a low-temperature transient EPR study (McConnel et al., 2004), which indicated that the formation of the 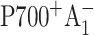 radical pair was not affected in the PsaB-side mutant, whereas it was significantly diminished in the PsaA-side mutant.
radical pair was not affected in the PsaB-side mutant, whereas it was significantly diminished in the PsaA-side mutant.
Recently, Cohen et al. (2004) constructed identical mutations at the corresponding eC-A3 and eC-B3 cofactors of PS I on both the PsaA-side and the PsaB-side in Synechocystis sp. PCC 6803. In the case of symmetric electron transfer along both branches, one would expect that this change would modify the properties of the primary electron acceptor and that one would observe similar physiological and spectroscopic changes in both mutants. However, the optical and EPR experiments revealed significant differences between the PsaA-branch and the PsaB-branch mutants, which were consistent with the assessment that ET occurs primarily along PsaA-branch in cyanobacteria (Cohen et al., 2004). This conclusion contrasts with studies on several eukaryotes (Boudreaux et al., 2001; Fairclough et al., 2001, 2003; Guergova-Kuras et al., 2001; Joliot and Joliot, 1999; Muhiuddin et al., 2001) that argue for significant PsaB-branch activity. Moreover, EPR measurements performed on similar A0 mutations in C. reinhardtii (Fairclough et al., 2001, 2003) were interpreted to indicate that the PsaA branch was not essential. These measurements, however, did not have a time resolution sufficient to detect changes in the first two electron transfer steps, which directly involve A0 and would be naturally affected by such point mutations: the primary charge separation (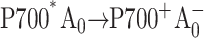 ) and the secondary electron transfer (
) and the secondary electron transfer (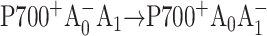 ). It is widely accepted (Brettel, 1997) that the formation of the radical pair
). It is widely accepted (Brettel, 1997) that the formation of the radical pair 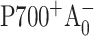 in wild-type PS I complexes occurs within 1–3 ps after the formation of the electronically excited special pair P700*. Since this intrinsic charge separation time is very short compared to the electronic excitation lifetime in antenna (the effective excitation trapping time is ∼20–30 ps in wild-type PS I), a direct measurement of this process is not feasible. However, the changes in the intrinsic charge separation rate may alter the effective excitation trapping time, which is readily detectable in an optical femtosecond pump-probe experiment. Most estimates of the timescale for the secondary
in wild-type PS I complexes occurs within 1–3 ps after the formation of the electronically excited special pair P700*. Since this intrinsic charge separation time is very short compared to the electronic excitation lifetime in antenna (the effective excitation trapping time is ∼20–30 ps in wild-type PS I), a direct measurement of this process is not feasible. However, the changes in the intrinsic charge separation rate may alter the effective excitation trapping time, which is readily detectable in an optical femtosecond pump-probe experiment. Most estimates of the timescale for the secondary 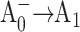 electron transfer step in wild-type PS I range from 10 to 50 ps (Brettel and Vos, 1999; Hastings et al., 1994b; Iwaki et al., 1995; Kumazaki et al., 1994a; Savikhin et al., 2001; White et al., 1996). The kinetics of the secondary electron transfer can be directly measured in an optical pump-probe experiment by monitoring the formation dynamics of
electron transfer step in wild-type PS I range from 10 to 50 ps (Brettel and Vos, 1999; Hastings et al., 1994b; Iwaki et al., 1995; Kumazaki et al., 1994a; Savikhin et al., 2001; White et al., 1996). The kinetics of the secondary electron transfer can be directly measured in an optical pump-probe experiment by monitoring the formation dynamics of 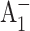 in the near ultraviolet (UV) region (Brettel and Vos, 1999), or by using a more sophisticated (open-closed) subtraction technique (Hastings et al., 1994b; Savikhin et al., 2001; White et al., 1996) to resolve the formation and decay kinetics of the
in the near ultraviolet (UV) region (Brettel and Vos, 1999), or by using a more sophisticated (open-closed) subtraction technique (Hastings et al., 1994b; Savikhin et al., 2001; White et al., 1996) to resolve the formation and decay kinetics of the 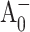 state.
state.
In this article, we applied optical femtosecond pump-probe techniques to study ET in two complimentary pairs of point mutants, in which Met688PsaA or Met668PsaB were replaced by Leu (Cohen et al., 2004) or Asn (R. O. Cohen, G. Shen, J. H. Golbeck, W. Xu, P. R. Chitnis, A. Valieva, A. van der Est, Y. Pushkar, and D. Stehlik, unpublished) in Synechocystis sp. PCC 6803. These Met residues are proposed to provide the axial ligands to the respective Mg2+ ions of the two A0 chlorophyll molecules. The Met ligand to Mg2+ is expected to be weak, and the possibility that no axial ligand exists also needs to be considered (Goldsmith et al., 1996). However, regardless of the nature of the interaction between the Met residues and the chlorophylls, their proximity to one another means that a mutation in the amino acid will likely alter the midpoint potential of the 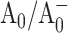 redox pair. This should result in a change in Gibbs free energy associated with both primary charge separation and the secondary electron transfer step due to a change in the Frank-Condon factor in the Marcus equation, which relates the rate of electron transfer to changes in Gibbs free energy and reorganization energy (Moser et al., 1992). We will show that the mutations on the PsaA side have a pronounced effect on the primary charge separation and especially on the secondary electron transfer kinetics, whereas the corresponding mutations on the PsaB side have a minimal effect on the ET process. These results support previous suggestions that forward electron transfer in cyanobacterial PS I under physiologically relevant conditions is asymmetric and occurs predominantly along the PsaA branch of cofactors.
redox pair. This should result in a change in Gibbs free energy associated with both primary charge separation and the secondary electron transfer step due to a change in the Frank-Condon factor in the Marcus equation, which relates the rate of electron transfer to changes in Gibbs free energy and reorganization energy (Moser et al., 1992). We will show that the mutations on the PsaA side have a pronounced effect on the primary charge separation and especially on the secondary electron transfer kinetics, whereas the corresponding mutations on the PsaB side have a minimal effect on the ET process. These results support previous suggestions that forward electron transfer in cyanobacterial PS I under physiologically relevant conditions is asymmetric and occurs predominantly along the PsaA branch of cofactors.
EXPERIMENTAL PROCEDURES
Sample preparation
The Met688PsaA-to-Leu688PsaA (M688LPsaA) and the Met668PsaB-to-Leu668PsaB (M668LPsaB) mutants were constructed in Synechocystis sp. PCC 6803 as described in Cohen et al., 2004). The Met688PsaA-to-Asn688PsaA (M688NPsaA) and the Met668PsaB-to-Asn668PsaB (M668NPsaB) mutants were constructed similarly (R. O. Cohen, G. Shen, J. H. Golbeck, W. Xu, P. R. Chitnis, A. Valieva, A. van der Est, Y. Pushkar, and D. Stehlik, unpublished). The mutant cells were grown photomixotrophically with 5 mM glucose in liquid culture under conditions of light-activated heterotrophic growth. Growth was monitored turbidometrically at 730 nm using a CARY-14 spectrophotometer modified for computerized data acquisition by On-Line Instruments. PS I trimers were isolated from thylakoid membranes using n-dodecyl-β-d-maltopyranoside and purified on two sucrose density gradients The trimeric PS I complexes were present in the bottom green band and resuspended in 50 mM Tris, pH 8.0, containing 0.03% n-dodecyl-β-d-maltopyranoside and 15% glycerol. The optical measurements were performed on the same three batches of PS I trimers studied in Cohen et al. (2004), which were isolated from independently grown cells. However, unlike the considerable variability noted in both the spin-polarized radical pair spectra and the triplet spectra of the M688LPsaA mutants (Cohen et al., 2004), the time-resolved optical spectra of 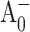 and
and 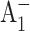 reported here yielded highly reproducible results.
reported here yielded highly reproducible results.
Ultrafast pump-probe spectroscopy
Excitation pulses (660 nm, ∼100 fs FWHM, 1 kHz repetition rate) were generated using a self-mode-locked Ti:sapphire laser, regenerative amplifier, optical parametric amplifier, and frequency doubler as described earlier (Savikhin et al., 2000). Transient sample absorption was probed with broadband continuum light pulses that were generated in a sapphire plate; cross-correlations between the pump and probe pulses were typically 100–200 fs FWHM. Continuum pulses were split into signal and reference beams, dispersed in an Oriel MS257 imaging monochromator operated at ∼3 nm bandpass, and directed onto separate Hamamatsu (Hamamatsu City, Japan) S3071 Si pin photodiodes. The probe, reference and pump pulse energies were measured in the Stanford Research Systems SR250 boxcar integrators, digitized, and processed in a computer. Noise performance was near shot noise-limited; the root mean square noise in absorption difference (ΔA) measurements was ∼3 × 10−5 for 1-s accumulation time. Operation in the annihilation-free regime was ensured by control experiments in which the pump power was varied; all experiments were performed at room temperature. In experiments with closed RC, a small fraction of 532 nm, 400 ns pulses from a Nd:YAG laser utilized in a femtosecond regenerative amplifier was used to optically close RC in PS I complexes; these pulses preceded pump/probe pulse pairs by ∼6 μs and were focused into the same spot where the pump and probe pulses intersected. Similar optical manipulation of the RC state has been performed previously using ambient continuous light (Savikhin et al., 2001). Since comparison between dynamics in PS I with closed and open RC was essential in some experiments, this optical switching automatically ensured amplitude normalization of pump-probe signals accumulated for species with open and closed RC.
All PS I samples for ultrafast pump-probe experiments exhibited ∼0.3 optical density at 660 nm and contained 40 mM sodium ascorbate and 20 μM phenazine methosulfate to ensure rapid recovery of the RC. Samples were housed in a spinning cell with 0.7 mm path length; the excitation density was ∼1.5 μJ/cm2 (1.5 nJ/pulse, ∼300 μm spot size). This yielded an excitation rate of 1 out of every ∼1000 Chls. The pump and probe polarizations were separated by 54.7°, to exclude anisotropy effects in the measured kinetics.
Steady-state spectroscopy
PS I samples for steady-state experiments contained 40 mM sodium ascorbate. As shown by Savikhin et al. (2000), samples in experiments conducted in total darkness contained predominantly open reaction centers; continuous illumination of the sample cell by a 3 V flashlight bulb yielded samples in which the reaction centers were almost exclusively closed. Steady-state absorption spectra of PS I samples with open and closed reaction centers were accumulated in a PerkinElmer (Wellesley, MA) Lambda 3B spectrophotometer modified according to Savikhin et al. (1999). The (P700+-P700) absorption difference spectra were then obtained by subtracting absorption spectrum of the sample with open RC from that with closed RC.
RESULTS
P700+-P700 difference spectra
The steady-state electronic absorption spectra (not shown) of PS I trimers from the M688LPsaA and M688NPsaA mutants and the M668LPsaB, and M668NPsaB mutants are nearly superimposable with the steady-state electronic absorption spectra of PS I trimers from the wild-type (WT) complexes in the Qy spectral region of Chl a (640–720 nm). Hence, the spectral distributions of antenna chlorophyll absorption wavelengths in the mutants are essentially the same as in the WT. The (P700+-P700) difference spectra of the mutants are presented in Fig. 1. The major photobleaching (PB), an ∼30 nm-wide band centered at ∼700 nm, has been conventionally attributed to the bleaching of the P700 absorption band, which arises due to the excitonic absorption of the special pair. This band is essentially the same in the WT and mutants, indicating that the changes around A0 have little effect on the properties of the special pair. The mutations also have little effect on the broad absorption band centered at ∼800 nm, which has been attributed to the absorption of the oxidized special pair (P700+).
FIGURE 1.

P700+-P700 absorption difference spectra of PS I complexes with mutations in the PsaA branch (A) and in the PsaB branch (B), along with respective WT PS I spectrum.
However, noticeable deviations from the WT spectra are observed in the region of the relatively narrow (FWHM ∼11 nm) absorption band centered at ∼690 nm, denoted as C690 in Savikhin et al. (2001) and tentatively ascribed to the absorption from the neutral monomeric chlorophyll a that emerges from P700 when one of the pigments in this special pair becomes oxidized. Although the C690 feature encounters relatively minor changes when the mutations are introduced near A0 in the PsaB branch (Fig. 1 B), the amplitude of this band decreases almost twofold when the mutations are introduced near A0 in the PsaA branch (Fig. 1 A). The sensitivity of this spectral feature to the change around A0 suggests that either the electronic properties of the P700+ in mutants are altered in a way that leads to a more uniform distribution of the hole over the two pigments in the special pair, or the C690 feature is derived to a large extent from other effects such as excitonic coupling of A0 with the rest of the pigments in the RC (Byrdin et al., 2002; Damjanovic et al., 2002; Witt et al., 2002), and/or is derived from an electrochromic shift of the absorption band of A0 (or accessory Chl a) due to the local electric field created by P700+. It is likely that various mechanisms contribute to the observed changes as they are not limited to the C690 feature alone. The integrated deviations from the P700+-P700 spectra of WT are 1.5 times larger in the PsaA-branch mutants than in the PsaB-branch mutants, suggesting that the eC-A3 chlorophyll is more strongly coupled to the rest of the pigments in the RC than is the eC-B3 chlorophyll. These optical properties, however, may not necessarily be reflected in differences in electron transfer.
The A0 electronic absorption spectrum
The absorption spectrum of A0 is not accessible directly due to significant spectral overlap with absorption spectra of the remaining Chl a pigments that comprise the RC cofactors and antenna. Hastings et al. (1994b) obtained an ( ) difference spectrum as an additional nondecaying component superimposed on the (P700+-P700) spectrum in time-resolved pump-probe profiles of PS I with a chemically reduced secondary acceptor, A1. This reduction blocks electron transfer from A0 and facilitates the formation of a long-living charge-separated state
) difference spectrum as an additional nondecaying component superimposed on the (P700+-P700) spectrum in time-resolved pump-probe profiles of PS I with a chemically reduced secondary acceptor, A1. This reduction blocks electron transfer from A0 and facilitates the formation of a long-living charge-separated state  which can then be observed in a conventional pump-probe experiment. Savikhin et al. (2001) proposed a method for measuring the (
which can then be observed in a conventional pump-probe experiment. Savikhin et al. (2001) proposed a method for measuring the ( ) spectrum by comparing pump-probe profiles of wild-type PS I with open and closed RC, and were able to obtain a spectrum similar to that observed by Hastings et al. (1994b), but without chemical treatment. In this article, we used the latter approach. The transient (
) spectrum by comparing pump-probe profiles of wild-type PS I with open and closed RC, and were able to obtain a spectrum similar to that observed by Hastings et al. (1994b), but without chemical treatment. In this article, we used the latter approach. The transient ( ) absorption difference spectra of PS I trimers from the M688LPsaA, M688NPsaA, M668LPsaB, and M668NPsaB mutants were obtained by exciting the samples at 660 nm, the blue edge of the core antenna spectrum, and sweeping the probe wavelength from 660 nm to 720 nm at fixed time delays. To isolate spectral changes from antenna processes, transient spectra for closed RC were subtracted from the corresponding spectra for open RC, resulting in (open-closed) absorption difference spectra (or ΔΔA spectra). Since the excitation trapping time is nearly independent of the P700 oxidation state (Klug et al., 1989; Nuijs et al., 1986; Owens et al., 1988; Savikhin et al., 2000; Shuvalov et al., 1986; Turconi et al., 1993), this procedure essentially cancels the antenna energy transfer dynamics and isolates spectral changes in the RC (Hastings et al., 1995; Savikhin et al., 2001). At early times, a detectable amount of the
) absorption difference spectra of PS I trimers from the M688LPsaA, M688NPsaA, M668LPsaB, and M668NPsaB mutants were obtained by exciting the samples at 660 nm, the blue edge of the core antenna spectrum, and sweeping the probe wavelength from 660 nm to 720 nm at fixed time delays. To isolate spectral changes from antenna processes, transient spectra for closed RC were subtracted from the corresponding spectra for open RC, resulting in (open-closed) absorption difference spectra (or ΔΔA spectra). Since the excitation trapping time is nearly independent of the P700 oxidation state (Klug et al., 1989; Nuijs et al., 1986; Owens et al., 1988; Savikhin et al., 2000; Shuvalov et al., 1986; Turconi et al., 1993), this procedure essentially cancels the antenna energy transfer dynamics and isolates spectral changes in the RC (Hastings et al., 1995; Savikhin et al., 2001). At early times, a detectable amount of the 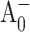 state is expected to form. It has been shown in Savikhin et al. (2001) that the
state is expected to form. It has been shown in Savikhin et al. (2001) that the 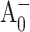 population maximizes at ∼8 ps. At that time, however, in a large fraction of RCs the electronic excitation is not yet trapped, and in some RCs the electron has already left
population maximizes at ∼8 ps. At that time, however, in a large fraction of RCs the electronic excitation is not yet trapped, and in some RCs the electron has already left 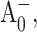 forming
forming  By subtracting profiles for closed RC from those for open RC, the contribution from the untrapped excitation can be cancelled. Additional subtraction of properly scaled profiles measured at long times (when all RC are in
By subtracting profiles for closed RC from those for open RC, the contribution from the untrapped excitation can be cancelled. Additional subtraction of properly scaled profiles measured at long times (when all RC are in 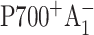 state) would also remove any contribution from the
state) would also remove any contribution from the 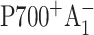 state (Savikhin et al., 2001). Since ET dynamics in the PsaB-side mutants M668LPsaB and M668NPsaB are very similar to the WT (to be discussed later), the transient absorption difference signal for these mutants were obtained at 8 ps and at 200 ps (Savikhin et al., 2001), and the procedure described above was used to isolate (
state (Savikhin et al., 2001). Since ET dynamics in the PsaB-side mutants M668LPsaB and M668NPsaB are very similar to the WT (to be discussed later), the transient absorption difference signal for these mutants were obtained at 8 ps and at 200 ps (Savikhin et al., 2001), and the procedure described above was used to isolate ( ) difference spectra. The PsaA-side mutants M688LPsaA and M688NPsaA exhibited substantially slower
) difference spectra. The PsaA-side mutants M688LPsaA and M688NPsaA exhibited substantially slower 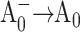 electron transfer, with the
electron transfer, with the 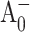 population maximized at ∼25 ps, and the (
population maximized at ∼25 ps, and the ( ) spectra were obtained by similar treatment of absorption difference spectra at 25 ps and at 650 ps.
) spectra were obtained by similar treatment of absorption difference spectra at 25 ps and at 650 ps.
Fitting the ( ) absorption difference spectra of the PsaB-side mutants as described in Savikhin et al. (2001) reveals that the positions and widths of the A0 absorption bands are respectively 685.2 nm and 11 nm FWHM in PS I complexes from the M668LPsaB mutant, and 686.6 nm and 10 nm FWHM in PS I complexes from the M668NPsaB mutant (not shown). Within the error of our experiment (∼1 nm), these parameters are essentially the same as measured earlier for PS I complexes from the WT (685.7 nm and 11 nm, Savikhin et al., 2001). The best fits to the (
) absorption difference spectra of the PsaB-side mutants as described in Savikhin et al. (2001) reveals that the positions and widths of the A0 absorption bands are respectively 685.2 nm and 11 nm FWHM in PS I complexes from the M668LPsaB mutant, and 686.6 nm and 10 nm FWHM in PS I complexes from the M668NPsaB mutant (not shown). Within the error of our experiment (∼1 nm), these parameters are essentially the same as measured earlier for PS I complexes from the WT (685.7 nm and 11 nm, Savikhin et al., 2001). The best fits to the ( ) absorption spectra of the PsaA-side mutants reveal that the A0 band positions and widths are respectively 688.6 nm and 11.6 nm FWHM in PS I complexes from the M688LPsaA mutant and 683.7 nm and 15.6 nm FWHM in PS I complexes from the M688NPsaA mutant (data not shown). The A0 band of the M688LPsaA mutant is therefore ∼3 nm red-shifted and that of the M688NPsaA mutant is ∼2 nm blue-shifted with respect to the A0 band position measured in the WT.
) absorption spectra of the PsaA-side mutants reveal that the A0 band positions and widths are respectively 688.6 nm and 11.6 nm FWHM in PS I complexes from the M688LPsaA mutant and 683.7 nm and 15.6 nm FWHM in PS I complexes from the M688NPsaA mutant (data not shown). The A0 band of the M688LPsaA mutant is therefore ∼3 nm red-shifted and that of the M688NPsaA mutant is ∼2 nm blue-shifted with respect to the A0 band position measured in the WT.
Probing formation of A1− by near-UV pump-probe spectroscopy
The formation of the semiquinone state of the secondary electron acceptor A1 can be monitored directly by its broad near-UV absorption band around 380–390 nm (Brettel, 1988; Brettel et al., 1986; Brettel and Vos, 1999; Lüneberg et al., 1994). The time-resolved absorption difference spectra of WT PS I with open and closed RCs were recorded at 390 nm after exciting at 660 nm (Fig. 2 A). The absorption difference profile for PS I complexes with open RCs in the 380–390 nm spectral region was previously obtained by Brettel and Vos (1999), and was described by two exponentials with time constants of 7 ps and 28 ps and a nondecaying offset. Brettel and Vos attributed the longer decay component (28 ps) to the electron transfer process from 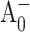 to A1, assuming that the 7 ps component reflected the spectral equilibration, the excitation trapping, and the primary charge separation. Since antenna kinetics were not independently monitored, it is unclear to what extent it may have been influenced by excitation annihilation. In our experiment, the pump power was reduced to the level at which no annihilation effects were observed in antenna kinetics, and the measured kinetics at 390 nm could be described accurately as a single exponent with a time constant of 30 ± 3 ps (and a nondecaying positive offset). This suggests that the 7 ps component observed by Brettel and Vos stems primarily from excitation annihilation processes. The ∼30 ps decay component, however, cannot be attributed entirely to electron transfer from
to A1, assuming that the 7 ps component reflected the spectral equilibration, the excitation trapping, and the primary charge separation. Since antenna kinetics were not independently monitored, it is unclear to what extent it may have been influenced by excitation annihilation. In our experiment, the pump power was reduced to the level at which no annihilation effects were observed in antenna kinetics, and the measured kinetics at 390 nm could be described accurately as a single exponent with a time constant of 30 ± 3 ps (and a nondecaying positive offset). This suggests that the 7 ps component observed by Brettel and Vos stems primarily from excitation annihilation processes. The ∼30 ps decay component, however, cannot be attributed entirely to electron transfer from 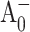 to A1, as the
to A1, as the 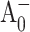 state is not formed immediately upon excitation in our experiment. Global analysis of the antenna decay signals reveals that effective trapping of excitation (i.e., formation of the
state is not formed immediately upon excitation in our experiment. Global analysis of the antenna decay signals reveals that effective trapping of excitation (i.e., formation of the  state) occurs with a characteristic time constant of 23–24 ps (Savikhin et al., 2000, 2001), and the observed 30 ps kinetics must be due to two consecutive steps—formation of
state) occurs with a characteristic time constant of 23–24 ps (Savikhin et al., 2000, 2001), and the observed 30 ps kinetics must be due to two consecutive steps—formation of 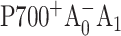 followed by the electron transfer to the secondary electron acceptor resulting in the formation of
followed by the electron transfer to the secondary electron acceptor resulting in the formation of 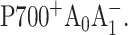 Moreover, the 390-nm signal at early times is dominated by the photobleaching of PS I antenna Chl a pigments, and the formation and decay of the reduced A0 pigment may also contribute to the signal. To simplify the analysis of the pump-probe profiles we used the fact that antenna decay is almost independent of the state of the reaction center (Hastings et al., 1994b; Klug et al., 1989; Owens et al., 1988; Savikhin et al., 1999; Shuvalov et al., 1986; Turconi et al., 1993). In Fig. 2 B, the ΔΔA profile was obtained by subtraction of the ΔA profile for closed RC from that for open RC (Fig. 2 A). As the result, the negative early time component which stems from excited antenna pigments was eliminated (Savikhin et al., 2001). The ΔΔA profile was then fitted within a simple sequential energy/electron transfer model shown below and similar to that of Savikhin et al. (2001):
Moreover, the 390-nm signal at early times is dominated by the photobleaching of PS I antenna Chl a pigments, and the formation and decay of the reduced A0 pigment may also contribute to the signal. To simplify the analysis of the pump-probe profiles we used the fact that antenna decay is almost independent of the state of the reaction center (Hastings et al., 1994b; Klug et al., 1989; Owens et al., 1988; Savikhin et al., 1999; Shuvalov et al., 1986; Turconi et al., 1993). In Fig. 2 B, the ΔΔA profile was obtained by subtraction of the ΔA profile for closed RC from that for open RC (Fig. 2 A). As the result, the negative early time component which stems from excited antenna pigments was eliminated (Savikhin et al., 2001). The ΔΔA profile was then fitted within a simple sequential energy/electron transfer model shown below and similar to that of Savikhin et al. (2001):
 |
where k1 is the effective excitation trapping rate, which encompasses the cumulative effects of antenna excitation equilibration, trapping at P700, back-transfer from P700* to the antenna complex and the primary charge separation; k2 is the intrinsic rate of the secondary electron transfer 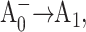 and k3 is the intrinsic rate of the following electron transfer step A1→FX. The terms of effective and intrinsic rates are used here as defined in Gatzen et al. (1996) and Schatz et al. (1988). The charge recombination time of the
and k3 is the intrinsic rate of the following electron transfer step A1→FX. The terms of effective and intrinsic rates are used here as defined in Gatzen et al. (1996) and Schatz et al. (1988). The charge recombination time of the 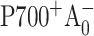 pair is very long (∼35 ns) and was not included into the model (Kleinherenbrink et al., 1994). In our simulations the effective excitation trapping rate k1 was fixed at (24 ps)−1 in accordance with Savikhin et al. (2001), the intrinsic electron transfer rate k3 was assumed to be long compared to the studied time window (Bock et al., 1989; Brettel, 1988, 1998; Sakuragi et al., 2002; Setif and Brettel, 1993; van der Est et al., 1994), and the ratio between the signals due to the formation of
pair is very long (∼35 ns) and was not included into the model (Kleinherenbrink et al., 1994). In our simulations the effective excitation trapping rate k1 was fixed at (24 ps)−1 in accordance with Savikhin et al. (2001), the intrinsic electron transfer rate k3 was assumed to be long compared to the studied time window (Bock et al., 1989; Brettel, 1988, 1998; Sakuragi et al., 2002; Setif and Brettel, 1993; van der Est et al., 1994), and the ratio between the signals due to the formation of 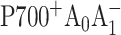 and
and 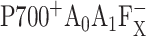 was set to 3:1 as measured in Brettel (1988) and Brettel et al. (1986). Best fit to the (open-closed) profile shown in Fig. 2 was obtained with k2 = (12.8 ps)−1; reasonable fits could be obtained with k2 = (8…15 ps)−1, which is consistent with our previous results derived from global analysis of the (open-closed) profiles in the Chl a absorption spectral region (Savikhin et al., 2001). Earlier estimates of the
was set to 3:1 as measured in Brettel (1988) and Brettel et al. (1986). Best fit to the (open-closed) profile shown in Fig. 2 was obtained with k2 = (12.8 ps)−1; reasonable fits could be obtained with k2 = (8…15 ps)−1, which is consistent with our previous results derived from global analysis of the (open-closed) profiles in the Chl a absorption spectral region (Savikhin et al., 2001). Earlier estimates of the 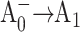 electron transfer kinetics in PS I, which have ranged from 20 to 50 ps (Brettel and Vos, 1999; Hastings et al., 1994b; Iwaki et al., 1995; Kumazaki et al., 1994a; White et al., 1996), may have been influenced by the relatively slow antenna excitation trapping at P700. Fig. 2 B also shows the modeled signal due to the formation of each of the intermediate states. The amplitude of the signal due to the formation of the
electron transfer kinetics in PS I, which have ranged from 20 to 50 ps (Brettel and Vos, 1999; Hastings et al., 1994b; Iwaki et al., 1995; Kumazaki et al., 1994a; White et al., 1996), may have been influenced by the relatively slow antenna excitation trapping at P700. Fig. 2 B also shows the modeled signal due to the formation of each of the intermediate states. The amplitude of the signal due to the formation of the  state at 390 nm cannot be derived independently and was a free parameter of the fit. The best fit resulted in a very small amplitude of the
state at 390 nm cannot be derived independently and was a free parameter of the fit. The best fit resulted in a very small amplitude of the  signal and as a consequence the dynamics of this state could not be reliably determined by the analysis of the 390 nm signal.
signal and as a consequence the dynamics of this state could not be reliably determined by the analysis of the 390 nm signal.
FIGURE 2.

Time-resolved absorption difference profiles for WT PS I complex excited at 660 nm and probed at 390 nm. Negative-going signals indicate photobleaching/stimulated emission. (A) Time-resolved absorption difference profiles obtained for PS I complexes with open and closed RC. (B) Time-resolved (open-closed) absorption difference profile, optimized fit to it using Eq. 1, and signal contributions due to the formation of 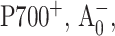 and
and 
In an alternative fit, the ΔA profile for PS I with open RC (Fig. 2 A) was fitted “as is”. In this case, the antenna signal was modeled as PB with amplitude equal to the initial amplitude in the experimentally measured profile (all of the signal at time zero comes from antenna), and its decay time was set to 24 ps (Savikhin et al., 2001). The optimized value of k2 in this fit turned out to be the same as in the previous fit to the (open-closed) profile.
The time-resolved (open-closed) profiles measured for PsaB-side mutants are almost superimposable on that of the WT as seen in Fig. 3 B, suggesting that the rate of secondary electron transfer k2 is unaffected by mutations on the PsaB branch of the RC. However, the 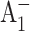 formation kinetics were dramatically altered in the PS I complexes in which the mutations were introduced on the PsaA branch of the RC (Fig. 3 A). The fits to the (open-closed) pump-probe profiles shown in Fig. 3 A revealed that the intrinsic time of the electron transfer from A0 to A1 is 112 ± 10 ps in the M688NPsaA and 100 ± 10 ps in the M688LPsaA mutants, which is almost an order of magnitude slower than secondary electron transfer in the WT.
formation kinetics were dramatically altered in the PS I complexes in which the mutations were introduced on the PsaA branch of the RC (Fig. 3 A). The fits to the (open-closed) pump-probe profiles shown in Fig. 3 A revealed that the intrinsic time of the electron transfer from A0 to A1 is 112 ± 10 ps in the M688NPsaA and 100 ± 10 ps in the M688LPsaA mutants, which is almost an order of magnitude slower than secondary electron transfer in the WT.
FIGURE 3.

Time-resolved (open-closed) absorption difference profiles for WT, M688LPsaA and M688NPsaA mutants (A), and for WT, M668LPsaB, and M668NPsaB mutants (B). All samples were excited at 660 nm and absorption differences were probed at 390 nm.
Probing the kinetics of A0− absorption at 690 nm
Absorption changes created by the formation and decay of  state are not easily accessible in conventional pump-probe experiments, as the A0 absorption overlaps with the absorption of the other RC cofactors and numerous antenna pigments. Hastings et al. (1994b) proposed a strategy for isolating the reaction center kinetics from the antenna kinetics by obtaining time-resolved absorption difference profiles for PS I core complexes with open and closed RC. This method was later used by Savikhin et al. (2000, 2001). According to Savikhin et al. (2001), the (open-closed) ΔΔA profile probed at 690 nm is especially informative as it closely follows the dynamics of formation and decay of the
state are not easily accessible in conventional pump-probe experiments, as the A0 absorption overlaps with the absorption of the other RC cofactors and numerous antenna pigments. Hastings et al. (1994b) proposed a strategy for isolating the reaction center kinetics from the antenna kinetics by obtaining time-resolved absorption difference profiles for PS I core complexes with open and closed RC. This method was later used by Savikhin et al. (2000, 2001). According to Savikhin et al. (2001), the (open-closed) ΔΔA profile probed at 690 nm is especially informative as it closely follows the dynamics of formation and decay of the 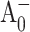 state. At this particular wavelength, the initial rise of PB is primarily due to the formation of the
state. At this particular wavelength, the initial rise of PB is primarily due to the formation of the 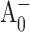 state, since the (
state, since the ( ) spectrum has a maximum PB at ∼686 nm (Hastings et al., 1994b; Savikhin et al., 2001). As
) spectrum has a maximum PB at ∼686 nm (Hastings et al., 1994b; Savikhin et al., 2001). As 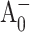 returns to neutral A0 state, the signal is superceded by the (P700+-P700) absorption difference signal, which is positive at 690 nm (Fig. 1). In the WT, the effective charge trapping time (∼24 ps) is longer than the subsequent intrinsic electron transfer time from A0 to A1 (∼13 ps). Therefore, the latter is reflected in the rise of the PB, and the 24 ps component appears as the decay of the PB in 690 nm (open-closed) kinetics. It was shown in Savikhin et al. (2001) that (open-closed) ΔΔA kinetics at 690 nm (and other wavelengths) can be described within a simple model similar to the one shown in Eq. 1.
returns to neutral A0 state, the signal is superceded by the (P700+-P700) absorption difference signal, which is positive at 690 nm (Fig. 1). In the WT, the effective charge trapping time (∼24 ps) is longer than the subsequent intrinsic electron transfer time from A0 to A1 (∼13 ps). Therefore, the latter is reflected in the rise of the PB, and the 24 ps component appears as the decay of the PB in 690 nm (open-closed) kinetics. It was shown in Savikhin et al. (2001) that (open-closed) ΔΔA kinetics at 690 nm (and other wavelengths) can be described within a simple model similar to the one shown in Eq. 1.
Fig. 4 B shows the (open-closed) ΔΔA profile measured at 690 nm for PS I complexes from the WT (Savikhin et al., 2001) and the M668LPsaB and M668NPsaB mutants. The only change in the case of the PsaB-side mutants is the level of the residual absorption, which is consistent with the lower (P700+-P700) absorption level at 690 nm in the mutants as shown in Fig. 1 B. Simple two-exponential fits to the ΔΔA profiles (Fig. 4 B, smooth lines) reveal a 10 ± 3 ps PB rise followed by a 24 ± 3 ps PB decay for all three curves, suggesting that the PsaB-side mutations do not influence ET in the PS I RC.
FIGURE 4.

Time-resolved (open-closed) absorption difference profiles for WT, M688LPsaA, and M688NPsaA mutants (A), and for WT, M668LPsaB, and M668NPsaB mutants (B). All samples were excited at 660 nm and absorption differences were probed at 690 nm.
As in the case of the 390 nm pump-probe profiles, dramatic differences were observed in the ΔΔA signals measured for the PsaA-side mutants at 690 nm (Fig. 4 A; notice the timescale change). Biexponential fits to these profiles reveal ∼11.4 ps and ∼10.4 ps PB rise times, combined with 130 ± 20 ps and 145 ± 20 ps PB decay times for the M688LPsaA and M688NPsaA mutants, respectively (Fig. 4 A, smooth lines). According to the simple model shown in Eq. 1, the longer PB decay at 690 nm observed for the PsaA-side mutants reflects a longer A0→A1 ET time, and is consistent with the pump-probe data obtained at 390 nm. The model in Eq. 1 also predicts that in the case when the A0→A1 ET time is longer than the effective charge-separation time, the PB rise-time at 690 nm should reflect the latter ∼24 ps process. However, the fits show that the PB rise times for the PsaA-side mutants are essentially the same as in the WT, i.e., ∼10 ps. This may be explained if one more intermediate radical pair state involving an accessory pigment as a primary electron acceptor (or donor) is included in the model shown in Eq. 1. Experimental evidence of such a scenario has been recently reported by Müller et al. (2003). On the other hand, the analysis of the (open-closed) kinetics relies on the assumption that the difference between the pump-probe profiles measured for the closed and open RC is entirely due to the presence of the electron transfer process in the latter. It is possible, however, that the observed ∼10 ps PB rise component in ΔΔA profile reflects dynamics of quenching of intermediate states in PS I complexes with closed RC that are not present in PS I complexes with open RC. The nature of the electronic excitation quenching in PS I complexes with closed RC is still unknown.
Global analysis of pump-probe kinetics
Pump-probe profiles for PS I complexes with open RC from all mutants were measured as a function of probe wavelength and analyzed globally to produce decay-associated spectra (DAS) as described in (Savikhin et al., 2000). The excitation (pump) wavelength was set to 660 nm and the probe wavelength was varied in the range 650–720 nm.
The pump-probe profiles for PS I complexes from the PsaB-side mutants were essentially the same as for complexes from the WT and could be described by four major components (Fig. 5 A) (Savikhin et al., 2000). The two shortest DAS components (∼500 fs and ∼2 ps) have been previously assigned to excitation equilibration in the PS I antenna, the ∼24 ps DAS component was identified as effective energy trapping by the RC, and the residual long-time (>1-ns) component was ascribed to the (P700+-P700) absorption difference. Our global analysis did not reveal any additional decay components that could reflect electron transfer from 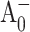 to A1, which is consistent with the finding that the latter process is faster than the effective excitation energy trapping process.
to A1, which is consistent with the finding that the latter process is faster than the effective excitation energy trapping process.
FIGURE 5.

Decay-associated spectra from global analyses of pump-probe data obtained for PS I complexes with open RCs. (A) WT (Savikhin et al., 2000). (B) M688LPsaA mutant shows five distinct components: 530 fs (•), 2.3 ps (▪), 30 ps (▴), 102 ps (○), and long (♦). (C) M688NPsaA mutant shows five distinct components: 530 fs (•), 2.3 ps (▪), 33 ps (▴), 210 ps (○), and long (♦).
Global fits to pump-probe profiles obtained for PS I complexes from the PsaA-side mutants were only possible with five distinct decay components (Fig. 5). Within the error of the experiment, the DAS for the two shortest components (530 fs and 2.3 ps) for the PsaA-side mutants are almost identical to the two shortest components found in the WT, and can be assigned to excitation equilibration among antenna pigments. The spectral shape of DAS of the ∼30 ps component is qualitatively similar to the ∼24 ps component found in WT and it can be attributed to the effective energy trapping rate, which is somewhat slower in the PsaA-side mutants. Similarly, we attribute the long (>1-ns) component to the (P700+-P700) absorption difference (compare to Fig. 1 A). Although there may be subtle differences in the DAS between the PsaA-side and PsaB-side mutants, the important difference is the presence of an additional kinetic component in the PsaA-side mutants. The DAS of the additional fifth (102- and 210 ps) component has a FWHM of 11–15 nm and is centered at 689 ± 1 and 684 ± 1 nm in the case of PS I complexes from the M688LPsaA and M688NPsaA mutants, respectively. Its spectral shape is consistent with the PB spectrum of a single Chl a molecule, and we attribute this component to the decay of the 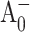 state, which reflects electron transfer from
state, which reflects electron transfer from 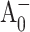 to A1. In the WT and PsaB-side mutants, the electron transfer from
to A1. In the WT and PsaB-side mutants, the electron transfer from 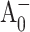 to A1 is too fast (∼13 ps) compared to the overall antenna decay (∼24 ps), and could not to be distinguished as a separate DAS component in the global analysis of pump-probe profiles.
to A1 is too fast (∼13 ps) compared to the overall antenna decay (∼24 ps), and could not to be distinguished as a separate DAS component in the global analysis of pump-probe profiles.
The lifetime of the fifth DAS component obtained by the global analysis of the M688NPsaA mutant data is significantly longer than A0→A1 electron transfer time extracted from the kinetics measured at 390 nm and from the (open-closed) kinetics measured at 690 nm. This discrepancy may be due to the presence of a measurable amount of not fully functional PS I complexes that may contain unconnected Chl a molecules with excited-state lifetimes significantly longer than 100 ps. In addition, rather large number of free parameters (all amplitudes) in the global fit and the inherent signal/noise ratio raise the error margins of the decay times obtained during global fit. In contrast, the 390 nm profiles as well as (open-closed) profiles are not as susceptible to the presence of unconnected Chls and corresponding fits require fewer free parameters.
DISCUSSION
The results described in this work indicate that mutations on the PsaA branch of PS I markedly alter the kinetics of the first steps of electron transfer as well as the spectral properties of the primary electron acceptor A0, whereas mutations on the PsaB-branch maintain kinetics and spectral properties that are essentially undistinguishable from the WT. The secondary electron transfer 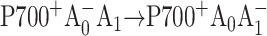 occurs with an intrinsic time of 100-150 ps in the M688LPsaA and M688NPsaA mutants, which is approximately an order of magnitude slower than the secondary electron transfer step in the WT, as determined recently by Savikhin et al. (2001), and it is at least 3 times slower than earlier estimates (Brettel and Vos, 1999; Hastings et al., 1994b; Iwaki et al., 1995; Kumazaki et al., 1994a; White et al., 1996). There are two possible explanations for this effect: 1), in wild-type PS I complexes ET occurs concurrently along both branches, but the mutation near A0 blocks the ET along the respective RC branch, and the observed ET rate difference in the PsaA- and PsaB-branch mutants is due to different ET rates along PsaA- and PsaB-branches in wild-type PS I; and 2), ET always occurs primarily along the PsaA-branch and its rate is affected by mutations near A0. In the following, we will test these scenarios against the experimental data in keeping with the assumption that symmetric mutations in the PsaA and PsaB branches cause comparable changes in the properties of the respective A0 chlorophyll.
occurs with an intrinsic time of 100-150 ps in the M688LPsaA and M688NPsaA mutants, which is approximately an order of magnitude slower than the secondary electron transfer step in the WT, as determined recently by Savikhin et al. (2001), and it is at least 3 times slower than earlier estimates (Brettel and Vos, 1999; Hastings et al., 1994b; Iwaki et al., 1995; Kumazaki et al., 1994a; White et al., 1996). There are two possible explanations for this effect: 1), in wild-type PS I complexes ET occurs concurrently along both branches, but the mutation near A0 blocks the ET along the respective RC branch, and the observed ET rate difference in the PsaA- and PsaB-branch mutants is due to different ET rates along PsaA- and PsaB-branches in wild-type PS I; and 2), ET always occurs primarily along the PsaA-branch and its rate is affected by mutations near A0. In the following, we will test these scenarios against the experimental data in keeping with the assumption that symmetric mutations in the PsaA and PsaB branches cause comparable changes in the properties of the respective A0 chlorophyll.
Scenario 1: ET occurs concurrently along both branches of the RC in the WT, but mutations on the PsaA and PsaB branches block transfer along the respective branch
In this case the two measured A0→A1 electron transfer rates (13 ps and ∼100 ps) characterize electron transfer processes along the PsaA and PsaB branches of RC, respectively. This interpretation also implies that both lifetime components must be present in the kinetics of the wild-type PS I. However, our analysis reveals a single 13 ps component in the kinetics of wild-type PS I complexes, which can be explained only if at least 80% of ET occurs along the PsaA branch. Given the noise level in our data, this argument cannot exclude a scenario in which up to 20% of ET proceeds along the PsaB branch with an A0→A1 lifetime of 100–200 ps.
Blocking ET along the PsaA or PsaB branch of RC would necessarily lead to the change in the intrinsic charge separation rate. If both branches are active in WT, the observed intrinsic charge separation rate rWT would be equal to a sum of intrinsic charge separation rates along the PsaA branch (rA) and the PsaB branch (rB): rWT = rA + rB. If the PsaA or PsaB branch were blocked in the mutants, the intrinsic charge separation rate would decrease and become rB and rA, respectively. The ratio rA/rB would define the relative yield of ET along the PsaA and PsaB branches in the WT. Since our data indicate that at least 80% of ET must occur along the PsaA branch, the rA must be at least 4 times larger than rB. Therefore, the intrinsic charge separation rates in PsaA- and PsaB-side mutants must differ drastically. According to several studies ET in PS I core complexes is essentially trap-limited (Beddard, 1998; Dorra et al., 1998; Hastings et al., 1994a; Holzwarth et al., 1993; Kumazaki et al., 1994b; Laible et al., 1994; Melkozernov et al., 1997; Turconi et al., 1993; White et al., 1996), or partly trap-limited (Byrdin et al., 2000, 2002; Holzwarth et al., 1998; Karapetyan et al., 1999; Trinkunas and Holzwarth, 1996). In the case of a trap-limited ET, drastic differences in intrinsic charge separation rate should lead to drastic changes in the effective excitation trapping time, which was not observed in our experiments.
Scenario 2: ET occurs primarily along the PsaA branch of the RC in wild-type and mutated PS I
The results of our experiments are most consistent with ET proceeding primarily along the PsaA branch in the wild-type and mutant PS I. Mutations in the PsaB branch of the RC have no effect on the effective electronic energy trapping time or on the 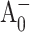 and
and 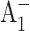 formation kinetics probed at 690 nm and 390 nm. The (
formation kinetics probed at 690 nm and 390 nm. The ( ) spectra for PS I with mutations around A0 on the PsaB side of the RC are the same as for wild-type PS I, suggesting that
) spectra for PS I with mutations around A0 on the PsaB side of the RC are the same as for wild-type PS I, suggesting that 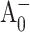 is formed on the PsaA side of the RC. At the same time, mutations around A0 on the PsaA side of the RC have a dramatic effect on electron transfer kinetics monitored at 390 nm and 690 nm. Moreover, the (
is formed on the PsaA side of the RC. At the same time, mutations around A0 on the PsaA side of the RC have a dramatic effect on electron transfer kinetics monitored at 390 nm and 690 nm. Moreover, the ( ) spectra for the two PsaA-side mutants differ from each other as well as from the spectrum measured for wild-type PS I. The latter can be easily explained under the assumption that ET transfer occurs primarily through the PsaA branch of the RC: optical properties of the A0 chlorophyll are sensitive to the local protein environment. We also observed a slight change in the effective excitation trapping time. The structure-based model of energy transfer in PS I described in (Savikhin et al., 2001) predicts that the observed increase of the effective excitation energy trapping time from 24 ps to 30 ps can be achieved by increasing the intrinsic charge separation time from 1.3 ps to 2.4 ps.
) spectra for the two PsaA-side mutants differ from each other as well as from the spectrum measured for wild-type PS I. The latter can be easily explained under the assumption that ET transfer occurs primarily through the PsaA branch of the RC: optical properties of the A0 chlorophyll are sensitive to the local protein environment. We also observed a slight change in the effective excitation trapping time. The structure-based model of energy transfer in PS I described in (Savikhin et al., 2001) predicts that the observed increase of the effective excitation energy trapping time from 24 ps to 30 ps can be achieved by increasing the intrinsic charge separation time from 1.3 ps to 2.4 ps.
Qualitatively, the conclusion that the forward electron transfer rate from 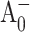 to A1 is slowed in the PsaA-side mutants, but not in the PsaB-side mutants, agrees with earlier transient EPR data (Cohen et al., 2004), which shows both a change in the spin polarization pattern of P700+
to A1 is slowed in the PsaA-side mutants, but not in the PsaB-side mutants, agrees with earlier transient EPR data (Cohen et al., 2004), which shows both a change in the spin polarization pattern of P700+ 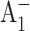 and the appearance of a triplet state formed by radical pair recombination. Both effects can be explained by an increase in the lifetime of
and the appearance of a triplet state formed by radical pair recombination. Both effects can be explained by an increase in the lifetime of 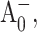 which would slow electron transfer to an extent that charge recombination via 3P700 can compete with forward electron transfer. The changes in the polarization pattern can be explained qualitatively as the result of singlet-triplet mixing due to an increase in the lifetime of the primary radical pair,
which would slow electron transfer to an extent that charge recombination via 3P700 can compete with forward electron transfer. The changes in the polarization pattern can be explained qualitatively as the result of singlet-triplet mixing due to an increase in the lifetime of the primary radical pair, 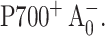 To explain the unexpected temperature dependency of the spin-polarization pattern, it was necessary to invoke a broad distribution of forward electron transfer rates such that when the temperature is lowered, the fraction that enters into charge recombination would increase. Simulations of the temperature-dependent EPR spectra assumed an “effective lifetime” of 2 ns for the limiting case of the long-lived
To explain the unexpected temperature dependency of the spin-polarization pattern, it was necessary to invoke a broad distribution of forward electron transfer rates such that when the temperature is lowered, the fraction that enters into charge recombination would increase. Simulations of the temperature-dependent EPR spectra assumed an “effective lifetime” of 2 ns for the limiting case of the long-lived 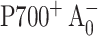 state (Cohen et al., 2004), which is considerably longer than the 100- to 200 ps lifetime measured for the lifetime of the
state (Cohen et al., 2004), which is considerably longer than the 100- to 200 ps lifetime measured for the lifetime of the 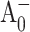 to A1 forward electron transfer (this work). However, our ultrafast optical measurements cannot exclude a possibility that in fraction of PsaA-side mutants the
to A1 forward electron transfer (this work). However, our ultrafast optical measurements cannot exclude a possibility that in fraction of PsaA-side mutants the 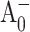 to A1 electron transfer lifetimes extend into the 1-ns range, which is beyond the time window of our experimental setup (Fig. 4 A). Thus, the results of these two independent studies lead to a consistent model that requires a slowing of electron transfer through A0 in the PsaA-side mutants.
to A1 electron transfer lifetimes extend into the 1-ns range, which is beyond the time window of our experimental setup (Fig. 4 A). Thus, the results of these two independent studies lead to a consistent model that requires a slowing of electron transfer through A0 in the PsaA-side mutants.
The kinetics of downhill electron transfer reactions are frequently parameterized using the semiempirical Moser-Dutton relationship (Moser et al., 1992):
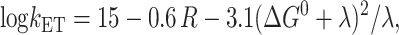 |
(2) |
where the ET rate constant kET is given in terms of R, the edge-to-edge separation between electron donor and acceptor; ΔG0 is the Gibbs free energy change for the electron transfer; and λ is the reorganization energy. Assuming that in mutants the distances between P700, A0, and A1, as well as the reorganization energies associated with ET, are not noticeably changed, the decrease in both charge separation and secondary electron transfer rates can be explained if the mutations on the PsaA branch increase the redox potential of 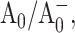 shifting both ET processes further from the “optimal” regime, where −ΔG0 matches the reorganizational energy λ that accompanies ET. In WT PSI, the Gibbs free energies ΔG0 for the initial charge separation and the A0→A1 electron transfer step are ∼−0.25 eV and −0.35 eV, respectively (Brettel and Leibl, 2001), and the corresponding edge-to-edge distances, R, are 12.3 Å and 6.5 Å (calculated from x-ray structure (Jordan et al., 2001) as described in Page et al. (1999)). Using Eq. 2 and assuming (13 ps)−1 intrinsic ET rate for the A0→A1 electron transfer step in WT, reorganization energy of the latter reaction can be derived to be 0.55 eV. The observed 10-fold decrease in the rate of this ET step in PsaA-side mutants may then be explained if the substitution of the axial ligand to A0 raises its oxidation redox potential by ∼0.25 eV. The increase in the
shifting both ET processes further from the “optimal” regime, where −ΔG0 matches the reorganizational energy λ that accompanies ET. In WT PSI, the Gibbs free energies ΔG0 for the initial charge separation and the A0→A1 electron transfer step are ∼−0.25 eV and −0.35 eV, respectively (Brettel and Leibl, 2001), and the corresponding edge-to-edge distances, R, are 12.3 Å and 6.5 Å (calculated from x-ray structure (Jordan et al., 2001) as described in Page et al. (1999)). Using Eq. 2 and assuming (13 ps)−1 intrinsic ET rate for the A0→A1 electron transfer step in WT, reorganization energy of the latter reaction can be derived to be 0.55 eV. The observed 10-fold decrease in the rate of this ET step in PsaA-side mutants may then be explained if the substitution of the axial ligand to A0 raises its oxidation redox potential by ∼0.25 eV. The increase in the 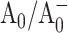 redox potential can also slow down the rate of the primary charge separation, provided the latter occurs in the “inverted” regime, where increase in −ΔG0 above λ leads to a lower ET rate, and would imply that the reorganizational energy for the charge separation is <0.25 eV (Marcus, 1956, 1960, 1964; Turró et al., 1996). The latter value is significantly lower than values 0.7–1.1 eV proposed for typical protein environments (Page et al., 2003). On the other hand, direct application of Eq. 2 to describe the kinetics of the primary charge separation in PS I is problematic—even in “optimal” regime this equation predicts ∼30 ns for ET between a donor and an acceptor that are 12.3 Å apart. Hence, it is very likely that an accessory Chl located between P700 and A0 acts as an intermediate in this ET process (Brettel, 1997; Brettel and Leibl, 2001; Klukas et al., 1999; Müller et al., 2003), and quantitative analysis of charge separation rate is impossible since an oxidation redox potential for the accessory pigment is unknown. The reorganizational energies, λ, of ET, as well as the distances, may be also affected by mutations around A0 and be additional factors leading to the observed changes in ET kinetics.
redox potential can also slow down the rate of the primary charge separation, provided the latter occurs in the “inverted” regime, where increase in −ΔG0 above λ leads to a lower ET rate, and would imply that the reorganizational energy for the charge separation is <0.25 eV (Marcus, 1956, 1960, 1964; Turró et al., 1996). The latter value is significantly lower than values 0.7–1.1 eV proposed for typical protein environments (Page et al., 2003). On the other hand, direct application of Eq. 2 to describe the kinetics of the primary charge separation in PS I is problematic—even in “optimal” regime this equation predicts ∼30 ns for ET between a donor and an acceptor that are 12.3 Å apart. Hence, it is very likely that an accessory Chl located between P700 and A0 acts as an intermediate in this ET process (Brettel, 1997; Brettel and Leibl, 2001; Klukas et al., 1999; Müller et al., 2003), and quantitative analysis of charge separation rate is impossible since an oxidation redox potential for the accessory pigment is unknown. The reorganizational energies, λ, of ET, as well as the distances, may be also affected by mutations around A0 and be additional factors leading to the observed changes in ET kinetics.
CONCLUSION
Our experiments provide additional evidence for asymmetric ET in cyanobacterial PS I, which was previously suggested by Cohen et al. (2004). The alteration in the ligand to A0 likely causes a change in its redox potential, which alters the kinetic of electron transfer to A1. According to our data, at least 80% of the electrons transferred in these RCs proceed along the PsaA branch. These results contrast with the finding that electron transfer in PS I from several eukaryotes proceeds with almost equal probability along both branches of the RC.
Acknowledgments
The authors thank Dr. Parag R. Chitnis for helpful discussions and Dr. Gaozhong Shen for advice in the growth of the mutant strains. Some of the experiments were performed using Ames Laboratory equipment under support of the Division of Chemical Sciences, Office of Basic Energy Sciences, and US Department of Energy. Ames Laboratory is operated by Iowa State University under Contract W-7405-Eng-82.
This work was supported by a grant from the National Science Foundation (MCB-0117079), and Purdue Research Foundation grant 6903680.
References
- Beddard, G. S. 1998. Excitations and excitons in photosystem I. Phil. Trans. Roy. Soc. London. 356:421–448. [Google Scholar]
- Bock, C. H., A. J. van der Est, K. Brettel, and D. Stehlik. 1989. Nanosecond electron transfer kinetics in photosystem I as obtained from transient EPR at room temperature. FEBS Lett. 247:91–96. [Google Scholar]
- Boudreaux, B., F. MacMillan, C. Teutloff, R. Agalarov, F. Gu, S. Grimaldi, R. Bittl, K. Brettel, and K. Redding. 2001. Mutations in both sides of the photosystem I reaction center identify the phylloquinone observed by electron paramagnetic resonance spectroscopy. J. Biol. Chem. 276:37299–37306. [DOI] [PubMed] [Google Scholar]
-
Brettel, K. 1988. Electron transfer from
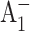 to an iron-sulfur center with t1/2 = 200 ns at room temperature in photosystem I. Characterization by absorption spectroscopy. FEBS Lett. 239:93–98. [Google Scholar]
to an iron-sulfur center with t1/2 = 200 ns at room temperature in photosystem I. Characterization by absorption spectroscopy. FEBS Lett. 239:93–98. [Google Scholar] - Brettel, K. 1997. Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta. 1318:322–373. [Google Scholar]
- Brettel, K. 1998. Electron transfer from acceptor A1 to the iron-sulfur cluster in Photosystem I measured with a time resolution of 2 ns. In Photosynthesis: Mechanisms and Effects. G. Garab, editor. Kluwer Academic, Dordrecht, The Netherlands. 611–614.
- Brettel, K., and W. Leibl. 2001. Electron transfer in photosystem I. Biochim. Biophys. Acta. 1507:100–114. [DOI] [PubMed] [Google Scholar]
- Brettel, K., P. Setif, and P. Mathis. 1986. Flash-induced absorption changes in photosystem I at low temperature: evidence that the electron acceptor A1 is vitamin K1. FEBS Lett. 203:220–224. [Google Scholar]
- Brettel, K., and M. H. Vos. 1999. Spectroscopic resolution of the picosecond reduction kinetics of the secondary electron acceptor A1 in photosystem I. FEBS Lett. 447:315–317. [DOI] [PubMed] [Google Scholar]
- Byrdin, M., P. Jordan, N. Krauss, P. Fromme, D. Stehlik, and E. Schlodder. 2002. Light harvesting in photosystem I: modeling based on the 2.5-Å structure of photosystem I from Synechococcus elongatus. Biophys. J. 83:433–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrdin, M., I. Rimke, E. Schlodder, D. Stehlik, and T. A. Roelofs. 2000. Decay kinetics and quantum yields of fluorescence in photosystem I from Synechococcus elongatus with P700 in the reduced and oxidized state: are the kinetics of excited state decay trap-limited or transfer-limited? Biophys. J. 79:992–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, R. O., G. Shen, J. H. Golbeck, W. Xu, P. R. Chitnis, A. Valieva, A. van der Est, Y. Pushkar, and D. Stehlik. 2004. Evidence for asymmetric electron transfer in cyanobacterial photosystem I: analysis of a methionine to leucine mutation of the ligand to the primary electron acceptor A0. Biochemistry. 43:4741–4754. [DOI] [PubMed] [Google Scholar]
- Damjanovic, A., H. M. Vaswani, P. Fromme, and G. R. Fleming. 2002. Chlorophyll excitations in photosystem I of Synechococcus elongatus. J. Phys. Chem. B. 106:10251–10262. [Google Scholar]
- Dorra, D., P. Fromme, N. V. Karapetyan, and A. R. Holzwarth. 1998. Fluorescence kinetics of photosystem I: multiple fluorescence components. In Photosynthesis: Mechanisms and Effects. G. Garab, editor. Kluwer Academic, Dordrecht, The Netherlands. 587–590.
- Fairclough, W. V., M. C. W. Evans, S. Purton, S. Jones, S. E. J. Rigby, and P. Heathcote. 2001. Site-directed mutagenesis of PsaA:M684 in Chlamydomonas reinhardtii. Proceedings of the 12th International Congress on Photosynthesis, PS2001. CSIRO, Melbourne, Australia.
- Fairclough, W. V., A. Forsyth, M. C. W. Evans, S. E. J. Rigby, S. Purton, and P. Heathcote. 2003. Bidirectional electron transfer in photosystem I: electron transfer on the PsaA side is not essential for phototrophic growth in Chlamydomonas. Biochim. Biophys. Acta. 1606:43–55. [DOI] [PubMed] [Google Scholar]
- Gatzen, G., M. G. Müller, K. Griebenow, and A. R. Holzwarth. 1996. Primary processes and structure of the photosystem II reaction center. 3. Kinetic analysis of picosecond energy transfer and charge separation processes in the D1–D2-cyt-b559 complex measured by time resolved fluorescence. J. Phys. Chem. 100:7269–7278. [Google Scholar]
- Golbeck, J., W. Xu, B. Zybailov, A. van der Est, J. Pushkar, S. Zech, D. Stehlik, and P. R. Chitnis. 2001. Electron transfer through the quinone acceptor in cyanobacterial Photosystem I. Proc. of the 12th International Congress on Photosynthesis, PS2001. Commonwealth Scientific and Industrial Research Organisation, Melbourne, Australia.
- Goldsmith, J. O., B. King, and S. G. Boxer. 1996. Mg coordination by amino acid side chains is not required for assembly and function of the special pair in bacterial photosynthetic reaction centers. Biochemistry. 35:2421–2428. [DOI] [PubMed] [Google Scholar]
- Guergova-Kuras, M., B. Boudreaux, A. Joliot, P. Joliot, and K. Redding. 2001. Evidence for two active branches for electron transfer in photosystem I. Proc. Natl. Acad. Sci. USA. 98:4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings, G., S. Hoshina, A. N. Webber, and R. E. Blankenship. 1995. Universality of energy and electron transfer processes in photosystem I. Biochemistry. 34:15512–15522. [DOI] [PubMed] [Google Scholar]
- Hastings, G., F. A. M. Kleinherenbrink, S. Lin, and R. E. Blankenship. 1994a. Time-resolved fluorescence and absorption spectroscopy of photosystem I. Biochemistry. 33:3185–3192. [DOI] [PubMed] [Google Scholar]
- Hastings, G., F. A. M. Kleinherenbrink, S. Lin, T. J. McHugh, and R. E. Blankenship. 1994b. Observation of the reduction and reoxidation of the primary electron acceptor in photosystem I. Biochemistry. 33:3193–3200. [DOI] [PubMed] [Google Scholar]
- Holzwarth, A. R., D. Dorra, M. G. Müller, and N. V. Karapetyan. 1998. Structure-function relationships and excitation dynamics in photosystem I. In Photosynthesis: Mechanism and Effects. G. Garab, editor. Kluwer Academic, Dordrecht, The Netherlands. 497–502.
- Holzwarth, A. R., G. Schatz, H. Brock, and E. Bittersmann. 1993. Energy transfer and charge separation kinetics in photosystem I. Part I: Picosecond transient absorption and fluorescence study of cyanobacterial photosystem I particles. Biophys. J. 64:1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki, M., S. Kumazaki, K. Yoshihara, T. Erabi, and S. Itoh. 1995. ΔG0 dependence of the rate constant of
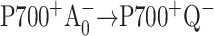 reaction in the quinone-reconstituted photosystem I reaction center. In Photosynthesis: From Light to Biosphere. P. Mathis, editor. Kluwer Academic, Dordrecht, The Netherlands. 147–150.
reaction in the quinone-reconstituted photosystem I reaction center. In Photosynthesis: From Light to Biosphere. P. Mathis, editor. Kluwer Academic, Dordrecht, The Netherlands. 147–150. - Joliot, P., and A. Joliot. 1999. In vivo analysis of the electron transfer within photosystem I: are the two phylloquinones involved? Biochemistry. 38:11130–11136. [DOI] [PubMed] [Google Scholar]
- Jordan, P., P. Fromme, H. T. Witt, O. Klukas, W. Saenger, and N. Krauss. 2001. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 411:909–917. [DOI] [PubMed] [Google Scholar]
- Karapetyan, N. V., A. R. Holzwarth, and M. Rögner. 1999. The photosystem I trimer of cyanobacteria: molecular organization, excitation dynamics and physiological significance. FEBS Lett. 460:395–400. [DOI] [PubMed] [Google Scholar]
- Kleinherenbrink, F. A. M., G. Hastings, B. P. Wittmershaus, and R. E. Blankenship. 1994. Delayed fluorescence from Fe-S type photosynthetic reaction centers at low redox potential. Biochemistry. 33:3096–3105. [DOI] [PubMed] [Google Scholar]
- Klug, D. R., L. B. Giorgi, B. Crystal, J. Barber, and G. Porter. 1989. Energy transfer to low energy chlorophyll species prior to trapping by P700 and subsequent electron transfer. Photosynth. Res. 22:277–284. [DOI] [PubMed] [Google Scholar]
- Klukas, O., W.-D. Schubert, P. Jordan, N. Krauss, P. Fromme, H. T. Witt, and W. Saenger. 1999. Photosystem I, an improved model of the stromal subunits PsaC, PsaD, and PsaE. J. Biol. Chem. 274:7351–7360. [DOI] [PubMed] [Google Scholar]
- Krauss, N., W. Hinrichs, I. Witt, P. Fromme, W. Pritzkow, Z. Dauter, C. Betzel, K. S. Wilson, H. T. Witt, and W. Saenger. 1993. Three-dimensional structure of system I of photosynthesis at 6 Å resolution. Nature. 361:326–331. [Google Scholar]
- Krauss, N., W. D. Schubert, O. Klukas, P. Fromme, H. T. Witt, and W. Saenger. 1996. Photosystem I at 4 Å resolution represents the first structural model of a joint photosynthetic reaction centre and core antenna system. Nat. Struct. Biol. 3:965–973. [DOI] [PubMed] [Google Scholar]
- Kumazaki, S., M. Iwaki, I. Ikegami, H. Kandori, K. Yoshihara, and S. Itoh. 1994a. Rates of primary electron transfer reactions in the photosystem I reaction center reconstituted with different quinones as the secondary acceptor. J. Phys. Chem. 98:11220–11225. [Google Scholar]
- Kumazaki, S., H. Kandori, H. Petek, I. Yoshihara, and I. Ikegami. 1994b. Primary photochemical processes in P700-enriched photosystem I particles: trap-limited excitation decay and primary charge separation. J. Phys. Chem. 98:10335–10342. [Google Scholar]
- Laible, P. D., W. Zipfel, and T. G. Owens. 1994. Excited state dynamics in chlorophyll-based antennae: The role of transfer equilibrium. Biophys. J. 66:844–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Lüneberg, J., P. Fromme, P. Jekow, and E. Schlodder. 1994. Spectroscopic characterization of PS I core complexes from thermophilic Synechococcus sp.: identical reoxidation kinetics of
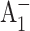 before and after removal of the iron-sulfur-clusters FA and FB. FEBS Lett. 338:197–202. [DOI] [PubMed] [Google Scholar]
before and after removal of the iron-sulfur-clusters FA and FB. FEBS Lett. 338:197–202. [DOI] [PubMed] [Google Scholar] - Marcus, R. A. 1956. On the energy of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys. 24:966–978. [Google Scholar]
- Marcus, R. A. 1960. Theory of oxidation-reduction reactions involving electron transfer. IV. A statistical-mechanical basis for treating contributions from solvent, ligands, and inert salt. Faraday Discuss. Chem. Soc. 29:21–31. [Google Scholar]
- Marcus, R. A. 1964. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 15:155–196. [Google Scholar]
- McConnel, M. D., V. M. Ramesh, I. Wyndham, A. van der Est, and A. N. Webber. 2004. Directionality of electron transport through Photosystem I of Chlamydomonas reinhardtii probed by transient electron paramagnetic resonance. In 13th International Congress of Photosynthesis, Program and Abstracts, August 29–September 3. Humana Press, Montreal, Canada. P2A–18.
- Melkozernov, A. N., H. Su, S. Lin, S. Bingham, A. N. Webber, and R. E. Blankenship. 1997. Specific mutations near the primary donor in photosystem I from Chlamydomonas reinhardtii alters trapping time and spectroscopic properties of P700. Biochemistry. 36:2898–2907. [DOI] [PubMed] [Google Scholar]
- Moser, C. C., J. M. Keske, K. Warncke, R. S. Farid, and P. L. Dutton. 1992. Nature of biological electron transfer. Nature. 355:796–802. [DOI] [PubMed] [Google Scholar]
-
Muhiuddin, I. P., P. Heathcote, S. Carter, S. Purton, S. E. J. Rigby, and M. C. W. Evans. 2001. Evidence from time resolved studies of the
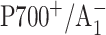 radical pair for photosynthetic electron transfer on both the PsaA and PsaB branches of the Photosystem I reaction centre. FEBS Lett. 503:56–60. [DOI] [PubMed] [Google Scholar]
radical pair for photosynthetic electron transfer on both the PsaA and PsaB branches of the Photosystem I reaction centre. FEBS Lett. 503:56–60. [DOI] [PubMed] [Google Scholar] - Müller, M. G., J. Niklas, W. Lubitz, and A. R. Holzwarth. 2003. Ultrafast transient absorption studies on photosystem I reaction centers from Chlamydomonas reinhardtii. 1. A new interpretation of the energy trapping and early electron transfer steps in photosystem I. Biophys. J. 85:3899–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuijs, A. M., V. A. Shuvalov, H. J. van Gorkom, J. J. Plijter, and L. N. M. Duysens. 1986. Picosecond absorbance difference spectroscopy on the primary reactions and the antenna-excited states in photosystem I particles. Biochim. Biophys. Acta. 850:310–318. [Google Scholar]
- Owens, T. G., S. P. Webb, R. S. Alberte, L. Mets, and G. R. Fleming. 1988. Antenna structure and excitation dynamics in photosystem I. I. Studies of detergent-isolated photosystem I preparations using time-resolved fluorescence analysis. Biophys. J. 53:733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, C. C., C. C. Moser, X. Chen, and P. L. Dutton. 1999. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 402:47–52. [DOI] [PubMed] [Google Scholar]
- Page, C. C., C. C. Moser, and P. L. Dutton. 2003. Mechanism for electron transfer within and between proteins. Curr. Opin. Chem. Biol. 7:551–556. [DOI] [PubMed] [Google Scholar]
-
Ramesh, V. M., K. Gibasiewicz, S. Lin, S. E. Bingham, and A. N. Webber. 2004. Bidirectional electron transfer in photosystem I: accumulation of
 in A-side or B-side mutants of the axial ligand to chlorophyll A0. Biochemistry. 43:1369–1375. [DOI] [PubMed] [Google Scholar]
in A-side or B-side mutants of the axial ligand to chlorophyll A0. Biochemistry. 43:1369–1375. [DOI] [PubMed] [Google Scholar] - Sakuragi, Y., B. Zybailov, G. Shen, A. D. Jones, P. R. Chitnis, A. van der Est, R. Bittl, S. Zech, D. Stehlik, J. H. Golbeck, and D. A. Bryant. 2002. Insertional inactivation of the menG gene, encoding 2-phytyl-1,4-naphthoquinone methyltransferase of Synechocystis sp. PCC 6803, results in the incorporation of 2-phytyl-1,4-naphthoquinone into the A1 site and alteration of the equilibrium constant between A1 and FX in Photosystem I. Biochemistry. 41:394–405. [DOI] [PubMed] [Google Scholar]
- Savikhin, S., W. Xu, P. R. Chitnis, and W. S. Struve. 2000. Ultrafast primary processes in PS I from Synechocystis sp. PCC 6803: roles of P700 and A0. Biophys. J. 79:1573–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Savikhin, S., W. Xu, P. Martinsson, P. R. Chitnis, and W. S. Struve. 2001. Kinetics of charge separation and
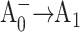 electron transfer in photosystem I reaction centers. Biochemistry. 40:9282–9290. [DOI] [PubMed] [Google Scholar]
electron transfer in photosystem I reaction centers. Biochemistry. 40:9282–9290. [DOI] [PubMed] [Google Scholar] - Savikhin, S., W. Xu, V. Soukoulis, P. R. Chitnis, and W. S. Struve. 1999. Ultrafast primary processes in photosystem I of the cyanobacterium Synechocystis sp. PCC 6803. Biophys. J. 76:3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz, G. H., H. Brock, and A. R. Holzwarth. 1988. Kinetic and energetic model for the primary processes in photosystem II. Biophys. J. 54:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setif, P., and K. Brettel. 1993. Forward electron transfer from phylloquinone A1 to iron-sulfur centers in spinach photosystem I. Biochemistry. 32:7846–7854. [DOI] [PubMed] [Google Scholar]
- Shuvalov, V. A., A. M. Nuijs, H. J. van Gorkom, H. W. J. Smit, and L. N. M. Duysens. 1986. Picosecond absorbance changes upon selective excitation of the primary electron donor P700 in photosystem I. Biochim. Biophys. Acta. 850:319–323. [Google Scholar]
- Trinkunas, G., and A. R. Holzwarth. 1996. Kinetic modeling of exciton migration in photosynthetic systems. 3. Application of genetic algorithms to simulations of excitation dynamics in three dimensional photosystem core reaction center complexes. Biophys. J. 71:351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turconi, S., G. Schweitzer, and A. R. Holzwarth. 1993. Temperature-dependence of picosecond fluorescence kinetics of a cyanobacterial photosystem-I particle. Photochem. Photobiol. 57:113–119. [Google Scholar]
- Turró, C., J. M. Zaleski, Y. M. Karabatsos, and D. G. Nocera. 1996. Bimolecular electron transfer in the Marcus inverted region. J. Am. Chem. Soc. 118:6060–6067. [Google Scholar]
- van der Est, A., C. Bock, J. Golbeck, K. Brettel, P. Setif, and D. Stehlik. 1994. Electron transfer from the acceptor A1 to the iron-sulfur centers in photosystem I as studied by transient EPR spectroscopy. Biochemistry. 33:11789–11797. [DOI] [PubMed] [Google Scholar]
- White, N. T. H., G. S. Beddard, J. R. G. Thorne, T. M. Feehan, T. E. Keyes, and P. Heathcote. 1996. Primary charge separation and energy transfer in the photosystem I reaction center of higher plants. J. Phys. Chem. 100:12086–12099. [Google Scholar]
- Witt, H., E. Schlodder, C. Teutloff, J. Niklas, E. Bordignon, D. Carbonera, S. Kohler, A. Labahn, and W. Lubitz. 2002. Hydrogen bonding to P700: Site-directed mutagenesis of threonine A739 of photosystem I in Chlamydomonas reinhardtii. Biochemistry. 41:8557–8569. [DOI] [PubMed] [Google Scholar]
- Xu, W., P. R. Chitnis, A. Valieva, A. van der Est, K. Brettel, M. Guergova-Kuras, Y. N. Pushkar, S. G. Zech, D. Stehlik, G. Shen, B. Zybailov, and J. H. Golbeck. 2003a. Electron transfer in cyanobacterial photosystem I: II. Determination of forward electron transfer rates of site-directed mutants in a putative electron transfer pathway from A0 through A1 to FX. J. Biol. Chem. 278:27876–27887. [DOI] [PubMed] [Google Scholar]
- Xu, W., P. R. Chitnis, A. Valieva, A. van der Est, Y. N. Pushkar, M. Krzystyniak, C. Teutloff, S. G. Zech, R. Bittl, D. Stehlik, B. Zybailov, G. Shen, and J. H. Golbeck. 2003b. Electron transfer in cyanobacterial photosystem I: I. Physiological and spectroscopic characterization of site-directed mutants in a putative electron transfer pathway from A0 through A1 to FX. J. Biol. Chem. 278:27864–27875. [DOI] [PubMed] [Google Scholar]