

Abstract
The unfolding pathway of two very similar tetrameric legume lectins soybean agglutinin (SBA) and Concanavalin A (ConA) were determined using GdnCl-induced denaturation. Both proteins displayed a reversible two-state unfolding mechanism. The analysis of isothermal denaturation data provided values for conformational stability of the two proteins. It was found that the ΔG of unfolding of SBA was much higher than ConA at all the temperatures at which the experiments were done. ConA had a Tg 18°C less than SBA. The higher conformational stability of SBA in comparison to ConA is largely due to substantial differences in their degrees of subunit interactions. Ionic interactions at the interface of the two proteins especially at the noncanonical interface seem to play a significant role in the observed stability differences between these two proteins. Furthermore, SBA is a glycoprotein with a GlcNac2Man9 chain attached to Asn-75 of each subunit. The sugar chain in SBA lies at the noncanonical interface of the protein, and it is found to interact with the amino acid residues in the adjacent noncanonical interface. These interactions further stabilize SBA with respect to ConA, which is not glycosylated.
INTRODUCTION
Small monomeric proteins have been studied extensively for characterization of their folding reactions. This is mostly related to the high degree of reversibility and two-state unfolding reaction displayed by them. Additionally, these proteins also fold/unfold fairly fast, making them highly suitable systems for kinetic analyses of the reactions involved therein. Also, the folding process of small, monomeric globular proteins is amenable to study by NMR spectroscopy. Although the vast majority of proteins in nature exist as oligomers, quantitative study of their unfolding reactions have been few. Delineation of the unfolding of oligomeric proteins is relatively challenging as compared to the monomeric proteins, where only intrasubunit interactions are involved whereas the oligomeric proteins are stabilized by intersubunit interactions as well (Bowie and Sauer, 1989; Neet and Timm, 1994). Studying the unfolding of multimeric proteins provides an insight into the role and the contribution of intersubunit interactions to their stability. For example, under standard conditions viz. at pH 7.6, 298 K, the stability of dimeric tryptophan aporepressor, was found to be 23.3 kcal/mol as compared to the dissociated monomer at pH range 3.5–6.0, whose stability was only 5.4 kcal/mol monomer (Matthews, 1993; Gittelman and Matthews, 1990). Thus, we see that dimerization increases the stability of the protein by almost two times. It is, however, worth mentioning here that many oligomeric proteins show only a two-state transition, where the folded oligomer and the unfolded constituent polypeptide exist as the equilibrium species at any given time during the unfolding reaction. This suggests that a significant amount of stability is imparted to the system via intersubunit interactions.
This article reports the unfolding reaction of two tetrameric legume lectins, soybean agglutinin (SBA) and Concanavalin A (ConA). Legume lectins are a group of oligomeric proteins that bind carbohydrates reversibly and specifically. Although they differ in their specificities, the orientation of their binding site and mechanism of ligand recognition remains almost the same. This is perhaps due to the fact that all legume lectins share a common tertiary structure (Manoj and Suguna, 2001) although their quaternary structures differ considerably. The tertiary structural fold in legume lectins is described as the “jelly roll” motif. This is a kind of a β-sandwich fold consisting of three antiparallel β-sheets (Prabu et al., 1999; Chandra et al., 2001). The mutual disposition of the three β-sheets is shown in Fig. 1 a. There is a six-stranded “back” β-sheet, a curved seven-stranded “front” β-sheet, and a five-stranded “top” β-sheet, which forms a roof above the other two. There are several loop regions in the motif, which hold the sheets together. There are two strong hydrophobic cores stabilizing the structure; one lies at the center of the three sheets and the other in the curvature of the front β-sheet. Our laboratory has been working on legume lectins as paradigms for the unfolding reactions of oligomeric protein (Mitra et al., 2002, 2003; Ahmad et al., 1998). Such reactions have been characterized to a considerable extent for this family of proteins that exists as dimers.
FIGURE 1.

(a) A typical legume lectin monomer. (b) Tetramer of soybean agglutinin showing the N-linked glycan in ball and stick models. (c) Tetramer of Concanavalin A. In both tetramers the interface type is marked by arrows.
Both lectins used in this study exist as tetramers under physiological conditions. There is one Ca2+ and one Mn2+ ion per subunit in both proteins (Hardman and Ainsworth, 1972; Olsen et al., 1997). Each subunit is capable of binding to sugar independently. Both proteins form dimer of dimers. The initial dimerization occurs by antiparallel side-by-side alignment of two flat six-stranded “back” β-sheets, one from each monomer, giving rise to the formation of a contiguous 12-stranded sheet forming the canonical dimer. These dimers associate in a back-to-back fashion to form the tetramer, although the relative orientations of the canonical dimers vary in the two proteins. (Fig. 1, b and c). Thus, both SBA and ConA tetramers have four interfaces of which two are canonical and two are noncanonical. Despite 45.6% sequence identity, similarity of their primary structure, and nearly completely superimposable tertiary structures, SBA tetramer has greater conformational stability and 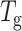 as compared to ConA. Detailed examination of their subunit interfaces provides a rationale of these observations.
as compared to ConA. Detailed examination of their subunit interfaces provides a rationale of these observations.
MATERIALS AND METHODS
Materials
Ultrapure guanidinium hydrochloride (GdnCl) was purchased from Sigma Chemical (St. Louis, MO). All other reagents used for the study were of highest purity available. Stock GdnCl solutions were prepared fresh, in 25 mM HEPES (N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid) buffer pH 7 containing 15 mM CaCl2 and MnCl2. The concentration of GdnCl was determined by refractive index as described by Pace (1990).
Protein purification
Soybean agglutinin
In a typical preparation, 250 g of soybean seeds were homogenized and defatted. The defatted dry meal was extracted with 20 mM phosphate buffer pH 7.4 containing 150 mM sodium chloride (PBS) for 12 h at 4°C under constant stirring. The extract was subjected to ammonium sulfate fractionation of 30%. The precipitate was removed by centrifugation at 8000 rpm for 30 min. The supernatant was again subjected to 65% ammonium sulfate fractionation. The precipitate was collected this time by centrifugation at 8000 rpm for 45 min. The precipitate was dissolved in a minimum amount of the buffer and extensively dialyzed against the same buffer. The dialyzed solution was centrifuged at 8000 rpm for 15 min and the clear supernatant was loaded to a lactosylamine Biogel P-150 column preequilibrated with buffer (Baues and Gray, 1977). The column was then washed extensively with PBS until the washings have A280 < 0.005. Elution was carried out in 0.2 M lactose in PBS. The concentration of the protein solution was determined from specific extinction coefficient of 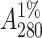 ∼ 12.8 for SBA (Lotan et al., 1974).
∼ 12.8 for SBA (Lotan et al., 1974).
Concanavalin A
Jack bean seeds (250 g) were soaked overnight in water. After the removal of the seed coat the seeds were cut into small pieces and left in a homogenization buffer (20 mM Tris, 150 mM sodium chloride, 1 mM Ca2+, and Mn2+, pH 7.4). The seeds were then homogenized in six volumes of homogenized buffer and centrifuged at 5000 rpm for 10 min. The pellet was discarded and the supernatant was subjected to ammonium sulfate fractionation of 65%. The precipitate was collected by centrifugation at 8000 rpm for 30 min. The pellet was resuspended in a minimum amount of buffer and extensively dialyzed against the same buffer. The dialyzed solution was centrifuged at 8000 rpm for 15 min and the clear supernatant was loaded onto a Sephadex G-75 column preequilibrated with buffer (Agrawal and Goldstein, 1967). The concentration of the protein solution was determined from specific extinction coefficient of 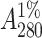 ∼ 12.4 for ConA (Mitra et al., 2002).
∼ 12.4 for ConA (Mitra et al., 2002).
Isothermal GdnCl-induced denaturation
Equilibrium unfolding studies as a function of guanidinium hydrochloride concentration was monitored using fluorescence spectroscopy and far-ultraviolet (UV) circular dichroism (CD). The fluorescence measurements were done on a Jobin Yvon Horiba fluorometer (Jobin Yvon (Spex division), Cedex, France) in a 1-cm water-jacketed cell using a protein concentration of 2 μM. The protein concentration used for all the experiments was 2 μM in terms of the monomer, unless otherwise mentioned. The excitation and emission wavelengths were fixed at 280 and 370 nm, respectively for SBA and at 280 and 323 nm for ConA. These wavelengths were determined by obtaining the maxima of the difference spectra of the native and unfolded forms of the respective proteins. This is very well seen in Fig. 2, a and c. The maximum difference between the native and the denatured spectra occurs at 370 nm for SBA and 323 nm for ConA. This has been indicated by a dashed line drawn at those wavelengths in the figure. In all experiments, the slit width was fixed at 3 and 5 nm for excitation and emission, respectively. All the CD experiments were done in a JASCO-J715 polarimeter (JASCO, Tokyo, Japan) in a 0.1-cm pathlength cell, with a slit width of 1 nm, response time of 4 s and scan speed of 50 nm/s. Each data point was an average of three accumulations. The data for SBA were collected at 222 nm and for ConA at 226 nm. The reversibility of unfolding was checked by refolding the unfolded protein by both dialyzing the unfolded protein in HEPES buffer containing 15 mM metal ions and diluting the unfolded protein from 7.8 M GdnCl to 0.5 M GdnCl or less. Eight and 12 GdnCl-induced isothermal denaturation curves were collected between the temperature range 273–323 K for SBA and ConA, respectively. A Julabo water bath was used to maintain the sample temperature within 0.1 K of the set temperature.
FIGURE 2.

Refolding of SBA monitored by (a) fluorescence and (b) CD. Refolding of ConA monitored by (c) fluorescence and (d) CD. The CD spectra are normalized with respect to protein concentration (2 μM) and pathlength of the cuvette used (0.1 cm). Fluorescent spectra are normalized to the highest intensity observed.
Gel filtration
Gel filtration experiments were done in a Bio-Gel P150 column (BIO-RAD laboratories, Richmond, CA). The column had a volume of 40 ml and void volume of ∼11 ml with blue dextran, at a flow rate of 5.0 ml/h, on the native protein and in the presence of different concentrations of the denaturant. Each time the column was equilibrated with the desired concentration of the denaturant before loading the protein at 293 K.
RESULTS
Both SBA and ConA showed >90% reversibility upon refolding of the unfolded protein obtained by GdnCl denaturation. To obtain the condition of total reversibility we have used different concentrations of metal ions and found that at 15 mM metal ion concentration the unfolding reaction is almost reversible for both proteins. Figs. 2 and 3 show the refolding of the two proteins monitored by spectroscopy (fluorescence and CD) and gel filtration, respectively.
FIGURE 3.

Refolding of SBA and ConA as observed by gel filtration. The protein concentration used for the refolding study was 40 μM. The protein sample was denatured in 6 M GdnCl and then renatured by dialyzing in 25 mM HEPES buffer for 12 h. The samples were then loaded to a P-150 Biogel column (40 ml bed volume; void volume was 11 ml as determined by blue dextran). The flow rate was maintained at 5 ml/h throughout the experiment. The gel filtration was conducted at 293 K.
The analysis of the denaturation curves was done based on the assumption that only two states, i.e., the native and the denatured, exist at equilibrium. This assumption may not be always true for oligomeric proteins and would depend upon the contribution of intersubunit interactions to the total protein stability. In the denaturation process, the formation of a partially folded intermediate is possible. The overall scheme for such a process can be described as:
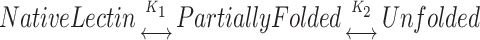 |
where K1 and K2 are the equilibrium constants for the formation of the partially unfolded and fully unfolded protein, respectively. Here, the intermediate could be a partially folded/unfolded tetramer or dimer or even a monomer.
Transitions that occur with the formation of intermediate(s) are associated with biphasic denaturation curves or nonsuperimposable transitions when different spectroscopic probes, monitoring different properties of the protein, are used. In this case the respective denaturation curves for both ConA and SBA monitored by fluorescence and far-UV CD were cooperative and superimposable (Fig. 4). This ruled out the possibility for a non-two-state process (Barrick and Baldwin, 1993). In addition to this, the overlay of the fluorescence spectra of the two proteins in different GdnCl concentrations (0–7.3 M range) shows the existence of an isoemissive point in both cases (Fig. 5). This suggests the existence of only two species in the transition (Gualfetti et al., 1999). Furthermore, analyses of the gel filtration profiles suggested that in the pretransition region both proteins elute in their native form and as an unfolded form in the posttransition region (Fig. 6). The unfolded form is unfolded monomers whose size is often bigger than that of the folded tetramer; this property is in general observed in proteins. In the transition region, different concentrations of the native and unfolded form are seen, which are almost similar to what is observed in the spectroscopic studies. Because it was not possible to do the gel filtration experiment at low protein concentrations of 2 μM, denaturation transition of the two proteins were studied by both fluorescence and CD in the presence of 40 μM protein concentration. It was observed that under this condition the transitions monitored by the two spectroscopic probes overlay (Fig. 7). This also strongly supports the two-state assumption. Although the proteins show concentration dependence in their profile they do not deviate from their two-state behavior (Fig. 8, b and c). At this point, only one scenario seems possible and that is N4 ⇔ 4U transition, i.e., the native tetramer dissociates to unfolded monomers. A general consequence of a two-state process is that the fraction of molecules populating the native state increases with protein concentration. This is very well evident from Fig. 8, b and c. The equilibrium constant for the above reaction may be given as:
 |
(1) |
where α is the fraction of folded protein molecules, n is the number of subunits in the oligomeric protein, and p is the protein concentration in n-mer equivalents.
FIGURE 4.

Overlap of GdnCl-induced unfolding profiles obtained using fluorescence (▽) and far UV-CD (•) at 298 K. The completely superimposable profiles confirm the two-state unfolding reaction for both proteins.
FIGURE 5.

Fluorescence emission spectra of SBA and ConA in different GdnCl concentrations (0–7.5 M range). The gradient is indicated by an arrow near the wavelength, which was used to monitor the isothermal melts. All the curves are equally spaced with respect to Gdncl concentration. The spectra are normalized to the highest intensity.
FIGURE 6.

Gel filtration profiles of SBA and ConA at different GdnCl concentrations. The denaturant concentration is indicated at the side of each profile. The protein concentrations used were 40 μM in each case. The optical densities were taken at 280 nm. The column used was a P-150 Biogel column (40 ml bed volume; void volume was 11 ml as determined by blue dextran). The flow rate was maintained at 5 ml/h throughout the experiment. The gel filtration was conducted at 293 K. During the experiment the column volume changed at the most by 0.75 ml at 3.5 M GdnCl, 2 ml at 4.9 M GdnCl, and 2.7 ml at 6.2 M GdnCl. Each time the elution volume was normalized with respect to the starting column volume.
FIGURE 7.

Overlap of GdnCl-induced unfolding profiles obtained using fluorescence (▽) and far UV-CD (•), with a protein concentration of 40 μM at 298 K. The superimposable curves indicate the two-state folding behavior of the transition.
FIGURE 8.

(a) GdnCl-induced unfolding of SBA (○) and ConA (□) at 298 K. The solid line passing through the points is the fit of Eq. 7 to the experimental data. (b) Protein concentration-dependent unfolding of ConA at 298 K, (○) 2 μM, and (▽) 10 μM. (c) Protein concentration-dependent unfolding of SBA at 298 K, (○) 2 μM, and (▽) 10 μM.
So in this case,
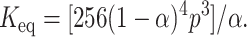 |
(2) |
ΔGu was calculated at various denaturant concentrations using the equation:
 |
(3) |
The free energy of unfolding of the two proteins at different temperatures is calculated according to the LEM model (Schellman, 1990). According to this model the changes in free energy, enthalpy, entropy, and heat capacity, which accompany the unfolding reaction, bear a linear relationship to the molar concentration of the denaturant, i.e.,
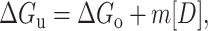 |
(4) |
(ΔGu represents the free energy of unfolding obtained in presence of a known concentration of denaturant and ΔGo is the free energy of unfolding in the absence of any denaturant).
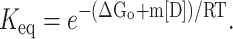 |
(5) |
By substituting Eq. 5 in Eq. 2 we get four roots for α, two of which are real and two imaginary. Out of the two real roots one is a positive fraction, which corresponds to the fraction unfolded in this investigation. The software MATHEMATICA was used to determine the desired root. The root is shown below:
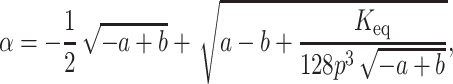 |
(6a) |
where
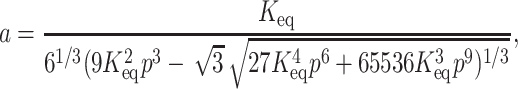 |
(6b) |
and
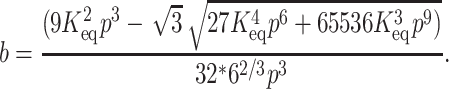 |
(6c) |
Finally, this root was used in the following equation and fitted to the normalized spectroscopic data obtained experimentally (fluorescence or CD).
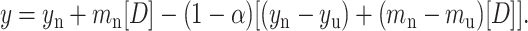 |
(7) |
From the fit of Eq. 7, the parameters of interest ΔGo, m, mu, mn, yn, and yu were obtained. The free energies of unfolding of ConA and SBA at each temperature thus obtained from the fit are listed in Table 1. The fit of the plot of fraction native against denaturant concentration is shown in Fig. 8 a.
TABLE 1.
Unfolding free-energy change for SBA and ConA at different temperatures obtained from denaturant-induced isothermal denaturation melts
| SBA
|
ConA
|
||||
|---|---|---|---|---|---|
| T(K) | ΔGH2O (kcal/mol) | −m (kcal/mol/K) | T(K) | ΔGH2O (kcal/mol) | −m (kcal/mol/K) |
| 281 | 56.4 ± 0.1 | 7.4 ± 1.2 | 280 | 26.4 ± 0.1 | 1.3 ± 0.1 |
| 283 | 57.4 ± 0.3 | 6.7 ± 0.5 | 285 | 28.1 ± 0.5 | 1.6 ± 0.1 |
| 288 | 58.5 ± 0.1 | 5.6 ± 0.5 | 291 | 29.7 ± 0.6 | 1.8 ± 0.1 |
| 293 | 58.4 ± 0.9 | 6.5 ± 0.3 | 294 | 29.1 ± 0.8 | 1.8 ± 0.2 |
| 295 | 59.9 ± 0.3 | 6.2 ± 0.1 | 298 | 29.3 ± 0.3 | 1.8 ± 0.1 |
| 298 | 59.2 ± 0.5 | 6.1 ± 0.1 | 300 | 30.2 ± 0.6 | 2.1 ± 0.1 |
| 300 | 60.0 ± 0.1 | 6.3 ± 0.1 | 303 | 30.9 ± 0.5 | 2.3 ± 0.3 |
| 303 | 60.1 ± 0.2 | 5.4 ± 0.4 | 306 | 30.7 ± 0.4 | 1.8 ± 0.1 |
| 306 | 59.9 ± 0.2 | 5.6 ± 0.6 | 312 | 29.3 ± 0.1 | 2.2 ± 0.2 |
| 313 | 59.3 ± 0.1 | 5.4 ± 0.1 | – | – | – |
| 318 | 56.7 ± 0.7 | 5.4 ± 0.1 | – | – | – |
| 323 | 56.1 ± 0.1 | 5.3 ± 0.2 | – | – | – |
The signal monitored was fluorescence in all cases. Each denaturation experiment was performed twice. The profiles from each duplicate were completely superimposable and consequently yielded almost identical results.
The unfolding of a protein is accompanied by the exposure of the hydrophobic core region, which is reflected in the change in heat capacity, Cp. To calculate the change in heat capacity (ΔCp) for the reaction, we used the method of Pace, where the free energies (ΔGo) calculated at different temperatures is fitted to the Gibbs-Helmholtz equation (Agashe and Udgaonkar, 1995; Nicholson, and Scholtz, 1996).
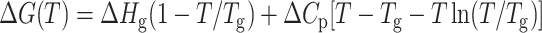 |
(8a) |
×
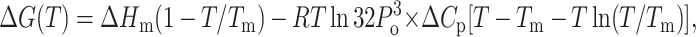 |
(8b) |
where, Tg is the temperature at which ΔG = 0 and Hg is the unfolding enthalpy at Tg; Tm is the melting temperature for the protein, and Hm is the enthalpy at Tm. Values of the parameters obtained from the fit of Eqs. 8a and 8b are summarized in Table 2.
TABLE 2.
Thermodynamic parameters of ConA and SBA analyzed on the basis of stability curves drawn for fitting Eqs. 8a and 8b
| Protein | ΔHg(kcal/mol) | ΔHm(kcal/mol) | Tg(K) | Tm(K) | ΔCp(kcal/mol/K) |
|---|---|---|---|---|---|
| SBA | 505.7 ± 25.2 | 400.8 ± 20.9 | 389.5 ± 5.6 | 368.7 ± 3.6 | 5.0 ± 0.6 |
| ConA | 318.4 ± 38.8 | 178.9 ± 24.7 | 371.7 ± 12.3 | 338.2 ± 6.5 | 4.2 ± 1.1 |
DISCUSSION
The GdnCl-induced denaturation curves for the lectins SBA and ConA were monitored by both fluorescence and far-UV CD. The analysis of unfolding curves of both proteins was done with the assumption of a simple two-state transition, as the denaturation profiles obtained from different spectroscopic means were completely superimposable (Figs. 4 and 7). In addition to this, the overlay of the fluorescence spectra of the two proteins in the pretransition, transition, and posttransition ranges shows the existence of an isoemissive point for both the proteins suggesting the presence of only two species in the unfolding reaction (Fig. 5). Furthermore, gel filtration profiles also showed no sign of an intermediate in the transition region (Fig. 6). Upon refolding the unfolded protein, fluorescence and CD spectra overlapped with that of the native protein, thus confirming total reversibility of the folding process (Fig. 2). Refolding experiments using gel filtration also confirmed the reversibility of the processes (Fig. 3).
Unfolding results in demetallization, which will introduce perturbation in the system, and prevent the protein from folding back to its native conformation, when attempts to refold in sparse metal ion conditions are made. However, if the system contains enough metal ions, the unfolded protein can get remetallized leading to recovery of the native state as indeed is observed in these studies.
One of the most important features of protein unfolding is the exposure of its hydrophobic core. This exposure is reflected in the positive change in heat capacity of the protein during the unfolding reaction. Thus, the change in heat capacity is directly proportional to the extent of unfolding and conversely the extent to which the apolar residues are buried in the native protein. Both SBA and ConA show a positive change in heat capacity upon unfolding, thus confirming the exposure of nonpolar surfaces to the polar aqueous environment. It was shown by Spolar et al. (1992) that the transfer of hydrocarbons from water to pure liquid state showed similar proportionality in the reduction of water-accessible surface area as is shown by the folding of globular proteins (Livingstone et al., 1991; Spolar et al., 1992; Ganesh et al., 1997). The theoretical calculation of ΔCp is done according to the equation given by Spolar:
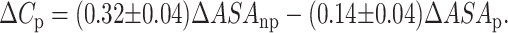 |
(9) |
We calculated the change in accessible surface area (ΔASA) using NACCESS (Hubbard, 1996; Manoj et al., 2000) for both proteins and it was found that they were very similar at 32,583.8 A2 and 33,739.7 A2 for SBA and ConA, respectively. The contribution of the polar and apolar regions (ΔASAp and ΔASAnp) in the above figures is ∼40 and 60%, respectively, for SBA. For ConA they are 48 and 52%, respectively. The proportion of ΔASA buried at the interface is 15% in SBA and 25% in ConA. ΔCp values obtained from Eq. 9 are 19.6 kcal/mol/K for SBA and 17.7 kcal/mol/K for ConA. The greater value for ΔCp for SBA suggests that the bulk of the buried hydrophobic core is greater in SBA when compared to ConA. This strong hydrophobic core may be a contributing factor for the higher stability of SBA (Table 1; Fig. 9). The percentage of hydrophobic residues in SBA is ∼50% as compared to ConA where it is ∼40%, the actual numbers being 512 and 448 for SBA and ConA, respectively. This suggests that the hydrophobic core is stronger in SBA than in ConA. The experimentally obtained values of ΔCp for SBA and ConA are 5.0 and 4.2 kcal/mol/K, respectively. Although the experimentally determined values of ΔCp for both lectins are lower than the calculated values they follow, the trend of the theoretically determined ones viz. values for SBA are greater than that for ConA. Why does a difference between the theoretically and experimentally determined values exist? This is perhaps a reflection of the fact that some proteins do not undergo complete unfolding, i.e., they do not attain the complete random coil structure (on which the theoretical calculations are based) upon denaturant-induced unfolding reactions. This has been previously observed in many oligomeric proteins (Backmann et al., 1998).
FIGURE 9.

Stability curve for SBA and ConA obtained from GdnCl-induced isothermal unfolding experiments done at different temperatures indicated in Table 1. The continuous line shows the fit of the points to Eq. 8a. The data points (○) for SBA, and (□) for ConA have been obtained using individual melts done at different temperatures.
It is clear from Tables 1 and 2 that in general SBA is more stable than ConA with higher Tg and ΔGo of unfolding at all temperatures at which the experiments were done. The Tg of SBA is ∼20 K higher than that of ConA. Furthermore, Figs. 6 and 8 a show that denaturation in SBA occurs at a higher GdnCl concentration compared to ConA. In fact, SBA starts denaturing at a concentration of ∼4.5 M GdnCl. At this concentration the denaturation of ConA is almost complete.
Although the main force driving protein stability is hydrophobic interactions, many mutagenesis studies have revealed that hydrogen bonds also play an important role in stabilizing proteins (Shirley et al., 1992; Pace, 1995, 2001; Pace et al., 1996; Myers and Pace, 1996). In this study we have used HBPLUS to determine the number of hydrogen bonds in both proteins (McDonald and Thornton, 1994). When we analyzed the relative strength of the bonds it was observed that the number of strong hydrogen bonds (donor acceptor distance D–A < 2.5 Å) were ∼18 times greater in SBA than in ConA, enhancing the stability of the SBA tetramer. The distributions of the hydrogen bond lengths in both proteins are shown in Fig. 10. The protein data bank (PDB) coordinates of 1cvn and 2sba have been used for the above and all other calculations.
FIGURE 10.

Plot of the number of hydrogen bonds as a function of the donor-acceptor distance in SBA and ConA.
It has been observed by experimental as well as computational studies that salt bridges play an important role in stabilizing the native as well as the intermediate states in various proteins, e.g., barnase, cold shock proteins, ribosomal protein S6 (Tissot et al., 1996; Perl et al., 2001; Stoycheva et al., 2003). This led us to examine the ionic interactions in SBA and ConA. Pairs of oppositely charged atoms with a distance of 4 Å or less between them were considered as salt bridges.
The amino acid composition of the two proteins show that the population of charged residues is 6% more in SBA than in ConA; the actual numbers being 240 and 224, respectively. The increase in charged residues in SBA probably results in an enhanced number of electrostatic interactions in SBA as compared to ConA. Contrary to expectations, the total number of ionic interactions are greater in ConA than in SBA. But, a further analysis of the interactions in the interface revealed that the noncanonical interface in SBA, was significantly stronger than in ConA. There were 10 intersubunit interactions across this interface in SBA (giving rise to a total of 20 in the tetramer), whereas in ConA only six such interactions (a total of 12 in the tetramer) were observed across the corresponding interface. The details of the interactions are summarized in Table 3. All the intersubunit interactions are shown in Figs. 11 and 12.
TABLE 3.
Ionic interactions at the noncanonical interface in SBA and ConA
| SBA | ConA |
|---|---|
| Lys-163 NZ–Asp'-169 OD2 | Asp-58 OD1–Arg'-60 NH1 |
| Asp-169 OD2–Lys'-163 NZ | Arg-60 NH1–Asp'-58 OD1 |
| Arg-185 NE–Asp'-192 OD1 | Glu-192 OE1–Lys'-114 NZ |
| Arg-185 NH1–Asp'-192 OD1 | Glu-192 OE2–Lys'-114 NZ |
| Arg-185 NH2–Asp'-192 OD1 | Lys-114 NZ–Glu'-192 OE2 |
| Arg-185 NH2–Asp'-192 OD2 | Lys-114 NZ–Glu'-192 OE1 |
| Asp-192 OD1–Arg'-185 NE | |
| Asp-192 OD1–Arg'-185 NH1 | |
| Asp-192 OD1–Arg'-185 NH2 | |
| Asp-192 OD2–Arg'-185 NH2 |
FIGURE 11.

(a) The noncanonical interface in ConA showing the intersubunit salt bridges. Six such salt bridges are formed per noncanonical interface so the tetramer has a total of 12 salt bridges. (b) Enlarged view of the area where the interactions take place. The salt bridges are shown by dashed lines. Only three pairs are shown in the figure. The other three are just the mirror images of the three shown. Figure was generated using ViewerLite42. All the interactions are listed in Table 3.
FIGURE 12.

(a) The noncanonical interface in SBA showing the intersubunit salt bridges. Ten such salt bridges are formed per noncanonical interface so the tetramer has a total of 20 salt bridges. (b) Enlarged view of the area where the interactions take place, which are marked by a square frame in panel a. The salt bridges in both the frames are identical with the only difference that the chain identity of the amino acids involved is reversed; i.e., the Arg-182 shown in the enlarged version is changed to Arg'-182 in the other window. The salt bridges are shown by dashed lines. Figure was generated using ViewerLite42. All the interactions are listed in Table 3.
The noncanonical interface in SBA is one of the strongest seen in legume lectins with respect to ionic interaction analysis (N. Mitra and A. Surolia, unpublished data). An analysis of dimeric interfaces by the graph theoretical approach shows the noncanonical interface in SBA to be one of the strongest interfaces studied by this method. Thus we see the presence of a very strong interface in SBA that helps explain the greater stability of SBA as observed experimentally.
Between SBA and ConA, the total area buried at the interface is greater in ConA. Despite this, SBA shows greater stability. Thus, we see that SBA makes up for the relatively smaller buried area at the interface by other interactions that strengthen the interface. This is in contrast to what was observed in dimeric ConA and WBA II. Here, ConA with a greater buried area at the interface was also found to be more stable (Mitra et al., 2002).
Another factor that probably accounts for the greater stability of SBA over ConA is the presence of an N-linked glycan in SBA. SBA has three potential glycosylation sites out of which only one, Asn-75, is glycosylated due to the perfect geometry of this site. It has been reported that glycosylation affects both the structural elements and the global stability of proteins. The N-linked glycans are large hydrophilic structures. The molecular weight of the N-linked glycan in SBA (GlcNac2Man9) is ∼1884. The volume of such a big carbohydrate moiety is ∼1800 Å3, excluding the solvation sphere. This size is therefore significant in comparison to the monomeric protein size, which is 30,000 Å3. If the solvent sphere is also taken into consideration, one can imagine the enormous stability imparted to the system due to its enlarged solvent cage as compared to ConA (Wyss and Wagner, 1996; O'Connor and Imperiali, 1996; Rudd and Dwek, 1997; Imperiali and O'Connor, 1999). N-linked glycan induced stability has also been shown in another legume lectin, Erythrina corallodendron lectin (Mitra et al., 2003).
By a careful analysis of the SBA structure (1g9f in the PDB database) we found that the glycan is found at the noncanonical interface of the protein. This gives rise to interactions between the glycan of one of the subunits with the adjacent noncanonical subunit. This glycan-mediated interaction, gives added stability to the noncanonical interface of the protein (Fig. 13). No such stabilizing interactions are expected in ConA because it is not glycosylated.
FIGURE 13.

(a) The interactions between the glycan of one subunit and the residues of the other subunit at the noncanonical interface in SBA. (b) The enlarged view of the interactions. The solid ones are hydrogen bond interactions and the dashed ones are the hydrophobic interactions. Figure was generated using ViewerLite42. Contacts of structural units (CSU) were derived with CSU software (Sobolev et al., 1999). The PDB structure 1g9f was used for the analyses of glycan protein interactions.
Thus, we see that two proteins with very similar secondary and tertiary structures differ significantly in their stabilities. These differences arise due to variations in the amino acid compositions of the two proteins, consequently affecting the hydrogen bonding and ionic interaction patterns, and thus introducing stabilizing interactions in SBA, not found in ConA. Additional stability is imparted to SBA by the presence of an N-linked glycan. Delineation of the unfolding pathway, especially the characterization of the unfolding pathway of deglycosylated SBA will tell us the extent to which the glycan stabilizes the protein.
Acknowledgments
S.S. and G.K. thank the Council of Scientific and Industrial Research, of India, for the award of Senior Research Fellowships. This work is supported by a grant from the Department of Biotechnology, government of India, to A.S.
References
- Agashe, V. R., and J. B. Udgaonkar. 1995. Thermodynamics of denaturation of barstar: evidence for cold denaturation and evaluation of the interaction with guanidine hydrochloride. Biochemistry. 34:3286–3299. [DOI] [PubMed] [Google Scholar]
- Agrawal, B. B., and I. J. Goldstein. 1967. Protein-carbohydrate interaction. VI. Isolation of concanavalin A by specific adsorption on cross- linked dextran gels. Biochim. Biophys. Acta. 147:262–271. [PubMed] [Google Scholar]
- Ahmad, N., V. R. Srinivas, G. B. Reddy, and A. Surolia. 1998. Thermodynamic characterization of the conformational stability of the homodimeric protein, pea lectin. Biochemistry. 37:16765–16772. [DOI] [PubMed] [Google Scholar]
- Backmann, J., G. Schafer, L. Wyns, and H. Bonisch. 1998. Thermodynamics and kinetics of unfolding of the thermostable trimeric adenylate kinase from the archaeon Sulfolobus acidocaldarius. J. Mol. Biol. 284:817–833. [DOI] [PubMed] [Google Scholar]
- Barrick, D., and R. L. Baldwin. 1993. Stein and Moore Award address. The molten globule intermediate of apomyoglobin and the process of protein folding. Protein Sci. 2:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baues, R. J., and G. R. Gray. 1977. Lectin purification on affinity columns containing reductively aminated disaccharides. J. Biol. Chem. 252:57–60. [PubMed] [Google Scholar]
- Bowie, J. U., and R. T. Sauer. 1989. Equilibrium dissociation and unfolding of the Arc repressor dimer. Biochemistry. 28:7139–7143. [DOI] [PubMed] [Google Scholar]
- Chandra, N. R., M. M. Prabu, K. Suguna, and M. Vijayan. 2001. Structural similarity and functional diversity in proteins containing the legume lectin fold. Protein Eng. 14:857–866. [DOI] [PubMed] [Google Scholar]
- Ganesh, C., A. N. Shah, C. P. Swaminathan, A. Surolia, and R. Varadarajan. 1997. Thermodynamic characterization of the reversible, two-state unfolding of maltose binding protein, a large two-domain protein. Biochemistry. 36:5020–5028. [DOI] [PubMed] [Google Scholar]
- Gittelman, M. S., and C. R. Matthews. 1990. Folding and stability of Trp aporepressor from Escherichia coli. Biochemistry. 29:7011–7020. [DOI] [PubMed] [Google Scholar]
- Gualfetti, P. J., O. Bilsel, and C. R. Matthews. 1999. The progressive development of structure and stability during the equilibrium folding of the alpha subunit of tryptophan synthase from Escherichia coli. Protein Sci. 8:1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman, K. D., and C. F. Ainsworth. 1972. Structure of Concanavalin A at 2.4-A resolution. Biochemistry. 11:4910–4919. [DOI] [PubMed] [Google Scholar]
- Hubbard, S. J. 1996. NACCESS, version 2.1.1 computer program. Department of Biomolecular Sciences, UMIST, Manchester, UK.
- Imperiali, B., and S. E. O'Connor. 1999. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 3:643–649. [DOI] [PubMed] [Google Scholar]
- Livingstone, J. R., R. S. Spolar, and M. T. Record, Jr. 1991. Contribution to the thermodynamics of protein folding from the reduction in water accessible nonpolar surface area. Biochemistry. 30:4237–4244. [DOI] [PubMed] [Google Scholar]
- Lotan, R., H. W. Siegelman, H. Lis, and N. Sharon. 1974. Subunit structure of soybean agglutinin. J. Biol. Chem. 249:1219–1224. [PubMed] [Google Scholar]
- Manoj, N., V. R. Srinivas, A. Surolia, M. Vijayan, and K. Suguna. 2000. Carbohydrate specificity and salt-bridge mediated conformational change in acidic winged bean agglutinin. J. Mol. Biol. 302:1129–1137. [DOI] [PubMed] [Google Scholar]
- Manoj, N., and K. Suguna. 2001. Signature of quaternary structure in the sequences of legume lectins. Protein Eng. 14:735–745. [DOI] [PubMed] [Google Scholar]
- Matthews, C. R. 1993. Pathways of protein folding. Annu. Rev. Biochem. 62:653–658. [DOI] [PubMed] [Google Scholar]
- McDonald, I. K., and J. M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- Mitra, N., N. Sharon, and A. Surolia. 2003. Role of N-linked glycan in the unfolding pathway of Erythrina corallodendron lectin. Biochemistry. 42:12208–12216. [DOI] [PubMed] [Google Scholar]
- Mitra, N., V. R. Srinivas, T. N. Ramya, N. Ahmad, G. B. Reddy, and A. Surolia. 2002. Conformational stability of legume lectins reflect their different modes of quaternary association: solvent denaturation studies on Concanavalin A and winged bean acidic agglutinin. Biochemistry. 41:9256–9263. [DOI] [PubMed] [Google Scholar]
- Myers, J. K., and C. N. Pace. 1996. Hydrogen bonding stabilizes globular proteins. Biophys. J. 71:2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neet, K. E., and D. E. Timm. 1994. Conformational stability of dimeric proteins: quantitative studies by equilibrium denaturation. Protein Sci. 3:2167–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, E. M., and J. M. Scholtz. 1996. Conformational stability of the Escherichia coli HPr protein: test of the linear extrapolation method and a thermodynamic characterization of cold denaturation. Biochemistry. 35:11369–11378. [DOI] [PubMed] [Google Scholar]
- O'Connor, S. E., and B. Imperiali. 1996. Modulation of protein structure and function by asparagines linked glycosylation. Chem. Biol. 3:803–812. [DOI] [PubMed] [Google Scholar]
- Olsen, L. R., A. Dessen, D. Gupta, S. Sabesan, J. C. Sacchettini, and C. F. Brewer. 1997. X-ray crystallographic studies of unique cross-linked lattices between four isomeric biantennary oligosachharides and soybean agglutinin. Biochemistry. 36:15073–15080. [DOI] [PubMed] [Google Scholar]
- Pace, C. N. 1990. Conformational stability of globular proteins. Trends Biochem. Sci. 15:14–17. [DOI] [PubMed] [Google Scholar]
- Pace, C. N. 1995. Evaluating contribution of hydrogen bonding and hydrophobic bonding to protein folding. Methods Enzymol. 259:538–554. [DOI] [PubMed] [Google Scholar]
- Pace, C. N. 2001. Polar group burial contributes more to protein stability than nonpolar group burial. Biochemistry. 40:310–313. [DOI] [PubMed] [Google Scholar]
- Pace, C. N., B. A. Shirley, M. McNutt, and K. Gajiwala. 1996. Forces contributing to the conformational stability of proteins. FASEB J. 10:75–83. [DOI] [PubMed] [Google Scholar]
- Perl, D., G. Holtermann, and F. X. Schmid. 2001. Role of chain termini for the folding transition state of cold shock protein. Biochemistry. 40:15501–15511. [DOI] [PubMed] [Google Scholar]
- Prabu, M. M., K. Suguna, and M. Vijayan. 1999. Variability in quaternary association of proteins with the same tertiary fold: a case study and rationalization involving legume lectins. Proteins. 35:58–69. [DOI] [PubMed] [Google Scholar]
- Rudd, P. M., and R. A. Dwek. 1997. Glycosylation: heterogeneity and 3D structure of proteins. Crit. Rev. Biochem. Mol. 32:1–100. [DOI] [PubMed] [Google Scholar]
- Schellman, J. A. 1990. Selective binding and solvent denaturation. Biophys. Chem. 37:121–140.2285775 [Google Scholar]
- Shirley, B. A., P. Stanssens, U. Hahn, and C. N. Pace. 1992. Contribution of hydrogen bonding to the conformational stability of ribonuclease T1. Biochemistry. 31:725–732. [DOI] [PubMed] [Google Scholar]
- Sobolev, V., A. Sorokine, J. Prilusky, E. E. Abola, and M. Edelman. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics. 15:327–332. [DOI] [PubMed] [Google Scholar]
- Spolar, R. S., J. R. Livingstone, and M. T. Record, Jr. 1992. Use of liquid hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry. 31:3947–3955. [DOI] [PubMed] [Google Scholar]
- Stoycheva, A. D., J. N. Onuchic, and C. L. Brooks. 2003. Effect of gatekeepers on the early folding kinetics of a model β-barrel protein. J. Chem. Phys. 119:5722–5729. [Google Scholar]
- Tissot, A. C., S. Vuilleumier, and A. R. Fersht. 1996. Importance of two buried salt-bridges in the stability and folding pathway of barnase. Biochemistry. 35:6786–6794. [DOI] [PubMed] [Google Scholar]
- Wyss, D. F., and G. Wagner. 1996. Structural role of sugars in glycoproteins. Curr. Opin. Biotechnol. 7:409–416. [DOI] [PubMed] [Google Scholar]