

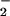
Abstract
The chemical origins of nitrated tyrosine residues (NT) formed in proteins during a variety of pathophysiological conditions remain controversial. Although numerous studies have concluded that NT is a signature for peroxynitrite (ONOO−) formation, other works suggest the primary involvement of peroxidases. Because metal homeostasis is often disrupted in conditions bearing NT, the role of metals as catalysts for protein nitration was examined. Cogeneration of nitric oxide (NO) and superoxide (O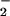 ), from spermine/NO (2.7 μM/min) and xanthine oxidase (1–28 μM O
), from spermine/NO (2.7 μM/min) and xanthine oxidase (1–28 μM O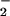 /min), respectively, resulted in protein nitration only when these species were produced at approximately equivalent rates. Addition of ferriprotoporphyrin IX (hemin) to this system increased nitration over a broad range of O
/min), respectively, resulted in protein nitration only when these species were produced at approximately equivalent rates. Addition of ferriprotoporphyrin IX (hemin) to this system increased nitration over a broad range of O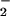 concentrations with respect to NO. Nitration in the presence of superoxide dismutase but not catalase suggested that ONOO− might not be obligatory to this process. Hemin-mediated NT formation required only the presence of NO
concentrations with respect to NO. Nitration in the presence of superoxide dismutase but not catalase suggested that ONOO− might not be obligatory to this process. Hemin-mediated NT formation required only the presence of NO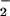 and H2O2, which are stable end-products of NO and O
and H2O2, which are stable end-products of NO and O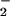 degradation. Ferrous, ferric, and cupric ions were also effective catalysts, indicating that nitration is mediated by species capable of Fenton-type chemistry. Although ONOO− can nitrate proteins, there are severe spatial and temporal constraints on this reaction. In contrast, accumulation of metals and NO
degradation. Ferrous, ferric, and cupric ions were also effective catalysts, indicating that nitration is mediated by species capable of Fenton-type chemistry. Although ONOO− can nitrate proteins, there are severe spatial and temporal constraints on this reaction. In contrast, accumulation of metals and NO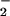 subsequent to NO synthase activity can result in far less discriminate nitration in the presence of an H2O2 source. Metal catalyzed nitration may account for the observed specificity of protein nitration seen under pathological conditions, suggesting a major role for translocated metals and the labilization of heme in NT formation.
subsequent to NO synthase activity can result in far less discriminate nitration in the presence of an H2O2 source. Metal catalyzed nitration may account for the observed specificity of protein nitration seen under pathological conditions, suggesting a major role for translocated metals and the labilization of heme in NT formation.
Keywords: nitrotyrosine‖nitric oxide‖peroxynitrite‖oxidation‖hemin
Nitrated tyrosine residues (NT) have been used as a marker of the involvement of nitric oxide (NO) and its related chemistry in a number of pathophysiological disorders including cardiovascular (1, 2) neurodegenerative and malignant conditions (1–5), such as septic shock (6), multiple sclerosis (7), Alzheimer's disease (8), and amyotrophic lateral sclerosis (9). In cancer the presence of NT often correlates with a poor prognosis (10), suggesting diagnostic implications.
Several mechanisms for NT formation have been proposed, and its origin remains controversial (11–16). Detection of NT in vitro following exposure to synthetic peroxynitrite (ONOO−) initially suggested that the reaction of NO with superoxide (O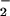 ) may be the primary source of NT (17, 18). Further studies demonstrated that cogeneration of NO and O
) may be the primary source of NT (17, 18). Further studies demonstrated that cogeneration of NO and O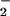 gave dissimilar results than bolus synthetic ONOO− (12, 19, 20). These disparities in nitration yields arose from further reactions of ONOO− with excess NO or O
gave dissimilar results than bolus synthetic ONOO− (12, 19, 20). These disparities in nitration yields arose from further reactions of ONOO− with excess NO or O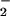 (21). Given the complexity of this seemingly simple reaction, much debate has ensued over the likelihood of NT formation under biologically relevant conditions (12, 14).
(21). Given the complexity of this seemingly simple reaction, much debate has ensued over the likelihood of NT formation under biologically relevant conditions (12, 14).
In addition to a ONOO−-mediated pathway, NT formation has been shown to result from the catalysis of nitrite oxidation by peroxidases, thus providing an alternate mechanism for its formation in vivo (22–24). Although leukocyte peroxidases likely account for a substantial amount of NT production in vivo, this mechanism fails to explain NT formation in the absence of inflammatory cells or peroxidases [i.e., a MPO−/− mouse model (25)].
NT has been shown to occur on distinct proteins rather than in an indiscriminate manner (26). This may result from variations in protein susceptibility to nitration due to conformational differences or to association with redox active metals (27, 28). We evaluated the relative efficacy of the synthetic ONOO−, NO/O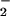 cogeneration, peroxidase, and redox active metal pathways to promote NT formation.
cogeneration, peroxidase, and redox active metal pathways to promote NT formation.
Materials and Methods
BSA (essentially globulin-free), ribonuclease A, ferriprotoporphyrin IX (hemin), superoxide dismutase (SOD; bovine erythrocytes), cytochrome c, horseradish peroxidase (HRP), catalase (Cat), hypoxanthine (HX), myoglobin (horse heart), 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS), FeSO4, diethylenetriaminepentaacetic acid (DTPA), dimethylformamide (DMF), β-amyloid (1–42) protein (Aβ), MnO2, and CuCl2 were purchased from Sigma-Aldrich. H2O2, NaHCO3, NaNO2, and FeCl3 were from Fisher Scientific. Other reagents were as follows: xanthine oxidase (XO; Roche, Nutley, NJ), myeloperoxidase (MPO; human PMN leukocytes; Calbiochem), dihydrorhodamine 123 (DHR; Molecular Probes), GFP (CLONTECH).
Stock solutions were prepared fresh daily at 100× in MilliQ filtered H2O unless otherwise noted. The assay buffer contained the metal chelator DTPA (50 μM) in calcium and magnesium-free Dulbecco's PBS (pH 7.4) ± 500 μM HX as indicated (HX buffer). Figures are representative of n ≥ 3 individual experiments.
Synthetic ONOO− was prepared by simultaneously mixing solutions of 0.5 M NaNO2 in 0.5 M HCl and 0.5 M H2O2, followed by rapid quenching in 1 M NaOH as described (29). The resulting basic solution was exposed to MnO2 to remove excess H2O2, which was reduced to <1% per mole of ONOO−. After filtering, aliquots were stored at −20°C for less than 2 weeks. Directly before use, the concentration was determined from the absorbance value at 302 nm (ɛ = 1,670 M−1⋅cm−1; ref. 30).
Instrumentation.
UV-visible spectroscopy was performed with a Hewlett–Packard 8452A diode-array spectrophotometer. Fluorescence measurements were acquired on a Perkin–Elmer HTS 7100 plate reader or an LS50B fluorimeter.
The NO/O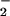 Reaction.
Reaction.
NO was produced by decomposition of the diazeniumdiolate (NONOate) spermine (SPER)/NO (a generous gift from J. A. Hrabie, National Cancer Institute–Frederick Cancer Research and Development Center, Frederick, MD). Dilutions were made in assay buffer from 100 mM stock solutions in 10 mM NaOH. The stock concentration was determined immediately before use by measuring the absorbance at 250 nm (ɛ = 8,000 M−1⋅cm−1; ref. 31). Superoxide was generated by XO-catalyzed degradation of HX (500 μM). The rate of production was assessed by reduction of cytochrome c (550 nm, Δɛ = 21,000 M−1⋅cm−1), as described (32).
The steady-state concentration of NO produced during SPER/NO degradation was determined electrochemically with a NO probe (World Precision Instruments, Sarasota, FL) controlled by a DUO18 amplifier and suspended into the assay cuvette. Signals were calibrated using argon-purged 100 mM phosphate solutions of saturated NO (Matheson, Montgomeryville, PA) following determination of NO concentration with ABTS (660 nm, ɛ = 12,000 M−1⋅cm−1; ref. 33). A steady-state NO concentration of 6–8 μM was achieved with 100 μM SPER/NO for >2 h (data not shown). The calculated rate of NO release from 100 μM SPER/NO based on the decomposition rate (pH 7.4, 37°C, t1/2 = 42 min; ref. 34) is 3.0 μM/min. An actual rate of 2.7 μM NO/min was determined by measuring oxymyoglobin (MbO2) oxidation (582 nm, Δε = 9,200 M−1⋅cm−1; ref. 35) in HX buffer.
Oxidation Assay.
Peroxynitrite, either produced synthetically or in situ by the NO/O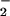 reaction, produces the fluorescent compound rhodamine (RH) via two-electron oxidation of DHR (36). Immediately before use, stock solutions of 50 mM DHR (10 mg) were prepared in DMF (0.6 ml) and diluted 1,000-fold into stock HX assay buffer. Various concentrations of XO (0.22–28 μM O
reaction, produces the fluorescent compound rhodamine (RH) via two-electron oxidation of DHR (36). Immediately before use, stock solutions of 50 mM DHR (10 mg) were prepared in DMF (0.6 ml) and diluted 1,000-fold into stock HX assay buffer. Various concentrations of XO (0.22–28 μM O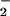 /min) were added to black-wall microtiter plates (Costar) on ice containing 200 μl of the DHR/HX buffer. After addition of 100 μM SPER/NO, the reactions were allowed to proceed for 1 h at 37°C with or without CO2 (20 mM NaHCO3 in 5% CO2, 95% air, incubator), and the fluorescence was measured at 570 nm following excitation at 500 nm. Under these conditions, production of the ONOO− scavenger urate from xanthine was determined spectrophotometrically (305 nm; ɛ = 8,030 M−1⋅cm−1; ref. 37) to be negligible.
/min) were added to black-wall microtiter plates (Costar) on ice containing 200 μl of the DHR/HX buffer. After addition of 100 μM SPER/NO, the reactions were allowed to proceed for 1 h at 37°C with or without CO2 (20 mM NaHCO3 in 5% CO2, 95% air, incubator), and the fluorescence was measured at 570 nm following excitation at 500 nm. Under these conditions, production of the ONOO− scavenger urate from xanthine was determined spectrophotometrically (305 nm; ɛ = 8,030 M−1⋅cm−1; ref. 37) to be negligible.
Western Blot and Dot Blot Analysis.
Protein samples (1.4 μg) were subjected to PAGE on either 10% or 4–20% gradient Tris-glycine acrylamide gels (Novex-Invitrogen, Carlsbad, CA). Following transfer to PDVF Immunolon P membranes (Millipore), samples were probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml; Upstate Biotechnologies, Waltham, MA). Bands were visualized with HRP-conjugated secondary antibodies (1:10,000; Sigma) and chemiluminescent substrate (Pierce). For dot-blot analysis, samples (protein or cell extract, 14 μg) were vacuum transferred to PDVF membranes by using a filtration manifold system (Minifold 1; Schleicher & Schuell), and the membrane was probed as above. Gel images were scanned using an AGFA (Wilmington, MA) DuoScan hiD scanner, and relevant bands were cropped to size by using PHOTOSHOP 5.0 (Adobe Systems, Mountain View, CA) with no further manipulation.
Nitration of BSA, ribonuclease A, GFP, and Aβ (all at 80 μg/ml) was examined following exposure to SPER/NO and XO as described above for DHR but in HX buffer alone. Further, the effects of SOD (50 nM), catalase (100 units/ml), hemin (2 μM), HRP (1 unit/ml; 100 nM), MPO (50 nM), and FeCl2 (5 μM without DTPA) on BSA nitration by the NO/O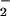 reaction were investigated. Nitration of BSA by synthetic ONOO− (0.6–20 μM, 30 min incubation) was also examined.
reaction were investigated. Nitration of BSA by synthetic ONOO− (0.6–20 μM, 30 min incubation) was also examined.
Nitration of BSA or Aβ by HRP (100 nM), MPO (50 nM), hemin (0.048–4.0 μM from 1 mM stock in 50% DMSO), or various metals (2–25 μM FeSO4, FeCl3, and CuCl2 without DTPA) was also examined in the presence of varying concentrations of NaNO2 and H2O2 (1–1,000 μM) in PBS assay buffer after a 1.5-h incubation at 37°C.
Cell Culture.
MCF-7 human breast carcinoma cells (American Type Culture Collection) were plated and grown to 90% confluency in RPMI medium 1640 (Life Technologies, Grand Island, NY) containing 10% FBS (HyClone). Cells were washed five times with PBS, incubated with serum-free RPMI medium 1640 and then were either pretreated with hemin (4–32 μM) for 1 h and washed five times or were coincubated with hemin during a 2-h exposure to SPER/NO (100 μM) + H2O2 (100 μM).
Protein cell extracts were made by suspending washed MCF-7 cells into cold PBS, spinning, and resuspending in lysis buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail; Calbiochem). Following 30 min incubation on ice, the samples were centrifuged at 14,000 × g, and the supernatant protein concentration was determined by the bichoncinic acid method (Pierce).
Results
A concentration-dependant NT signal was detected following BSA exposure to synthetic ONOO− (Fig. 1), consistent with previous studies (38, 39). The lower limit of detection with this system was 1 μM synthetic ONOO−. Bolus addition of synthetic ONOO− was shown to not accurately reflect the resultant chemistry of this species under biological conditions (19). Therefore, nitration elicited by the reaction between NO and O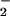 was examined using 100 μM SPER/NO, which releases NO at 2.7 μM NO/min, and various concentrations of XO to generate O
was examined using 100 μM SPER/NO, which releases NO at 2.7 μM NO/min, and various concentrations of XO to generate O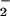 fluxes of 0.22–28 μM/min. The SPER/NO concentration was selected to approximate biological production (40). Under these conditions, DHR oxidation to RH resulted in the characteristic bell-shaped fluorescence curve (36), with maximal oxidation occurring when the rates of NO and O
fluxes of 0.22–28 μM/min. The SPER/NO concentration was selected to approximate biological production (40). Under these conditions, DHR oxidation to RH resulted in the characteristic bell-shaped fluorescence curve (36), with maximal oxidation occurring when the rates of NO and O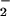 formation were approximately equivalent (Fig. 2D). A similar DHR oxidation profile was observed at a constant rate of O
formation were approximately equivalent (Fig. 2D). A similar DHR oxidation profile was observed at a constant rate of O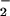 production (1.8 μM/min) with increasing SPER/NO (6.3–1600 μM; data not shown).
production (1.8 μM/min) with increasing SPER/NO (6.3–1600 μM; data not shown).
Figure 1.

Immunoblot demonstrating nitration of BSA by bolus ONOO−. Synthetic ONOO− (0.6–20 μM) was added to 1 ml of PBS containing DPTA (50 μM) and BSA (80 μg/ml), followed by 30 min incubation at 37°C. BSA (1.4 μg) was loaded to each lane and subjected to PAGE on 10% Tris-glycine acrylamide gels. Bands, representing BSA (66 kDa), were probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml) and HRP-conjugated secondary antibodies (1:10,000).
Figure 2.

Oxidative and nitrative profile elicited by co-generation of NO and O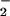 ± hemin or Fe2+. SPER/NO (100 μM; 2.7 μM NO/min), HX (500 μM), XO (O
± hemin or Fe2+. SPER/NO (100 μM; 2.7 μM NO/min), HX (500 μM), XO (O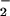 fluxes of 0.22–28 μM/min), and BSA (80 μg/ml) or DHR (50 μM) were added to 200 μl of PBS containing DTPA (50 μM) in a black-walled microtiter plate. After a 1.5-h incubation at 37°C, NT Western blot analysis (A–C) or rhodamine formation (D; λex/em 500/570) was measured. A and D, SPER/NO + XO; B, SPER/NO + XO + hemin (1 μM); C, SPER/NO + XO + Fe2+ (5 μM, without DTPA).
fluxes of 0.22–28 μM/min), and BSA (80 μg/ml) or DHR (50 μM) were added to 200 μl of PBS containing DTPA (50 μM) in a black-walled microtiter plate. After a 1.5-h incubation at 37°C, NT Western blot analysis (A–C) or rhodamine formation (D; λex/em 500/570) was measured. A and D, SPER/NO + XO; B, SPER/NO + XO + hemin (1 μM); C, SPER/NO + XO + Fe2+ (5 μM, without DTPA).
Under similar conditions, Western blot analysis revealed immunoreactivity for NT on BSA (Fig. 2A) only at points corresponding to maximal DHR oxidation (Fig. 2D). The chemistry of ONOO− can be augmented in the presence of CO2 (41). However, CO2 resulted in only a slight enhancement of both DHR oxidation and BSA nitration (data not shown). These data illustrate that nitration results from the NO/O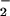 reaction only under very specific reactant fluxes.
reaction only under very specific reactant fluxes.
Disruption of metal homeostasis in vivo may accompany generation of high levels of NO, O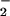 , and other reactive oxygen species (ROS) (42). Therefore, the effects of either hemin or free Fe2+ on nitration during simultaneous generation of NO and O
, and other reactive oxygen species (ROS) (42). Therefore, the effects of either hemin or free Fe2+ on nitration during simultaneous generation of NO and O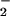 were examined. As shown in Fig. 2B, hemin substantially increased both the yield and range of O
were examined. As shown in Fig. 2B, hemin substantially increased both the yield and range of O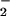 fluxes that resulted in BSA nitration. Similar patterns were observed with ribonuclease A, GFP, and Aβ under these conditions (data not shown), indicating a general susceptibility of tyrosine-containing proteins to heme-mediated nitration. Conversely, addition of Fe2+ shifted the point of maximal nitration to 50% lower O
fluxes that resulted in BSA nitration. Similar patterns were observed with ribonuclease A, GFP, and Aβ under these conditions (data not shown), indicating a general susceptibility of tyrosine-containing proteins to heme-mediated nitration. Conversely, addition of Fe2+ shifted the point of maximal nitration to 50% lower O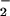 relative to NT formation from NO/O
relative to NT formation from NO/O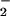 alone (Fig. 2C).
alone (Fig. 2C).
Because metals are known to catalyze ONOO−-mediated nitration (43), SOD and catalase were used to determine whether hemin-facilitated nitration was driven by ONOO−. At the NO and O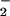 fluxes giving maximal NT and RH signals, addition of SOD (3.5 μg/ml, ≈7 units), which catalytically converts O
fluxes giving maximal NT and RH signals, addition of SOD (3.5 μg/ml, ≈7 units), which catalytically converts O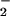 into H2O2 (32), resulted in complete abolishment of nitration (Fig. 3). In the presence of hemin, however, SOD enhanced nitration, indicating that H2O2 may be required. This finding was verified by loss of signal with addition of catalase. Similar nitration patterns were seen with the heme enzymes HRP (Fig. 3) and MPO (data not shown).
into H2O2 (32), resulted in complete abolishment of nitration (Fig. 3). In the presence of hemin, however, SOD enhanced nitration, indicating that H2O2 may be required. This finding was verified by loss of signal with addition of catalase. Similar nitration patterns were seen with the heme enzymes HRP (Fig. 3) and MPO (data not shown).
Figure 3.

Immunoblot demonstrating nitration of BSA by co-generation of NO/O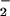 under various conditions. SPER/NO (100 μM), HX (500 μM), XO (O
under various conditions. SPER/NO (100 μM), HX (500 μM), XO (O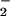 fluxes of 1.75 μM/min), and BSA (80 μg/ml) were added to 200 μl of PBS containing DTPA (50 μM) in a black-walled microtiter plate. SPER/NO and XO concentrations were chosen that corresponded to maximal DHR oxidation (Fig. 2D) indicating optimal ONOO− formation. In addition, each well contained SOD (3.5 μg/ml), hemin (2 μM), catalase (100 units/ml), or HRP (100 nM, 1 unit/ml), as indicated. After a 1.5-h incubation at 37°C, PAGE (1.4 μg of BSA) and Western blot analysis were preformed. The bands represent BSA (66 kDa) run on a single gel with control lanes not shown.
fluxes of 1.75 μM/min), and BSA (80 μg/ml) were added to 200 μl of PBS containing DTPA (50 μM) in a black-walled microtiter plate. SPER/NO and XO concentrations were chosen that corresponded to maximal DHR oxidation (Fig. 2D) indicating optimal ONOO− formation. In addition, each well contained SOD (3.5 μg/ml), hemin (2 μM), catalase (100 units/ml), or HRP (100 nM, 1 unit/ml), as indicated. After a 1.5-h incubation at 37°C, PAGE (1.4 μg of BSA) and Western blot analysis were preformed. The bands represent BSA (66 kDa) run on a single gel with control lanes not shown.
The similarities between the peroxidase and hemin profiles raised the possibility that hemin may result in nitration independent of ONOO− due to build-up of end-products from the generation of NO and O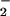 , such as NO
, such as NO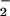 and H2O2 (22). NT was observed with various combinations of these reactants (Fig. 4). BSA nitration was evident with reactant concentrations of 1 μM, which are within physiologic or pathologic ranges. Nitration by hemin required both NO
and H2O2 (22). NT was observed with various combinations of these reactants (Fig. 4). BSA nitration was evident with reactant concentrations of 1 μM, which are within physiologic or pathologic ranges. Nitration by hemin required both NO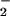 and H2O2 but was more dependent on the NO
and H2O2 but was more dependent on the NO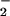 than H2O2 concentration (Fig. 4B). Thus, the rate-limiting step in nitration involves oxidation of NO
than H2O2 concentration (Fig. 4B). Thus, the rate-limiting step in nitration involves oxidation of NO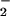 .
.
Figure 4.

Immuno dot-blot demonstrating nitration of BSA from NO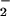 /H2O2/hemin. BSA (80 μg/ml), NaNO2 (1–100 μM), H2O2 (1–100 μM), and hemin (0.25–4 μM) were added to 1 ml of PBS containing DTPA (50 μM), followed by a 1.5-h incubation at 37°C. Each dot represents 14 μg of protein vacuum transferred to a PDVF membrane and probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml) and HRP-conjugated secondary antibodies (1:10,000). A and B represent separate membranes and therefore intensity differences between them should not be directly compared.
/H2O2/hemin. BSA (80 μg/ml), NaNO2 (1–100 μM), H2O2 (1–100 μM), and hemin (0.25–4 μM) were added to 1 ml of PBS containing DTPA (50 μM), followed by a 1.5-h incubation at 37°C. Each dot represents 14 μg of protein vacuum transferred to a PDVF membrane and probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml) and HRP-conjugated secondary antibodies (1:10,000). A and B represent separate membranes and therefore intensity differences between them should not be directly compared.
The nitration signal on Aβ observed in the presence of hemin/H2O2/NO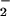 was enhanced by the addition of an equivalent amount of BSA (Fig. 5). These data suggest that BSA, by virtue of its ability to bind hemin (44), may facilitate nitration of neighboring proteins. This hypothesis was further examined with intact human MCF-7 carcinoma cells. Similar to purified proteins, hemin coincubation with H2O2 (100 μM) and SPER/NO (≈200 μM NO
was enhanced by the addition of an equivalent amount of BSA (Fig. 5). These data suggest that BSA, by virtue of its ability to bind hemin (44), may facilitate nitration of neighboring proteins. This hypothesis was further examined with intact human MCF-7 carcinoma cells. Similar to purified proteins, hemin coincubation with H2O2 (100 μM) and SPER/NO (≈200 μM NO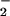 ), conditions that would not be expected to produce ONOO−, catalyzed nitration of cellular proteins (Fig. 6). Nitration was also observed following addition of H2O2 and SPER/NO subsequent to preincubation of cells with hemin followed by vigorous washing. These data indicate that nitration was catalyzed not only by free hemin but also by hemin that was either adherent or internalized. BSA slightly increased the level of NT signal from MCF-7 cells during coincubation but substantially reduced nitration if preincubated (data not shown). This indicates that the association of hemin with BSA is sufficiently strong to compete with binding to MCF-7 proteins, and therefore, the availability of hemin is reduced following BSA removal by washing.
), conditions that would not be expected to produce ONOO−, catalyzed nitration of cellular proteins (Fig. 6). Nitration was also observed following addition of H2O2 and SPER/NO subsequent to preincubation of cells with hemin followed by vigorous washing. These data indicate that nitration was catalyzed not only by free hemin but also by hemin that was either adherent or internalized. BSA slightly increased the level of NT signal from MCF-7 cells during coincubation but substantially reduced nitration if preincubated (data not shown). This indicates that the association of hemin with BSA is sufficiently strong to compete with binding to MCF-7 proteins, and therefore, the availability of hemin is reduced following BSA removal by washing.
Figure 5.

Immunoblot demonstrating nitration of Aβ ± BSA and hemin. Aβ (80 μg/ml), NaNO2 (100 μM), and H2O2 (100 μM) were added to 1 ml of PBS containing DTPA (50 μM) and hemin (1 or 0.5 μM) and BSA (80 μg/ml), as indicated. After a 1.5-h incubation at 37°C each sample was subjected to PAGE on 4–20% Tris-glycine acrylamide gel followed by membrane transfer and was probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml) and HRP-conjugated secondary antibodies (1:10,000).
Figure 6.

Immuno dot-blot demonstrating nitration of cellular proteins by hemin, H2O2, and SPER/NO. MCF-7 cells in culture (90% confluent) were exposed for 2 h to 100 μM H2O2 and 100 μM SPER/NO in serum-free media. Cells were either pretreated (for 1 h) with hemin (4–32 μM) and washed five time, or cotreated with hemin. Cell proteins were extracted, vacuum transferred (14 μg per dot) to a PDVF membrane, and probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml) and HRP-conjugated secondary antibodies (1:10,000).
Finally, because proteins such as BSA and Aβ are capable of binding free metals (45), the ability of different metal ions to catalyze nitration by H2O2 and NO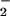 was examined. Ferric, ferrous, and cupric (Cu2+) ions readily nitrated BSA in a dose-dependant manner (Fig. 7).
was examined. Ferric, ferrous, and cupric (Cu2+) ions readily nitrated BSA in a dose-dependant manner (Fig. 7).
Figure 7.

Immunoblot demonstrating nitration of BSA by free metals. CuCl2, FeCl3, or FeSO4 at 2 or 25 μM was added to 1 ml of BSA solution (80 μg/ml PBS) containing NaNO2 (100 μM) + H2O2 (100 μM). After a 2-h incubation at 37°C, each sample (1.4 μg of BSA per lane) was subjected to PAGE on 10% Tris-glycine acrylamide gels followed by membrane transfer and was probed with rabbit polyclonal 3-nitrotyrosine antibodies (0.5 μg/ml) and HRP-conjugated secondary antibodies (1:10,000). Bands represent BSA (66 kDa) run on a single gel.
Discussion
Peroxynitrite is often presented as the sole source of NT in vivo (46). This simple and initially attractive concept has been substantiated by the fact that bolus synthetic ONOO− can readily nitrate proteins (ref. 38; Fig. 1). However, the complexity of the chemistry resulting from simultaneous generation of NO and O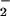 is emphasized by the dramatic reduction in NT yield at non-stoichiometric reactant amounts (Fig. 2A). Intuitively, it would be anticipated that excess O
is emphasized by the dramatic reduction in NT yield at non-stoichiometric reactant amounts (Fig. 2A). Intuitively, it would be anticipated that excess O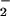 or NO would completely convert the other reactant to ONOO− by the near diffusion limited nature of this reaction (Eq. 1; k = 4.3–6.7 × 109 M−1⋅s−1; ref. 47). Thus, for example, the nitration yield with respect to increasing O
or NO would completely convert the other reactant to ONOO− by the near diffusion limited nature of this reaction (Eq. 1; k = 4.3–6.7 × 109 M−1⋅s−1; ref. 47). Thus, for example, the nitration yield with respect to increasing O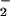 should reach a plateau at the equivalence point with NO. The dramatic decrease in the yield of nitrated protein observed when the concentration of NO or O
should reach a plateau at the equivalence point with NO. The dramatic decrease in the yield of nitrated protein observed when the concentration of NO or O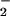 is in excess demonstrates the requirement for specific conditions and reactant fluxes of NO/O
is in excess demonstrates the requirement for specific conditions and reactant fluxes of NO/O .
.
The small window of nitration seen over a broad range of reactant concentrations results from the secondary reactions of ONOO−. Protonation of ONOO− produces peroxynitrous acid (ONOOH), which decomposes to yield the reactive intermediates that mediate oxidation and nitration (Eq. 2; ref. 48). Under excess NO, intermediates such as NO2 (Eq. 3) can react further to yield N2O3 (Eq. 5), or ONOOH may react directly with NO (29) to form NO2/N2O3 (Eqs. 4 and 5). If NO is in excess, nitration of tyrosine (or tyrosyl radical produced by oxidation from NO2 or ⋅OH) will be in direct competition with formation of N2O3 (1.1 × 109 M−1⋅s−1; ref. 21), which subsequently leads to nitrosating, rather than nitrating or oxidizing, chemistry (Eq. 5). On the other hand, one must similarly conclude that O scavenges either ONOO− or NO2 (Eq. 7; ref. 29) because nitration was quenched during excess production of this radical.
scavenges either ONOO− or NO2 (Eq. 7; ref. 29) because nitration was quenched during excess production of this radical.
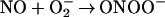 |
1 |
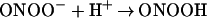 |
2 |
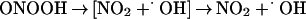 |
3 |
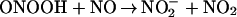 |
4 |
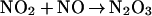 |
5 |
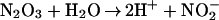 |
6 |
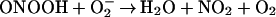 |
7 |
It is interesting to note that under excess O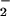 while significant DHR was oxidized, nitration was not observed (Fig. 2 A and D). This indicates that nitration and oxidation are mediated by different species, as suggested (49).
while significant DHR was oxidized, nitration was not observed (Fig. 2 A and D). This indicates that nitration and oxidation are mediated by different species, as suggested (49).
Given the challenges, even under ideal conditions, of achieving NO/O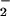 mediated NT formation, it seems unlikely that in the complex setting of cellular biology that ONOO− should be considered the major source of NT in vivo, and alternative biochemical pathways become necessary. The well studied peroxidase oxidation of NO
mediated NT formation, it seems unlikely that in the complex setting of cellular biology that ONOO− should be considered the major source of NT in vivo, and alternative biochemical pathways become necessary. The well studied peroxidase oxidation of NO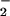 to NO2 has been proposed as the principle source of nitration (50). This system has the advantage over the intermediacy of ONOO− by using the more chemically stable substrates NO
to NO2 has been proposed as the principle source of nitration (50). This system has the advantage over the intermediacy of ONOO− by using the more chemically stable substrates NO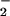 and peroxide (hydrogen or alkyl), lifting the temporal and spatial constraints of reactant production allowing accumulation over time. In fact Pfeiffer et al. (20) demonstrated that cytokine-activated macrophages produce O
and peroxide (hydrogen or alkyl), lifting the temporal and spatial constraints of reactant production allowing accumulation over time. In fact Pfeiffer et al. (20) demonstrated that cytokine-activated macrophages produce O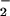 maximally at 1–3 h, whereas NO production was not maximally observed until 6–8 h after stimulation.
maximally at 1–3 h, whereas NO production was not maximally observed until 6–8 h after stimulation.
Myleoperoxidase has been shown to nitrate proteins through a one-electron oxidation of NO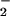 to NO2 and has been proposed to be the major source of this alteration (50). This is quite reasonable because leukocytes are often present in conditions bearing the signature of NT. Both HRP (Fig. 3) and MPO (data not shown) generate NT from NO/O
to NO2 and has been proposed to be the major source of this alteration (50). This is quite reasonable because leukocytes are often present in conditions bearing the signature of NT. Both HRP (Fig. 3) and MPO (data not shown) generate NT from NO/O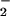 , but only after conversion of each reactant to NO
, but only after conversion of each reactant to NO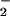 and H2O2, respectively.
and H2O2, respectively.
Although peroxidases unmistakably account for significant amounts of NT formation in vivo, this mechanism may not adequately explain the nitrated proteins detected in a MPO−/− mouse model (25) or under acute pathologic conditions void of inflammatory cells, such as ischemia reperfusion injury to the brain (51). Brennan et al. (50) have recently shown, using an EPO−/−/MPO−/− mouse inflammatory model, that leukocyte peroxidase-dependant formation of the peroxidase activity markers bromo- and chloro-tyrosine does not necessarily parallel formation of NT.
Production of NT is associated with many chronic neurodegenerative diseases such as Alzheimer's disease (8), multiple sclerosis (7), Parkinson's disease, and amyotrophic lateral sclerosis (9). A feature common to these diseases is the presence of redox active metals and concomitant increases in oxidative damage (52). Hemin-facilitated nitration of BSA occurred across a broad range of NO/O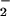 fluxes (Fig. 2B) as a result of end-product formation, for example by SOD, rather than via ONOO− catalysis (Fig. 3). Multiple tyrosine-containing proteins are nitrated by hemin, NO
fluxes (Fig. 2B) as a result of end-product formation, for example by SOD, rather than via ONOO− catalysis (Fig. 3). Multiple tyrosine-containing proteins are nitrated by hemin, NO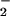 and H2O2 (Figs. 4 and 5; data not shown), indicating that high yield nitration is not specific to peroxidase alone.
and H2O2 (Figs. 4 and 5; data not shown), indicating that high yield nitration is not specific to peroxidase alone.
Hemin/BSA complexes are representative of peroxidase chemistry in general but also may serve as a model for conditions where hemes are translocated. Chronic NO exposure has been shown to result in liberation of prosthetic heme (53). This mobilization of heme in the cell could easily serve as a catalyst for NT formation in the presence of H2O2 and NO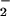 derived from NO (Fig. 6). Free heme also up-regulates expression of hemeoxygenase-1 (HO-1; ref. 54), which is responsible for heme degradation. A resultant product of this regulatory mechanism is free iron, which is also capable of mediating nitration reactions (Fig. 7). Therefore, dysregulation of heme proteins and iron homeostasis, as seen under pathologic conditions such as hemolysis, hemorrhage, and metabolic and neurodegenerative disorders, may explain the associated increase in NT observed with these diseases.
derived from NO (Fig. 6). Free heme also up-regulates expression of hemeoxygenase-1 (HO-1; ref. 54), which is responsible for heme degradation. A resultant product of this regulatory mechanism is free iron, which is also capable of mediating nitration reactions (Fig. 7). Therefore, dysregulation of heme proteins and iron homeostasis, as seen under pathologic conditions such as hemolysis, hemorrhage, and metabolic and neurodegenerative disorders, may explain the associated increase in NT observed with these diseases.
One obvious explanation is that heme and Fe2+/Fe3+ are simply catalyzing ONOO−-mediated oxidation. Although this may be a minor, if any, contribution to the total NT yield, it should be emphasized that in the case of NO/O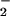 and hemin, SOD increased the effect, whereas catalase abolished the signal. Thus, rather than ONOO−-mediated nitration, this is indicative of a peroxidase-type mechanism, whereby H2O2 reacts with heme to form the ferryl π-cation radical species, which subsequently oxidizes NO
and hemin, SOD increased the effect, whereas catalase abolished the signal. Thus, rather than ONOO−-mediated nitration, this is indicative of a peroxidase-type mechanism, whereby H2O2 reacts with heme to form the ferryl π-cation radical species, which subsequently oxidizes NO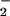 to form NO2, as seen with myeloperoxidase (Eqs. 8 and 9; ref. 50).
to form NO2, as seen with myeloperoxidase (Eqs. 8 and 9; ref. 50).
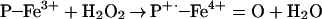 |
8 |
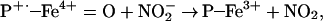 |
9 |
where P is porphyrin. This is consistent with the fact that heme/BSA complexes possess peroxidase activity and can also oxidize phenolic compounds (55).
In a complex cellular environment, the question arises as to the effect of hemin on nitration of multiple proteins. Coincubation of hemin with BSA and Aβ resulted in nitration of both proteins (Fig. 5). The differential intensity may be attributed to 21 tyrosine residues in BSA compared with 1 in Aβ. Under these conditions the relative nitration of Aβ by hemin was increased, suggesting that the tight association of hemin with BSA (55) could serve as a catalyst for nitration of other proteins through a diffusible intermediate, presumably NO2. Additionally, a robust NT signal was observed following treatment of cells with hemin or hemin/BSA (Fig. 6). Cellular nitration was achieved after hemin treatment followed by extensive washing, demonstrating limited temporal constraints in contrast to in situ ONOO− formation. This finding provides important insight into conditions of hemolysis, hemorrhage, and interstitial bleeding where heme may accumulate before recruitment of NO and O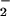 /H2O
/H2O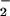 -generating inflammatory cells to the sight of injury. This suggests that translocation of heme can serve as a local catalyst, as well as forming adducts on neighboring proteins. This substantiates our previous findings, in which NO2, but not ONOO−, was shown to effectively migrate into cells to nitrate intracellular GFP (19).
-generating inflammatory cells to the sight of injury. This suggests that translocation of heme can serve as a local catalyst, as well as forming adducts on neighboring proteins. This substantiates our previous findings, in which NO2, but not ONOO−, was shown to effectively migrate into cells to nitrate intracellular GFP (19).
Because the coordinated iron of hemin was able to oxidize NO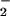 and effectively nitrate proteins, it was important to determine the efficacy of free transition metals. This process may have potential importance in the pathogenesis of several neurodegenerative disorders, especially in Alzheimer's disease, where metals are a prominent etiologic feature (52). Iron and copper levels have been measured as high as 0.4 and 1.0 mM, respectively, in the brains of Alzheimer's patients (56). NT formation on BSA in the presence of NO
and effectively nitrate proteins, it was important to determine the efficacy of free transition metals. This process may have potential importance in the pathogenesis of several neurodegenerative disorders, especially in Alzheimer's disease, where metals are a prominent etiologic feature (52). Iron and copper levels have been measured as high as 0.4 and 1.0 mM, respectively, in the brains of Alzheimer's patients (56). NT formation on BSA in the presence of NO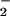 and H2O2 was catalyzed by free ferrous, ferric, and cupric ion (Fig. 7). These ions most likely react with peroxide to form a metal-oxo complex or ⋅OH through Fenton-type chemistry (57, 58). These products can oxidize NO
and H2O2 was catalyzed by free ferrous, ferric, and cupric ion (Fig. 7). These ions most likely react with peroxide to form a metal-oxo complex or ⋅OH through Fenton-type chemistry (57, 58). These products can oxidize NO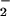 into the nitrating species NO2.
into the nitrating species NO2.
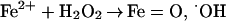 |
10 |
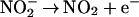 |
11 |
Although there were differences in the relative efficacy of each ion to catalyze nitration, this could be attributed to differences in solubility or additionally to possible variations in preference for different protein ligand environments. In direct contrast to hemin, nitration by Fe2+ during the NO/O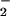 reaction resulted in nitration only at limited fluxes (Fig. 2 B and C). It is interesting to note that the O
reaction resulted in nitration only at limited fluxes (Fig. 2 B and C). It is interesting to note that the O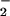 concentration that resulted in maximal NT formation in the presence of Fe2+ is shifted to a lower O
concentration that resulted in maximal NT formation in the presence of Fe2+ is shifted to a lower O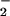 flux compared with NO/O
flux compared with NO/O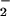 alone (Fig. 2 A and C). This can be explained by one of two mechanisms: either an increase in ONOO−-mediated nitration by metal catalysis (43) or through optimal conditions to facilitate a Haber-Weiss reaction (reduction of Fe3+ by O
alone (Fig. 2 A and C). This can be explained by one of two mechanisms: either an increase in ONOO−-mediated nitration by metal catalysis (43) or through optimal conditions to facilitate a Haber-Weiss reaction (reduction of Fe3+ by O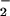 ) for maximal catalytic oxidation of NO
) for maximal catalytic oxidation of NO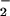 . This finding suggests that Eqs. 10 and 11 are predominant in vivo because NO
. This finding suggests that Eqs. 10 and 11 are predominant in vivo because NO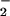 and H2O2 can nitrate proteins. The restricted nitration profile of Fe2+ as compared with heme during NO/O
and H2O2 can nitrate proteins. The restricted nitration profile of Fe2+ as compared with heme during NO/O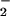 cogeneration (Fig. 2 B and C) may be a result of the differential chemistry of free versus coordinated iron, as well as kinetic differences of catalytic and noncatalytic substrates.
cogeneration (Fig. 2 B and C) may be a result of the differential chemistry of free versus coordinated iron, as well as kinetic differences of catalytic and noncatalytic substrates.
In conclusion, this evaluation of protein nitration, using low, biologically relevant fluxes of NO, supports other work that suggests that nitration from ONOO− is a highly unlikely event in biological systems (12, 19). Further, we propose that any species capable of Fenton-type chemistry, such as peroxidases, free heme, or metal ions, will mediate tyrosine nitration, providing an attractive alternative to the narrow constraints placed on the ONOO− reaction. Due to the rather ubiquitous nature of heme proteins, NO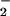 and H2O2, it seems likely that metal-mediated nitration has broader applicability to pathophysiological systems.
and H2O2, it seems likely that metal-mediated nitration has broader applicability to pathophysiological systems.
Abbreviations
- SPER/NO
spermine/NO adduct
- Aβ
β-amyloid (1–42)
- DTPA
diethylenetriaminepentaacetic acid
- DHR
dihydrorhodamine
- hemin
ferriprotoporphyrin IX
- HX
hypoxanthine
- MPO
myeloperoxidase
- NT
nitrated tyrosine residues
- O
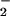
superoxide
- SOD
superoxide dismutase
- XO
xanthine oxidase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wattanapitayakul S K, Weinstein D M, Holycross B J, Bauer J A. FASEB J. 2000;14:271–278. doi: 10.1096/fasebj.14.2.271. [DOI] [PubMed] [Google Scholar]
- 2.Ferdinandy P, Danial H, Ambrus I, Rothery R A, Schulz R. Circ Res. 2000;87:241–247. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 3.Oyama J, Shimokawa H, Momii H, Cheng X, Fukuyama N, Arai Y, Egashira K, Nakazawa H, Takeshita A. J Clin Invest. 1998;101:2207–2214. doi: 10.1172/JCI986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo S, Toyokuni S, Tsuruyama T, Ozeki M, Tachibana T, Echizenya M, Hiai H, Onodera H, Imamura M. Cancer Lett. 2002;179:87–93. doi: 10.1016/s0304-3835(01)00859-x. [DOI] [PubMed] [Google Scholar]
- 5.Mendes R V, Martins A R, de Nucci G, Murad F, Soares F A. Histopathology. 2001;39:172–178. doi: 10.1046/j.1365-2559.2001.01189.x. [DOI] [PubMed] [Google Scholar]
- 6.Fukuyama N, Takebayashi Y, Hida M, Ishida H, Ichimori K, Nakazawa H. Free Radical Biol Med. 1997;22:771–774. doi: 10.1016/s0891-5849(96)00401-7. [DOI] [PubMed] [Google Scholar]
- 7.Liu J S, Zhao M L, Brosnan C F, Lee S C. Am J Pathol. 2001;158:2057–2066. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj E K, Jones P K, Ghanbari H, Wataya T, Shimohama S, et al. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 9.Shaw P J, Williams R. Amyotroph Lateral Scler Other Motor Neuron Disorders. 2000;1, Suppl. 2:S61–S67. [PubMed] [Google Scholar]
- 10.Ekmekcioglu S, Ellerhorst J, Smid C M, Prieto V G, Munsell M, Buzaid A C, Grimm E A. Clin Cancer Res. 2000;6:4768–4775. [PubMed] [Google Scholar]
- 11.Pfeiffer S, Schmidt K, Mayer B. J Biol Chem. 2000;275:6346–6352. doi: 10.1074/jbc.275.9.6346. [DOI] [PubMed] [Google Scholar]
- 12.Pfeiffer S, Mayer B. J Biol Chem. 1998;273:27280–27285. doi: 10.1074/jbc.273.42.27280. [DOI] [PubMed] [Google Scholar]
- 13.Pfeiffer S, Lass A, Schmidt K, Mayer B. FASEB J. 2001;15:2355–2364. doi: 10.1096/fj.01-0295com. [DOI] [PubMed] [Google Scholar]
- 14.Reiter C D, Teng R J, Beckman J S. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein S, Czapski G, Lind J, Merenyi G. J Biol Chem. 2000;275:3031–3036. doi: 10.1074/jbc.275.5.3031. [DOI] [PubMed] [Google Scholar]
- 16.Sawa T, Akaike T, Maeda H. J Biol Chem. 2000;275:32467–32474. doi: 10.1074/jbc.M910169199. [DOI] [PubMed] [Google Scholar]
- 17.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin J C, Smith C D, Beckman J S. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 18.Ischiropoulos H, Zhu L, Beckman J S. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 19.Espey M G, Xavier S, Thomas D D, Miranda K M, Wink D A. Proc Natl Acad Sci USA. 2002;99:3481–3486. doi: 10.1073/pnas.062604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeiffer S, Lass A, Schmidt K, Mayer B. J Biol Chem. 2001;276:34051–34058. doi: 10.1074/jbc.M100585200. [DOI] [PubMed] [Google Scholar]
- 21.Koppenol W H, Moreno J J, Pryor W A, Ischiropoulos H, Beckman J S. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 22.van der Vliet A, Eiserich J P, Halliwell B, Cross C E. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 23.Wu W, Chen Y, Hazen S L. J Biol Chem. 1999;274:25933–25944. doi: 10.1074/jbc.274.36.25933. [DOI] [PubMed] [Google Scholar]
- 24.Sampson J B, Ye Y, Rosen H, Beckman J S. Arch Biochem Biophys. 1998;356:207–213. doi: 10.1006/abbi.1998.0772. [DOI] [PubMed] [Google Scholar]
- 25.Takizawa S, Aratani Y, Fukuyama N, Maeda N, Hirabayashi H, Koyama H, Shinohara Y, Nakazawa H. J Cereb Blood Flow Metab. 2002;22:50–54. doi: 10.1097/00004647-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Aulak K S, Miyagi M, Yan L, West K A, Massillon D, Crabb J W, Stuehr D J. Proc Natl Acad Sci USA. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Souza J M, Daikhin E, Yudkoff M, Raman C S, Ischiropoulos H. Arch Biochem Biophys. 1999;371:169–178. doi: 10.1006/abbi.1999.1480. [DOI] [PubMed] [Google Scholar]
- 28.Pignatelli B, Li C Q, Boffetta P, Chen Q, Ahrens W, Nyberg F, Mukeria A, Bruske-Hohlfeld I, Fortes C, Constantinescu V, et al. Cancer Res. 2001;61:778–784. [PubMed] [Google Scholar]
- 29.Koppenol W H, Kissner R, Beckman J S. Methods Enzymol. 1996;269:296–302. doi: 10.1016/s0076-6879(96)69030-2. [DOI] [PubMed] [Google Scholar]
- 30.Beckman J S, Wink D A, Crow J P. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. New York: Wiley; 1996. pp. 61–70. [Google Scholar]
- 31.Maragos C M, Morley D, Wink D A, Dunams T M, Saavedra J E, Hoffman A, Bove A A, Isaac L, Hrabie J A, Keefer L K. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 32.McCord J M, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 33.Nims R W, Cook J C, Krishna M C, Christodoulou D, Poore C M, Miles A M, Grisham M B, Wink D A. Methods Enzymol. 1996;268:93–105. doi: 10.1016/s0076-6879(96)68012-4. [DOI] [PubMed] [Google Scholar]
- 34.Keefer L K, Nims R W, Davies K M, Wink D A. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 35.Feelisch M, Kubitzzek K, Werringloer J. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. New York: Wiley; 1996. pp. 455–478. [Google Scholar]
- 36.Kooy N W, Royall J A, Ischiropoulos H, Beckman J S. Free Radical Biol Med. 1994;16:149–156. doi: 10.1016/0891-5849(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 37.Squadrito G L, Cueto R, Splenser A E, Valavanidis A, Zhang H, Uppu R M, Pryor W A. Arch Biochem Biophys. 2000;376:333–337. doi: 10.1006/abbi.2000.1721. [DOI] [PubMed] [Google Scholar]
- 38.Beckman J S, Chen J, Ischiropoulos H, Crow J P. Methods Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 39.Haddad I Y, Ischiropoulos H, Holm B A, Beckman J S, Baker J R, Matalon S. Am J Physiol. 1993;265:L555–L564. doi: 10.1152/ajplung.1993.265.6.L555. [DOI] [PubMed] [Google Scholar]
- 40.Joshi M S, Lancaster J R, Jr, Liu X, Ferguson T B., Jr Nitric Oxide. 2001;5:561–565. doi: 10.1006/niox.2001.0369. [DOI] [PubMed] [Google Scholar]
- 41.Lymar S V, Jiang Q, Hurst J K. Biochemistry. 1996;35:7855–7861. doi: 10.1021/bi960331h. [DOI] [PubMed] [Google Scholar]
- 42.Darley-Usmar V, Halliwell B. Pharm Res. 1996;13:649–662. doi: 10.1023/a:1016079012214. [DOI] [PubMed] [Google Scholar]
- 43.Beckman J S, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin J C, Tsai M. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- 44.Grinberg L N, O'Brien P J, Hrkal Z. Free Radical Biol Med. 1999;27:214–219. doi: 10.1016/s0891-5849(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Akilesh S, Wilcox D E. Inorg Chem. 2000;39:3057–3064. doi: 10.1021/ic000036s. [DOI] [PubMed] [Google Scholar]
- 46.Nakazawa H, Fukuyama N, Takizawa S, Tsuji C, Yoshitake M, Ishida H. Free Radical Res. 2000;33:771–784. doi: 10.1080/10715760000301291. [DOI] [PubMed] [Google Scholar]
- 47.Czapski G, Goldstein S. Free Radical Biol Med. 1995;19:785–794. doi: 10.1016/0891-5849(95)00081-8. [DOI] [PubMed] [Google Scholar]
- 48.Pryor W A, Squadrito G L. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 49.Beckman J S. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 50.Brennan M L, Wu W, Fu X, Shen Z, Song W, Frost H, Vadseth C, Narine L, Lenkiewicz E, Borchers M T, et al. J Biol Chem. 2002;277:17415–17427. doi: 10.1074/jbc.M112400200. [DOI] [PubMed] [Google Scholar]
- 51.Hirabayashi H, Takizawa S, Fukuyama N, Nakazawa H, Shinohara Y. Brain Res. 2000;852:319–325. doi: 10.1016/s0006-8993(99)02117-4. [DOI] [PubMed] [Google Scholar]
- 52.Campbell A, Smith M A, Sayre L M, Bondy S C, Perry G. Brain Res Bull. 2001;55:125–132. doi: 10.1016/s0361-9230(01)00455-5. [DOI] [PubMed] [Google Scholar]
- 53.Kim Y M, Bergonia H A, Muller C, Pitt B R, Watkins W D, Lancaster J R., Jr Adv Pharmacol. 1995;34:277–291. doi: 10.1016/s1054-3589(08)61092-3. [DOI] [PubMed] [Google Scholar]
- 54.Ryter S W, Tyrrell R M. Free Radical Biol Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 55.Monzani E, Bonafe B, Fallarini A, Redaelli C, Casella L, Minchiotti L, Galliano M. Biochim Biophys Acta. 2001;1547:302–312. doi: 10.1016/s0167-4838(01)00192-3. [DOI] [PubMed] [Google Scholar]
- 56.Lovell M A, Robertson J D, Teesdale W J, Campbell J L, Markesbery W R. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 57.Wink D A, Wink C B, Nims R W, Ford P C. Environ Health Perspect. 1994;102, Suppl. 3:11–15. doi: 10.1289/ehp.94102s311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wink D A, Nims R W, Saavedra J E, Utermahlen W E, Jr, Ford P C. Proc Natl Acad Sci USA. 1994;91:6604–6608. doi: 10.1073/pnas.91.14.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]