

Abstract
The spread of acquired immune deficiency syndrome has increasingly become a great concern owing largely to the failure of chemotherapies. The G48V is considered the key signature residue mutation of HIV-1 protease developing with saquinavir therapy. Molecular dynamics simulations of the wild-type and the G48V HIV-1 protease complexed with saquinavir were carried out to explore structure and interactions of the drug resistance. The molecular dynamics results combined with the quantum-based and molecular mechanics Poisson-Boltzmann surface area calculations indicated a monoprotonation took place on D25, one of the triad active site residues. The inhibitor binding of the triad residues and its interaction energy in the mutant were similar to those in the wild-type. The overall structure of both complexes is almost identical. However, the steric conflict of the substituted valine results in the conformational change of the P2 subsite and the disruption of hydrogen bonding between the −NH of the P2 subsite and the backbone −CO of the mutated residue. The magnitude of interaction energy changes was comparable to the experimental Ki data. The designing for a new drug should consider a reduction of steric repulsion on P2 to enhance the activity toward this mutant strain.
INTRODUCTION
The spread of acquired immune deficiency syndrome (AIDS) has constantly threatened the world because the disease leads to a significant loss of morbidity and mortality. Unfortunately, chemotherapy for the disease has, in many cases, failed to achieve complete viral suppression (Deeks, 2003). This relies on the fact that the human immunodeficiency virus (HIV) develops resistance to antiretroviral drugs by genetic mutation. The development of novel drug targets and HIV vaccine is promising but the results of those studies remain far from the clinical stage. Understanding the mutations that confer resistance to available drugs is thus an urgent issue in HIV chemotherapy.
The HIV type-1 protease (HIV-1 PR) is an important target for AIDS chemotherapy. This viral protein cleaves the gag and pol nonfunctional polypeptide into functional proteins essential for maturation of infectious HIV particles (Debouck et al., 1987). The protein is a homodimer. Each protein monomer consists of 99 amino acids (Meek et al., 1989). From x-ray data (Fig. 1 B), the substrate/inhibitor binding site is located at the dimer interface (Hong et al., 1996, 2000; Jaskolski et al., 1991; Krohn et al., 1991; Swain et al., 1990; Vondrasek and Wlodawer, 2002). As a member of the aspartyl protease family, HIV-1 PR is composed of the conserved sequences, so-called the binding triads: D25-T26-G27 and D25′-T26′-G27′, of which D25 and D25′ are known as the active site residues. These two ionizable residues play a major role in the catalytic reaction.
FIGURE 1.

Schematic representation of saquinavir (A) and the wild-type HIV-1 protease-saquinavir complex (B). According to a conventional classification of the protease subsites, the binding pockets are designated by the inhibitor side chains P1, P2, P3, P1′, and P2′.
Because of the therapeutically important enzyme, structural and functional studies have been carried out to gain understanding of molecular mechanisms of the proteolytic cleavage process (Hyland et al., 1991; Northrop, 2001; Okimoto et al., 1999; Scott and Schiffer, 2000; Smith et al., 1996). The size and the availability of high-resolution x-ray structures of HIV-1 PR are amenable for molecular dynamics (MD) technique to investigate the relationship of structure, dynamics, and function of the enzyme (Collins et al., 1995; Harte et al., 1992, 1990; Levy and Caflisch, 2003; Piana et al., 2002; Scott and Schiffer, 2000) as well as to serve as a test system for developing computational methodology (Piana et al., 2004, 2001; Wang and Kollman, 2000; York et al., 1993a). The MD approach has provided insightful information on the enzyme-substrate interactions and binding conformations, the protonation states of the active site residues, the role of the flexible flap and the binding waters, and drug resistance. Characterizations of structural intermediates have been made useful for rational drug design (Randolph and DeGoey, 2004; Roberts et al., 1990; Rodriguez-Barrios and Gago, 2004).
Saquinavir (SQV, Fig. 1 A), a peptidomimetic protease inhibitor, is clinically used to treat infected HIV patients. The inhibitor containing a nonhydrolyzable hydroxyethylene isostere was designed based on the transition state structure in the enzyme-substrate complex. Combination of PR and reverse transcriptase inhibitors appears to be a highly effective treatment against HIV (Boucher, 1996). The PR inhibitor blocks the maturation step of the HIV life cycle, which is the crucial stage in the formation of new viral particles. Nevertheless, the current cure with SQV has introduced several resistant variants of HIV-1 PR, some of which can dramatically reduce drug susceptibility (Vondrasek and Wlodawer, 2002). G48V and L90M are considered as the primary mutations commonly occurring in vivo or in vitro (Eberle et al., 1995; Vaillancourt et al., 1999). These “signature” residue mutations can be associated with a dramatic decrease in drug susceptibility. According to Ki values, the G48V, L90M, and G48V/L90M mutants decrease saquinavir sensitivity by 13.5-, 3-, and 419-fold with respect to that of the wild-type (wt) protease (Ermolieff et al., 1997).
Among common mutations associated with antiretroviral drug resistance, G48V is a unique mutation characteristically generated by SQV. In a view of substituted-type residue, glycine was replaced by a bulkier side-chain residue. The steric conflict of the mutant should involve in a destabilization of the complex. Although several x-ray structures of the HIV-1 PR provided valuable information on the inhibitor binding, this is not the case for the primary resistance to SQV. The crystal structure of the G48V complex is not yet available. With the aid of the available x-ray data, the molecular modeling techniques offer an opportunity to investigate the structural basis of the mutant enzyme (Prabu-Jeyabalan et al., 2003; Swain et al., 1990).
The missing hydrogens in the structural data have led to studies of the ionization state of the active site residues D25/D25′ (Smith et al., 1996; Wang et al., 1996; Wlodawer and Vondrasek, 1998; Yamazaki et al., 1994). This subject is important for drug design in a way to optimize the interactions of the inhibitor with the enzyme. Different protonation models were found depending upon the local environment of the enzyme-inhibitor complex. The single protonation at one of the two acidic residues has been most commonly observed with the binding of the hydroxylethylene-based inhibitors (Baldwin et al., 1995; Chen and Tropsha, 1995; Hyland et al., 1991; Smith et al., 1996; Wang et al., 1996; Wlodawer and Vondrasek, 1998). From NMR experiments, the neutral D25/D25′ side chain (diprotonation) was determined in the presence of inhibitor diol groups (Yamazaki et al., 1994), whereas the dianionic form (unprotonation) was observed in the free enzyme (Smith et al., 1996; Wang and Kollman, 2000).
In this study, we employed a computational approach to access information regarding molecular structure and dynamics of the G48V HIV-1 protease conferring to saquinavir resistance. The MD simulations were carried out for the wt and the G48V HIV-1 protease complexed with saquinavir in explicit aqueous solution. The study of the protonation state of the HIV-1 PR complexed with SQV has been carried out before exploring the structure and dynamic data of the signature resistance. The density functional theory (DFT), ONIOM, and molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) methods have been performed to identify the protonation model of the active site residues. The MM/PBSA approach offers an efficient computation for calculating the binding free energy of biomolecules (Kollman et al., 2000; Srinivasan et al., 1998; Wang and Kollman, 2000). The method has been extensively used to study protein-ligand complexes. The quantum-based approach, DFT and ONIOM, has been useful in providing accurate energy information of the interested region. In particular, the hybrid quantum mechanical/molecular mechanical (QM/MM) method, ONIOM (our own N-layered integrated molecular orbital and molecular mechanics), developed by Morokuma, has been extended from small molecules to biological applications (Friesner and Beachy, 1998; Morokuma, 2002; Prabhakar et al., 2004; Torrent et al., 2002). Its efficiency has been improved over the years. Simply, the concept of the ONIOM approach is partitioning a large molecular system into onion skin-like layers, and applying the quantum mechanics and molecular mechanics methods to the defined different parts (Morokuma, 2002). In the partitioned system, the high-level quantum computations engage the essential part of the central activity, whereas the lower-level energy calculations take into account the contribution of the remaining region. The comparison of MD results of the two systems provides insightful details of how the G48V mutant is associated with saquinavir resistance. The study provided fundamental principles on the molecular mechanism of inhibitor binding and resistance, which will be useful for designing an anti-HIV inhibitor to combat AIDS.
METHODS
Before starting the MD simulations, the problem of the protonated state of the active site residues was addressed, since such information cannot be directly obtained from the x-ray data. Our approach consists of molecular orbital energy calculations and solvent continuum free-energy calculations. However, structural data of the complexes, particularly the structure of the G48V-SQV, are not available, and the quantum-based computation for the whole enzyme-inhibitor complex is not feasible. A strategy is developed. First, MD simulations of the four protonation states for the wt and for the G48V complexes were performed to obtain the protonation models, which were subsequently subjected to calculate interaction and binding energies. Details of the methodology are described as follows.
Initial structure
The x-ray structure of the wt HIV-1 PR complexed with Ro 31-8959, saquinavir, (Protein Data Bank code 1HXB; 2.3 Å resolution) was used as a starting model. All missing hydrogens of the protein were added using the LEaP module in the AMBER 7 software package (Case et al., 2002). The protonation state of the ionizable residues, the C- and the N-termini, except for D25/25′, was assigned based on the predicted pKa values at pH 7. The pKas of ionizable residues were calculated based on the Poisson-Boltzmann free-energy calculations (see the pKa prediction). The results concluded that all Lys, Arg, Glu, Asp, and the terminal groups are charged, whereas His was in the neutral form. Protonation states of D25/25′ were explicitly assigned into four different ionizable states, including unprotonation, monoprotonation (each site of D25 and D25′), and diprotonation (protonated at both aspartyl residues). For the wt study, the simulated systems were labeled as wt-unpro, wt-mono25, wt-mono25′, and wt-dipro, respectively.
The starting structure and force-field parameters for the inhibitor were obtained as follows. Hydrogens were added to the x-ray coordinates of SQV (1HXB) by taking into account the hybridization of the covalent bonds. Geometric optimization was subsequently performed at the Hartree-Fock level with 6-31G** basis functions to adjust the bond-length involving hydrogens. Then, the RESP fitting procedure was employed to calculate partial atomic charges of the inhibitor (Cornell et al., 1993). Force-field parameters of the inhibitor were assigned based on the atom types of the Cornell et al. (1995) force-field model. Gaussian 98 (Frisch et al., 2002) was used to optimize the molecular structure, generate electrostatic potentials, and calculate ab initio energies. Partial charge generation and assignment of the force field were performed using the Antechamber suite (Wang et al., 2001).
The preparation of the initial structure for the simulation of the G48V mutant-SQV complex was similar to that of the wt complex. The comparative model of the mutant was constructed based on 1HXB because the three-dimensional structure of the G48V-SQV complex is not available. It should be noted that the x-ray structure of the double mutant, G48V/L90M-SQV complex (1FB7) could be considered as an alternative template. However, the x-ray coordinates of the second monomer of the double mutant are not available. Thus, 1HXB is considered to be more appropriate as a template. The simulated systems of the mutant complex consist of four protonated states, which were defined similar to those of the wt complex, i.e., unprotonation (mt-unpro), monoprotonations (mt-mono25 and mt-mono25′), and diprotonation (mt-dipro).
The next step was to incorporate the solvent and counterions into the models previously prepared. The crystallographic waters were also included in the simulations. Each model was solvated with the TIP3P waters (Jorgensen et al., 1983) and neutralized by the counterions using the LEaP module. The total number of the TIP3P waters in the periodic box for all systems was in a range of 9100–9900 molecules.
Molecular dynamics simulations
Energy minimization and MD simulations were carried out using the SANDER module of AMBER 7 (Case et al., 2002) with the Cornell force field (Cornell et al., 1995). The whole systems were subjected to energy minimization within a range of 200–5000 steepest descent steps to avoid bad contacts. It should be noted that position-restrained minimizations of some particular regions were carried out for systems that clashed during minimization because of incidental overlay of atoms. This procedure was repeated until there was no sign of bad contacts. The resulting protein structure was compared with the before-minimized structure. Root mean-square displacement (RMSD) for nonhydrogen atoms of the compared protein structures showed that there were no RMSDs exceeding 0.1 Å in all systems.
The MD simulation was performed employing the periodic boundary condition with the NPT ensemble. A Berendsen coupling time of 0.2 ps was used to maintain the temperature and pressure of the systems (Berendsen et al., 1984). The SHAKE algorithm (Ryckaert et al., 1977) was employed to constrain all bonds involving hydrogens. The simulation time step of 2 fs was used. All MD simulations were run with a 12 Å residue-based cutoff for nonbonded interactions and the particle-mesh Ewald method was used for an adequate treatment of long-range electrostatic interactions (York et al., 1993a).
The simulation consists of thermalization, equilibration, and production phases. Initially, the temperature of the system was gradually heated from 0 to 298 K during the first 60 ps. Then, the systems were maintained at 298 K until MD reached 400 ps of the simulation. Finally, the production phase started from 400 ps to 1 ns of the simulation. The convergence of energies, temperature, pressure, and global RMSD was used to verify the stability of the systems. The MD trajectory was collected every 0.1 ps. The 600 ps trajectory of the production phase was used to calculate the average structure. All MD simulations were carried for 1 ns. Analysis of all MD trajectories i.e., RMSD, distances, torsion angles, etc. was carried out using the CARNAL and Ptraj modules of AMBER 7. The geometry and stereochemistry of the protein structure were validated using PROCHECK (Laskowski et al., 1996). In summary, a total of eight systems for the MD simulations were carried out.
Graphic visualization and presentation of protein structures were done using RasMol, Swiss-Pdb Viewer (Guex and Peitsch, 1997), WebLab Viewer (Accelrys, San Diego, CA), and MolScript (Kraulis, 1991).
The pKa prediction
An assumption used for assigning the protonation state of the ionizable residues in the simulations was inspected by the prediction of the pKa values. The method estimates the pKa shift by calculating the electrostatic free energy of ionizable residues in the neutral and the charge states in solution (Antosiewicz et al., 1994). The computations were done by solving finite different Poisson-Boltzmann equations implemented in the University of Houston Brownian Dynamics program (Davis et al., 1991). The protocols describe as follows. Polar hydrogens were added to the x-ray model using Insight II (Accelrys, San Diego, CA). For generating electrostatic potentials, the model was then placed in a 65 × 65 × 65 dimension with a grid spacing of 2.5 Å. The focusing technique was additionally employed using finer grid spacing of 1.2, 0.75, and 0.25 Å for a cubic dimension of 15, 15, and 20, respectively (Antosiewicz et al., 1994; Yang et al., 1993). Atomic radii and charges available in the University of Houston Brownian Dynamics program were originally derived from optimized potentials for liquid simulations and CHARMm 22 parameter sets (Brooks et al., 1983; Jorgensen and Tirado-Rives, 1988). The 1.4 Å probe radius with a resolution of 500 dots/atomic-sphere was used. The calculations employed a solvent dielectric of 80 with 150 mM ionic strength, and a temperature of 298 K. A dielectric constant of HIV-1 PR was examined by varying to 1, 4, and 20. We found that a protein dielectric constant of 20 produced the best pKa prediction. A dielectric constant of 1 and 4 yielded unusual pKa values due to an overestimation of electrostatic potentials. This phenomenon is thoroughly discussed in an early work (Antosiewicz et al., 1994).
Protonation state of the HIV-1 PR
In an evaluation of the protonation state of D25/D25′, we employed three different approaches: density functional theory, ONIOM, and MM/PBSA methods. The DFT and ONIOM methods provide the interaction energy of the complex, whereas the MM/PBSA calculates the binding free energy (ΔGbinding). It should be noted that properties of the system should be analyzed using the structure ensemble from the MD trajectory. However, quantum chemical methods are too expensive to calculate such enormous structural data. Alternatively, the statistically averaged structure obtained from the 600 ps production phase of the MD trajectory was chosen as the studied model.
The DFT method
In each protonation model, the cluster consists of the two triad residues, D25-T26-G27 and D25′-T26′-G27′, and SQV (see Fig. 3 A). Atoms that are not within the selected part were removed. To reduce possible terminal-charge effect, both ends of the triad fragments were capped by CH3NH− and −COCH3 groups. Geometric optimization was performed for the added atoms. The energy for the model was computed using a single-point calculation method with mixed basis sets. B3LYP/6-31 + G** was defined explicitly on the carboxylate oxygens of D25 and D25′, and B3LYP/6-31G** was assigned on all the remaining atoms. The quantum calculations were carried out using the program Gaussian 98 (Frisch et al., 2002).
FIGURE 3.

ΔEcpx between the triad residues and saquinavir of the wt and the G48V complexes for the dipro, mono25, and mono25′ systems.
A general form of the interaction energy calculation of the enzyme-inhibitor complex (ΔEEI) is the subtraction of the energy of the complex (EEI) from that of the free enzyme (EE) and that of the free inhibitor (EI).
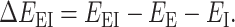 |
(1) |
In this case, the model system contains a cluster of the two triads and SQV. Thus, interaction energy of the complex (ΔEcluster) was estimated by
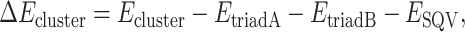 |
(2) |
where Ecluster is the total energy of the cluster, and EtriadA, EtriadB, and ESQV are the total energy of the isolated triads of chain A and B and the unbound SQV, respectively.
The ONIOM method
To account for an effect of the protein environment, the QM/MM ONIOM method was used. Here, the three-layers approach (ONIOM3) was performed to reduce the boundary effect at the QM/MM junction but maintain considerably accurate energy information. The method is described as follows. The model of the HIV-1 PR-SQV complex was divided into three regions: A, B, and C (see the Appendix). The ONIOM layers were represented by inner (A), intermediate (A + B), and real (A + B + C). The inner layer, the “hot spot” region, consisting of D25, D25′, and SQV, was treated at the high-level of quantum chemical calculations using density functional theory (B3LYP/6-31G**). The intermediate layer contains a total of 36 residues including D25-D30, I47-F53, P80-I84, and L90. These residues are located within a 5 Å distance from SQV. This intermediate layer was treated with the semiempirical method using PM3. Lastly, the real layer includes the entire enzyme. The molecular mechanic method using universal force field (UFF) was applied to this layer. All calculations based on the ONIOM approach were carried out using the program Gaussian 98 (Frisch et al., 2002).
Hence, the total energy obtained from the ONIOM3 calculations (EONIOM3) herein can be defined by (see the Appendix):
 |
(3) |
For the overlap regions, the subtraction energy terms are introduced to substitute the low-level energy calculations with the high-level one. Therefore, the total interaction energy (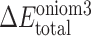 [ABC]) between SQV and the enzyme using the ONIOM3 method can be expressed as independent energy components from each layer as follows:
[ABC]) between SQV and the enzyme using the ONIOM3 method can be expressed as independent energy components from each layer as follows:
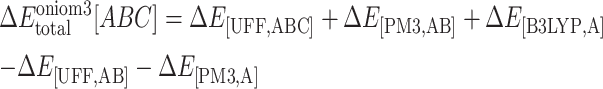 |
(4) |
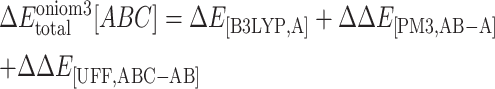 |
(5) |
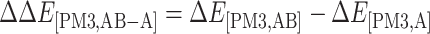 |
(6) |
 |
(7) |
where 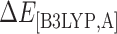 is the interaction energy in the region A evaluated at the B3LYP/6-31G(d,p) level.
is the interaction energy in the region A evaluated at the B3LYP/6-31G(d,p) level. 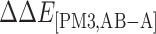 is the interaction energy contributed from the region B evaluated at the PM3 level, and
is the interaction energy contributed from the region B evaluated at the PM3 level, and  is the interaction energy contributed from the region C evaluated at the UFF molecular mechanics.
is the interaction energy contributed from the region C evaluated at the UFF molecular mechanics.
The MM/PBSA method
In general, the free energy of the inhibitor binding, ΔGbinding, is obtained from the difference between the free energy of the receptor-ligand complex (Gcpx), and the unbound receptor (Grec) and ligand (Glig) as follows:
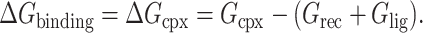 |
(8) |
The MM/PBSA approach calculates ΔGbinding on the basis of a thermodynamic cycle. Therefore, Eq. 8 can be approximated as
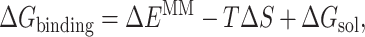 |
(9) |
where ΔEMM is related to the enthalpic changes in the gas phase upon binding and obtained from molecular mechanics van der Waals and electrostatic energies, TΔS involves the entropy effect, and ΔGsol is the free energy of solvation. The ΔGsol is composed of the electrostatic and nonpolar contributions (Srinivasan et al., 1998), and therefore can be expressed as
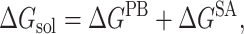 |
(10) |
where ΔGPB is calculated using a continuum solvent model with Poisson-Boltzmann solution (Gilson et al., 1987), and ΔGSA is estimated from the solvent-accessible surface area (SASA) (Sitkoff et al., 1994).
The ΔGbinding was obtained using the MM/PBSA module in the program AMBER 7, which interfaces the program DelPhi 4 (Rocchia et al., 2001). To calculate electrostatic free energy of solvation, the grid resolution of 0.5 Å with the boundary conditions of Debye-Huckel potentials was employed. Atomic charges were taken from the Cornell force field (Cornell et al., 1995). The water and protein dielectric was set to 80 and 4, respectively. The SASA was calculated using a 1.4 Å probe radius. Atomic radii were taken from the PARSE parameter set (Sitkoff et al., 1994). The nonpolar free energy of solvation was obtained by 0.00542 × SASA + 0.92 kcal mol−1 (Sitkoff et al., 1994).
In the study, the contribution of the entropy (TΔS) was not included. An estimation of the entropy effect from normal mode analysis requires the high computation demands. The effect should be very small because all system models are very similar. In addition, we considered the relative values of the binding free energy.
RESULTS
Hydrogen bonding in the binding site
One of the most important HIV-1 PR-SQV interactions is the formation of the hydrogen bond at the active site. This observation is estimated from the x-ray data by the close proximity between the terminal side chain of the two aspartyl residues and the hydroxylethylene isostere moiety (−OH) of the inhibitors. Due to the lack of hydrogen position in the structure, the pattern of this typical hydrogen bond is investigated here. Therefore, it is necessary to monitor the active site structure of the MD results and determine the most preferential interactions.
The MD snapshot of the dipro, mono25, mono25′, and unpro systems for the wt and the G48V complexes is illustrated in Fig. 2, A–H. The D25/D25′ side chains and the −OH of SQV of all protonated states, except for the unprotonation, occupies the positions suitable for the formation of the hydrogen bond. The distal separation between the active site residues and the hydroxylethylene isostere of SQV maintains similarity to the x-ray structure. The structure of the wt-unpro and the mt-unpro provide the worst scenario of the complex (Fig. 2, D and H). A majority of MD data shows that the active site residues adopted to a conformation completely different from the x-ray data. Hence, the wt-unpro and mt-unpro systems are not an appropriate state for the formation of the HIV-1 PR-SQV complex.
FIGURE 2.

MD snapshots showing interactions in the binding pocket of the wt and G48V for various protonation states: diprotonation (dipro), monoprotonation of D25 (mono25), monoprotonation of D25′ (mono25′), and unprotonation (unpro). Saquinavir (SQV) and the active site residues D25 and D25′ of the enzyme are shown in stick mode. Chain A and B of the enzyme are colored as blue and green.
In comparing the four protonated states, the binding pattern of the −OH of SQV to the D25/D25′ was remarkably different. Nevertheless, when comparing the wt with the mutant structure of the same protonated state, the binding pattern at the active site was similar. The results allow drawing a simple scheme of the hydrogen bond pattern at the active site, which is illustrated in Fig. 2, I–L.
Protonation state of the wild-type complex
From the pKa prediction, the D25/D25′ in the free wt HIV-1 PR exhibited different protonation states depending on the examined pHs of 4, 5, 6, and 7 because the assay of the HIV-1 PR is measured between pH 5 and 6, and in the basic solution the enzyme precipitates (Wang et al., 1996). However, the dianionic form was the most apparent state between pH 6 and 7. The protonation state at pH 4 and 5 remains inconclusive. In this study, these results were not fully understood. It is possibly associated with the used parameters (atomic charges and radii, dielectric constant, etc.) and the studied model. This awaits further investigation.
Table 1 shows the resulting energy calculated from DFT, ONIOM, and MM/PBSA approaches. Here, the term “the stabilization energy” is used for a simpler and more straightforward definition. The negative sign of the stabilization energy suggests that the protonation model of wt-dipro, wt-mono25, and wt-mono25′ systems are energetically favorable. From the full quantum DFT treatment, the ΔEcluster of wt-mono25 compared to that of wt-dipro and wt-mono25′ was lower by 7.62 and 17.22 kcal mol−1, respectively. The lowest ΔEcpx from the ONIOM3 method also took place on the protonation model of the wt-mono25. The energy difference was 14.13 and 16.63 kcal mol−1 more stable than that of the wt-dipro and of the wt-mono25′, respectively. From the MM/PBSA method, the stabilization energy of the wt-mono25 system was slightly lower in energy than the other two states. Apparently, all the three approaches showed that the monoprotonation at D25 was the most energetically favorable state.
TABLE 1.
The stabilization energy (kcal mol−1)
| DFT
|
ONIOM3
|
MM/PBSA
|
|
|---|---|---|---|
| System | ΔEcluster | ΔEcpx | ΔGcpx |
| wt-dipro | −59.32 | −64.22 | −91.62 |
| wt-mono25 | −66.94 | −78.35 | −94.44 |
| wt-mono25′ | −49.72 | −61.72 | −92.91 |
The stabilization energy shown in Table 1 was decomposed into individual energy components (Table 2) for analyzing quantitatively essential interactions of the different protonation models. From the DFT method, an order of the ΔEtriadA+SQV ranging from the lowest value was wt-dipro < wt-mono25 < wt-mono25′. The sum of ΔEtriadA+SQV and ΔEtriadB+SQV was used to account for interactions of SQV to the triad residues. Still the wt-dipro was the first preferential protonation model with the lowest energy of −34.19 kcal mol−1. Considering the sum of ΔEtriadA+SQV and ΔEtriadB+SQV of wt-mono25, the energy difference was 7.51 kcal mol−1 greater than that of wt-dipro. On the other hand, its ΔEtriadA,B value accounting for the triad-triad interactions has significantly gained by 13.16 kcal mol−1 over the wt-dipro system. These data suggested the interactions between the triads were essential to the complex stability.
TABLE 2.
Decomposition of the stabilization energy (kcal mol−1)
| DFT
|
ONIOM3
|
MM/PBSA
|
||||||
|---|---|---|---|---|---|---|---|---|
| System | ΔEtriadA+SQV | ΔEtriadB+SQV | ΔEtriadA,B | ΔE[B3LYP,A] | ΔE[PM3,AB-A] | ΔE[UFF,ABC-AB] | ΔEMM | ΔGsol |
| wt-dipro | −23.81 | −10.38 | −27.64 | −11.45 | −36.66 | −16.11 | −109.42 | 17.81 |
| wt-mono25 | −12.27 | −14.41 | −40.80 | −33.63 | −28.34 | −16.38 | −109.83 | 15.39 |
| wt-mono25′ | −6.33 | −6.88 | −41.53 | −18.33 | −27.48 | −15.91 | −109.88 | 16.98 |
ΔEtriadA+SQV or ΔEtriadB+SQV account for interactions of the triad residues of chain A or B to SQV, and ΔEtriadA,B accounts for interactions between the triad residues of chain A and those of chain B.
ΔE[B3LYP,A] accounts for interactions of D25, D25′, and SQV.
ΔE[PM3, AB-A] represents interactions of SQV and the 5 Å surrounding residues, excluding D25 and D25′, and ΔE[UFF,ABC-AB] represents interactions of SQV and the remaining residues.
The ONIOM3 results showed the lowest ΔE[B3LYP,A] took place on wt-mono25. It should be noted that ΔE[B3LYP,A] has considered the interactions of SQV to D25 and to D25′, and between D25/D25′. This is still the case when accounting for interactions of the 5 Å neighboring residues to SQV (ΔE[PM3,AB-A]). The ΔE[UFF,ABC-AB] of the three states are almost equivalent. There was no significant change of the stabilization energy with an addition of ΔE[UFF,ABC-AB]. Thus, the effect of the protein environment at the long distance-range interactions is negligible.
In the MM/PBSA method, there was no substantial difference of ΔEMM for all three systems, whereas the ΔGsol of the wt-mono25 revealed the lowest values. This observation suggests the solvation free-energy values dominate the contribution of the stabilization energy. The molecular mechanics energy term cannot discriminate the protonation model.
Interaction energy at the catalytic site: the wild-type versus G48V
We have illustrated previously that the quantum-based approach gave promising results to the characterization of the complex model. Here, the approach has been further exploited by calculating the enzyme-inhibitor interaction energy of the G48V complex. The interaction energy of the triads/SQV in the wt complex, ΔEcluster, previously obtained using the DFT method (Table 1) was used for a comparison.
The results illustrated in Fig. 3 suggested that in the G48V-SQV complex, the lowest ΔEcluster also took place at the monoprotonated D25 model. The interaction energy for both the wt and the G48V in the mono25 was ∼8 and 17 kcal·mol−1 lower than those from the dipro and the mono25′, respectively. At the same protonation state, the ΔEcluster of the G48V was no essentially different from that of the wt. This conclusion followed from the fact that the thermal fluctuation at room temperature of ∼0.6 kcal·mol−1, equivalent to 1 kT, where T is 300 K and k is the Boltzmann constant. Thus the interactions of the two triad residues and SQV remain unchanged by the G48V mutation.
Those results discussed previously lead us to conclude that the monoprotonation at D25 is the most energetically favorable state. Therefore, further comparison and discussion for both the wt and the mutant complex were focused only on the results of monoprotonation D25.
The energies and RMSD plots (Fig. 4) demonstrated a well-behaved MD simulation for both the wt and G48V-SQV systems. After 400 ps, the RMSD fluctuates 1.27–1.78 Å for the wt and 1.29–1.80 Å for the mutant. The fluctuation is <0.5 Å over the entire production phase. This structural fluctuation is not uncommon in the typical MD simulation of protein, indicating the reliable equilibration of the system in this study. In Table 3, the low RMSD values calculated from 100 snapshot structures taken from the production phase suggested the structures in each set were similar to each other. This allows useful information to be extracted from the MD trajectories. Analysis of some statistic quantities such as structure and dynamics was performed from the trajectories of wt-mono25 and mt-mono25 systems.
FIGURE 4.

Plots of the energies (A) and RMSDs (B) versus the simulation time for the wt and G48V-SQV complex. The obtained RMSD was computed using the structure at t = 0 as a reference.
TABLE 3.
Mean global RMSD values calculated from a set of 100 snapshot structures of the 600 ps production phase
| 〈RMSD〉(Å)
|
|||||
|---|---|---|---|---|---|
| Dipro | Mono25 | Mono25′ | Unpro | ||
| wt | Backbone | 0.90 ± 0.13 | 0.86 ± 0.12 | 0.87 ± 0.11 | 0.83 ± 0.11 |
| Heavy atoms | 1.56 ± 0.22 | 1.45 ± 0.17 | 1.48 ± 0.18 | 1.39 ± 0.15 | |
| G48V | Backbone | 0.83 ± 0.11 | 0.86 ± 0.13 | 0.85 ± 0.12 | 0.86 ± 0.12 |
| Heavy atoms | 1.39 ± 0.18 | 1.52 ± 0.22 | 1.51 ± 0.21 | 1.47 ± 0.19 | |
Structural similarity between the wild-type and the mutant
The global backbone RMSD of the x-ray versus the average MD structure of the wt of 1.04 Å indicates that overall main-chain structures of the complex in the crystal state and in solution are similar. In addition, comparison of the two average MD structures between the wt and the G48V mutant resulted in a backbone RMSD of 0.70 Å. This suggests that the tertiary structure of the G48V mutant was insignificantly different from the native enzyme.
Detailed analysis of RMSD per residue is illustrated in Fig. 5. One can see that most regions of the enzyme, except for the flexible loop of the first subunit, exhibited a small difference in backbone conformation of the wt enzyme with respect to the mutant. Particularly, RMSDs of E21–D30 residues, covering the triad sequence of the enzyme, were in a range from 0.11 to 0.36 Å. This indicates no essential structure alteration of the main-chain hydroxyethylene isostere of SQV and the surrounding residues. Thus, the contacts between the hydroxyl of SQV and the active site residues were independent of the amino acid substitution at residue 48. This result supported the evidence discussed previously that interaction energies of the two triad residues and saquinavir were not significantly changed by mutation of G48V.
FIGURE 5.

Backbone RMSD between the wild-type and the G48V structure.
Structural difference between the wild-type and the mutant
In Fig. 5, the apparent difference between the wt and the mutant HIV-1 PR-SQV complexes was observed around the protein flap (residues 46–55). In this region, overall RMSD values of chain A were relatively greater than those of chain B (Fig. 6). The structure of the flap B of the G48V is similar to that of the wt enzyme, whereas the β-hairpin structure of the flap A of the mutant does not superimpose well. Surprisingly, the tip of flap A of the G48V-SQV complex shifted slightly toward the twofold axis of symmetry of HIV-1 PR. However, distance separations between the tips, residues 49–51 in the mutant, were not significantly different from that in the wt complex.
FIGURE 6.

HIV-1 PR flap structures of the wt (green) and the G48V (yellow and blue). Some selected residues and saquinavir are presented in stick mode. Hydrogen atoms of the enzyme are not shown for simplification.
Flexibility and conformational changes of SQV subsites
Flexibility and conformational changes of the inhibitor side chains P1, P1′, P2, P2′, and P3 were investigated in terms of torsion angle fluctuations of χP1, χP1′, χP2, χP2′, and χP3, respectively. From Fig. 7 A, an oscillation of all dihedral angles throughout the simulations, which was no greater than ±10°, suggested that in the wt complex, all SQV side chains undergo a narrow range of dynamic fluctuation. In other words, the inhibitor side chains were inflexible and retained their starting conformation during the course of the MD trajectory. In the case of the G48V mutant, all torsion angles except for χP2 adopted the values similar to that of the wt (Fig. 7 B). This indicated that the conformations of these subsites are unchanged. However, a remarkable shift in χP2 implied a rotation of the P2 subsite starting from ∼−70° to its equilibrium value ∼90°. This suggested a substantial rotation of the P2 of SQV in the G48V (Fig. 8). In addition, the ±40° fluctuation of χP2 in the mutant larger than that of the wt indicated that the P2 side chain looses its rigidity. The rearrangement of the P2 was related to the event of the flap motion as describe previously. Moreover, it involves a decrease of the strength of the hydrogen bond between the mutated residue and the P2 subsite (described in the next topic). The overall change in terms of torsion angles is supposed to explain a decrease of saquinavir sensitivity.
FIGURE 7.

Fluctuation of χP1, χP1′, χP2, χP2′, and χP3 corresponding to the dihedral angles of the inhibitor side chains P1, P1′, P2, P2′, and P3, respectively.
FIGURE 8.

Conformational change of SQV at the P2 side chain. The rotation of the P2 subsite and the hydrogen bonds are illustrated.
Decrease of hydrogen bond strength
The MD results show that the conformational difference in SQV subsites between the wt and the G48V complexes was located at the P2 side chain. Among residues surrounding the P2 subsite, position 48 is critical to conformational change of the inhibitor subsite. Direct contacts from the backbone oxygen of the mutated residue (O(48)) to the side-chain amide group (HNP2) of P2, and to the backbone amide proton (HNbb) of the inhibitor are illustrated by Fig. 8.
The O(48)-HNbb(SQV) distance was, on average, 1.95 Å for the wt and extends to 2.25 Å in the G48V complex (Fig. 9 A). An increase of the distance is an evidence of decreasing the hydrogen bond strength.
FIGURE 9.

Distance trajectories of hydrogen bonding involving the CO backbone of residue 48.
A loss of the hydrogen bond interactions of residue 48 to SQV in the mutant complex was confirmed by a very large distance between O(48) and HNP2(SQV) (Fig. 9 B). The wt complex shows the close proximity between O(48) and HNP2(SQV). On the other hand, it is clear that the O(48)-HNP2 (SQV) distance in the mutant was not in a range of hydrogen bonding. The rotation of the P2 side chain as described previously is the cause of the disruption of the hydrogen bond of O(48)-HNP2 (SQV).
Analysis of these MD results indicates that the mutation at position 48 decreases the capability of inhibitor binding. A reduction of the interactions was estimated by calculating the ab initio energy. The ΔEG48-SQV decreased ∼3.5 kcal·mol−1 with respect to the ΔEV48-SQV. The magnitude of changes in the interaction energy of V48-SQV supports the experimental Ki data that explain a small decrease (13.5-fold) of saquinavir sensitivity.
DISCUSSION
The MD simulations were carried out to compare structure and dynamics of the wt and the G48V HIV-1 PR-saquinavir complex. In addition, the MD simulations for the four protonation systems were carried out to obtain structural models before an evaluation of the ionization form of the active site residues. The study of protonation state of the HIV-PR-SQV complex was achieved with extensive energy calculations using the DFT, ONIOM, and MM/PBSA methods. Based on our data, both quantum chemical and molecular dynamics free-energy calculations confirm that the protonation model of wt-mono25 is the most energetically favorable case.
The results were in agreement with NMR studies of the hydroxyethylene isostere inhibitors, pepstatin and KNI-272, complexed with the enzyme (Smith et al., 1996; Wang et al., 1996). In this study, the protonation takes place only on the D25 side chain. Although the experimental data are not available for the HIV-1 PR-SQV complex, the structure of KNI-272 is almost identical to that of saquinavir. They share the most common features of drug specificity, including the capability of binding between the central hydroxylethylene isostere of the protease inhibitor and the catalytic residues of the enzyme.
Recently, the protonation state of the HIV-1 PR-SQV complex was studied using quantum and free-energy perturbation methods (Lepsik et al., 2004; Nam et al., 2003). The interaction energy of SQV and the active-site residues were obtained based on the model taken from the x-ray structure. The protonation model proposed from those studies was also monoprotonated D25.
It is worth nothing that the quantum-based method is a promising tool for the study of receptor-ligand complexes. The determination of the protonation state of the HIV-PR active site residues is achievable on the basis of the QM results. In particular, the ONIOM method has extended the limitation of system size by the pure QM method. As demonstrated by the ONIOM results, the enzyme-inhibitor interactions of the catalytic region and the 5 Å surrounding residues are important for stabilizing the complex. The effect of the long-distance range on the interaction energy was not dominant at the MM level. Nevertheless, the method should be used with some proper care. The effect of the boundary resulting from incompatibility between molecular orbital wave functions in the QM part and the MM region may drive unrealistic energy values (Morokuma, 2002).
An effect of solvent environment, which remains unobvious in quantum approach, has been fulfilled with the calculations of binding free energy. The binding free energy of wt-mono25 obtained from MM/PBSA was ∼2 kcal mol−1 lower than that of the other states (Table 1). The difference was contributed from ΔGsol rather than ΔEMM. Not surprisingly, ΔEMM of the three protonation model were almost equivalent (Table 2). Among all three systems, the only difference, that is the ionizable groups of D25 and D25′, cannot be well described by the empirical force-field energy. In addition, the influence of the solvent to the protonation site was trivial on the basis of the radial distribution function plot (Wittayanarakul et al., 2005). The radial distribution function of the hydroxylethylene oxygen of SQV showed an exclusion of water molecules from the protonation site. In this case, the energy information obtained from DFT and ONIOM were reliable. We anticipate that a method for the correction of the MM energy term by the QM treatment in MM/PBSA approach will be valuable for biomolecular research.
The binding pattern at the active site of the wt complex was similar that of the G48V as shown by the very low RMSD of residues E21–D30. Importantly, the interaction energy of the triad residues to SQV was insignificantly different between the wt and the G48V complexes. The results indicated the signature residue mutation developed in the primary resistance does not influence the interactions at the active site.
Significant changes were located at the protein flaps. Moreover, the flap structure of chain A was different from that of chain B. This is caused by different interactions of the enzyme to the asymmetric inhibitor. Particularly, the perturbation adopts heavily on the flap conformation of chain A rather than chain B. The slide of flap A in the G48V mutant seems to overcome a potential steric conflict caused by the substituted valine. As shown in Fig. 6, position 48 was in close contact with the F53 side chain of the enzyme and the P2 and P3 groups of SQV. Since the substituted valine of the mutant cannot be entirely accommodated in the hydrophobic pocket due to steric conflict of the dimethyl groups with the F53 and P3 side chains, the flap, therefore, shifted toward the symmetric axis of the enzyme. This rearrangement additionally destabilizes hydrogen bonding between the backbone CO of residue 48 of the enzyme and the P2 subsite of the inhibitor. In addition to the flap movement, the side chain of hydrophobic F53 became solvent-exposed to avoid steric clashes with V48 and Met-46, whereas an orientation of F53′ in flap B of the mutant was not changed dramatically.
The role of the HIV-1 PR mutation at the flexible flap has been considerably debated about whether it would facilitate the binding reaction or reduce stability of the inhibitor, or both (Ermolieff et al., 1997; Hong et al., 1997; Maschera et al., 1996). The crystal structure of the double mutant G48V/L90M complexed with SQV revealed side-chain rearrangement of the P2 subsite and the F53 of the enzyme similar to this study (Hong et al., 2000). Particularly, the missing of hydrogen bonding between the P2 subsite and the backbone C=O of residue 48 was also found in the x-ray structure of the double mutant. The crystal structure of G48H complexed with peptidic inhibitor U-89360E reported a decrease of flap mobility to stabilize the ligand (Hong et al., 1997).
Apparently the conformational change of the P2 subsite was an influence of steric conflict of the mutation at position 48. Importantly, it reduced saquinavir susceptibility to the mutant by interrupting the hydrogen bond interactions. Thus, a new drug with reduced steric repulsion on P2 could be designed to enhance the activity toward this mutant strain.
The x-ray structural data provided relevant information, insight into the molecular mechanism of HIV resistance to the protease inhibitor. Dynamic details of the full-atomic representation of the complex were required for further investigation. Therefore, MD simulation offers a good opportunity to fulfill basic information that could be useful in understanding the drug resistance mechanism and helpful in designing an anti-HIV inhibitor.
CONCLUSIONS
Molecular dynamics simulations of the wt and the G48V HIV-1 protease complexed with SQV were carried out to investigate the molecular basis of drug resistance. The MD results combined with quantum chemical calculations extend the capability of molecular modeling methods to study some biological systems, of which structural information is limited. This study showed that both complexes form the monoprotonation on D25. Overall tertiary structure of the wt and the mutant protease was not significantly altered. Particularly, structure and interactions at the central active site remain unchanged. However, conformational differences between the wt and the G48V mutant were on the protein flap. The conformational change of the P2 subsite decreased the strength of hydrogen bonding of the backbone CO of the residue 48 to SQV. The change in interaction energies was comparable to the experimental Ki data. These observations provide useful information for designing potent HIV-1 PR inhibitors.
Acknowledgments
Financial support by the Thailand Research Fund and the generous supply of computer time by the Austrian-Thai Center for Computer-Assisted Chemical Education and Research (Bangkok, Thailand) are gratefully acknowledged.
APPENDIX: ONIOM CALCULATIONS
The key interactions centered on SQV and the catalytic residues were treated at a high level of calculations, whereas the environmental effect of the entire protein was calculated at a lower level of calculations. In this study, three-layered ONIOM (ONIOM3) was employed (Morokuma, 2002).
On the basis of the ONIOM3 method shown in Fig. 10, the total energy of the system can be obtained from five independent calculations as
 |
(11) |
or
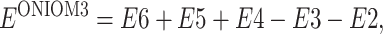 |
(12) |
where real denotes the entire system, of which the energy (E6) is calculated at the low level. For the intermediate layer, the energy is computed at both the medium (E5) and low (E3) level. For the inner layer, the energy is obtained at both high (E4) and medium (E2) level.
FIGURE 10.

Schematic representation of the three-layer ONIOM extrapolation scheme.
As shown by Fig. 11, the structure of the HIV-1 PR-SQV complex was partitioned into three parts represented by inner layer (A), intermediate layer (A + B), and real layer (A + B + C). The atoms of the enzyme and the inhibitor defined to each layers were described in the Methods section.
FIGURE 11.

Schematic representation for the structure of HIV-1PR-SQV with the three partitioned layers.
To obtain the interaction energy of the HIV-1 PR/SQV complex (ΔEcpx), one can be expressed as
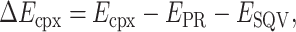 |
(13) |
where Ecpx, EPR, and ESQV are the total energy of the HIV-1 PR/SQV complex, HIV-1 PR and SQV, respectively.
Kitiyaporn Wittayanarakul and Ornjira Aruksakunwong contributed equally to this work.
References
- Antosiewicz, J., J. A. Mccammon, and M. K. Gilson. 1994. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 238:415–436. [DOI] [PubMed] [Google Scholar]
- Baldwin, E. T., T. N. Bhat, S. Gulnik, B. Liu, I. A. Topol, Y. Kiso, T. Mimoto, H. Mitsuya, and J. W. Erickson. 1995. Structure of HIV-1 protease with KNI-272, a tight-binding transition-state analog containing allophenylnorstatine. Structure. 3:581–590. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Boucher, C. 1996. Rational approaches to resistance: using saquinavir. AIDS. 10 (Suppl. 1) :S15–S19. [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Case, D. A., J. C. D. Pearlman, T. Cheatham III, J. Wang, W. Ross, C. Simmerling, T. Darden, T. Merz, R. Stanton, A. Cheng, J. Vincent, M. Crowley, V. Tsui, H. Gohlke, R. Radmer, Y. Duan, J. Pitera, I. Massova, G. Seibel, U. C. Singh, P. Weiner, and P. A. Kollman. 2002. AMBER 7. University of California, San Francisco, CA.
- Chen, X., and A. Tropsha. 1995. Relative binding free energies of peptide inhibitors of HIV-1 protease: the influence of the active site protonation state. J. Med. Chem. 38:42–48. [DOI] [PubMed] [Google Scholar]
- Collins, J. R., S. K. Burt, and J. W. Erickson. 1995. Flap 0pening in HIV-1 protease simulated by activated molecular-dynamics. Nat. Struct. Biol. 2:334–338. [DOI] [PubMed] [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, and P. A. Kollman. 1993. Application of RESP charges to calculate conformational energies, hydrogen-bond energies, and free-energies of solvation. J. Am. Chem. Soc. 115:9620–9631. [Google Scholar]
- Davis, M. E., J. D. Madura, B. A. Luty, and J. A. McCammon. 1991. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian Dynamics Program. Comput. Phys. Comm. 62:187–197. [Google Scholar]
- Debouck, C., J. G. Gorniak, J. E. Strickler, T. D. Meek, B. W. Metcalf, and M. Rosenberg. 1987. Human immunodeficiency virus protease expressed in Escherichia coli exhibits autoprocessing and specific maturation of the gag precursor. Proc. Natl. Acad. Sci. USA. 84:8903–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks, S. G. 2003. Treatment of antiretroviral-drug-resistant HIV-1 infection. Lancet. 362:2002–2011. [DOI] [PubMed] [Google Scholar]
- Eberle, J., B. Bechowsky, D. Rose, U. Hauser, K. von der Helm, L. Gurtler, and H. Nitschko. 1995. Resistance of HIV type 1 to proteinase inhibitor Ro 31–8959. AIDS Res. Hum. Retro. 11:671–676. [DOI] [PubMed] [Google Scholar]
- Ermolieff, J., X. Lin, and J. Tang. 1997. Kinetic properties of saquinavir-resistant mutants of human immunodeficiency virus type 1 protease and their implications in drug resistance in vivo. Biochemistry. 36:12364–12370. [DOI] [PubMed] [Google Scholar]
- Friesner, R. A., and M. D. Beachy. 1998. Quantum mechanical calculations on biological systems. Curr. Opin. Struct. Biol. 8:257–262. [DOI] [PubMed] [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, J. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, and others. 2002. Gaussian 98. Gaussian, Inc. Pittsburgh, PA.
- Gilson, M. K., K. Sharp, and B. Honig. 1987. Calculating electrostatic interactions in bio-molecules: method and error assessment. J. Comput. Chem. 9:327–335. [Google Scholar]
- Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- Harte, W. E., Jr., S. Swaminathan, and D. L. Beveridge. 1992. Molecular dynamics of HIV-1 protease. Proteins. 13:175–194. [DOI] [PubMed] [Google Scholar]
- Harte, W. E., Jr., S. Swaminathan, M. M. Mansuri, J. C. Martin, I. E. Rosenberg, and D. L. Beveridge. 1990. Domain communication in the dynamical structure of human immunodeficiency virus 1 protease. Proc. Natl. Acad. Sci. USA. 87:8864–8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, L., A. Treharne, J. A. Hartsuck, S. Foundling, and J. Tang. 1996. Crystal structures of complexes of a peptidic inhibitor with wild-type and two mutant HIV-1 proteases. Biochemistry. 35:10627–10633. [DOI] [PubMed] [Google Scholar]
- Hong, L., X. C. Zhang, J. A. Hartsuck, and J. Tang. 2000. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: insights into the mechanisms of drug resistance. Protein Sci. 9:1898–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, L., X. J. Zhang, S. Foundling, J. A. Hartsuck, and J. Tang. 1997. Structure of a G48H mutant of HIV-1 protease explains how glycine-48 replacements produce mutants resistant to inhibitor drugs. FEBS Lett. 420:11–16. [DOI] [PubMed] [Google Scholar]
- Hyland, L. J., T. A. Tomaszek Jr., and T. D. Meek. 1991. Human immunodeficiency virus-1 protease. 2. Use of pH rate studies and solvent kinetic isotope effects to elucidate details of chemical mechanism. Biochemistry. 30:8454–8463. [DOI] [PubMed] [Google Scholar]
- Jaskolski, M., A. G. Tomasselli, T. K. Sawyer, D. G. Staples, R. L. Heinrikson, J. Schneider, S. B. Kent, and A. Wlodawer. 1991. Structure at 2.5-Å resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene-based inhibitor. Biochemistry. 30:1600–1609. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Jorgensen, W. L., and J. Tirado-Rives. 1988. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- Kollman, P. A., I. Massova, C. Reyes, B. Kuhn, S. H. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case, and T. E. Cheatham 3rd. 2000. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. 1991. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- Krohn, A., S. Redshaw, J. C. Ritchie, B. J. Graves, and M. H. Hatada. 1991. Novel binding mode of highly potent HIV-proteinase inhibitors incorporating the (R)-hydroxyethylamine isostere. J. Med. Chem. 34:3340–3342. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., J. A. Rullmannn, M. W. MacArthur, R. Kaptein, and J. M. Thornton. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 8:477–486. [DOI] [PubMed] [Google Scholar]
- Lepsik, M., Z. Kriz, and Z. Havlas. 2004. Efficiency of a second-generation HIV-1 protease inhibitor studied by molecular dynamics and absolute binding free energy calculations. Proteins. 57:279–293. [DOI] [PubMed] [Google Scholar]
- Levy, Y., and A. Caflisch. 2003. Flexibility of monomeric and dimeric HIV-1 protease. J. Phys. Chem. B. 107:3068–3079. [Google Scholar]
- Maschera, B., G. Darby, G. Palu, L. L. Wright, M. Tisdale, R. Myers, E. D. Blair, and E. S. Furfine. 1996. Human immunodeficiency virus. Mutations in the viral protease that confer resistance to saquinavir increase the dissociation rate constant of the protease-saquinavir complex. J. Biol. Chem. 271:33231–33235. [DOI] [PubMed] [Google Scholar]
- Meek, T. D., B. D. Dayton, B. W. Metcalf, G. B. Dreyer, J. E. Strickler, J. G. Gorniak, M. Rosenberg, M. L. Moore, V. W. Magaard, and C. Debouck. 1989. Human immunodeficiency virus 1 protease expressed in Escherichia coli behaves as a dimeric aspartic protease. Proc. Natl. Acad. Sci. USA. 86:1841–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morokuma, K. 2002. New challenges in quantum chemistry: Quests for accurate calculations for large molecular systems. Philos. Transact. A Math. Phys. Eng. Sci. 360:1149–1164. [DOI] [PubMed] [Google Scholar]
- Nam, K. Y., B. H. Chang, C. K. Han, S. G. Ahn, and K. T. No. 2003. Investigation of the protonated state of HIV-1 protease active site. Bull. Kor. Chem. Soc. 24:817–823. [Google Scholar]
- Northrop, D. B. 2001. Follow the protons: a low-barrier hydrogen bond unifies the mechanisms of the aspartic proteases. Acc. Chem. Res. 34:790–797. [DOI] [PubMed] [Google Scholar]
- Okimoto, N., T. Tsukui, M. Hata, T. Hoshino, and M. Tsuda. 1999. Hydrolysis mechanism of the phenylalanine-proline peptide bond specific to HIV-1 protease: Investigation by the ab initio molecular orbital method. J. Am. Chem. Soc. 121:7349–7354. [Google Scholar]
- Piana, S., D. Bucher, P. Carloni, and U. Rothlisberger. 2004. Reaction mechanism of HIV-1 protease by hybrid CarParrinello/classical MD simulations. J. Phys. Chem. B. 108:11139–11149. [Google Scholar]
- Piana, S., P. Carloni, and U. Rothlisberger. 2002. Drug resistance in HIV-1 protease: flexibility-assisted mechanism of compensatory mutations. Protein Sci. 11:2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piana, S., D. Sebastiani, P. Carloni, and M. Parrinello. 2001. Ab initio molecular dynamics-based assignment of the protonation state of pepstatin A/HIV-1 protease cleavage site. J. Am. Chem. Soc. 123:8730–8737. [DOI] [PubMed] [Google Scholar]
- Prabhakar, R., D. G. Musaev, I. V. Khavrutskii, and K. Morokuma. 2004. Does the active site of mammalian glutathione peroxidase (GPx) contain water molecules? An ONIOM study. J. Phys. Chem. B. 108:12643–12645. [Google Scholar]
- Prabu-Jeyabalan, M., E. A. Nalivaika, N. M. King, and C. A. Schiffer. 2003. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J. Virol. 77:1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph, J. T., and D. A. DeGoey. 2004. Peptidomimetic inhibitors of HIV protease. Curr. Top. Med. Chem. 4:1079–1095. [DOI] [PubMed] [Google Scholar]
- Roberts, N. A., J. A. Martin, D. Kinchington, A. V. Broadhurst, J. C. Craig, I. B. Duncan, S. A. Galpin, B. K. Handa, J. Kay, A. Krohn, and others. 1990. Rational design of peptide-based HIV proteinase inhibitors. Science. 248:358–361. [DOI] [PubMed] [Google Scholar]
- Rocchia, W., E. Alexov, and B. Honig. 2001. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 105:6507–6514. [Google Scholar]
- Rodriguez-Barrios, F., and F. Gago. 2004. HIV protease inhibition: Limited recent progress and advances in understanding current pitfalls. Curr. Top. Med. Chem. 4:991–1007. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- Scott, W. R., and C. A. Schiffer. 2000. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Struct. Fold. Des. 8:1259–1265. [DOI] [PubMed] [Google Scholar]
- Sitkoff, D., K. A. Sharp, and B. Honig. 1994. Accurate calculation of hydration free-energies using macroscopic solvent models. J. Phys. Chem. 98:1978–1988. [Google Scholar]
- Smith, R., I. M. Brereton, R. Y. Chai, and S. B. H. Kent. 1996. Ionization states of the catalytic residues in HIV-1 protease. Nat. Struct. Biol. 3:946–950. [DOI] [PubMed] [Google Scholar]
- Srinivasan, J., T. E. Cheatham, P. Cieplak, P. A. Kollman, and D. A. Case. 1998. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 120:9401–9409. [Google Scholar]
- Swain, A. L., M. M. Miller, J. Green, D. H. Rich, J. Schneider, S. B. Kent, and A. Wlodawer. 1990. X-ray crystallographic structure of a complex between a synthetic protease of human immunodeficiency virus 1 and a substrate-based hydroxyethylamine inhibitor. Proc. Natl. Acad. Sci. USA. 87:8805–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent, M., T. Vreven, D. G. Musaev, K. Morokuma, O. Farkas, and H. B. Schlegel. 2002. Effects of the protein environment on the structure and energetics of active sites of metalloenzymes. ONIOM study of methane monooxygenase and ribonucleotide reductase. J. Am. Chem. Soc. 124:192–193. [DOI] [PubMed] [Google Scholar]
- Vaillancourt, M., D. Irlbeck, T. Smith, R. W. Coombs, and R. Swanstrom. 1999. The HIV type 1 protease inhibitor saquinavir can select for multiple mutations that confer increasing resistance. AIDS Res. Hum. Retroviruses. 15:355–363. [DOI] [PubMed] [Google Scholar]
- Vondrasek, J., and A. Wlodawer. 2002. HIVdb: a database of the structures of human immunodeficiency virus protease. Proteins. 49:429–431. [DOI] [PubMed] [Google Scholar]
- Wang, J. M., W. Wang, and P. A. Kollman. 2001. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 222:U403–U403 (Abstr). [Google Scholar]
- Wang, W., and P. A. Kollman. 2000. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 303:567–582. [DOI] [PubMed] [Google Scholar]
- Wang, Y. X., D. I. Freedberg, T. Yamazaki, P. T. Wingfield, S. J. Stahl, J. D. Kaufman, Y. Kiso, and D. A. Torchia. 1996. Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272. Biochemistry. 35:9945–9950. [DOI] [PubMed] [Google Scholar]
- Wittayanarakul, K., O. Aruksakunwong, P. Sompornpisut, V. Sanghiran-Lee, V. Parasuk, S. Pinitglang, and S. Hannongbua. 2005. Structure, dynamics and solvation of HIV-1 protease/saquinavir complex in aqueous solution and their contributions to drug resistance: molecular dynamic simulations. J. Chem. Inf. Comput. Sci. In press. [DOI] [PubMed]
- Wlodawer, A., and J. Vondrasek. 1998. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 27:249–284. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T., L. K. Nicholson, D. A. Torchia, P. Wingfield, S. J. Stahl, J. D. Kaufman, C. J. Eyermann, C. N. Hodge, P. Y. S. Lam, Y. Ru, and others. 1994. NMR and x-ray evidence that the HIV protease catalytic aspartyl groups are protonated in the complex formed by the protease and a nonpeptide cyclic urea-based inhibitor. J. Am. Chem. Soc. 116:10791–10792. [Google Scholar]
- Yang, A. S., M. R. Gunner, R. Sampogna, K. Sharp, and B. Honig. 1993. On the calculation of Pk(a)S in proteins. Proteins. 15:252–265. [DOI] [PubMed] [Google Scholar]
- York, D. M., T. A. Darden, and L. G. Pedersen. 1993a. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: a comparison of the Ewald and truncated list methods. J. Chem. Phys. 99:8345–8348. [Google Scholar]