

Abstract
The molecular structure of the brush-border of enterocytes has been investigated since the 1980s, but the dynamics of this highly specialized subcellular domain have been difficult to study due to its small size. To perform a detailed analysis of the dynamics of cytoskeleton proteins in this domain, we developed two-photon fluorescence recovery after photobleaching and a theoretical framework for data analysis. With this method, fast dynamics of proteins in the microvilli of the brush border of epithelial intestinal cells can be measured on the millisecond timescale in volumes smaller than 1 μm3. Two major proteins of the cytoskeleton of the microvilli, actin and myosin 1a (Myo1a; formerly named brush border myosin I), are mobile in the brush-border of Caco-2 cells, an enterocyte-like cellular model. However, the mobility of actin is very different from that of Myo1a and they appear to be unrelated (diffusion coefficient of 15 μm2 s−1 with a mobile fraction of 60% for actin, and 4 μm2 s−1 with a mobile fraction of 90% for Myo1a). Furthermore, we show for the first time, in vivo, that the dynamics of Myo1a in microvilli reflect its motor activity.
INTRODUCTION
The understanding of the molecular mechanism involved in the morphogenesis and the dynamics of the actomyosin cytoskeleton have been investigated in vivo in large cellular domains by photoactivation of fluorescence or fluorescence recovery after photobleaching, i.e., FRAP (McGrath et al., 1998; Tardy et al., 1995; Tyska and Mooseker, 2002). However, a quantitative analysis of myosin dynamics has never been made directly in very small structures like membrane protrusions. To perform a detailed analysis of the dynamics of cytoskeleton proteins in these structures, we combined two-photon FRAP and a theoretical framework for interpreting the fluorescence relaxation curves.
Two-photon FRAP allows one to confine the photobleaching to a 1 μm high region, in contrast to one-photon FRAP, where the whole illumination cone of the sample is photobleached (Brown et al., 1999). Therefore, it is possible to study with a better axial resolution the mobility of proteins in small structures like protrusions or microvilli.
The principle of one- or two-photon FRAP techniques consists of locally switching off the fluorescence (photobleaching) over a well-defined region and measuring the time course of molecules returning into this region. Quantitative information about the nature (diffusion, velocity-driven movement, etc.) and the kinetics (diffusion coefficients or velocities) of molecular movements are determined from fitting recovery curves with appropriate analytical results or numerical simulations. The interpretation of results primarily depend on the geometry of the sample, the nature of the molecular movements, and the photobleaching conditions considered in the model. Concerning the geometry, classical analytical solutions were given by Axelrod et al. (1976) in the case of infinite two-dimensional diffusion or convection, and by Brown et al. (1999) in the case of infinite three-dimensional diffusion with two-photon photobleaching. Theoretical efforts have been made to model more complex geometries, like cylindrical surfaces (Wolf, 1992), or geometries with obstacles (Schram et al., 1994; Olveczky and Verkman, 1998). As to the nature of movements, an effort has also been made to model complex behaviors, like several diffusive components (Gordon et al., 1995; Periasamy and Verkman, 1998), anomalous diffusion (Feder et al., 1996; Periasamy and Verkman, 1998), percolation (Coelho et al., 1997), and exchange between immobile and freely diffusing molecules (Tardy et al., 1995).
Yet there have been few models for experimental conditions that differ from the standard conditions described by Axelrod et al. (1976) (an exception is the modeling of uniform disk photobleaching, Lopez et al., 1988). Notably there are, to our knowledge, no models for photobleaching experiments deviating from the ideal hypotheses: no diffusion during the photobleaching pulse and no photobleaching during the recovery. However, these two assumptions are only valid in a limited number of cases depending on the nature of the sample and the parameters of the FRAP procedure. In particular, they are most often not verified in typical FRAP experiments on living cells. Our strategy is to develop a method to analyze two-photon FRAP experiments with numerical simulations taking into account the deviations from these two key assumptions.
This method was applied to two-photon FRAP experiments to study the dynamics of the brush border at the apical surface of enterocytes. The dynamics of actin and a nonconventional myosin were examined in stable plasma membrane protrusions; i.e., the microvilli that make up the brush-border. Each microvillus contains a core of actin filaments organized in bundles by actin binding proteins such as villin, fibrin, and espin (Bartles et al., 1998; Coudrier et al., 1988). The bundles are tethered to the plasma membrane via ezrin and a helical array of cross bridges containing an unconventional myosin, myosin 1a (Myo1a, previously called brush-border myosin I). Myo1a consists of an N-terminal motor domain, a neck domain with three IQ motifs that bind calmodulin, and a basic tail that may act as a membrane binding domain (Garcia et al., 1989). In the intact intestine the size of the microvilli depend on diet. In addition, the cytoskeletal proteins turn over every 24 h, but the general structure of the brush border remains intact (Stidwill et al., 1984). Similar to unstable protrusions the shape and length of microvilli may depend on key parameters that control the dynamics of actin and proteins that tether the actin bundle to the plasma membrane. We have recently determined key parameters concerning the dynamics of ezrin in microvilli (Coscoy et al., 2002), and we report in this article the analysis of the dynamics of actin and Myo1a in the microvilli of Caco-2 cells, an enterocyte-like cellular model. This approach shows that the dynamics of Myo1a are independent of that of actin, and that these dynamics depend on the motor activity of the myosin itself.
MATERIALS AND METHODS
Cellular clones and cell culture
EGFP-actin was expressed from a recombinant plasmid generously given by B. Imhof (Ballestrem et al., 1998). Recombinant EGFP-Myo1a was obtained by inserting cMyo1a cDNA (Genbank accession number U04049) downstream of the oligonucleotide (AGCTTCGCCACCGCC) in the HindIII-EcoRI restriction sites of the pEGFP vector. This construct led to a 12-amino-acids linker (SGLRSRQASPPP) between the EGFP protein and cMyo1a. Recombinant EGFP-Myo1aΔ730 was obtained by inserting the nucleotide sequence encoding Myo1aΔ730 (aa 730–1040 from Myo1a sequence) downstream of the oligonucleotide (AGCTTGCCCACCGA) in the HindIII-EcoRI restriction sites of the pEGFP vector. This EGFP-Myo1aΔ730 construct results in an 11-amino-acids insertion (SGLRSRQACPP) between EGFP and Myo1a. Transfections and isolation of stable transformations were performed as described in Durrbach et al. (1996). For FRAP experiments, cells were grown at 37°C under 10% CO2 in DMEM (Gibco-BRL Life Technologies, Paisley, Scotland) supplemented with 10% fetal calf serum (Gibco-BRL Life Technologies), and 0.7 mg/ml Geneticin (Gibco-BRL Life Technologies). Cells were plated at high confluency (5 × 105 cells/cm2) on glass coverslips (Nunc, Roskilde, Denmark) coated with 32 mg/cm2 of rat collagen type I (Harbor Bio-Product, Norwood, MA). Cells were confluent within one day and were grown for a total of 5–6 days to allow the brush-border to develop. Cells were incubated overnight with 10 mM sodium butyrate before experiments.
Cellular distribution and characteristics of EGFP-proteins in Caco-2 cellular clones
EGFP-actin forms a typical cortical network underneath the basolateral plasma membrane and bundles in the microvilli of Caco-2 cells (derived from a human colonic adenocarcinoma, Fig. 1 A). The filamentous actin bundles are also labeled with a rhodamine-phalloidin conjugate after fixation and permeabilization of the cells (Fig. 1 B). To establish whether the fusion protein, EGFP-actin, was fully functional in Caco-2 cells, we analyzed its sensitivity to jasplakinolide. This drug induces the recruitment of monomeric actin in filamentous structures and thus decreases the amount of monomeric actin available in the cytosol. Monomeric actin is recovered in a soluble fraction obtained by treating isolated brush-border from Caco-2 cells expressing EGFP-actin with 1% Triton X 100 (Fig. 1 G, column S). The amount of EGFP-actin in the soluble fraction (S) decreased when Caco-2 cells were treated with jasplakinolide before cell fractionation indicating that EGFP-actin is able to polymerize (Fig. 1 G).
FIGURE 1.

Cellular distribution of EGFP-fusion proteins in Caco-2 cells. (A–F) The distribution of EGFP-actin (A), EGFP-Myo1a (C), and EGFP-Myo1aΔ730 (E) in fully differentiated Caco-2 clones was compared to the distribution of filamentous actin (B, D, and F) decorated with rhodamine phalloidin conjugate in x,z optical confocal sections. (Scale bar represents 13 μm.) (G–H) Isolated brush-border from cells expressing EGFP-actin (G), EGFP-Myo1a, and EGFP-Myo1aΔ730 (H) were analyzed after extraction with 1% Triton X-100 and centrifugation (30 min at 300,000 g). Twenty micrograms of each fraction were then probed after Western blot with anti-GFP antibody. G shows that EGFP-actin from total brush-border fractions (BB) is detected in both the Triton insoluble (P) and soluble fractions (S) and shifts to the Triton insoluble fraction (P) after treatment with 1 μM jasplakinolide. H shows that 10 mM ATP releases EGFP-Myo1a from the Triton insoluble fraction, whereas EGFP-Myo1aΔ730 (tail domain) is mainly Triton soluble regardless of the ATP concentration.
The chicken EGFP-Myo1a is distributed in the microvilli and at the lateral plasma membrane domain together with actin filaments (Fig. 1, C and D). This cellular distribution is similar to that of the endogenous Myo1a and is in agreement with the cellular distribution of Myo1a in human small and large intestine (Heintzelman et al., 1994). Furthermore, this fusion protein demonstrates characteristics similar to endogenous protein. Its dissociation from brush border isolated from Caco-2 cells requires the combination of detergent and ATP as has been previously shown for EGFP-Myo1a and endogenous Myo1a in Caco-2 cells (Fig. 1 H) (Peterson and Mooseker, 1992). EGFP-Myo1aΔ730 is distributed in the microvilli together with actin filaments and in the basolateral cytoplasmic domain of Caco-2 cells (Fig. 1, E and F), in agreement with the cellular distribution of the human tail domain expressed in LLCPK1 cells, or the chicken tail without tag expressed in Caco-2 cells (Peterson and Mooseker, 1992; Durrbach et al., 2000). In contrast to EGFP-Myo1a the dissociation of EGFP-Myo1aΔ730 from isolated brush border requires only detergent (Fig. 1 H).
Drug treatments
In some experiments, cells were treated with 1 μM jasplakinolide (Molecular Probes, Eugene, OR) for 10 min before measurements to inhibit the dynamics of actin. In other experiments, cells were treated with 20 mM butanedione monoxime (BDM) in culture medium for 10 min to inhibit the ATPase activity of myosin. ATP depletion was achieved by inhibiting mitochondria metabolism with 10 μM antimycin A and glycolysis with 10 mM 2-deoxyglucose (Sigma, St. Louis, MO). After 20 min of this treatment, cells were analyzed for their intracellular ATP, ADP, and AMP content by high-pressure liquid chromatography (Bacallao et al., 1994). Intracellular ATP is reduced by ∼40% under our experimental conditions (data not shown).
Two-photon FRAP setup
Two-photon FRAP experiments were carried out with a modified Fluoview confocal scanner (Olympus, Tokyo, Japan) coupled with a modified Olympus IX70 inverted microscope. Fluorophores were excited by a Ti:Sapphire laser (Tsunami; Spectra Physics, Mountain View, CA) providing 70-fs pulses with an 80-MHz repetition rate. The laser power sequence required for FRAP experiments was achieved using an electro-optical modulator (model 350-50, Conoptics, Danbury, CT), with relatively short transition times (< 30 μs). A three-step sequence was generated, using a low laser power for fluorescence observation (Pobs = 5 mW at the sample), and a high power for the photobleaching pulse (Ppulse = 30 mW), as follows:
Step 1: a 1 s illumination at Pobs to assess the initial fluorescence level.
Step 2: a 0.2 s illumination at Ppulse to photobleach the molecules.
Step 3: a 10 s illumination at Pobs, to measure the fluorescence recovery.
A home-written program using Labview 5.1 software and a 500 kHz A/D computer board (PCI-MIO-16E-4, National Instruments, Austin, TX) were used to control the beam position, the modulator transmission, and the acquisition of the fluorescence signal from the Fluoview photomultipliers (R928, Hamamatsu, Hamamatsu City, Japan). EGFP fluorescence was excited with the femtosecond laser at 890 nm and collected using a high numerical aperture objective (63× oil immersion, NA = 1.25, PlanNeofluar, Zeiss, Jena, Germany) via a 650-nm dichroic filter and a 510/20-nm bandpass filter (HQ510/20, Chroma Technologies, Brattleboro, VT). Under the above conditions, the excitation volume that determines the point spread function (PSF) could be approximated by a three-dimensional Gaussian excitation intensity distribution with axial symmetry, with 1/e full width dw = 0.6 μm and dz = 1.5 μm in the focal plane and along the optical axis, respectively.
Data acquisition and analysis
FRAP experiments were done using a single-shot method. Instead of repeating the same laser power sequence on the same region of the sample as it is usually done for in vitro experiments, we performed a single acquisition on one spot at a time and then moved to another spot (see Results).
Each individual fluorescence curve was then normalized by its own prebleach intensity (i.e., the fluorescence averaged over the 1 s prebleach period). The final recovery curve was obtained by calculating the mean of all of the individual acquisitions. Fluorescence was sampled at regular time intervals (usually 50 μs) and then averaged in real-time over a larger time interval (usually 11.2 ms) which sets the time resolution for the measurements. For analysis, the points were binned so that final averaged points were evenly distributed on a logarithmic timescale. This logarithmic binning ensures that the statistical weight of long timescale points does not dominate over short timescales, and that the fitting procedure is evenly sensitive throughout the time domains.
Numerical simulation
Considering the ratio of the transversal diameter of the PSF (0.6 μm) and that of a microvillus (0.1 μm), the motion of proteins within a microvillus was modeled to be unidimensional as in Coscoy et al. (2002). Indeed, any movement in the transverse section of the microvillus at a given height could not be measured here. For data fitting (see Figs. 7 and 10) and to take into account the closed-end of the microvillus, we used reflective boundary conditions set 1 μm away from the center of the PSF and a long axis (79 μm) to simulate the open boundary at the bottom of the microvillus (see Fig. 2). For all theoretical demonstrations (Figs. 6 and 7), the PSF was placed at the center of the axis far from the boundaries (40 μm), simulating an infinite axis situation.
FIGURE 7.

Numerical simulations of diffusion processes with observation photobleaching. (A) Numerical simulations of the recovery curve after a photobleaching pulse (bottom) and of the baseline curve without pulse bleaching (top) are represented for different values D = 1, 5, 20, and 80 μm2 s−1, and mobile fractions of 100%. (B) Numerical simulations of the recovery (bottom) and baseline (top) curves are represented for two different mobile fractions and diffusion coefficients (D = 20 μm2 s−1, mobile fraction = 50% and D = 2 μm2 s−1, mobile fraction = 80%, dashed and continuous lines, respectively). Note that the two curves intersect under these conditions.
FIGURE 10.

Effect of ATP depletion on the mobility of EGFP-actin, EGFP-Myo1a, and EGFP-Myo1aΔ730. Recovery curves were acquired before and after ATP depletion, and the ratio (recovery curve after ATP depletion)/(recovery curve before) is represented for EGFP-Myo1a (•), EGFP-actin (○), and EGFP-Myo1aΔ730 (▵). (Mean of 407, 140, and 149 curves, respectively.)
FIGURE 2.

Geometry used for numerical simulations of two-photon FRAP experiments. On the left is a representation of a microvillus and the PSF (not to scale); on the right is the one-dimensional axis used for numerical computations with reflective boundary conditions. The PSF (I2(x)) is projected on the axis and placed at 1 μm from one end of the axis for experimental data fitting. Axis length (L) was taken as 80 μm.
FIGURE 6.

Numerical simulations of fluorescence depletion. Concentration axial profiles of unbleached molecules during the photobleaching pulse are represented at different times (between 0 and 400 ms) for two extreme cases, a low mobility protein with D = 0.2 μm2 s−1 (A), and a high mobility protein with D = 80 μm2 s−1 (B). The dotted line represents the PSF. (Left inserts) The profiles were normalized so that their full width at half-maximum is the same, showing the evolution of the shape of the profile independently of their amplitude. (Right inserts) Evolution of the full width at half-maximum as a function of photobleaching pulse duration. Calculated fluorescence evolution during the photobleaching pulse and the recovery phase are represented for different photobleaching pulse durations (Tp = 2, 35, 200, and 500 ms; arrows indicate increasing photobleaching times), for D = 0.2 μm2 s−1 (with Tp < τd) (C) and D = 80 μm2 s−1 (with τd < Tp). (D) The values of characteristic diffusion times τd (τd = 5 s for the low mobility protein and 0.012 s for the high mobility protein assuming a 1 μm2 surface), of the characteristic photobleaching time τb-bl (= 0.026 s from Fig. 3), and of the photobleaching time used in our experiments shown in Figs. 7–10 (Td = 0.2 s), are indicated on the time axis for comparison with the cases shown in Fig. 2. The recovery curves for different photobleaching pulse durations (C, D), with normalized amplitudes are shown in E: D = 0.2 μm2 s−1 and F: D = 80 μm2 s−1. The vertical bar corresponds to the starting point of our experimental measurements on microvilli (see Figs. 7–10).
Protein mobility can be due to various phenomena (diffusion, motor-driven movement, flow), but in the absence of any prior knowledge of them, we made the assumption that the movement was purely diffusive. An immobile fraction was included in the model to simulate contributions to the recovery curves from immobile or very slowly moving molecules.
The protein mobility was then modeled by two equations: a one-dimensional diffusion equation with an additional term to take into account the non-uniform photobleaching process for mobile molecules, and an equation representing the photobleaching kinetics of immobile molecules,
 |
(1) |
where Cm is the concentration of mobile fluorescent molecules (unbleached), Cim is the concentration of immobile fluorescent molecules, and D is the diffusion coefficient. The value α(x) = kb I2(x), the photobleaching rate of the fluorophore, is proportional to the squared intensity distribution I2(x) (I(x) is squared because of two-photon excitation). The value of the photobleaching constant kb was experimentally estimated for the excitation power used during the photobleaching phase (Ppulse) and during the recovery phase (Pobs). Fluorescence decays of fixed cells were measured and then fit by a single exponential (see Results and Fig. 4). Characteristic decay times of the fluorescence signal integrated over the PSF gave: Kb (FRAP) = 38.46 s−1 and Kb (obs.) = 0.28 s−1.
FIGURE 4.

Photobleaching kinetics and characteristic times for immobilized EGFP. Normalized EGFP fluorescence decay upon photobleaching was measured at a pulse power Ppulse = 30 mW (♦) and observation power Pobs = 5 mW (○ and •) on immobilized EGFP-fused proteins: actin (•,♦) and Myo1a (○) in glutaraldehyde-fixed cells. Fluorescence decay at Pobs is fitted with a single exponential function with or without an offset (offset value = 0.3). Decay at Ppulse was also fit to an exponential (in the inset), with the same offset. The insert shows the same curves on a logarithmic timescale to reveal the fast kinetics of photobleaching, and illustrates the departure from exponential behaviors. Best exponential fits are shown with an offset (continuous lines).
The total concentration of unbleached molecules is calculated by: C(x,t) = f Cm(x,t) + (1 − f) Cim(x,t), with f the mobile fraction. We took I(x) (and thus α(x)) as a Gaussian function to represent the axial illumination distribution. This spatial dependence of the photobleaching rate makes it difficult to calculate an analytical solution so we used numerical simulations to produce theoretical fluorescence recovery curves. The algorithm used was based on the centered finite difference method (Woolfson and Pert, 1999) and the source code was written in C. The spatial axis was discrete with 0.02-μm steps dx and the time interval dt between two steps of the simulation was adjusted by dt = dx2/2D.
Simulated fluorescence curves were obtained after or without a photobleaching pulse, to show the recovery of the baseline. The fluorescence intensity at each time step was computed from
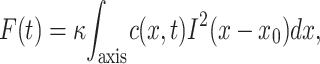 |
(2) |
where κ is an arbitrary constant. Because photobleaching occurs during the relaxation, a correction procedure is usually applied, which consists of the normalization of the fluorescence signal by the baseline curve (McGrath et al., 1998). This scheme is mathematically based on the assumption that all molecules bleach at the same rate. This is unfortunately rarely the case, except when the excitation intensity is uniform. This is obviously not the case here, as in most FRAP experiments. Because a mathematically exact normalization is too complex, we used a differential representation to fit the data (see Results). In this representation, the final curve is obtained by subtracting the raw fluorescence curve from the baseline curve. This difference curve truly reflects the response to the bleach-pulse perturbation, and will necessarily monotonously vanish at times longer than the diffusive relaxation time. In contrast, the raw fluorescence signal, somehow misleadingly called the recovery signal, is eventually dominated by samplewide bleaching at long enough times, and can decay. Experimental fluorescence recovery curves were then fit with simulated curves to estimate a diffusion coefficient D and a mobile fraction for each experiment.
RESULTS
Theoretical analysis of FRAP experiments
Re-evaluation of the classical hypotheses
FRAP experiments are based on measuring fluorescence recovery as an indication of the movement of fluorophores after a perturbation by photobleaching. A general assumption made for the interpretation of these experiments is that no movement occurs during the photobleaching pulse and no photobleaching occurs during the recovery phase.
We can formulate these assumptions with two simple hypotheses (see Fig. 3, lane a):
H1. The duration of the photobleaching pulse Tp is much shorter than the characteristic transport time over the bleached region (Tp ≪ τd ). Movements can be neglected during the photobleaching pulse under these conditions in the sense that there is no exchange between photobleached molecules in the excitation volume and fluorescent molecules outside this volume. This also implies that the photobleaching pulse duration must be much shorter than the observation time (Tp ≪ Tobs).
H2. During the recovery phase the characteristic photobleaching time when the fluorophores are exposed to the observation power is much longer than the observation time (τb-obs ≫ Tobs), so that no photobleaching occurs during the recovery of fluorescence. In that case, the fluorescence intensity during the recovery proportionally reflects the number of fluorescent molecules coming back into the illumination volume.
FIGURE 3.

Comparison of the timescales involved in FRAP experiments. In an experiment with a bleaching pulse of duration, τp, followed by a relaxation observation time, τobs, the physically relevant timescales are: τd the characteristic time of diffusive relaxation in the excitation volume, and the characteristic times τb-bl and τbl-obs of the photobleaching process caused by the high power of the bleaching pulse (Ppulse) and by the moderate power (Pobs) used for observing the relaxation, respectively. In the ideal situation (line a), the working hypotheses, often implicitly made but most often not realized in FRAP experiments, are that (H1) no diffusion occurs during the bleaching pulse (Tp < τd) and (H2) the bleaching is negligible during the time taken to assess the relaxation (Tobs < τb-obs). In the situation of the present work (line b), which is likely to be relevant in most FRAP experiments on cells, bleaching and relaxation cannot be temporally separated: diffusion is not negligible during the bleaching pulse (τd < Tp), and bleaching occurs during the time devoted to assess the relaxation (τb-obs < Tobs).
We tried to comply with the H1 hypothesis by choosing a photobleaching power of 30 mW that was high enough to allow a brief photobleaching pulse but small enough to avoid excessive excitation (saturation) in two-photon FRAP (Brown et al., 1999). Photobleaching of EGFP occurs under these conditions with a typical time (τ1/2) of 50 ms (Fig. 4). This τ1/2 is still long compared to the typical time of protein diffusion in aqueous solution (∼3 ms for a 1 μm2 surface for EGFP in the cytoplasm; Brown et al., 1999; Swaminathan et al., 1997). We are thus in the situation shown on Fig. 3, lane b, and cannot satisfy H1. One strategy is to perform numerous repetitions with pulses shorter than the shortest expected diffusion time. The typical recovery time of our proteins in microvilli is on the order of seconds, so this type of strategy (with typically 1000 repetitions) would require that the microvilli themselves do not move significantly relative to the size of the focal volume over several minutes. Unfortunately, we observe that the microvilli move at a typical rate of 0.1 μm/10 s, precluding the use of the repetitive strategy (S. Coscoy, unpublished data). Furthermore, repetitive photobleaching/recovery sequences can progressively lead to a persistent depletion in the total concentration of unbleached molecules (see below) if the repetition time is shorter than the largest recovery time. Since the latter time constant was not known a priori, repetitive FRAP could yield misleading results. A related drawback of making repetitive acquisition is that information on the immobile fraction is lost, because its recovery time is virtually infinite. Indeed, the immobile molecules will be photobleached after the first FRAP sequence and only mobile molecules will contribute to the fluorescence recovery, making it appear that there is no immobile fraction. We thus chose to use single-shot FRAP with a photobleaching pulse of 200 ms and 30 mW resulting in a large photobleaching depth (∼80%), and developed a numerical approach to take into account the deviations from the H1 hypothesis.
To comply with the H2 hypothesis, the observation power should be as low as possible. However, to obtain a good enough signal/noise ratio, a minimal power is required, and that power depends on the concentration of the fluorophore (Brown et al., 1999; Piston et al., 1992). In our case, the minimal observation power for EGFP-actin or EGFP-Myo1a in microvilli was 5 mW. The amount and rate of photobleaching at the observation power Pobs (referred to as observational photobleaching) was estimated on immobilized proteins. As expected, fluorescence recovery in living cells exhibited a reduced apparent photobleaching rate due to the continuous exchange of unbleached proteins into the excitation volume. The rate of photobleaching did not depend on the EGFP fusion protein used (Fig. 4). For observation times longer than 1 s, observational photobleaching with 5 mW laser power could not be neglected. Therefore, the H2 hypothesis was not satisfied and we were in the situation shown in Fig. 3 b. Hence, fluorescence photobleaching during the recovery had to be taken into account in our numerical simulations. The total fluorescence loss due to observational photobleaching on fixed cells was equal to 10% after 1 s, and occurred with a characteristic decay time (τ1/2) of 10 s. The exponential fit was not perfect, however, and showed a mismatch (Fig. 4).
Spatial depletion in concentration
Because the H1 hypothesis was not met under our experimental conditions, we measured the influence of the photobleaching pulse duration on the recovery curve of a soluble protein in solution. Increasing the photobleaching pulse duration from 2 ms to 135 ms resulted in a 30-fold increase in the recovery half-time (τ1/2, Fig. 5). The EGFP molecules are diffusing with a characteristic diffusion time of ∼3 ms under our experimental conditions (diffusion coefficient of 80 μm2 s−1, Brown et al., 1999) . This time is shorter than the duration of the photobleaching pulse, leading to a depletion of unbleached molecules in the vicinity of the laser-beam focus. The net result is a decrease in the apparent diffusion coefficient because unbleached molecules must diffuse over a larger distance to reach the illumination volume and be detected. The insert in Fig. 5 shows that if the curves are normalized in time the three curves are superimposed, indicating the diffusion dynamics of EGFP have qualitatively not changed. The recovery timescale has just been shifted.
FIGURE 5.

Experimental observation of spatial concentration depletion. Effect of the photobleaching pulse duration (τp) on the normalized fluorescence recovery of freely diffusing EGFP in aqueous solution. Increasing τp simply shifts the relaxation to longer timescales: τp = 2 ms (•), τp = 35 ms (○), and τp = 135 ms (♦). Fluorescence relaxation essentially depends on the relative time t/τp as shown by the superimposition on a single master curve of the relaxation kinetics after time normalization (inset).
Numerical simulations were introduced to develop these interpretations. Molecules were allowed to diffuse freely along a one-dimensional axis representing the microvillus, with a known probability of photobleaching (see Material and Methods). The time evolution of the system can then be described by the equation
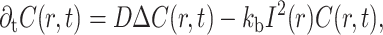 |
(3) |
where C is the concentration of unbleached molecules, r is the position, D is the diffusion coefficient, I(r) is the illumination intensity distribution, and kb is the photobleaching constant of the fluorophore. The first term on the right-hand side represents the diffusive relaxation of fluorophores, whereas the second term represents the fluorescence decay due to photobleaching. The immobile fraction was also taken into account as described in Materials and Methods. The boundary conditions were chosen depending on the purpose of the calculation. For the present theoretical analysis, the boundaries of the axis were far enough (40 μm) from the center of the PSF to simulate an infinite axis. However, for experimental data the PSF was placed 1 μm away from a reflective wall to simulate the closed-end of the microvillus, the other end of the axis being far from the PSF (79 μm).
The concentration of unbleached fluorophores during the photobleaching pulse was calculated and is shown in Fig. 6, A and B, for two different diffusion coefficients. In the case of a slowly diffusing molecule (D = 0.2 μm2 s−1), the concentration profile of unbleached fluorophores is getting deeper with longer photobleaching periods, but the profile width does not exceed the width of the point spread function (PSF) (Fig. 6 A). In the case of a highly mobile molecule (D = 80 μm2 s−1), the profile is greatly enlarged with increasing photobleaching pulse duration (Fig. 6 B) and extends beyond the PSF profile, but the shape remains constant (left insert). It is interesting to note that the photobleaching depth is lower in this case because the refilling of the illuminated region by quickly moving unbleached molecules counterbalances the loss in fluorescence due to photobleaching.
The simulated fluorescence curves corresponding to the concentration profiles are shown in Fig. 6, C and D, for different photobleaching pulse durations (2, 35, 200, and 500 ms) and two diffusion coefficients, D = 0.2 μm2 s−1 and D = 80 μm2 s−1, respectively. The curves represent the fluorescence during the photobleaching pulse and the recovery phase. Two situations can be distinguished: Tp < τd or Tp > τd. In our experiments, the bleaching pulse duration (Tp = 200 ms) is such that either situation can occur depending on the protein mobility. If proteins have a slow mobility (τd > Tp), recovery curves essentially do not depend on the photobleaching pulse duration (Fig. 6 E). On the contrary, for more mobile proteins (τd < Tp), recovery curves do shift to longer timescales for increased photobleaching pulse duration (Fig. 6 F). In a more general way, relaxation times shorter than the photobleaching pulse duration are suppressed, thus leading to a reduced apparent mobility. This is a major source of artifacts in the interpretation of FRAP data in general.
In summary, we have shown that:
The depth of photobleaching decreases with increasing diffusion coefficients. Interestingly, this photobleaching depth can be used to estimate D.
The concentration profile of unbleached molecules widens if the bleaching time Tp is larger than the diffusion time τd within the illumination volume. This decreases the apparent mobility of the protein and will be referred to as spatial concentration depletion.
Observational photobleaching
Because the H2 hypothesis is not satisfied under our experimental conditions, observational photobleaching has to be taken into account in the analysis. This process causes a fluorescence loss, at a rate that is proportional to the probability of photobleaching a chromophore during its residence time within the excitation volume. This probability is therefore controlled by the ratio of the mean residence time to the photobleach time for a fixed molecule. The amplitude loss will thus be maximal for EGFP in fixed cells (10% loss in 1 s, see Fig. 4) and virtually zero for fast moving molecules. In a previous study, McGrath et al. (1998) used a uniform normalization algorithm correct for the loss of fluorescence signal between time-lapse images. However, this approach is not valid here since we perform FRAP experiments in a way that photobleaching is not uniform over the sample.
Photobleaching is usually modeled as a first-order process. To assess the bleaching rate, we assessed the rate of fluorescence decay for fixed samples over four decades in time. Unfortunately, a simple exponential function does not fit the data well. A better fit is obtained with a non-zero asymptotic value at long times (Fig. 4). For times shorter than 10 s, the best exponential fit (τ = 13 s) matches the experimental data with a maximal relative error of 10%. Therefore, an exponential function was included in our simulations.
Interestingly, simulations in which slow diffusion is combined with observational bleaching yield non-monotonous recovery curves (Fig. 7 A). Experimentally, this was only observed for longer observation times (data not shown). The fluorescence recovery was represented as the difference between the baseline fluorescence curve (without a prior photobleaching pulse) and the recovery curve. This difference curve truly represents the fluorescence response after the bleach pulse perturbation, and the response monotonously vanishes (see Materials and Methods).
Conclusions of the theoretical analysis
The method proposed in this report makes use of numerical simulations to resolve two major difficulties related to the interpretation of FRAP data, namely spatial depletion that leads to reduced apparent mobility, and observational bleaching that yields apparently incomplete and possibly nonmonotonous recovery. Photobleaching during the bleaching pulse is modeled as a first-order process, in a spatially dependent manner, and fluorescence recovery is computed by taking observational bleaching into account. This approach makes it possible to analyze FRAP experiments even when the classical hypotheses H1 and H2 are not satisfied. This approach is valid for one-photon FRAP experiments on two-dimensional samples (like transmembrane proteins), and in three dimensions for two-photon FRAP as well, since the excitation and detection volumes overlap. Fig. 7 B shows examples of simulations of two processes with different mobile fractions and diffusion coefficients. In that particular case (different D and mobile fractions), the two recovery curves intersect.
Dynamics of cytoskeleton proteins in brush border microvilli
Actin and Myo1a display distinct recovery kinetics in the microvilli of Caco-2 cells
Caco-2 cells spontaneously differentiate at confluence into cells exhibiting many of the morphological and functional characteristics of mature enterocytes. Cells were grown for 3–6 days after confluence, at which time they exhibit stable microvilli between 2- and 2.4-μm long. Two-photon FRAP was used to study the dynamics of EGFP-actin and EGFP-Myo1a in these stable microvilli. The fluorescence recovery curves for EGFP-actin and EGFP-Myo1a are distinct with a longer characteristic time of recovery for EGFP-Myo1a (Fig. 8 A). The baseline corrected recovery curves were fit from numerical simulations giving a D = 15 μm2 s−1 for EGFP-actin with a mobile fraction of 60%, and D = 4 μm2 s−1 for EGFP-Myo1a with a mobile fraction of 90% (Fig. 8 B; error brackets are shown in Fig. 8, B and C). As may be expected, the two recovery curves intersect each other in a way that is similar to simulations of two situations with different diffusion coefficients and different mobile fractions (see Fig. 7 B).
FIGURE 8.

Mobility of EGFP-Myo1a and EGFP-actin in the brush-border of Caco-2 cells. (A) The recovery curves with (bottom) or without (baseline, top) a photobleaching pulse are shown for EGFP-Myo1a (○) and EGFP-actin (•). (B) The difference of these two curves (baseline minus recovery) is shown for EGFP-Myo1a (○). Data fit with simulated curves are shown (continuous lines), with D = 4 μm2 s−1 (best fit), D = 3 μm2 s−1 (curve above the data) and D = 5 μm2 s−1 (curve under the data) and a mobile fraction of 90% (mean ± SD of 273 curves for EGFP-Myo1a and 201 curves for EGFP-actin). (C) Same experimental curve as in B but with different values of the mobile fraction for the simulated curves: 90% for the best fit, 80% for the curve above the data, and 100% for the curve under the experimental data. (D) Data are shown for both EGFP-Myo1a (○) and EGFP-actin (•) with simulated curves (only best-fit curves are shown here). The EGFP-actin curve was fit with D = 15 μm2 s−1 and a mobile fraction of 60%.
The dynamics of the actin cytoskeleton depend on the equilibrium between filamentous (F-actin) and monomeric actin (G-actin) sequestered by proteins such as thymosin β4. Data collected in the presence of an actin filament stabilizer, jasplakinolide, give similar results to fixed cells (Fig. 9). This confirms that the diffusive fraction we measure (60%) corresponds mainly to sequestered monomeric actin in the microvilli. These observations suggest that under these experimental conditions (0.01–10 s with a small focal volume) the dynamics of monomeric actin but not that of the more slowly moving F-actin are being monitored by two-photon FRAP.
FIGURE 9.

Effect of jasplakinolide treatment on EGFP-actin mobility. The difference between the baseline curve and recovery curve is represented for EGFP-actin before (○) and after (•) jasplakinolide treatment, and for immobile actin in fixed cells (▵). (Mean of 69 curves cells expressing EGFP-actin treated with jasplakinolide.)
The dynamics of Myo1a depends on its motor activity
The mobility of EGFP-Myo1a is not directly related to the mobility of monomeric actin which has a larger diffusion coefficient, or to that of F-actin which is immobile on the timescale of the recovery measurements. To determine whether the mobility of EGFP-Myo1a depends on its motor activity, we examined the influence of depleting ATP which is required for myosin motor activity. The fluorescent recovery amplitude of EGFP-Myo1a decreased by 20% after depletion of the intracellular ATP whereas the fluorescent recovery of EGFP-actin remained unchanged under the same conditions (Fig. 10). This suggests that the processes that lead to the recovery of myosin and actin are not coupled, in agreement with the fact that they show different mobilities under nonperturbing conditions. The recovery of EGFP-Myo1a was also slightly reduced when cells were treated with BDM, a drug that inhibits the ATPase activity of some myosin proteins including Myo1a (Cramer and Mitchison, 1995) (Fig. 11 A). As a control, measurements show that the dynamics of a nonfunctional Myo1a where the motor domain is deleted, EGFP-Myo1aΔ730, are not sensitive to ATP depletion (in agreement with the lack of an ATPase domain; Fig. 10), or the BDM treatment (Fig. 11 A). As may be expected, the recovery of EGFP-Myo1aΔ730 is identical to that of WT-Myo1a after BDM treatment. According to numerical simulations, the diffusion coefficient after suppression of the motor activity or its inhibition is estimated at 2.8 μm2 s−1 with a mobile fraction of 84%. Both the diffusion coefficient and the mobile fraction decrease in a statistically significant manner when the motor activity of EGFP-Myo1a is inhibited or deleted. This suggests that the change in mobility observed after BDM treatment of EGFP-Myo1a, or for the EGFP-Myo1aΔ730 deletion mutant, is due to loss of the motor activity.
FIGURE 11.

Influence of the motor activity of EGFP-Myo1a on its mobility. The difference between the baseline curve and recovery curve is represented for EGFP-Myo1a (○), EGFP-Myo1a after BDM treatment (•), and EGFP-Myo1aΔ730 (▵). (A) Fits for EGFP-Myo1a (•) with D = 4 μm2 s−1, and mobile fraction 90%, and for EGFP-Myo1aΔ730 (○) with D = 2.8 μm2 s−1 and a mobile fraction 84% are shown in B (mean ± SD of 207 curves for cells expressing EGFP-Myo1aΔ730, and 112 for cells expressing EGFP-Myo1a before and after BDM treatment).
DISCUSSION
The two-photon FRAP instrument combined with the numerical method presented here allows us to measure the millisecond dynamics of proteins in volumes smaller than 1 μm3. Two-photon FRAP was conducted because it allows for a precise interpretation of the recovery data due to the strict overlap between the fluorescence excitation volume and the detection volume. This is not the case with one-photon excitation, and thus makes quantitative interpretations more complicated aside from proteins within the simple two-dimensional geometry of the cell membrane. As a consequence, we believe that many FRAP experiments (especially one-photon) are often over- or mis-interpreted.
Methodological aspects
Up to now two-photon FRAP has been mainly used in cell biology as a semi-quantitative tool, based on early experimental work by Peters et al. (1974) and on the mathematically rigorous framework defined by the seminal work of Axelrod et al. (1976). This work is based on the explicit assumptions that the bleaching pulse can be made short enough to neglect any diffusion during it (H1), and that the excitation power used to measure relaxation causes negligible photobleaching (H2). Most methodological variations of this work remain within this initial framework, although adding novel ingredients such as numerical simulations (see Meyvis et al., 1999 for a review), Fourier transform approach (Tsay and Jacobson, 1991), various geometries, continuous photobleaching rather than pulse/relax scheme (Cutts et al., 1995; Kubitscheck et al., 1998; Braeckmans et al., 2003), effect of receptor binding (Sprague et al., 2004), and adaptation to confocal microscopy (Braeckmans et al., 2003) have been done. To our knowledge, only our previous work (Coscoy et al., 2002), and two other methods modeling continuous bleaching along lines (Kubitscheck et al., 1998), or on single points (Cutts et al., 1995; Wachsmuth et al., 2004), explicitly treat the full diffusion-bleaching equation. Since the H1 and H2 hypotheses are often not valid, we based the present modeling effort on keeping the diffusion and the fluorescence bleaching terms together in the equation that describes the temporal variation of fluorescence intensity. This allows one to extract estimates of the mobility of fluorophores even if these two hypotheses are not satisfied. Since the photobleaching rate is not a constant but a function of the excitation intensity, the equation cannot be solved analytically, except in simple boundary condition cases, making numerical simulations necessary.
To our knowledge, spatial concentration depletion has not been studied in terms that practically apply to the scheme of FRAP experiments as performed here. An analytical approach has been used to describe the steady-state regime in a spherical geometry (Mertz, 1998), but the case of a transient regime with a Gaussian excitation volume has not been treated. Another approach has been proposed to model the depletion starting from a fixed trap, but does not apply to the fluorescence recovery problem (Peng et al., 2003). The consequence of spatial concentration depletion on recovery curves is clearly illustrated here by experiments on EGFP in aqueous solution (Fig. 5). These experiments show a shift in the recovery curves that can be qualitatively interpreted as follows: the concentration relaxation spectrum of the diffusion process is cut off at all timescales shorter than the duration of the bleaching pulse. The qualitative results reported in the present article concerning the spatial depletion are summarized as follows:
The concentration profile widens with increased bleaching pulse duration Tp, thus leading to underestimated apparent diffusion coefficients.
The photobleaching depth is bound by a maximal value obtained with immobile fluorophores, and shallower bleaching depths occur with increases in the diffusion coefficient, D. Notably with sufficient care, the photobleaching depth can be used to assess D.
Fluorescence recovery does not reflect the number of fluorophores returning to the detection volume, up to a point where non-monotonous recovery curves can be obtained.
Exact interpretations are impossible without a detailed analysis of the timescales involved in the photobleaching and diffusion processes.
Our simulations were done for one-dimensional diffusion but the basic framework can be extended in three dimensions, as shown in the experiments with EGFP.
Biological aspects
The modeling work reported here has been carried out to provide quantitative information on the molecular movements of cytoskeletal proteins in the cellular environment. The level of refinement to which the modeling has been developed is not aimed at extracting absolute D values, but rather at allowing for precise comparisons between the dynamics of proteins in non-homogeneous, and geometrically complex environments. The motivation is that the determination of the motions of proteins, their turnover rates and their residence time, in conjunction with pharmacological perturbations and molecular mutations, should provide insight into protein function. Our earlier work provided the first example of this kind of approach in a study of the dynamics of ezrin in the microvilli of renal epithelial cells (Coscoy et al., 2002). Two other important proteins in the microvilli are actin and Myo1a (formerly called brush-border myosin I), which are naturally expected to require each other for their function. Nevertheless, the exact role of Myo1a is not known.
Our data suggest that 60% of EGFP-actin is in a fast moving soluble G-actin pool, whereas the other 40% is in an immobile F-actin fraction. Some of the immobile actin is likely F-actin which can undergo treadmilling-based movement (velocity ∼0.1 μm s−1) but this slow motion would not be detected under our experimental conditions. The resulting G-/F-actin ratio in microvilli is similar to ratios determined in other cytoplasmic domains using biochemical approaches (Weber et al., 1992) but in this case is measured in a precise location in intact cells. The experiments conducted here were typically limited to a maximum recovery time of 10 s. A previous analysis of the dynamics of EGFP-actin by one-photon FRAP with a different timescale (5–80 s), in a different epithelial cell line (LLCPK1), has suggested two types of turnover—i.e., fast and slow (Tyska and Mooseker, 2002). However, these data cannot be compared to this work for four main reasons:
The photobleaching pulse duration is not known in these experiments, having an unknown consequence on the spectrum of recovery times.
The geometry of one-photon excitation is such that the photobleached volume is extended along the optical axis, thus potentially increasing the effective time for recovery.
The timescale of our experiments is much shorter (10 s with 11 ms time resolution instead of 80 s with 3 s resolution), and the consequences on the observational bleaching are likely to be very different. A meaningful comparison would require that observational bleaching be quantified in the work of Tyska and Mooseker (2002).
The slow and fast components reported by Tyska and Mooseker (2002) might be due to spatial depletion. Indeed, 30–40% of fluorescence has already been recovered within the first 5 s, and this might encompass most of the dynamics of G-actin. In contrast, diffusion coefficients assessed here for actin in Caco-2 cells are similar to those determined in endothelial cells (3.1–5.8 μm2 s−1) and in LLCPK1 renal cells (10 μm2 s−1) using the same two-photon FRAP method (Coscoy et al., 2002; McGrath et al., 1998).
The data suggest that the dynamics of Myo1a are uncoupled from that of actin and are dependent on the Myo1a motor domain. Indeed, treating with BDM or complete deletion of the motor domain result in the same modification to the recovery curves. It is important to point out that Myo1a has slow biochemical kinetics and supports actin movement in vitro with an average speed as slow as 0.08 μm s−1 (Collins et al., 1990). However, the introduction of a convective term in numerical simulations, with such low velocities (from 0.03 to 0.1 μm s−1), did not modify recovery curves in a detectable way on the timescale of our experiments (data not shown). The difference observed in terms of diffusion coefficients and mobile fractions between wild-type EGFP-Myo1a and Myo1a without motor activity (D = 2.8 μm2 s−1, mobile fraction of 84% versus D = 4 μm2 s−1, mobile fraction of 90%) may specifically reflect the motor activity within microvilli.
Similar to EGFP-actin, the one-photon FRAP work on LLCPK1 cells by Tyska and Mooseker (2002) reported two kinetic components (slow and fast rates) for EGFP-Myo1a. In addition, they did not observe any difference in the dynamics of WT EGFP-Myo1a and a tail deletion mutant similar to Myo1aΔ730. The discrepancy between these observations and ours might be due to different experimental conditions and cell types (see the four reasons detailed above). This is especially significant here because endogenous Myo1a has never been detected in the microvilli of LLCPK1 cells and may not play a functional role in that cell type.
Altogether our studies show for the first time in vivo that the dynamics of Myo1a in the intestinal microvilli are dependent on its ATPase activity and on the integrity of its motor domain. These results take the first step toward understanding the function of Myo1a within the highly specialized architecture of the brush-border.
Acknowledgments
We thank Patricia Bassereau, Jacques Prost, and Daniel Louvard for sharing important ideas and Véronique Bastouill for contributing some programs.
F.W. and S.C. were supported by fellowships from the Association de la Recherche Contre le Cancer. C.B. was supported by fellowships from the Institut Curie and the European Molecular Biology Organisation. This work was supported by grants from the Institut Curie, the Fondation de la Recherche Médicale, the Association de la Recherche Contre le Cancer, and the Centre National de la Recherche Scientifique to F.A.
Francois Waharte's present address is Institut de Biologie de Lille, Lab. “Imagerie Cellulaire Fonctionnelle”, 1 rue du Prof Calmette, 59021 Lille Cedex (France).
Claire M. Brown's present address is University of Virginia, Dept. of Cell Biology, 1300 Jefferson Park Ave., Charlottesville, VA 22908.
References
- Axelrod, D., D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacallao, R., A. Garfinkel, S. Monke, G. Zampighi, and L. J. Mandel. 1994. ATP depletion: a novel method to study junctional properties in epithelial tissues. I. Rearrangement of the actin cytoskeleton. J. Cell Sci. 107:3301–3313. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., B. Wehrle-Haller, and B. A. Imhof. 1998. Actin dynamics in living mammalian cells. J. Cell Sci. 111:1649–1658. [DOI] [PubMed] [Google Scholar]
- Bartles, J., L. Zheng, A. Li, A. Wierda, and B. Chen. 1998. Small espin: a third actin-bundling protein and potential forked protein ortholog in brush border microvilli. J. Cell Biol. 143:107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckmans, K., L. Peeters, N. N. Sanders, S. C. De Smedt, and J. Demeester. 2003. Three-dimensional fluorescence recovery after photobleaching with the confocal scanning laser microscope. Biophys. J. 85:2240–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, E. B., E. S. Wu, W. Zipfel, and W. W. Webb. 1999. Measurement of molecular diffusion in solution by multiphoton fluorescence photobleaching recovery. Biophys. J. 77:2837–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, F. P., W. L. Vaz, and E. Melo. 1997. Phase topology and percolation in two-component lipid bilayers: a Monte Carlo approach. Biophys. J. 72:1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, K., J. R. Sellers, and P. Matsudaira. 1990. Calmodulin dissociation regulates brush border myosin I (110-kD-calmodulin) mechanochemical activity in vitro. J. Cell Biol. 110:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscoy, S., F. Waharte, A. Gautreau, M. Martin, D. Louvard, P. Mangeat, M. Arpin, and F. Amblard. 2002. Molecular analysis of microscopic ezrin dynamics by two-photon FRAP. Proc. Natl. Acad. Sci. USA. 99:12813–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudrier, C., D. Kerjaschki, and D. Louvard. 1988. Cytoskeleton organization and submembranous interactions in intestinal and renal brush borders. Kidney Int. 34:309–320. [DOI] [PubMed] [Google Scholar]
- Cramer, L. P., and T. J. Mitchison. 1995. Myosin is involved in postmitotic cell spreading. J. Cell Biol. 131:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutts, L. S., P. A. Roberts, J. Alder, M. C. Davies, and C. D. Melia. 1995. Determination of localised diffusion coefficients in gels using confocal scanning microscopy. J. Microsc. 180:131–139. [Google Scholar]
- Durrbach, A., K. Collins, P. Matsudaira, D. Louvard, and E. Coudrier. 1996. Brush border myosin-I truncated in the motor domain impairs the distribution and the function of endocytotic compartments in an hepatoma cell line. Proc. Natl. Acad. Sci. USA. 93:7053–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrbach, A., G. Raposo, D. Tenza, D. Louvard, and E. Coudrier. 2000. Truncated brush border myosin I affects membrane traffic in polarized epithelial cells. Traffic. 1:411–424. [DOI] [PubMed] [Google Scholar]
- Feder, T. J., I. Brust-Mascher, J. P. Slattery, B. Baird, and W. W. Webb. 1996. Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys. J. 70:2767–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, A., E. Coudrier, J. Carboni, J. Anderson, J. Vandekerkhove, M. Mooseker, D. Louvard, and M. Arpin. 1989. Partial deduced sequence of the 110-kD-calmodulin complex of the avian intestinal microvillus shows that this mechanoenzyme is a member of the myosin I family. J. Cell Biol. 109:2895–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, G. W., B. Chazotte, X. F. Wang, and B. Herman. 1995. Analysis of simulated and experimental fluorescence recovery after photobleaching. Data for two diffusing components. Biophys. J. 68:766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzelman, M. B., T. Hasson, and M. S. Mooseker. 1994. Multiple unconventional myosin domains of the intestinal brush border cytoskeleton. J. Cell Sci. 107:3535–3543. [DOI] [PubMed] [Google Scholar]
- Kubitscheck, U., P. Wedekind, and R. Peters. 1998. Three-dimensional diffusion measurements by scanning microphotolysis. J. Microsc. 192:126–138. [DOI] [PubMed] [Google Scholar]
- Lopez, A., L. Dupou, A. Altibelli, J. Trotard, and J. F. Tocanne. 1988. Fluorescence recovery after photobleaching (FRAP) experiments under conditions of uniform disk illumination. Critical comparison of analytical solutions, and a new mathematical method for calculation of diffusion coefficient D. Biophys. J. 53:963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. L., Y. Tardy, C. F. Dewey, Jr., J. J. Meister, and J. H. Hartwig. 1998. Simultaneous measurements of actin filament turnover, filament fraction, and monomer diffusion in endothelial cells. Biophys. J. 75:2070–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyvis, T. K. L., S. C. De Smedt, P. Van Oostveldt, and P. Demeester. 1999. Fluorescence recovery after photobleaching: a versatile tool for mobility and interaction measurements in pharmaceutical research. Pharm. Res. 16:1153–1161. [DOI] [PubMed] [Google Scholar]
- Mertz, J. 1998. Molecular photodynamics involved in multi-photon excitation fluorescence microscopy. Eur. Phys. J. D. 3:53–66. [Google Scholar]
- Olveczky, B. P., and A. S. Verkman. 1998. Monte Carlo analysis of obstructed diffusion in three dimensions: application to molecular diffusion in organelles. Biophys. J. 74:2722–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, H., S. H. Park, P. Argyrakis, H. Taitelbaum, and R. Kopelman. 2003. Dynamics of the depletion zone at a finite-sized imperfect trap in two dimensions: photobleaching experiments and simulations. Phys. Rev. E. 68:061102. [DOI] [PubMed] [Google Scholar]
- Periasamy, N., and A. S. Verkman. 1998. Analysis of fluorophore diffusion by continuous distributions of diffusion coefficients: application to photobleaching measurements of multicomponent and anomalous diffusion. Biophys. J. 75:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, R., J. Peters, K. H. Tews, and W. Bahr. 1974. A microfluorimetric study of translational diffusion in erythrocyte membranes. Biochem. Biophys. Acta. 367:282–294. [DOI] [PubMed] [Google Scholar]
- Peterson, M. D., and M. S. Mooseker. 1992. Characterization of the enterocyte-like brush border cytoskeleton of the C2BB clones of the human intestinal cell line, Caco-2. J. Cell Sci. 102:581–600. [DOI] [PubMed] [Google Scholar]
- Piston, D., E. Wu, and W. Webb. 1992. Three-dimensional diffusion measurements in cells by two-photon excitation fluorescence photobleaching recovery. Biophys. J. 61:A34. [Google Scholar]
- Schram, V., J. F. Tocanne, and A. Lopez. 1994. Influence of obstacles on lipid lateral diffusion: computer simulation of FRAP experiments and application to proteoliposomes and biomembranes. Eur. Biophys. J. 23:337–348. [DOI] [PubMed] [Google Scholar]
- Sprague, B. L., R. L. Pego, D. A. Stavrena, and J. G. McNally. 2004. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 86:3473–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stidwill, R. P., T. Wysolmerski, and D. R. Burgess. 1984. The brush border cytoskeleton is not static: in vivo turnover of proteins. J. Cell Biol. 98:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan, R., C. Hoang, and A. Verkman. 1997. Photobleaching recovery and anisotropy decay of GFP-S65T in solution and cells: cytoplasmic viscosity probed by GFP translational and rotational diffusion. Biophys. J. 72:1900–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardy, Y., J. L. McGrath, J. H. Hartwig, and C. F. Dewey. 1995. Interpreting photoactivated fluorescence microscopy measurements of steady-state actin dynamics. Biophys. J. 69:1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay, T. T., and K. A. Jacobson. 1991. Spatial Fourier analysis of video photobleaching measurements: principles and optimization. Biophys. J. 60:360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyska, M. J., and M. S. Mooseker. 2002. Myo1a (brush border myosin I) dynamics in the brush border of LLC-PK1–CL4 cells. Biophys. J. 82:1869–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsmuth, M., T. Weidemann, G. Müller, U. Hoffmann-Roher, T. A. Knoch, W. Waldeck, and J. Langowski. 2004. Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys. J. 84:3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, A., V. T. Nachimias, C. Pennise, M. Pring, and D. Safer. 1992. Interaction of thymosin β4 with muscle and platelet actin: implications for actin sequestration in resting platelets. Biochemistry. 31:6179–6185. [DOI] [PubMed] [Google Scholar]
- Wolf, D. E. 1992. Theory of fluorescence recovery after photobleaching measurements of cylindrical surfaces. Biophys. J. 61:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson, M. M., and G. J. Pert. 1999. An Introduction to Computer Simulation. Oxford University Press, Oxford, UK.