

Abstract
Using molecular dynamics simulations, we studied the mode of association of the cell-penetrating peptide penetratin with both a neutral and a charged bilayer. The results show that the initial peptide-lipid association is a fast process driven by electrostatic interactions. The homogeneous distribution of positively charged residues along the axis of the helical peptide, and especially residues K46, R53, and K57, contribute to the association of the peptide with lipids. The bilayer enhances the stability of the penetratin helix. Oriented parallel to the lipid-water interface, the subsequent insertion of the peptide through the bilayer headgroups is significantly slower. The presence of negatively charged lipids considerably enhances peptide binding. Lateral side-chain motion creates an opening for the helix into the hydrophobic core of the membrane. The peptide aromatic residues form a π-stacking cluster through W48/R52/W56 and F49/R53, protecting the peptide from the water phase. Interaction with the penetratin peptide has only limited effect on the overall membrane structure, as it affects mainly the conformation of the lipids which interact directly with the peptide. Charge matching locally increases the concentration of negatively charged lipids, lateral lipid diffusion locally decreases. Lipid disorder increases, through decreased order parameters of the lipids interacting with the penetratin side chains. Penetratin molecules at the membrane surface do not seem to aggregate.
INTRODUCTION
Cell-penetrating peptides (CPPs) have been proposed as carriers for cellular uptake of various molecules, including proteins, oligonucleotides, and drugs. The homeodomain of Antennapedia is able to translocate through a cellular membrane (Joliot et al., 1991; Le Roux et al., 1993), mainly thanks to its third helix (Derossi et al., 1994, 1996), consisting of residues 43–58. The so-called pAntp peptide, or penetratin, was proposed as a vehicle for intracellular delivery of hydrophilic cargo molecules (Prochiantz, 1996, 1998; Pooga et al., 1998; Braun et al., 2002). Penetratin was the first member of an increasingly large family of natural or synthetic CPPs (Lindgren et al., 2000; Langel, 2002). Its uptake mechanism is to date not clear. Most membrane-associating peptides are amphipathic and translocate across membranes through either pore formation (Cornut et al., 1993; Bechinger, 1997; Matsuzaki, 1998) or in a potential-dependent manner (Maduke and Roise, 1993; Leenhouts et al., 1996). Charged residues in these peptides are mostly lysines. Molecular dynamics simulations of the N-terminal region of human surfactant protein-B (SP-B1–25) showed partial diffusion of the N-terminal part of the peptide into the hydrophobic phase of a bilayer on a nanosecond timescale (Kaznessis et al., 2002). The membrane-interacting residues in SP-B1–25 are hydrophobic, whereas the C-terminal charged residues remain within the lipid interface. The N-terminal end of penetratin is mostly hydrophobic, although not to the same extent. Its fluctuating helical hydrophobic moment (calculated over a seven-residue window), related to its DNA-binding ability, differentiates penetratin from the amphipathic helical peptide family (Thorén et al., 2000; Drin et al., 2001a; Binder and Lindblom, 2003a). Since cellular penetratin uptake occurs both at 37°C and 4°C, internalization through endocytosis was excluded (Derossi et al., 1994), whereas cellular import of reverse forms of various CPPs suggested that it is a receptor-independent process (Brugidou et al., 1995; Derossi et al., 1996; Mitchell et al., 2000; Wender et al., 2000). The nontoxic penetratin translocates across cellular membranes without membrane damage (Terrone et al., 2003), even when coupled to drugs (Hosotani et al., 2002), suggesting direct interaction with membrane lipids (Berlose et al., 1996; Derossi et al., 1996; Thorén et al., 2000; Terrone et al., 2003; Vivès et al., 2003). However, the mode of interaction and of lipid reorganization that allow for internalization without leakage is not known. Formation of inverted lipid micelles was suggested (Derossi et al., 1994, 1998; Prochiantz, 1996), implying that the peptide remains in an aqueous environment. Such a mechanism would facilitate the transport of hydrophilic compounds linked to the peptide. According to recent reports, penetratin internalization might at least partially occur through an energy-dependent process, involving endocytosis (Richard et al., 2003; Thorén et al., 2003; Terrone et al., 2003; Console et al., 2003; Vivès et al., 2003). On the other hand, the uptake of other arginine-rich peptides does not seem to be affected by endocytosis inhibitors (Suzuki et al., 2002; Console et al., 2003), suggesting the existence of different competing pathways for CPPs (Hällbrink et al., 2001; Vivès et al., 2003).
The penetratin sequence (RQIKIWFQNRRMKWKK) consists of 16 residues out of which four lysines and three arginines. Electrostatic interactions are therefore likely to play a key role in the association process (Christiaens et al., 2002). However, no single residue critical for membrane interaction could be identified (Drin et al., 2001a). Negatively charged lipids promote the transfer of penetratin from a hydrophilic to a hydrophobic environment (Dom et al., 2003), and electrostatic effects probably contribute not only to binding, but further to peptide translocation (Binder and Lindblom, 2003a). Penetratin is not sufficiently hydrophobic to insert deeply into phospholipid model membranes (Drin et al., 2001a; Brattwall et al., 2003). This suggests that charge neutralization is required for a deeper insertion of the peptide into the hydrophobic core of the membrane (Dom et al., 2003; Console et al., 2003). The contribution of the hydrophobic residues, and especially of the aromatic tryptophan and phenylalanine residues is probably crucial for internalization. Mutation of either tryptophan decreases internalization, whereas double substitution completely inhibits peptide internalization (Derossi et al., 1994; Fischer et al., 2000; Drin et al., 2001b; Christiaens et al., 2002; Lindberg et al., 2003).
We selected molecular dynamics (MD) simulations as a method to investigate the atomic interactions that lie at the basis of the penetratin-membrane association and internalization. MD is a well-established methodology for the study of the dynamics of proteins (Berendsen, 1996; Karplus, 2002), membranes (Tieleman et al., 1997), and peptide-membrane interactions (La Rocca et al., 1999; Shepherd et al., 2003) that enables a direct observation of dynamical events (Berendsen, 2001). The penetratin-membrane association was studied by careful analysis of the obtained trajectories. We examined the residues responsible for the peptide-lipid binding, the local environment around the peptide, the behavior of peptide aromatic residues, possible peptide aggregation, the effect of K/R→A mutations, and the penetratin structure in solution. The simulation results are summarized under the Discussion. The computational details are described in the following section.
COMPUTATIONAL DETAILS
Molecular dynamics
MD simulations were performed with the Gromacs 3.1.4 package (Berendsen et al., 1995; Lindahl et al., 2001), using the Gromos96 43a2 force field (Van Gunsteren et al., 1996), with an extension for the lipid phosphate and acyl chain parameters (Berger et al., 1997). The peptide structure was taken by cutting residues 43–58 (the 3rd helix) from the NMR structure of the Antennapedia homeodomain (pdb-code 1ahd (Billeter et al., 1993), model 1). The C-terminus was capped with an amine group. Peptide-membrane simulations were prepared by placing the peptide horizontally on top of an equilibrated bilayer box, at a distance of ∼2 nm from the bilayer surface; the axis normal to the bilayer plane was extended to include the peptide and the formed vacuum was filled with water molecules, resulting in ∼4500 water molecules in a box ∼6 × 6 × 8.5 nm3. Single point charge water was used (Berendsen et al., 1981). An equilibrated bilayer of 128 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) molecules was taken as the starting point (Tieleman et al., 1999), with at random 8 POPC molecules in each layer exchanged for the negatively charged alternatives 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG), or 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-L-serine] (POPS). Coordinates of the phosphate groups and acyl chains were kept in this process, adding water molecules in a formed vacuum (POPA) or deleting them upon overlap with the new headgroup function. All systems, including the bilayer systems without the peptide yet present, were made electrostatically neutral by adding the required amount of counterions Na+ or Cl− and then submitted to an energy minimization, 10 ps position restraint MD and 2 ns free MD before the production run was started. Weak coupling to a temperature (300 K) and pressure (isotropic, 1.0 bar) bath was employed (Berendsen et al., 1984), using coupling constants of 0.1 and 1.0 ps, respectively. Coulomb interactions were treated with fast particle mesh Ewald (Essman et al., 1995), using a grid spacing of 0.12 nm and fourth-order interpolation. A spherical cutoff of 1.0 nm was applied to the Van der Waals interactions. Bond lengths were constrained using the LINCS algorithm (Hess et al., 1997). Equations of motion for the water atoms were solved analytically with the SETTLE algorithm (Miyamoto and Kollman, 1992). Dummy atoms were used to limit high-frequency vibrations involving hydrogen atoms (Feenstra et al., 1999). A time step of 4 fs was employed, removing center of mass motion every step and updating the neighbor list every 10 steps. Table 1 shows a list of the performed simulations. The simulations were performed on our own linux cluster consisting of five dual-CPU machines (eight Athlon 2000+ (AMD, Sunnyvale, CA) and two Xeon processors (Intel, Santa Clara, CA)).
TABLE 1.
List of the performed simulations
| Composition of the simulation system*
|
|||||||
|---|---|---|---|---|---|---|---|
| Name | POPC | POPA | POPG | POPS | Penetratin | Start† | Length |
| P-PG | 112 | 0 | 16 | 0 | 1 | Equilibration | 230 ns |
| P-PC | 128 | 0 | 0 | 0 | 1 | Equilibration | 240 ns |
| P-PA | 112 | 16 | 0 | 0 | 1 | Equilibration | 50 ns |
| P-PS | 112 | 0 | 0 | 16 | 1 | Equilibration | 50 ns |
| P-PCα | 128 | 0 | 0 | 0 | 1 | Equilibration | 40 ns |
| P-PCβ | 128 | 0 | 0 | 0 | 1 | Equilibration | 30 ns |
| P2-PG | 112 | 0 | 16 | 0 | 2 | 100 ns P-PG | 25 ns |
| P3-PG | 112 | 0 | 16 | 0 | 3 | 25 ns P2-PG | 50 ns |
| P52A,55A-PG | 112 | 0 | 16 | 0 | 1‡ | 100 ns P-PG | 20 ns |
| P53A,57A-PG | 112 | 0 | 16 | 0 | 1§ | 100 ns P-PG | 20 ns |
| P-WAT | 0 | 0 | 0 | 0 | 1 | Equilibration | 120 ns |
All systems contain water and required counterions.
See Computational Details.
Penetratin (R52A/K55A).
Penetratin (R53A/K57A).
Simulations P52A, 55A-PG, P53A, 57A-PG, and P2-PG were started from a snapshot of the P-PG simulation at t = 100 ns. P3-PG was started from the end point of P2-PG, at t = 25 ns. Mutations were performed by mutating the residue in question into an alanine, filling a formed vacuum with water molecules and adding the required amount of counterions. For simulations P2-PG and P3-PG penetratin molecules were added at exactly the same position as in the initial system preparation, removing overlapping water molecules and updating the amount and type of counterions. These “continuation” simulations were submitted to the same energy minimization and position restraint MD, but no 2-ns free equilibration MD was performed.
Analyses
The axis of the penetratin helix is determined using a rotational least-squares fitting method mapping the Cα's of the helix onto itself, but one residue out of phase, i.e., residue i is mapped onto residue i + 1 (a screw-transform superimposes the two helices) (Christopher et al., 1996). A quaternion-based method is used to find the helical axis as the axis of rotation necessary to superimpose these atoms (Mackay, 1984).
The orientation of the aromatic residues is described in terms of two order parameters (Tieleman et al., 1998). SN is defined as  with θ the angle between the bilayer normal and the normal to the plane of the aromatic ring. SL is defined in the same way but with θ the angle between the bilayer normal and the vector from Cγ to Cζ or Cζ2 for Phe and Trp, respectively. For a more in-depth description, we refer to Fig. 2 of Tieleman et al. (1998).
with θ the angle between the bilayer normal and the normal to the plane of the aromatic ring. SL is defined in the same way but with θ the angle between the bilayer normal and the vector from Cγ to Cζ or Cζ2 for Phe and Trp, respectively. For a more in-depth description, we refer to Fig. 2 of Tieleman et al. (1998).
FIGURE 2.

Minimal distance between the hydrogen bond donor atoms of the penetratin side chains and any of the phosphorus atoms of the lipid bilayer.
The lipids used in these computations are POPC, POPG, POPA, and POPS. POPC is effectively neutral, whereas the others carry a negative charge. The lipid subgroups used for analysis (see Figs. 5 and 6) are the headgroup functions (e.g., choline for POPC, glycerol for POPG), the phosphate group, the two carbonyl groups that start the sn1 and sn2 acyl chains, and the double bond of the sn2 chain. For the calculation of the order parameters, the numbering goes from the first CH2–CH2 bond after the carbonyl groups to the end of the acyl chain.
FIGURE 5.

Atom density profile, calculated over a time frame of 100–230 ns, as a function of distance in the z-coordinate to the bilayer center. Density profiles for both POPG (A) and POPC (B) lipids are plotted. Solid lines indicate the peptide-containing layer, dashed lines are for the layer without peptide, mirrored in the center of the bilayer. Color coding is black for headgroup functions (choline or glycerol), red for phosphate, green for carbonyl, and blue for double-bond groups. Penetratin density is indicated with the cyan solid line, scaled (divided by 7) in A.
FIGURE 6.

(A) Number of lipids interacting with penetratin, as defined in Computational Details. (B) Average distance to the bilayer center (in the z-coordinate), for penetratin and selected lipid subgroups. Color coding corresponds to Fig. 5, with interacting phosphate, carbonyl, and double-bond groups in brown, dark green, and magenta, respectively.
Lipids are defined as interacting with penetratin when any of their atoms is within a cylinder, parallel to the z axis, with diameter 1 Å and height 1 nm from any of the peptide atoms.
The programs to calculate 1), the axis of a helix, 2), the orientation of aromatic residues, and 3), lipids that are under a peptide have been written by the author (M.F.L.) and are available upon request.
RESULTS
A number of simulations were performed, as listed in Table 1. The long simulations P-PC and P-PG, with neutral and 12.5% negatively charged bilayer were used for all major analyses. The P-PCα and P-PCβ simulations were used to test the peptide-membrane association for a neutral bilayer, using a different initial orientation of the peptide (rotated 60 and 120 degrees about the axis of the helix). The P-PA and P-PS simulations were used to investigate the influence a different negatively charged lipid has on the overall peptide-lipid interactions. As no significant difference in orientation was found during membrane binding with respect to the reference P-PC and P-PG simulations, these simulations show the same characteristics as their P-PC and P-PG counterparts, unless stated otherwise. Moreover, as the results from both these simulations were also similar, all figures refer to the P-PG simulations, unless stated otherwise in the figure caption. Once the peptide was well docked in the bilayer, we took a key frame of the P-PG simulation and performed additional simulations: P52A, 55A-PG and P53A, 57A-PG are simulations with R52A/K55A and R53A/K57A mutations, respectively. Aggregation of penetratin was investigated by adding additional penetratin peptides, yielding simulations P2-PG and P3-PG.
Penetratin-membrane association
Fig. 1 shows the approach of penetratin toward the membrane surface, as the z-component of the distance between the average coordinates of the peptide and the phosphorus atoms of the half-side lipid bilayer, that is closest to the peptide. The P-PG simulation (Table 1) in Fig. 1 A shows a rapid penetratin-membrane association that takes place during the equilibration phase of the simulation, as the mean distance decreases from 2 to <1 nm within 2 ns. It takes about a third of the remaining 230 ns of the simulation to reach an equilibrium between peptide and membrane (Fig. 1, B and C), when penetratin has docked between the hydrophilic headgroups of the lipids. The concomitant secondary structure evolution of the peptide (data not shown) shows that penetratin forms a stable α-helix when bound to the lipid bilayer.
FIGURE 1.

Penetratin-membrane distance and interaction energy for the P-PG (A–C) and P-PC (D–F) simulations. (A, B, D, and E) Distance (z-coordinate) between the mean coordinates of penetratin and the phosphorus atoms of the lipid bilayer half interacting with the peptide. Data are given for both the equilibration (A and D) and production runs (B and E), separated at t = 0 ns. (C and F) Peptide-membrane interaction energy, split into the Coulomb and Lennard-Jones contributions.
The peptide-lipid attraction is mainly due to electrostatic interactions, as illustrated in Fig. 1, C and F, showing both the electrostatic Coulomb and nonelectrostatic Lennard-Jones (LJ) contribution. Although both energy components decrease steadily, the electrostatic energy decreases faster. The increased peptide-membrane interaction is accompanied by a decrease of the peptide-water interaction (data not shown). A combined examination of Fig. 1, B and C, shows that although initial binding is associated with a large increase in Coulomb interaction, once equilibrium is reached, the electrostatic component remains constant, but the total interaction is increased by a decrease in Lennard-Jones energy: hydrophobic interactions become predominant, through interactions of the peptide with neutral atoms of the lipid chains.
Fig. 1, D and E, show the penetratin-membrane distance for the simulation of the peptide association with a neutral POPC membrane (P-PC). Peptide-membrane association is significantly slower in this case as binding is 40 ns longer than the equilibration phase, compared to P-PG. Interaction energies are similar to those in Fig. 1 C, but equilibrium is reached only after 160 ns. A slightly more favorable electrostatic energy component, and a deeper positioning of the peptide in the bilayer, is detected. The structure of the peptide bound to a neutral bilayer remains α-helical.
To characterize the peptide orientation during its approach toward the bilayer, we plotted the minimal distance between the nitrogen atoms of the seven positively charged penetratin residues and any of the lipid phosphorus atoms (Fig. 2). This figure identifies residues K46, R53, K57, and K58 as those accounting for the mutual electrostatic binding. The first three residues are located on the same side of the helical peptide (see also Fig. 3 B); K58 is located at the C-terminal end of the peptide and has therefore higher mobility. Fig. 3 A shows binding of penetratin to a phospholipid bilayer, from left to right in three concatenated snapshots. Whereas the left-hand image shows the initial structure before equilibration, the center image—taken at the end of the equilibration phase—shows K46, R53, and K57 directed toward the bilayer surface, making hydrogen bonds and salt bridges with the lipids. The right-hand image shows the position of penetratin in the lipid bilayer, as recorded toward the end of the simulation. Throughout this process, penetratin remains horizontal, as shown by the angle between the axis of the helix and the normal to the bilayer plane (Fig. 4 A), with the positively charged residues K46, R53, and K57 directed toward the membrane surface. Fig. 4 A shows that during practically the entire P-PG simulation, penetratin remains parallel, or slightly tilted, to the membrane surface. The interaction of penetratin with a neutral lipid membrane occurs through the C-terminal end of the peptide, and during the initial binding penetratin is perpendicular to the bilayer surface (Fig. 4 C). Subsequent rotations perpendicular to and about the helix axis enable interactions of the same residues K46, R53, and K57 with lipids. Once the peptide is settled in the membrane, the longitudinal vector (the helix axis) assumes a value of ∼80° (90° for P-PC). A steady value of 75° (Fig. 4 B) is found (60° for P-PC, Fig. 4 D) for the angle between the z-axis and the lateral vector, excluding further rotation about the helix axis. Fig. 3 B shows the position of the helix in the membrane, with the z-axis as in Fig. 3 A.
FIGURE 3.

(A) Binding of penetratin to a lipid bilayer. Snapshots are taken from the P-PG simulation at t = −1.85 ns, t = −50 ps, and t = 200 ns (concatenated left to right); only peptide and membrane are plotted. The arrow indicates the z axis and the normal to the bilayer plane. Membrane lipids are drawn in wireframe, the peptide, charged and aromatic residues only, with ball-and-sticks. Color coding for lipids: P, violet; O, red; N, blue; other, gray; color coding for the peptide: Lys and Arg, blue; Trp and Phe, orange. The figure was prepared with MOLSCRIPT (Kraulis, 1991). (B) Edmundson wheel representation of penetratin. The figure is plotted such that the orientation in the z-coordinate corresponds to A, i.e., the Cα of Q50 is located deepest in the membrane.
FIGURE 4.

Alignment of penetratin with the bilayer surface during the P-PG (A and B) and P-PC (C and D) simulations. (A and C) Longitudinal alignment: angle between the axis of the helix (N→C) and the normal of the bilayer plane. (B and D) Lateral alignment: angle between the vector from F49Cα to the geometric center of I47Cα and N51Cα, and the normal of the bilayer plane.
In the P-PG simulation, the membrane consists of 112 neutral POPC lipids, and 16 negatively charged POPG lipids, corresponding to a 7:1 POPC/POPG ratio. We observed only minor preferential hydrogen bond formation between penetratin and the negatively charged lipids, as these interactions are comparable to those with neutral POPC lipids. The same applies to the interaction energy, after correction of the Coulomb and Lennard-Jones contribution for the relative percentage of each lipid molecule.
Influence on the bilayer structure
Fig. 5 shows the density profile for the lipid bilayer halves in the presence and absence of penetratin, averaged over a time frame of 100–230 ns. The density peaks are broader and deeper for the peptide-containing layer (solid lines), for both lipid headgroup function, phosphate, carbonyl, and double-bond groups (for definitions see Computational Details), due to the presence of penetratin, docked between the lipid headgroups. The overall bilayer structure shows little change, as the peak maxima are not significantly shifted, but peaks are broadened as some lipids are pushed toward the core of the membrane by the bound penetratin. This applies in particular to the lower end of the phosphate and carbonyl group densities of POPG. The peak broadening is more pronounced for the POPG glycerol group, which is also clearly shifted, than for the POPC choline group. The POPG headgroups (glycerol and phosphate), including those which are not in the peptide-containing layer, are set deeper in the membrane core than the neutral POPC lipids.
Fig. 6 B shows a plot of the average mass-weighted distance (z-coordinate) to the membrane center of selected groups of the bilayer half containing the peptide, and of penetratin. Subtraction of the red phosphate line from the turquoise penetratin line yields Fig. 1 B. Fig. 6 A represents the number of lipids interacting with the peptide as defined by our criteria in Computational Details. This number is fluctuating around 15, corresponding to ∼25% of the bilayer half, including lipids whose acyl chains are partly under the peptide, but whose headgroups are beside it. During the first 10 ns of the simulation, when penetratin is still at an average distance of >5 Å from the bilayer surface, the interacting lipids are at roughly the same distance from the membrane center as the entire layer. Penetratin binding to the bilayer surface immediately affects the lipids under the peptide, as they are significantly pushed down toward the lipid core. This applies not only to the groups that directly interact with the peptide, i.e., the phosphate or carbonyl groups, but also to the double bonds further away.
The conformation of lipids is usually described by deuterium order parameters SCD, measured by 2H-NMR experiments, or calculated from the lipid acyl chain C–C dihedral angles (Merz and Roux, 1996). Fig. 7 shows the order parameters for the sn2 and sn1 acyl chains (Fig. 7, A and B, respectively). The shape of the order parameter curves resembles that obtained from 2H-NMR experiments, with a plateau near the carbonyl groups and increasing random structure toward the bilayer core, including the zig-zag appearance for the unsaturated oleoyl chain and the dip due to the sp2 instead of sp3 hybridization around the double bond (Seelig and Seelig, 1980). The order parameters calculated for the peptide-interacting lipids (dashed lines and open diamonds) are shifted downward over the entire range of the acyl chain, showing that the presence of penetratin introduces disorder over the entire length of the lipids.
FIGURE 7.

Calculated deuterium order parameters for the lipid acyl chains. (A) sn2 Oleoyl chain. (B) sn1 Palmitoyl chain. Solid lines and circles are order parameters calculated over all lipids, dashed lines and open diamonds denote the order parameters for the lipids that are interacting with penetratin. Order parameters were calculated over the entire simulation length.
Fig. 8 A illustrates how penetratin affects lipid diffusion, by showing all (x, y) coordinate combinations of the phosphorus atoms during the time frame between 100 and 230 ns. Whereas the xy-plane for the nonpeptide binding layer would show a uniform distribution (data not shown), the presence of penetratin (red dots, Cα atoms) strongly decreases the free motion of the lipids. The right-hand side residues of the peptide, including K46, R53, and K57, which correspond to the right-hand side in Fig. 3 B, decrease lipid motion more than the left-hand side residues. This helix's right-hand side further includes aromatic residues F49 and W56, but not W48. We also notice a preference for negatively charged lipids to reside at this side of the peptide, demonstrating their preferential interaction with the positively charged penetratin side chains. The solid bars in Fig. 8 B show restricted motion of the penetratin side chains, which applies not only to the lipid-binding residues K46, R53, and K57, but also to the solvent-directed residues, e.g., W48, R52, K55, and W56.
FIGURE 8.

(A) Scatter plot showing the diffusion of the lipid phosphate groups in the peptide-binding membrane layer. The (x, y) coordinates are plotted, with black and blue dots indicating a POPC and POPG phosphorus atom, respectively, and red dots a penetratin Cα atom. Coordinates are collected over the time frame 100–230 ns, with an increment of 100 ps. All coordinates are given relative to penetratin; during the simulation the lateral diffusion constant D of penetratin is ∼(1.8 ± 8.3) × 10−11 m2/s. This value, although taken from the simulation of a single penetratin molecule, agrees with experimental values (Andersson et al., 2004). Penetratin orientation in this figure, is with R43 at the lower left and the associating residues K46, R53, and K57 to the right, corresponding to a top view of Fig. 3 B. (B) Root mean-square fluctuation of penetratin side chains, in solution (thin-dashed bars), during initial binding to the bilayer (thick-dashed bars), and in complex with phospholipids (solid bars). The RMSF values are calculated from the P-WAT simulation, from the third added penetratin in the P3-PG simulation, and from the P-PG simulation, respectively.
Behavior of the penetratin tryptophan residues
The orientation of aromatic rings can be described by the orientational parameters SN and SL, taken relative to the normal to the bilayer plane, where SN relates to the normal to the aromatic ring, and SL describes the vector from Cγ through the ring. When S = 1, this vector is aligned with the normal of the bilayer plane, whereas S =  means orthogonality (see Computational Details). We plotted the orientational order parameters for the three aromatic residues of penetratin in Fig. 9, A and B. The tryptophan residues (top and bottom) seem more mobile than phenylalanine (center), which is more or less restricted to a combination of
means orthogonality (see Computational Details). We plotted the orientational order parameters for the three aromatic residues of penetratin in Fig. 9, A and B. The tryptophan residues (top and bottom) seem more mobile than phenylalanine (center), which is more or less restricted to a combination of 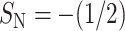 and
and  An increase of either SN or SL means a concomitant decrease of either SL or SN, respectively, as both parameters cannot simultaneously be equal to 1. However, the reverse is not true and a decrease in one orientational parameter does not imply an increase in the other. For example, at t ≈ 205 ns, SL increases for both W48 and F49 and SN has to decrease. Also, the larger value of SN for W56 after ∼60 ns induces a restricted value of
An increase of either SN or SL means a concomitant decrease of either SL or SN, respectively, as both parameters cannot simultaneously be equal to 1. However, the reverse is not true and a decrease in one orientational parameter does not imply an increase in the other. For example, at t ≈ 205 ns, SL increases for both W48 and F49 and SN has to decrease. Also, the larger value of SN for W56 after ∼60 ns induces a restricted value of  for SL. From these figures, we can conclude that both tryptophans adopt a tilted orientation, whereas phenylalanine is stacked parallel to the membrane surface. The average mass-weighted distances, plotted as the z-coordinate, between the three aromatic residues and the interacting lipid phosphate groups are shown in Fig. 9 C. The greater mobility of W56 in SN (Fig. 9 A) is due to its large distance from the lipid surface, whereas a combined investigation of Fig. 9, A–C, shows a strong correlation between the motions of W48 and F49.
for SL. From these figures, we can conclude that both tryptophans adopt a tilted orientation, whereas phenylalanine is stacked parallel to the membrane surface. The average mass-weighted distances, plotted as the z-coordinate, between the three aromatic residues and the interacting lipid phosphate groups are shown in Fig. 9 C. The greater mobility of W56 in SN (Fig. 9 A) is due to its large distance from the lipid surface, whereas a combined investigation of Fig. 9, A–C, shows a strong correlation between the motions of W48 and F49.
FIGURE 9.

(A, B, and C) Orientation and position of the aromatic residues W48 (top), F49 (center), and W56 (bottom). (A and B) Orientational parameters SN and SL, respectively. (C) Average distance (z-coordinate) between the aromatic ring systems and the phosphate groups of peptide-associated lipids. (D) Distance of the cation-π interactions between aromatic residues W48, F49, W56 and either R52 or R53. (Top) W48-R52; (center) F49-R53; (bottom) W56-R52. The distance is measured from the geometric center of the two arginine Nζ atoms to the center of the aromatic 6-ring group.
The π-clouds of an aromatic ring system are subject to cation-π interactions or XH-π hydrogen bonding, with X corresponding to C, N, or O (Meyer et al., 2003). Fig. 9 D illustrates the distances between the aromatic rings and the positively charged residue that is closest in space, i.e., R52 for W48 and W56, and R53 for F49. We find a mutually exclusive connection from R52 to either W48 or W56, with preference for W48. The shorter minimal distance between R52 and W48 corresponds to a parallel cation-π stacking compared to a perpendicular stacking through NH-π hydrogen bonding for R52–W56. As penetratin remains α-helical throughout the simulation, both tryptophan residues remain spatially close to R52, as they are only one helical turn apart (Fig. 3 B). Stacking of R53 and F49 is further found during the entire simulation (Fig. 9 D). This connection is only broken at the very beginning and during the last 35 ns of the simulation, and the behavior of the orientational parameter SL for F49 can be accounted for by the R53/F49 NH-π stacking. The R53-F49 connection is stronger as no other alternative exists, whereas R52 can be stacked to either W48 or to W56. The R52-W48 link is strongest when both SN and SL fluctuate between 0 and 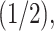 whereas a shift of this connection to R52-W56 is associated with an antiparallel orientation of
whereas a shift of this connection to R52-W56 is associated with an antiparallel orientation of 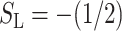 for both W48 and W56. At t ≈ 205 ns a persistent hydrophobic cluster is formed above the peptide by W48 and F49, inducing a steep jump in the z-coordinate. This cluster disrupts the R53-F49 association, which is compensated by a CH-π stacking of F49/W48. Together these results explain the link between W48 and F49 on the one hand, and W48 and W56 on the other.
for both W48 and W56. At t ≈ 205 ns a persistent hydrophobic cluster is formed above the peptide by W48 and F49, inducing a steep jump in the z-coordinate. This cluster disrupts the R53-F49 association, which is compensated by a CH-π stacking of F49/W48. Together these results explain the link between W48 and F49 on the one hand, and W48 and W56 on the other.
Association between penetratin peptides at the bilayer surface
A second penetratin molecule was added to the P-PG simulation at t = 100 ns, yielding simulation P2-PG, to investigate any association of penetratin molecules docked to the lipid membrane. After 25 ns of simulation, when the second peptide was docked to the bilayer, a third penetratin was added, yielding simulation P3-PG.
Binding of the second penetratin molecule occurred onto the bilayer side where no peptide was yet present (we apply periodic boundary conditions), with the same residues K46, R53, and K57 (and K58) accounting for initial peptide-lipid binding. However, binding of a third penetratin was significantly slower, resembling the P-PC simulation, due to prior neutralization of POPG negative charges by the bound penetratin peptides. The third peptide associates first with the lipids through its C-terminal end, perpendicular to the lipid bilayer, as observed for the neutral bilayer P-PC simulation. This binding occurs at a distance from the first and second peptides, and no peptide-peptide interaction is observed.
Binding of penetratin variants to the lipid bilayers
Fig. 3 B shows that the pairs of charged residues R52/K55 and R53/K57 are located at different sides of the helical peptide. Double mutants were synthesized to study the contributions of these residues to penetratin interactions with model membranes and cells (Christiaens et al., 2004). We looked at the effect of these mutations upon penetratin interactions with the lipid bilayer, by applying the R52A/K55A and R53A/K57A mutations to the P-PG simulation at t = 100 ns, and pursuing the simulations for 20 ns (simulations P52A, 55A-PG and P53A, 57A-PG).
The structure, position, and orientation of the peptides were not affected by the mutations. The α-helical structure was retained, and the lipid structure did not change significantly, in agreement with the experimental data (Christiaens et al., 2004).
Table 2 shows the interaction energies of the mutant peptides both with the membrane, expressed as separate electrostatic (Coulomb) and hydrophobic (Lennard-Jones) energies, and with the aqueous phase. The R/K→A mutations have little effect on the LJ energy, whereas the Coulomb energy was strongly affected. Peptide-membrane interactions increase for the R52A/K55A mutant, whereas they decrease for the R53A/K57A mutant. The peptide-solvent interaction decreases slightly for the R53A/K57A mutant, and more strongly for the R52A/K55A mutant.
TABLE 2.
Relevant interaction energies for the simulations involving mutations
| Energy (kJ/mol)
|
|||
|---|---|---|---|
| Peptide-membrane
|
|||
| System | Coulomb | LJ | Peptide-water Coulomb + LJ |
| P-PG | −1550 ± 69 | −717 ± 37 | −834 ± 56 |
| P52A,55A-PG | −1689 ± 73 | −748 ± 52 | −602 ± 58 |
| P53A,57A-PG | −1337 ± 63 | −671 ± 38 | −736 ± 49 |
Energies were averaged either over the last 10 ns of the trajectory (P52A,55A-PG, P53A,57A-PG), or over the last 10 ns before the mutation was introduced (P-PG). LJ, Lennard-Jones.
As residues R52 and K55 are solvent-accessible, mutations to an alanine strongly decrease the electrostatic contribution to the interaction energy, and consequently the peptide-solvent interaction. This is compensated by stronger hydrophobic interaction with membrane lipids underneath the peptide, resulting in an overall decrease in peptide-membrane energy. For the R53A/K57A mutation, as both residues are involved in initial peptide-lipid binding, mutations to an alanine disrupt the favorable side chains to phosphate interaction, accounting for a strong increase in the Coulomb energy. The peptide-water interaction is only slightly affected, as the mutated residues are located between the peptide and the membrane.
Penetratin structure in water
The original penetratin peptide is α-helical, as it originates from the third helix of the Antennapedia homeodomain. Circular dichroism measurements show that penetratin in buffer is mostly random, and that its α-helical content increases upon binding to phospholipid vesicles (Persson et al., 2001, 2003; Magzoub et al., 2003; Christiaens et al., 2004). We performed a 120-ns simulation of the helical penetratin to investigate its stability in aqueous solution. The peptide assumes a random conformation in water, in agreement with the experimental observations. The α-helical structure remains stable enough during the beginning of the simulation (∼10 ns) to prevent unfolding during the bilayer simulations, before lipid association. Moreover, our P-PC simulation shows that the presence of a neutral bilayer has a stabilizing effect on the helix, preventing unfolding of the peptide during the first 40 ns, before binding to the bilayer (Fig. 1 E). The simulation of the penetratin structure in an aqueous solution, using a cluster analysis based on backbone root mean-square deviation, shows a largest-populated cluster of structures with an unfolded C-terminus. W48, W56, and R53 form a π-cluster through R53/W56 cation-π and R53/W48 NH-π interactions. The other residues are all solvent-directed.
The association with phospholipids significantly lowers the freedom of motion of the peptide residues. Fig. 8 B shows the root mean-square fluctuation (RMSF) of the individual residues for the peptide in solution (thin dashed bars) and for the P-PG simulation (solid bars). For the peptide in water, the lower RMSF on the N-terminal side suggests that this end of the peptide remains α-helical, as is confirmed by the secondary structure evolution (data not shown). In contrast, when bound to a membrane, the peptide retains little flexibility, even for the terminal residues R43 and K58. The flexibility of a peptide approaching the membrane, such as the third peptide in the P3-PG simulation, is intermediate (thick dashed bars).
SUMMARY AND DISCUSSION
In this computational approach we showed that negatively charged lipids significantly increase the speed of association between penetratin and a lipid membrane, as peptide-lipid association occurs within a few nanoseconds for a partially negatively charged membrane, whereas it is slower by an order of magnitude for a neutral bilayer. Penetratin approaches a charged lipid bilayer horizontally, compared to a perpendicular approach toward a neutral bilayer. Peptide-lipid association occurs through formation of salt bridges between the positively charged residues K46, R53, and K57 and the lipid phosphate groups. Tryptophan fluorescence studies previously showed the importance of peptide positively charged residues for the initial binding to negatively charged vesicles, since double R/K→A mutations involving the residues K46/R52/R53/K55/K57 significantly decreased the binding affinity (Christiaens et al., 2002, 2004). Our results, which identify three of these residues as the ones responsible for initial binding, account for both the lower binding affinity of mutants including double mutations of binding residues, and for the relatively higher binding affinity for the R52A/K55A variant. Attraction between penetratin and bilayer lipids is bidirectional, i.e., penetratin is not only attracted toward the membrane, but a few lipids are further partially pulled out of the bilayer. However, as the displacement of a lipid out of a bilayer is an energetically unfavorable process (Cevc and Marsh, 1987; Marrink et al., 1998), and established hydrogen bonds are maintained, penetratin comes close to the bilayer. No preference for negatively charged phospholipids POPG, POPA, and POPS was found during initial binding.
After initial binding, penetratin is oriented parallel to the bilayer surface. Subsequent docking of the peptide between the lipid headgroups involves only side chain movement. This process takes ∼100 ns for a (negatively) charged bilayer and up to 200 ns for a neutral bilayer. In peptide uptake experiments, using a mixed POPC/POPG bilayer, an increase in neutral/charged vesicles ratio from 1:1 to 3:1 is found to decrease internalization by a factor of 2 (Terrone et al., 2003). This is in agreement with our simulations, where the absence of a negatively charged phospholipid headgroup slows down peptide insertion by increased repulsion with penetratin. In addition, this repulsion is also responsible for the deeper insertion of penetratin in a neutral bilayer, an effect also observed in the binding of magainin to lipid membranes (Wieprecht et al., 1999). Kinetics of binding and insertion decrease as POPA > POPG > POPS > POPC.
The peptide remains fully α-helical during the entire process, with the Trp residues oriented toward the solvent. No rotation around the helical axis occurs during docking. The abundance of aromatic residues in membrane proteins has long been observed (Von Heijne, 1997), together with their propensity to reside at membrane interfaces (Yau et al., 1998). A role of Trp as translocation determinant of peptides has been proposed (Schiffer et al., 1992), and mutation of both tryptophans in penetratin was found to abolish internalization (Derossi et al., 1994). Trp fluorescence quenching and oriented circular dichroism studies showed that Trp residues in penetratin remain close to the water-lipid interface (Magzoub et al., 2003; Christiaens et al., 2004). Although the preference of tryptophan for a membrane interface is generally attributed to an amphipathic and dipolar interaction, its flat and rigid shape limits its access to the hydrophobic core of the membrane, whereas its π-electronic structure favors its position at the interface (Yau et al., 1998). Previous studies have suggested a solvent-exposed role for both tryptophans in penetratin (Fragneto et al., 2000; Magzoub et al., 2002). Our results show that both tryptophans lie preferentially at the interface, in a slightly tilted orientation, acting as a shield between the peptide and the aqueous phase above. R52 forms mutually exclusive π-stacking interactions with W48 (cation-π) and W56 (NH-π hydrogen bond), with a strong preference for W48. A NH-π hydrogen bond interaction exists also between F49 and R53. When R53 forms hydrogen bonds with the phosphate groups of two lipids, F49 becomes part of the W48-R52 cluster through CH-π hydrogen bonds to W48. The neighboring residues W48 and F49 were previously proposed to belong to a structural motif contributing to peptide internalization (Le Roux et al., 1993). This hypothesis is supported by our simulations showing that these residues can assume a π T-stacking conformation. However, the W48/R52 and F49/R53 pairs seem more relevant, and thus might account for decreased internalization of mutants lacking either R52 or R53 (Christiaens et al., 2004).
Cellular uptake experiments previously demonstrated the crucial role of arginines in cell-penetrating peptides (Mitchell et al., 2000; Thorén et al., 2003). Our results further stress the importance of arginines R52 and R53 for penetratin-membrane interactions, with the latter being mainly responsible for the initial electrostatic binding, where it can form bidentate hydrogen bonds with lipid phosphate groups, whereas the former contributes to the subsequent insertion of the peptide into the lipid bilayer and possibly to its translocation through its cation-π interaction with both tryptophans. Although quantum-mechanical in nature, cation-π interactions can be described using classical mechanical force fields (Donini and Weaver, 1998). Cation-π interactions, especially between arginine and tryptophan, significantly contribute to the structure and function of biomolecules (Ma and Dougherty, 1997; Gallivan and Dougherty, 1999). In most proteins, aromatic residues forming cation-π interactions are found at the protein surface (Flocco and Mowbray, 1994). This is also observed for the third helix of the Antennapedia homeodomain (Billeter et al., 1993), i.e., penetratin, where W48 and R52 are in a stacking conformation between the protein and DNA. A parallel configuration of these residues, with the NH2 groups of arginine still available for hydrogen bonding, is energetically favorable (Mitchell et al., 1994; Gallivan and Dougherty, 1999), in agreement with the results of our calculations for the R52-W48 combination. The lower probability for the R52-W56 stacking might be due to destabilizing effects of the three lysines K55, K57, and K58 surrounding W56. In conclusion, we find that the aromatic residues do not contribute to the initial binding, but rather to the subsequent insertion of the peptide between the bilayer headgroups, when they shield the peptide from the aqueous phase. As W48, F49, and W56 behave as aromatic π-acceptors, whereas R52 and R53 act as donors, this shield is stretched over the entire length of the peptide through cation-π interactions and NH-π hydrogen bonding. The unique combination of hydrophobicity with (weak) hydrogen-bond donor and acceptor capacities might be the key parameter for the transition from a hydrophilic to a hydrophobic phase. A W→F mutation would still be functional, though less efficient. The W48F mutation would shift the cation-π interaction from W48/R52 to W56/R52, i.e., from the center of the peptide toward its C-terminal end. In contrast, the W56F mutation strengthens the W48/R52 interaction, as F56 would be a less favorable alternative, in agreement with the crucial role of W48 for the internalization process (Derossi et al., 1994).
The preferred orientation of penetratin is almost parallel to the bilayer, at a slight angle (from C to N) of 80–90° with the normal to the bilayer surface, as observed before (Magzoub et al., 2003), and as could be expected for cationic peptides (Zhang et al., 2001). Under this angle, W48 inserts deeper into the bilayer than W56, in agreement with previous reports (Lindberg et al., 2003; Salamon et al., 2003). Trp fluorescence-quenching measurements supported a deeper insertion of the W56F compared to the W48F variant, whereas in the R52A/K55A and R53A/K57A mutants Trp residues inserted deeper into the lipids compared to wild-type penetratin (Christiaens et al., 2004). The R52A/K55A double mutation has a twofold effect: removal of the W48/R52 cation-π interaction favors insertion of both tryptophan residues into the membrane hydrophobic phase, whereas the repulsion of penetratin and water is increased by the mutation of the solvent-directed residues R52 and K55 to an alanine. In the R53A/K57A double mutant, where two of the positively charged residues interacting with the lipid phosphate headgroups are mutated to an alanine, binding might occur through residues R52 and K55. However, the deeper insertion that was found for the R52A/K55A compared to the R53A/K57A mutant (Christiaens et al., 2004) suggests similar initial peptide-lipid binding, where both tryptophans remain solvent-accessible. Such a mode of binding might involve residues R43/K46/Q50. In both double mutants, decreased interaction of W56 with the surrounding lysines promotes its insertion into the hydrophobic membrane phase. In a neutral membrane (simulations P-PC and P3-PG) we find that W56 inserts deeper than W48, whereas the peptide remains parallel to the membrane surface. This is possible due to an increased freedom of motion for W56 as it is surrounded by positively charged residues that might otherwise interact with negatively charged lipids, preventing passage of the solvent-accessible W56 into the lipid bilayer core.
When the peptide docks between the lipid headgroups, the positively charged residues K46, R53, and K57, which were responsible for initial binding, do not penetrate further into the bilayer, but rather move sideways, together with Q50 and N51. The lipid-peptide hydrogen bonds remain intact, lipid headgroups move along, and the hydrophobic membrane core is exposed to the helical peptide surface. Upon deeper insertion into the membrane core, the charge of R52 might be compensated through a bidentate hydrogen bond with an additional lipid phosphate group, thereby enhancing the hydrophobicity of the surrounding tryptophan residues, and of the peptide as a whole. The lipid phase is condensed, and lipids are pushed down, with shifted and broader density peaks compared to the inner membrane. This effect is more significant for the negatively charged lipids, than for POPC. With respect to the inner membrane, positively charged POPC choline groups are directed outward, whereas their neutral POPG glycerol counterparts are directed inward. The presence of penetratin decreases the order of the lipid acyl chains in contact with the peptide, as deuterium order parameter curves are shifted down. The differences are short-ranged, and lipids that are not in the immediate vicinity of penetratin are not affected by its presence. Association with penetratin, moreover, stabilizes the membrane bilayer, through hydrogen bonding with nonsolvent-directed penetratin side chains. A study of penetratin diffusion showed that a negatively charged lipid surface, but not a neutral one, restricted motional flexibility (Andersson et al., 2004). The presence of negatively charged lipids around penetratin supports a mechanism involving charge compensation, whereas lipid association around the peptide might account for the lack of calcein leakage upon peptide translocation (Christiaens et al., 2004).
The complete internalization process occurs on a millisecond timescale (Hällbrink et al., 2001; Terrone et al., 2003), and is therefore out of range of conventional MD simulations. Moreover, although internalization does not need a transmembrane potential (Thorén et al., 2000), such a potential is required to obtain a physiologically relevant process (Drin et al., 2001a; Kramer and Wunderli-Allenspach, 2003; Terrone et al., 2003). We are currently investigating the effect a transbilayer potential has on the structure and positioning of penetratin in a bilayer.
Although these results clearly identify the charged residues that lie at the basis of the peptide-membrane association, and hence confirm the importance of electrostatics for initial binding, there are additional effects that play a role as well. The classical hydrophobic effect can be understood as the release of the hydration shell around the peptide upon membrane incorporation. This is an entropy-driven process (Tanford, 1973) that is strongly dependent on temperature (Gill and Wadsö, 1976) and as such was found not to be the dominating contribution to penetratin binding (Binder and Lindblom, 2003b). The classical hydrophobic effect should not be confused with the so-called nonclassical hydrophobic effect (Seelig and Ganz, 1991), where an exothermic binding heat is found due to favorable peptide-lipid and lipid-lipid interactions (Wieprecht et al., 1999). Our simulations show an alteration in the conformation of the lipid headgroups and an increase in the lipid packing density. Both effects have also been observed using surface plasmon resonance and impedance measurements (Binder and Lindblom, 2003b). A third, enthalpically favorable, effect is the transition to a more ordered (α-helical or β-sheet) conformation upon membrane binding (White and Wimley, 1998). According to our simulations, penetratin is mainly unfolded in water, whereas it becomes 60% α-helical when associated with lipids. This increased stability of the α-helical structure is due to both backbone hydrogen bond shielding at the lipid/water interface by R52, W48, and W56, and to the decreased dielectric constant in the hydrophobic membrane core. These effects lower the hydration of backbone NH and CO groups, resulting in a more stable and less hydrophilic helical structure (Avbelj et al., 2000; García and Sanbonmatsu, 2002; Roccatano et al., 2002).
We found no aggregation of penetratin peptides at the membrane surface, as reported in solution (Magzoub et al., 2002; Christiaens et al., 2004), suggesting that the negatively charged bilayer does not induce cooperative binding. A theoretical study of protein adsorption on a mixed membrane shows that the negatively charged lipids in the lipid bilayer adjust their local concentration to achieve optimal charge matching with the protein or peptide (May et al., 2000). Subsequent binding of additional peptides in the vicinity of the first one is significantly less favorable due to lateral repulsive interactions between the adsorbed peptides and the effective charge reversal as solvent-directed positive charges of the peptide are not matched by negatively charged lipids. A penetratin peptide, when bound to a bilayer, interacts with ∼15–20 lipids, suggesting that above a molar peptide:lipid ratio of 0.05 free peptides remain in the aqueous phase, as observed for the circular dichroism spectra of penetratin associated with 70:30 PC/PS vesicles (Christiaens et al., 2004). The lack of cooperation and aggregation reported here does not exclude a cooperative effect in the final phase of the internalization, as only the initial steps of this process were studied. Further internalization might involve cooperation between several penetratin peptides, to reduce surface tension and enable cell-penetrating peptides, and its cargo, to enter the membrane. Membrane thinning was shown for alamethicin (He et al., 1996) and many other peptide-lipid systems (Ludtke et al., 1995; Tieleman et al., 1998; Heller et al., 2000; Chen et al., 2003); at low concentrations, the peptides adsorb onto the bilayer surface, whereas above a critical lipid-dependent concentration, a fraction of the peptides insert into the membrane. Penetratin might induce membrane thinning when entering the hydrophobic inner bilayer and dragging phospholipids along. Motion of the phospholipid headgroups together with penetratin secures the current phase separation consisting of lipid headgroups that could tightly encompass any hydrophilic cargo, resulting in minimal leakage. This mechanism would include flip-flop of certain phospholipids (Matsuzaki et al., 1996), but excludes the formation of pores as with some antimicrobial peptides (Zasloff, 2002). Our simulations showed favorable interactions between penetratin and the hydrophobic core of the bilayer, which together with hydrogen bonding to negatively charged lipids, might destabilize the phospholipid bilayer, finally resulting in membrane translocation of the penetratin peptide.
Acknowledgments
M.F.L. acknowledges support from European Economic Community contract NPRN-CT-2001-0242 “Messenger proteins: mechanisms of action and biological significance”. B.C. was supported by a grant from the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen).
References
- Andersson, A., J. Almqvist, F. Hagn, and L. Mäler. 2004. Diffusion and dynamics of penetratin in different membrane mimicking media. Biochim. Biophys. Acta. 1661:18–24. [DOI] [PubMed] [Google Scholar]
- Avbelj, F., P. Luo, and R. L. Baldwin. 2000. Energetics of the interaction between water and the helical peptide group and its role in determining helix propensities. Proc. Natl. Acad. Sci. USA. 97:10786–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechinger, B. 1997. Structure and functions of channel-forming peptides: magainins, cecropins, melittin, and alamethicin. J. Membr. Biol. 156:197–211. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C. 1996. Bio-molecular dynamics comes of age. Science. 271:954–955. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C. 2001. Reality simulation: Observe while it happens. Science. 294:2304–2305. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 181:3684–3690. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. Van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht. 331–342.
- Berendsen, H. J. C., D. Van der Spoel, and R. Van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- Berger, O., O. Edholm, and F. Jähnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlose, J. P., O. Convert, D. Derossi, A. Brunissen, and G. Chassaing. 1996. Conformational and associative behaviours of the third helix of antennapedia homeodomain in membrane-mimetic environment. Eur. J. Biochem. 242:372–386. [DOI] [PubMed] [Google Scholar]
- Billeter, M., Y. Q. Qian, G. Otting, M. Müller, W. J. Gehring, and K. Wütrich. 1993. Determination of the NMR solution structure of an antennapedia homeodomain-DNA complex. J. Mol. Biol. 234:1084–1097. [DOI] [PubMed] [Google Scholar]
- Binder, H., and G. Lindblom. 2003a. Charge-dependent translocation of the trojan peptide penetratin across lipid membranes. Biophys. J. 85:982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder, H., and G. Lindblom. 2003b. Interaction of the trojan peptide penetratin with anionic lipid membranes: a calorimetric study. Phys. Chem. Chem. Phys. 5:5108–5117. [Google Scholar]
- Brattwall, C. E., P. Lincoln, and B. Nordén. 2003. Orientation and conformation of cell-penetrating peptide penetratin in phospholipid vesicle membranes determined by polarized-light spectroscopy. J. Am. Chem. Soc. 125:14214–14215. [DOI] [PubMed] [Google Scholar]
- Braun, K., P. Peschke, R. Pipkorn, S. Lampel, M. Wachsmuth, W. Waldeck, E. Friedrich, and J. Debus. 2002. A biological transporter for the delivery of peptide nucleic acids (PNAs) to the nuclear compartment of living cells. J. Mol. Biol. 318:237–243. [DOI] [PubMed] [Google Scholar]
- Brugidou, J., C. Legrand, J. Méry, and A. Rabié. 1995. The retro-inverso form of a homeobox-derived short peptide is rapidly internalised by cultured neurones: A new basis for an efficient intracellular delivery system. Biochem. Biophys. Res. Commun. 214:685–693. [DOI] [PubMed] [Google Scholar]
- Cevc, G., and D. Marsh. 1987. Phospholipid Bilayers: Physical Principles and Models. John Wiley & Sons, New York.
- Chen, F.-Y., M.-T. Lee, and H. W. Huang. 2003. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys. J. 84:3751–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaens, B., J. Grooten, M. Reusens, A. Joliot, M. Goethals, J. Vandekerckhove, A. Prochiantz, and M. Rosseneu. 2004. Membrane interaction and cellular internalization of penetratin peptides. Eur. J. Biochem. 271:1187–1197. [DOI] [PubMed] [Google Scholar]
- Christiaens, B., S. Symoens, S. Vanderheyden, Y. Engelborghs, A. Joliot, A. Prochiantz, J. Vandekerckhove, M. Rosseneu, and B. Vanloo. 2002. Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes. Eur. J. Biochem. 269:2918–2926. [DOI] [PubMed] [Google Scholar]
- Christopher, J. A., R. Swanson, and T. O. Baldwin. 1996. Algorithms for finding the axis of a helix: Fast rotational and parametric least-squares methods. Comput. Chem. 20:339–345. [DOI] [PubMed] [Google Scholar]
- Console, S., C. Marty, C. García-Echeverría, R. Schwendener, and K. Ballmer-Hofer. 2003. Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J. Biol. Chem. 278:35109–35114. [DOI] [PubMed] [Google Scholar]
- Cornut, I., E. Thiaudière, and J. Dufourcq. 1993. The amphipathic helix in cytotoxic peptides. In The Amphipathic Helix. R. M. Epand, editor. CRC Press, Boca Raton, FL. 173–219.
- Derossi, D., S. Calvet, A. Trembleau, A. Brunissen, G. Chassaing, and A. Prochiantz. 1996. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 271:18188–18193. [DOI] [PubMed] [Google Scholar]
- Derossi, D., G. Chassaing, and A. Prochiantz. 1998. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 8:84–87. [PubMed] [Google Scholar]
- Derossi, D., A. H. Joliot, G. Chassaing, and A. Prochiantz. 1994. The third helix of the antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 269:10444–10450. [PubMed] [Google Scholar]
- Dom, G., C. Shaw-Jackson, C. Matis, O. Bouffioux, J. Picard, A. Prochiantz, M.-P. Mingeot-Leclercq, R. Brasseur, and R. Rezsohazy. 2003. Cellular uptake of antennapedia penetratin peptides is a two-step process in which phase transfer precedes a tryptophan-dependent translocation. Nucleic Acids Res. 31:556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donini, O., and D. F. Weaver. 1998. Development of modified force field for cation-amino acid interactions: Ab initio-derived empirical correction terms with comments on cation-π interactions. J. Comput. Chem. 19:1515–1525. [Google Scholar]
- Drin, G., H. Demene, J. Temsamani, and R. Brasseur. 2001a. Translocation of the pAntp peptide and its amphipathic analogue AP-2AL. Biochemistry. 40:1824–1834. [DOI] [PubMed] [Google Scholar]
- Drin, G., M. Mazel, P. Clair, D. Mathieu, M. Kaczorek, and J. Temsamani. 2001b. Physico-chemical requirements for cellular uptake of pAntp peptide: Role of lipid-binding affinity. Eur. J. Biochem. 268:1304–1314. [DOI] [PubMed] [Google Scholar]
- Essman, U., L. Perela, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh ewald method. J. Chem. Phys. 103:8577–8592. [Google Scholar]
- Feenstra, K. A., B. Hess, and H. J. C. Berendsen. 1999. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20:786–798. [DOI] [PubMed] [Google Scholar]
- Fischer, P. M., N. Z. Zhelev, S. Wang, J. E. Melville, R. F. Hraeus, and D. P. Lane. 2000. Structure-activity relationship of truncated and substituted analogues of the intracellular delivery vector penetratin. J. Pept. Res. 55:163–172. [DOI] [PubMed] [Google Scholar]
- Flocco, M. M., and S. L. Mowbray. 1994. Planar stacking interactions of arginine and aromatic side-chains in proteins. J. Mol. Biol. 235:709–717. [DOI] [PubMed] [Google Scholar]
- Fragneto, G., F. Graner, T. Charitat, P. Dubos, and E. Bellet-Amalric. 2000. Interaction of the third helix of antennapedia homeodomain with a deposited phospholipid bilayer: A neutron reflectivity structural study. Langmuir. 16:4581–4588. [Google Scholar]
- Gallivan, J. P., and D. A. Dougherty. 1999. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA. 96:9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García, A. E., and K. Y. Sanbonmatsu. 2002. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA. 99:2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S. J., and I. Wadsö. 1976. An equation of state describing hydrophobic interactions. Proc. Natl. Acad. Sci. USA. 73:2955–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hällbrink, M., A. Florén, A. Elmquist, M. Pooga, T. Bartfai, and Ü. Langel. 2001. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta. 1515:101–109. [DOI] [PubMed] [Google Scholar]
- He, K., S. J. Ludtke, W. T. Heller, and H. W. Huang. 1996. Mechanism of alamethicin insertion into lipid bilayers. Biophys. J. 71:2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller, W. T., A. J. Waring, R. I. Lehrer, T. A. Harroun, T. M. Weiss, L. Yang, and H. W. Huang. 2000. Membrane thinning effect of the β-sheet antimicrobial protegrin. Biochemistry. 39:139–145. [DOI] [PubMed] [Google Scholar]
- Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1473. [Google Scholar]
- Hosotani, R., Y. Miyamoto, K. Fujimoto, R. Doi, A. Otaka, N. Fukii, and M. Imamura. 2002. Trojan p16 peptide suppresses pancreatic cancer growth and prolongs survival in mice. Clin. Cancer Res. 8:1271–1276. [PubMed] [Google Scholar]
- Joliot, A. H., A. Triller, M. Volovitch, C. Pernelle, and A. Prochiantz. 1991. Alpha-2,8-polysialic acid is the neuronal surface receptor of antennapedia homeobox peptide. New Biol. 3:1121–1134. [PubMed] [Google Scholar]
- Karplus, M. 2002. Molecular dynamics simulations of biomolecules. Acc. Chem. Res. 35:321–322. [DOI] [PubMed] [Google Scholar]
- Kaznessis, Y. N., S. Kim, and R. G. Larson. 2002. Specific mode of interaction between components of model pulmonary surfactants using computer simulations. J. Mol. Biol. 322:569–582. [DOI] [PubMed] [Google Scholar]
- Kramer, S. D., and H. Wunderli-Allenspach. 2003. No entry for TAT(44–57) into liposomes and intact MDCK cells: Novel approach to study membrane permeation of cell-penetrating peptides. Biochim. Biophys. Acta. 1609:161–169. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- La Rocca, P., P. C. Biggin, D. P. Tieleman, and M. S. P. Sansom. 1999. Simulation studies of the interaction of antimicrobial peptides and lipid bilayers. Biochim. Biophys. Acta. 1462:185–200. [DOI] [PubMed] [Google Scholar]
- Langel, Ü. editor. 2002. Cell Penetrating Peptides. CRC Press, Boca Raton, FL.
- Le Roux, I., A. Joliot, E. Bloch-Gallego, A. Prochiantz, and M. Volovitch. 1993. Neurotrophic activity of the antennapedia homeodomain depends on its specific DNA-binding properties. Proc. Natl. Acad. Sci. USA. 90:9120–9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenhouts, J. M., Z. Torok, V. Mandieau, E. Goormaghtigh, and B. de Kruijff. 1996. The N-terminal half of a mitochondrial presequence peptide inserts into cardiolipin-containing membranes. FEBS Lett. 388:34–38. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., B. Hess, and D. Van der Spoel. 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- Lindberg, M., H. Biverstahl, A. Graslund, and L. Maler. 2003. Structure and positioning comparison of two variants of penetratin in two different membrane mimicking systems by NMR. Eur. J. Biochem. 270:2055–2063. [DOI] [PubMed] [Google Scholar]
- Lindgren, M., M. Hällbrink, A. Prochiantz, and Ü. Langel. 2000. Cell-penetrating peptides. Trends Pharmacol. 21:99–103. [DOI] [PubMed] [Google Scholar]
- Ludtke, S., K. He, and H. Huang. 1995. Membrane thinning caused by magainin 2. Biochemistry. 34:16764–16769. [DOI] [PubMed] [Google Scholar]
- Ma, J. C., and D. A. Dougherty. 1997. The cation-π interaction. Chem. Rev. 97:1303–1324. [DOI] [PubMed] [Google Scholar]
- Mackay, A. L. 1984. Quaternion transformation of molecular orientation. Acta Cryst. Sect. A. 40:165–166. [Google Scholar]
- Maduke, M., and D. Roise. 1993. Import of a mitochondrial presequence into protein-free phospholipid vesicles. Science. 260:364–367. [DOI] [PubMed] [Google Scholar]
- Magzoub, M., L. E. Eriksson, and A. Graslund. 2002. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: Effects of surface charge and peptide concentration. Biochim. Biophys. Acta. 1563:53–63. [DOI] [PubMed] [Google Scholar]
- Magzoub, M., L. E. Eriksson, and A. Graslund. 2003. Comparison of the interaction, positioning, structure induction and membrane perturbation of cell-penetrating peptides and non-translocating variants with phospholipid vesicles. Biophys. Chem. 103:271–288. [DOI] [PubMed] [Google Scholar]
- Marrink, S.-J., O. Berger, D. P. Tieleman, and F. Jähnig. 1998. Adhesion forces of lipids in a phospholipid membrane studied by molecular dynamics simulations. Biophys. J. 74:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki, K. 1998. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta. 1376:391–400. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., O. Murase, H. Tokuda, N. Fujii, and K. Miyajima. 1996. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 35:11361–11368. [DOI] [PubMed] [Google Scholar]
- May, S., D. Harries, and A. Ben-Shaul. 2000. Lipid demixing and protein-protein interactions in the adsorption of charged proteins on mixed membranes. Biophys. J. 79:1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz, K. M., Jr., and B. Roux. editors. 1996. Biological Membranes: A Molecular Perspective from Computation and Experiment. Birkhäuser, Boston, MA.
- Meyer, E. A., R. K. Castellano, and F. Diederich. 2003. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed Engl. 42:1210–1250. [DOI] [PubMed] [Google Scholar]
- Mitchell, D. J., D. T. Kim, L. Steinman, C. G. Fathman, and J. B. Rothbard. 2000. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56:318–325. [DOI] [PubMed] [Google Scholar]
- Mitchell, J. B. O., C. L. Nandi, I. K. McDonald, J. M. Thornton, and S. L. Price. 1994. Amino/aromatic interactions in proteins: Is the evidence stacked against hydrogen bonding? J. Mol. Biol. 239:315–331. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., and P. A. Kollman. 1992. SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- Persson, D., P. E. G. Thorén, M. Herner, P. Lincoln, and B. Nordén. 2003. Application of a novel analysis to measure the binding of the membrane-translocating peptide penetratin to negatively charged liposomes. Biochemistry. 42:421–429. [DOI] [PubMed] [Google Scholar]
- Persson, D., P. E. G. Thorén, and B. Nordén. 2001. Penetratin-induced aggregation and subsequent dissociation of negatively charged phospholipid vesicles. FEBS Lett. 505:307–312. [DOI] [PubMed] [Google Scholar]
- Pooga, M., U. Soomets, M. Hällbrink, A. Valkna, K. Saar, K. Rezaei, U. Kahl, J. Hao, S. Wiesenfeld-Hallin, T. Hökfelt, T. Bartfai, and Ü. Langel. 1998. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 16:857–861. [DOI] [PubMed] [Google Scholar]
- Prochiantz, A. 1996. Getting hydrophilic compounds into cells: lessons from homeopeptides. Curr. Opin. Neurobiol. 6:629–634. [DOI] [PubMed] [Google Scholar]
- Prochiantz, A. 1998. Peptide nucleic acid smugglers. Nat. Biotechnol. 16:819–820. [DOI] [PubMed] [Google Scholar]
- Richard, J. P., K. Melikov, E. Vivès, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik, and B. Lebleu. 2003. Cell-penetrating peptides: a reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 278:585–590. [DOI] [PubMed] [Google Scholar]
- Roccatano, D., G. Colombo, M. Fioroni, and A. E. Mark. 2002. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study. Proc. Natl. Acad. Sci. USA. 99:12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamon, Z., G. Lindblom, and G. Tollin. 2003. Plasmon-wave-guide resonance and impedance spectroscopy studies of the interaction between penetratin and supported lipid bilayer membranes. Biophys. J. 84:1796–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer, M., C.-H. Chang, and F. J. Stevens. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5:213–214. [DOI] [PubMed] [Google Scholar]
- Seelig, J., and P. Ganz. 1991. Nonclassical hydrophobic effect in membrane binding equilibria. Biochemistry. 30:9354–9359. [DOI] [PubMed] [Google Scholar]
- Seelig, J., and A. Seelig. 1980. Lipid conformation in model membranes and biological membranes. Q. Rev. Biophys. 13:19–61. [DOI] [PubMed] [Google Scholar]
- Shepherd, C. M., H. J. Vogel, and D. P. Tieleman. 2003. Interactions of the designed antimicrobial peptide MB21 and truncated dermaseptin S3 with lipid bilayers: molecular-dynamics simulations. Biochem. J. 370:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T., S. Futaki, M. Niwa, S. Tanaka, K. Ueda, and Y. Sugiura. 2002. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 277:2437–2443. [DOI] [PubMed] [Google Scholar]
- Tanford, C. editor. 1973. The Hydrophobic Effect. Wiley and Sons, New York.
- Terrone, D., S. L. Sang, L. Roudaia, and J. R. Silvius. 2003. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry. 42:13787–13799. [DOI] [PubMed] [Google Scholar]
- Thorén, P. E. G., D. Persson, P. Isakson, M. Goksör, A. Önfelt, and B. Nordén. 2003. Uptake of analogs of penetratin, TAT(48–60) and oligoarginine in live cells. Biochem. Biophys. Res. Commun. 307:100–107. [DOI] [PubMed] [Google Scholar]
- Thorén, P. E. G., D. Persson, M. Karlsson, and B. Nordén. 2000. The antennapedia peptide penetratin translocates across lipid bilayers—the first direct observation. FEBS Lett. 482:265–268. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., L. R. Forrest, M. S. P. Sansom, and H. J. C. Berendsen. 1998. Lipid properties and the orientation of aromatic residues in OmpF, influenza M2, and alamethicin systems: Molecular dynamics simulations. Biochemistry. 37:17554–17561. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., S.-J. Marrink, and H. J. C. Berendsen. 1997. A computer perspective of membranes: Molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta. 1331:235–270. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., M. S. P. Sansom, and H. J. C. Berendsen. 1999. Alamethicin helices in a bilayer and in solution: molecular dynamics simulations. Biophys. J. 76:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Hochschulverlag AG, Zürich, Switzerland.
- Vivès, E., J. P. Richard, C. Rispal, and B. Lebleu. 2003. TAT peptide internalization: seeking the mechanism of entry. Curr. Protein. Pept. Sci. 4:125–132. [DOI] [PubMed] [Google Scholar]
- Von Heijne, G. editor. 1997. Membrane Protein Assembly. Chapman & Hall, Austin, TX.
- Wender, P. A., D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman, and J. B. Rothbard. 2000. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. USA. 97:13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, S. H., and W. C. Wimley. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1376:339–352. [DOI] [PubMed] [Google Scholar]
- Wieprecht, T., M. Beyermann, and J. Seelig. 1999. Binding of antibacterial magainin peptides to electrically neutral membranes: thermodynamics and structure. Biochemistry. 38:10377–10387. [DOI] [PubMed] [Google Scholar]
- Yau, W.-M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature. 415:389–395. [DOI] [PubMed] [Google Scholar]
- Zhang, L., A. Rozek, and R. E. W. Hancock. 2001. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 276:35714–35722. [DOI] [PubMed] [Google Scholar]