

Abstract
The role of electrostatic interactions in the assembly of a native protein structure was studied using fragment complementation. Contributions of salt, pH, or surface charges to the kinetics and equilibrium of calbindin D9k reconstitution was measured in the presence of Ca2+ using surface plasmon resonance and isothermal titration calorimetry. Whereas surface charge substitutions primarily affect the dissociation rate constant, the association rates are correlated with subdomain net charge in a way expected for Coulomb interactions. The affinity is reduced in all mutants, with the largest effect (260-fold) observed for the double mutant K25E+K29E. At low net charge, detailed charge distribution is important, and charges remote from the partner EF-hand have less influence than close ones. The effects of salt and pH on the reconstitution are smaller than mutational effects. The interaction between the wild-type EF-hands occurs with high affinity (KA = 1.3 × 1010 M−1; KD = 80 pM). The enthalpy of association is overall favorable and there appears to be a very large favorable entropic contribution from the desolvation of hydrophobic surfaces that become buried in the complex. Electrostatic interactions contribute significantly to the affinity between the subdomains, but other factors, such as hydrophobic interactions, dominate.
INTRODUCTION
The native conformations of proteins are governed by noncovalent interactions. However, the specific contributions of hydrogen bonds, electrostatic and van der Waals interactions, and the hydrophobic effect remain to be clarified. Many studies have demonstrated that protein stability can be increased by optimizing the Coulomb interactions among charged groups on the protein surface (see, for instance, Akke and Forsén, 1990; Dahlke Ojennus et al., 2003; Grimsley et al., 1999; Hendsch and Tidor, 1999; Makhatadze et al., 2004; Marti and Bosshard, 2003; Perl et al., 2000; Schwehm et al., 2003; Spector et al., 2000). Loladze and Makhatadze (2002) show that although surface charge-charge interactions are not essential for protein folding and stability, the stability can be modulated by charge substitutions. One may anticipate that the contribution of electrostatic interactions to the free energy of folding depends not only on the distribution of ionizable groups within the protein structure but also on the net charge that they produce. In general, increased net charge (positive or negative) leads to higher solubility in water, but may reduce the stability due to electrostatic repulsions within the folded state. Electrostatic interactions are also important for guiding the correct folding by disfavoring alternative structures, as in Fos-Jun heterodimerization (O'Shea et al., 1992) and calmodulin subdomain pairing (Linse et al., 2000).
A general problem with stability studies is that the role of electrostatic interactions under native conditions is inferred by extrapolation from very non-physiological conditions, sometimes even involving such high concentrations of GuHCl that all electrostatic interactions are attenuated. The extrapolation may be necessary, because otherwise only a very small fraction of the protein is present in the unfolded state. Under physiological conditions, the folding/unfolding equilibrium is often strongly shifted toward the folded state and the equilibrium constant between the two states cannot be determined with any appreciable level of accuracy. Much of our knowledge about factors governing protein stability, and the relative importance of different contributions, is hence based on extrapolations from conditions under which both the native and unfolded states are significantly populated, for example at elevated temperature or at very high concentration of denaturant. Here we will explore an alternative method to study the influence of electrostatic interactions under physiological conditions.
The fragment complementation approach allows the contributions of different kinds of noncovalent interactions to be measured at the condition of interest, including physiological conditions and the conditions under which the protein is maximally stable. By this approach, a reconstitution equilibrium is measured, i.e., the complex formation between subdomain fragments. For this purpose, we have chosen a small (Mr ≈ 8500) Ca2+-binding protein of the EF-hand family (Nakayama et al., 1992)—calbindin D9k (Fig. 1). This protein can be reconstituted with high affinity from two fragments representing its two EF-hand helix-loop-helix subdomains (Berggård et al., 2001; Finn et al., 1992). The protein has no disulfide bonds and the three-dimensional structure is known to high resolution (Svensson et al., 1991). A recent crystal structure of a three-dimensional domain-swapped dimer shows that calbindin reconstituted from its two EF-hands has the same structure as intact calbindin (Håkansson et al., 2001), except at the region connecting the subdomains. A strong correlation was found in previous work between the stability of the intact protein and the affinity between its two subdomains (Berggård et al., 2001). This implies that fragment complementation studies may be valuable in assessing the relative importance of different noncovalent interactions.
FIGURE 1.

Calbindin D9k in Ca2+-loaded form. prepared from the PDB file 4ICB (Svensson et al., 1992) using MOLMOL software (Koradi et al., 1996). EF1 is shown in gold and EF2 is shown in silver. (A) Ribbon diagram with the side chains of all charged and mutated residues displayed. Lys side chains are in blue, Asp in magenta, Glu in red, and others in black. Mutated residues are labeled with their identity in the wild-type protein. (B) Space-filling model with mutated residues shown in the same colors as in A.
Ionizable amino acids are abundant in calbindin D9k (Fig. 1) and include both positive and negative charges. The pKa values of all ionizable groups are measured under a range of solution conditions (Kesvatera et al., 1996, 1999, 2001b). As many as 30 out of the 75 residues are charged at neutral pH, producing a net charge of −7. These features make this protein highly suitable for studies of electrostatic interactions in proteins, for example through amino acid substitutions and changes in the solution conditions. It is known that electrostatic interactions are important for the function of this protein, which involves the binding of two Ca2+ ions (Kesvatera et al., 1994; Linse et al., 1988, 1991;) and that repulsive interactions among surface charges reduce the global stability of the protein (Akke and Forsén, 1990).
In the present study, wild-type calbindin D9k as well as five different mutants with surface charge substitutions in EF-hand 1 were used in the reconstitution of the protein. The effect of changes in the electrostatic properties of either the protein itself or the surrounding dielectric solution on the affinity between the two EF-hands in calbindin D9k was measured using surface plasmon resonance (SPR) technology and isothermal titration calorimetry (ITC). Our data show that for highly charged subdomains, the association is correlated with net charge, whereas at zero net charge, the detailed distribution of charges is important for the affinity between subdomains.
MATERIALS AND METHODS
Materials
All chemicals were of the highest grade commercially available. The CM5 sensor chips and amine-coupling kit containing NHS, EDC, and ethanolamine hydrochloride were from Biacore AB (Uppsala, Sweden) as well as PDEA used for the thiol coupling. The surfactant TWEEN 20 was from Riedel de Haen (Seelze, Holland). EDTA disodium salt is from Merck (Darmstadt, Germany).
Protein mutagenesis and purification
Mutant proteins derived from bovine calbindin D9k were expressed in Escherichia coli from a synthetic gene. The gene for calbindin with the P43M substitution was first moved from the runaway plasmid pRCB1 (Brodin et al., 1986) into a modified Pet3a plasmid with NdeI and SacI cloning sites (PetSac) using PCR to introduce a SacI cloning site and keep the NdeI site. In parallel, the gene for P43M was extended using PCR to include a Gly-Gly-Cys extension (P43M-GGC) and cloned into the PetSac plasmid. Mutations were then introduced into P43M in PetSac by Quik-Change mutagenesis (Stratagene, San Diego, CA), and proteins were purified as described (Johansson et al., 1990). The mutants are named after their substitution using one-letter codes for amino acids. Hence, in addition to the substitution Pro43→Met, E17Q+D19N contains the substitution Glu17→Gln and Asp 19→Asn, A15D+P20G contains Ala15→Asp and Pro20→Gly, K25Q contains Lys25→Gln, K12Q contains Lys12→Gln, and K25E+K29E contains Lys25→Glu and Lys29→Glu. The homogeneity of the purified proteins was confirmed by agarose gel electrophoresis, SDS-polyacrylamide gel electrophoresis, and 1H NMR spectroscopy.
Agarose gel electrophoresis
Agarose gel electrophoresis was carried out in sodium barbitone buffer, 2 mM EDTA, pH 8.6, using a 1% agarose gel. The protein was visualized by staining with Coomassie blue.
1H NMR spectroscopy
1H NMR spectra were recorded on a Varian Unity Plus 600 MHz spectrometer (Varian, Palo Alto, CA) at 27°C. Purified intact proteins were dissolved in H2O with 10% D2O in the presence of three equivalents of Ca2+ at pH 7.
CD spectroscopy
Far UV CD spectra were recorded on a JASCO J-720 spectrometer (JASCO, Tokyo, Japan) in a 1-mm quartz cuvette between 250 and 190 nm, with a bandwidth of 1 nm, a step of 1 nm, a scan rate of 10 nm/min, and an 8-s response time.
CNBr cleavage and fragment purification
Each mutant was cleaved by CNBr to produce EF-hand fragments EF1 and EF2. The two fragments from the wild-type protein have different net charges (−1 for EF1 and −6 for EF2 when in apo-form), and can be separated from each other using ion exchange chromatography in the presence of EDTA (Berggård et al., 2001; Finn et al., 1992). In the calcium bound state, the two EF-hands interact so tightly that they resist separation by this method. Cleavage of the calbindin D9k mutants at the methionine residues at positions 0 and 43 with CNBr and purification of the resulting fragments with ion-exchange chromatography were performed as follows. Fifty milligrams purified protein was dissolved in 1.2 mL MilliQ H2O (Millipore, Billerica, MA) and put on ice. TFA, 4.8 mL, was added gradually with gentle shaking. The protein/TFA solution was added to 0.6 g CNBr on ice, and N2 gas was bubbled through for 5 min. The sample was sealed and left at room temperature overnight. Cleavage of P43M-GGC yields wild-type EF1 and EF2-GGC. Cleavage of the other mutants yields mutated EF1 and wild-type EF2. The volume of the mixture was reduced by evaporation using a Buchi rotavapor (Buchi, Flawil, Switzerland) and the mixture was redissolved in 30 ml of 5 mM EDTA. The pH was adjusted to 7.5 using 1 M TRIS base (final TRIS concentration was ∼24 mM). The sample was pumped onto a 1.5 × 12 cm DEAE-Sephacel column pre-equilibrated in 10 mM TRIS/HCl, 1 mM EDTA, pH 7.5. The fragments were eluted with a linear NaCl gradient from 0.05 M to 0.40 M. The mutated EF1 fragments (residues 1–43) have net charges ranging from +1 to −5 without calcium and elute earlier in the gradient than EF2 (residues 44–75). The elution order of EF2 and residual intact protein differs among the mutants, but in all cases EF1 elutes before residual uncleaved protein. EF2 eluted in two separate peaks corresponding to monomeric and dimeric material. The identity and purity of the fragments was readily confirmed by agarose gel electrophoresis because EF2 and EF1 have different net charges, and differ also from the remaining uncleaved proteins (Fig. 2 A). The isolated fragments were analyzed by MALDI-TOF mass spectrometry (Swegene Proteomics Centre, Lund University). The concentrations of fragment stock solutions were determined by amino acid analysis after acid hydrolysis (Biomedical Centre, Uppsala University).
FIGURE 2.

(A) Agarose gel electrophoresis in 75 mM sodium barbitone buffer, pH 8.4, with 2 mM EDTA, on DEAE-Sephacel fractions after CNBr cleavage for the mutant K12Q. Fractions 36–41 were pooled as EF1, fractions 57–67 as residual uncleaved protein, and fractions 82–89 as EF2. (B) Chemistry of the EF2-GGC immobilization using the ligand-thiol disulfide-exchange method.
Surface plasmon resonance studies
The interaction between EF1 and EF2 was studied by surface plasmon resonance technology, using the Biacore3000 apparatus. Different flow buffers were used depending on the pH. As flow buffer for physiological conditions, 10 mM HEPES/NaOH, pH 7.4, with 0.15 M NaCl, 0.005% TWEEN 20, 0.02% NaN3, and 2 mM CaCl2 was used. The same buffer was used to monitor the salt concentration effect on the reconstitution, the only difference being the varying amount of NaCl. AMP was used instead of HEPES for the measurements at pH 9 and 9.5 and MES was used for pH 5.5 and 6.5 whereas HEPES was used for pH 8 and all other components were identical to the pH 7.4 buffer. The small amount of TWEEN 20 in the buffers was used to prevent clogging of the tubes in the Biacore apparatus and prevent unspecific binding. All buffers were filtrated through sterile 0.22 μm filters before use and degassed for 30 min. The built-in thermostat kept the temperature at 25°C during all experiments.
Immobilization of EF2 to the sensor chip was performed through two different couplings techniques, amine coupling and ligand thiol disulfide exchange coupling. 10 mM HEPES/NaOH at pH 7.4, 0.15 M NaCl, 0.005% TWEEN20, 0.02% NaN3, and 3.4 mM EDTA was used as flow buffer during coupling. Amine coupling was performed as described earlier (Berggård et al., 2001). Ligand thiol coupling (Fig. 2 B) was performed at a constant flow rate of 5 μl/min. Equal volumes of 0.1 M NHS and 0.4 M EDC were first mixed, and 25 μl of the mixture was allowed to flow over the sensor-chip surface to activate the carboxymethylated dextran (5 min). Twenty microliters of a PDEA solution, made by dissolving 4.5 mg PDEA in 205 μl 0.1 M borate buffer at pH 8.5, were injected to introduce a reactive disulphide group onto carboxyl groups of the sensor chip. EF2-GGC at 0.01 or 0.1 mg/ml in 70 μl 10 mM sodium formate buffer at pH 4.3 was then injected over the sensor chip. The C-term Cys of EF2-GGC was used to create a covalent link between the immobilized EF2-GGC and the dextrane matrix of the sensor chip (Fig. 2 B). Deactivation of the excess reactive disulphides on the chip was done by injecting 20 μl of 50 mM L-cysteine with 1 M NaCl in 100 mM formate buffer at pH 4.3. At least one of the four flow cells on each sensor chip was reserved for a blank immobilization with no protein in the coupling step and was used as reference. Chips with immobilized EF2 were generally used for less than two weeks. Amine coupling was used only for a control experiment on the reconstitution with EF1 wild-type in physiological conditions, whereas the rest of the measurements were done by immobilizing EF2-GGC by thiol disulfide exchange coupling.
Association and dissociation experiments
The kinetics of the reconstitution reaction was studied at constant flow rates of 10 μl/min and 30 μl/min with no significant change in the rate constants obtained. Therefore flow rate of 10 μl/min was used in subsequent experiments. The association of EF1 to the immobilized EF2 and the following dissociation was studied at several different EF1 concentrations ranging from 0.275 nM to 140 nM for the amine coupling and from 5 nM to 164 nM for the ligand thiol coupling. Protein stock solutions were diluted using the flow buffer and 300 μl was injected during the association phase which was followed for 30 min. The dissociation process was followed until >85% of the bound EF1 had dissociated, with the time required depending on the particular solution condition or EF1 mutant used. After each experimental cycle, the surfaces were regenerated by injecting 10 mM EDTA, pH 8, for 5 min to remove residual-associated fragments.
Control experiments
Control experiments were performed to confirm both the specificity of the binding and the stability of the immobilized EF2. In particular, 300 μl of 100 mM wild-type calbindin D9k without any performed cleavage was flowed for 2 h onto the chip directly after a previous association was performed as described before. No binding was detected as a consequence of the injection, indicating that the intact protein does not interact with the reconstituted one; moreover, the dissociation rate of the reconstituted protein during the injection of intact calbindin D9K was not affected at all, indicating no interaction between intact protein and immobilized EF2. The time-stability of the immobilized EF2 was also checked to exclude any effects that might arise from very slow dissociation of coupled EF2 from the sensor chip over the range of time of a measurement. Running buffer without dissolved EF1 was flowed over all of the flow cells of the sensor chip for 3.5 h and no variation of the signal was detected, indicating that the amount of the immobilized protein is constant. Immobilization is performed using buffer with EDTA to avoid close coupling of two EF2-GGC as a homodimer, which may happen in the presence of Ca2+.
During the coupling procedure, EF2-GGC is subjected to pH 7.4 in flow buffer with EDTA for ∼5–15 min before changing to buffer with Ca2+. EF2-GGC contains one potential deamidation site (Asn56), and deamidation is catalyzed at basic pH and more rapid for flexible Ca2+-free EF-hands. A small amount of the purified EF2-GGC was therefore incubated in flow buffer with EDTA for up to two days at pH 7.6 and 8.6, and analyzed on agarose gels. These gels separate similar peptides according to net charge, and show maximum 5% deamidated EF2-GGC after two days. Hence EF2-GGC is not deamidated during the time frame of our study.
Data analysis
The data were evaluated using Levenberg-Marquardt nonlinear least-square method. The data analysis was made using the software KaleidaGraph (Synergy Software, Reading, PA). The association reaction studied by the use of SPR occurs between EF2 immobilized to the sensor chip surface and EF1 in solution. EF1 with or without mutations is in constant flow during the association phase, and EF1-EF2 complex formation leads to a change in refractive index of the sensor-chip surface, which is reported continuously in terms of response units (RU). The response is proportional to the total mass of reconstituted molecules onto the chip surface. The dissociation process is initiated by change to a constant flow of protein-free buffer, and a decrease in the SPR signal corresponds to EF1 dissociating from the immobilized EF2 into the solution. In addition to the response changes due to association and dissociation, the signal changes abruptly when protein injection starts or ends, due to mechanical disturbances. The first and the last few minutes of each phase are therefore omitted in the fitting procedure. The dissociation and the association data were fitted separately.
The dissociation of the complex can be modeled as a first-order reaction,
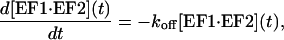 |
(1) |
where koff is the dissociation rate constant. Equation 1 can be analytically solved by integration, yielding an exponential decrease for the complex concentration as a function of time, which leads to the equation
 |
(2) |
Since the instrumental response R(t) is assumed to be linear with the complex concentration (Stenberg et al., 1991), Eq. 2 can be transformed into the following equation for fitting to dissociation data,
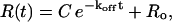 |
(3) |
where C is equal to R (0) − R0. R0 is the baseline value. The SPR response approaches R0 when t approaches infinity. For each case, 4–12 repeat measurements were performed to yield an average koff and its standard deviation.
During the association phase, the complex concentration at the sensor chip surface is also affected by the rate of dissociation koff, resulting in a more complicated kinetics that can be written as
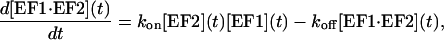 |
(4) |
where kon is the association rate constant. The consumption of EF1 can be shown to be negligible therefore [EF1] can be approximated as constant and the total concentration of EF2 is the sum of the free immobilized EF2 and EF2 in complex with EF1. Equation 4 can be solved analytically, and with the boundary of [EF1·EF2] (t = 0) = 0, the solution is
 |
(5) |
The equation curve for the fitting of association data is then
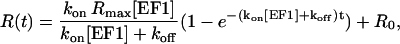 |
(6) |
where Rmax indicates the maximum SPR response that would be expected if all the active immobilized EF2 molecules would interact with EF1. Rmax would, in principle, be constant for all association experiments run on one particular surface of immobilized EF2. However, instabilities in the instrument and the deterioration of the immobilized protein with time and number of experiments may cause variations in Rmax. Six to twelve measurements were performed for each condition and an average kon and standard deviation was obtained.
Isothermal titration calorimetry
Isothermal titration calorimetry was carried out using a VP-ITC instrument from MicroCal (Northhampton, MA) at constant temperature (25°C) in 2 mM HEPES/NaOH buffer, pH 7.4 with 1 mM CaCl2. In addition, 0, 20, 50, 150, or 400 mM NaCl was present in the buffer for the salt-dependence experiments and 150 mM NaCl for the experiments with EF1 mutants. Deionized water was used in the reference cell. All solutions were thoroughly degassed by stirring under vacuum before use. For the salt-dependence experiments, 39 μM EF2 was titrated into 1.2 μM EF1 in the sample cell (1.4 ml). Two to three titrations were carried out at each NaCl concentration. For each titration, a 2 μl injection was followed by 24 injections of 9 μl each into the sample cell. The mixture was allowed to react for 4 min between injections. The baselines of the raw data were adjusted manually before integration. Heats due to injection and dilution were obtained by titrating additional EF2 after the saturation of EF1. For the titrations with EF1 mutants, 160 μM EF2 was titrated into 2–6 μM EF1 mutants with a 1 μl injection followed by 24 injections of 5 μl each into the sample cell. Two titrations were carried out for each EF1 mutant and the experimental procedure was otherwise identical to that of the salt-dependence experiments.
ΔH° was obtained by global fits of a 1:1 binding model to all titration curves for the same condition using MATLAB 6.5 (The MathWorks, Natick, MA). Because the experiments were designed for ΔH° determination and not affinity determination, the binding constants were fixed to values obtained from the SPR measurements. The first injection has been omitted in the data analysis. The errors in ΔH° were obtained using a Monte Carlo analysis.
Assuming ideal behavior, the standard free energy change ΔG° and the standard entropy change ΔS° can be calculated from the equilibrium constant and ΔH° using Eqs. 7–9, which are valid at equilibrium.
 |
(7) |
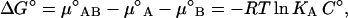 |
(8) |
 |
(9) |
KA is the equilibrium constant in molar units, and ° denotes standard state (C° = 1 M).
RESULTS
Mutagenesis and nomenclature
The design goal was to probe the role of electrostatic interactions in the assembly of a protein from its subdomains. Site-specific substitutions involving six residues of EF-hand 1 of calbindin D9k were introduced to produce five mutants. The residues were chosen to represent surface positions both close to and far from the interface with EF-hand 2. Three of the substituted residues (A15, E17, D19) are in the loop of the N-terminal calcium-binding site; one (K12) is in α-helix I, and two (K25 and K29) in helix II. The substitutions were introduced in a mutated version of the minor-A form of bovine calbindin D9k in which Met43 substitutes for Pro43 in the loop between the two EF-hands. The mutants are named after their substitutions (in addition to P43M) using one-letter amino acid codes. The net charge at neutral pH is altered by +2 units for E17Q+D19N, by −1 unit for each of A15D+P20G, K25Q and K12Q, and by −4 units for K25E+K29E. This last mutant has a drastic charge change (from +1 to −3 in the presence of calcium) that should invoke significant net repulsion of the negatively charged partner subdomain EF2. Intact proteins with the A15D+P20G, and E17Q+D19N substitutions have been studied before (Akke and Forsén; 1990; Johansson et al., 1991; Linse et al., 1988, 1991), whereas the other three mutants were designed specifically for this study. The extra P20G substitution provides A15D+P20G with a Ca2+ affinity that is only twofold reduced compared to wild-type (Johansson et al., 1991). The backbone carbonyls of E17 and D19 ligate the Ca2+ ion in site I, whereas the side chains are surface-exposed. The Ca2+ affinity for E17Q+D19N is 10-fold reduced relative to wild-type (Linse et al., 1991), yielding a KD of 1 μM, which means that E17Q+D19N is 99.95% saturated in 2 mM CaCl2. For the other mutants we expect smaller effects or even an increase in calcium affinity, based on earlier studies of surface charge mutants (Linse et al., 1988; Svensson et al., 1991).
Protein purification, CNBr cleavage, and isolation of fragments
The protocol for the wild-type (wt) protein (Johansson et al., 1990) was used for the purification of all intact mutants, except that the salt concentrations for elution had to be adjusted according to the net charge of the protein. The solution structure has been solved for A15D+P20G (Johansson et al., 1993), and found essentially identical to the parent protein, even in the calcium loop. NMR assignments reveal that the structure is essentially retained in E17Q+D19N (Linse et al., 1991). 1H NMR spectra in H2O for K12Q, K25Q, and K25E+K29E are very similar to the spectrum of the wild-type substitute P43M (Fig. 3). This close similarity includes all the outshifted NH and methyl protons, indicating that no major structural rearrangements have occurred due to the surface charge substitutions. In the gel filtration of the intact protein with the GGC extension, both the monomer and a higher aggregate were obtained. Only the monomeric peak was used for further purification and CNBr cleavage to produce EF2-GGC. All purified mutant proteins were cleaved by CNBr and fragments separated used ion exchange in EDTA followed by a desalting step. Typical yields of pure fragment were 16–19 mg EF1 and 8–12 mg EF2 when 50-mg purified intact protein was cleaved. MALDI-TOF mass spectroscopy was performed for all isolated fragments confirming that there was no further degradation beyond the cleavage at methionine. EF2-GGC shows only one peak that agrees with the expected mass. Each EF1-peptide shows a major peak that agrees with retained Met0 on the N-terminus and a homoserine on the C-terminus (as should be the result of the CNBr reaction). In addition there are two minor peaks, one with 18-Da higher mass corresponding to Met0 plus homoserine lactone, and one with 131-Da lower mass corresponding to the loss of Met0 and homoserine on C-terminus. CD spectra of the various EF1 mutants at 20 μM in the presence of 2 mM CaCl2 show that under these conditions, the helical content is the same (data not shown).
FIGURE 3.

1H NMR spectra in H2O for calbindin D9k P43M, and the charge mutants K12Q, K25Q, and K25E+K29E. The charge mutants also contain the P43M substitution.
Surface plasmon resonance studies
Immobilization of EF2-GGC using the ligand thiol-coupling method (Fig. 2) was more efficient than immobilization of EF2 using the amine-coupling method. Coupling of >∼1000 RU was difficult using amine coupling, most likely because this method disfavors a negatively charged protein or fragment. Apo EF2 has a formal charge of −6 at neutral pH and requires very low pH to become positively charged. Although much higher coupling levels of EF2-GGC were easily achieved using the ligand thiol-coupling method, the procedure was controlled to reach ∼2000 RU. The maximum signal during EF1-binding experiments was ∼200 RU for thiol coupled surfaces and <50 RU with amine-coupled surfaces, indicating that a larger percentage of coupled EF2 was active for binding to EF1 using the ligand thiol method.
EF1 wt was injected over surfaces with thiol-coupled EF2-GGC to study the association kinetics over a range of EF1 concentrations. This mode of analysis was chosen based on the known dimerization properties of the two fragments. EF1 has the higher homodimerization constant (Julenius et al., 2002) and may form homodimers when immobilized due to local high concentration on the sensor-chip surface. Therefore, homodimer dissociation becomes the rate-limiting step for association of EF2 with immobilized EF1 (Berggård et al., 2001). This problem is not encountered when EF2 is immobilized because EF1 is injected in concentrations that are orders-of-magnitude below the inverse of its association constant (Berggård et al., 2001; Julenius et al., 2002). Injection of EF1 was interrupted after 30 min, after which buffer was flowed over the surfaces to study the dissociation kinetics for up to 28 h. Under physiological conditions, the dissociation was only ∼85% complete after 28 h. However, instabilities in the instrument made it difficult to study the dissociation process for longer periods of time. Binding and dissociation was observed in buffer with 2 mM CaCl2 (Figs. 4 and 5 A). Equations 5 and 3 for a simple 1:1 binding model were very well fitted to the association and dissociation phase data, respectively (Fig. 4). The affinity we obtain from the rate constants is slightly lower than that obtained previously using amine coupling (Berggård et al., 2001). A test experiment using amine coupling produced roughly the same results for wt EF1 as previously obtained (data not shown). No binding was observed in the presence of EDTA (Fig. 5 A).
FIGURE 4.

Typical SPR sensorgrams for the association (A) and dissociation (B) of 1.2 nM EF1 wt with EF2-GGC immobilized. Experimental data from one replicate (shaded) are shown together with least-squares fitted curves (solid).
FIGURE 5.

SPR sensorgrams (experimental data from one replicate) for the association and dissociation of EF1 wild-type in the presence of Ca2+ or EDTA (A) and EF1 surface charge mutants in the presence of Ca2+ (B–F) to immobilized EF2-GGC. The first 3500 s are shown for 50 nM (solid) and 10 nM (shaded) EF1. The arrow indicates the point where protein injection is interrupted and replaced with buffer flow.
Effects of point mutations in EF1 on protein reconstitution (SPR)
Five mutants with charge substitutions in the EF1 subdomain were used to study the role of electrostatic interactions in the reconstitution of calbindin D9k (Table 1, Fig. 5). The data show that the kinetics of the reconstitution is strongly affected by the charge substitutions. All mutants show higher dissociation rate constants than wt. For the mutants E17Q+D19N, K25Q, and K12Q, a time of 25 h was sufficient for following the complete dissociation process. The mutants A15D+P20G and K25E+K29E were almost totally dissociated after 3 h. The highest dissociation rate constants (∼10−3 s−1) were found for A15D+P20G and K25E+K29E, which are neutral or negatively charged in the presence of calcium. These mutant EF1s dissociate ∼100-fold faster than EF1 wt. In all tested cases, the mutations affect the dissociation rate constant much more than the association rate constant. The highest association rate constant (only twofold higher than wt) is found for E17Q+D19N, which has a formal net charge of +3 at neutral pH, opposite to the charge of EF2. The lowest association rate constant is found for K25E+K29E (threefold lower than for wt). The affinity between EF1 and EF2, as deduced from the rate constants, is lower for all the mutants compared to wt. The lowest affinity between EF1 and EF2 was found for K25E+K29E (260-fold lower than wt), whereas it was reduced 100-fold for A15D+P20G and 25-fold for K25Q relative to wt. Only modest effects on the affinity were seen in E17Q+D19N (twofold lower than wt, which is approximately the size of the errors) and K12Q (threefold lower than wt).
TABLE 1.
Rate and equilibrium constants, and thermodynamic parameters, for the reconstitution of EF1 wt and mutants with immobilized EF2-GGC (at physiological conditions of pH 7.4, 150 mM NaCl, 25°C)
| Net EF1 charge* | 10log kon | 10log koff | 10log KA | ΔH°/kJ mol−1 | TΔS°/kJ mol−1 | |
|---|---|---|---|---|---|---|
| Wild-type | +1 | 5.1 ± 0.2 | −5.0 ± <0.1 | 10.1 ± 0.2 | −29.3 ± 0.8 | 28.3 ± 1.9 |
| E17Q+D19N | +3 | 5.4 ± 0.1 | −4.4 ± <0.1 | 9.8 ± 0.1 | −46.7 ± 1.1 | 9.3 ± 1.6 |
| K12Q | 0 | 4.9 ± 0.2 | −4.7 ± 0.3 | 9.6 ± 0.5 | −50.9 ± 1.1 | 3.9 ± 3.9 |
| K25Q | 0 | 4.8 ± 0.3 | −4.0 ± 0.1 | 8.7 ± 0.4 | −36.0 ± 1.1 | 13.6 ± 3.4 |
| A15D+P20G | 0 | 5.1 ± 0.2 | −3.0 ± <0.1 | 8.1 ± 0.2 | −48.8 ± 1.3 | −2.6 ± 2.4 |
| K25E+K29E | −3 | 4.6 ± 0.1 | −3.1 ± 0.1 | 7.7 ± 0.2 | −35.0 ± 1.4 | 8.9 ± 2.5 |
The net charge of the mutant is given as the formal charge at pH 7.4 with Ca2+ bound.
Effects of point mutations in EF1 on protein reconstitution (ITC)
ITC was used to investigate the effect of mutations on the enthalpic and entropic contributions to the free energy of binding (Fig. 6). Calorimetric titrations were performed in 2 mM HEPES/NaOH pH 7.4 with 1 mM CaCl2 and 150 mM NaCl. Due to the very high affinity between EF1 and EF2, ITC could not be used to determine the affinity, as this would require such low concentrations that the signal would be negligible. For all the mutants, the standard enthalpy of binding was more favorable than for wild-type EF1. This suggests that the unfavorable effect on binding free energy ΔG° as observed for all mutants is due to the entropic component, TΔS°, being much less favorable, or even unfavorable, offsetting the gain in enthalpy.
FIGURE 6.

ITC data for 39 μM EF2 wt titrated into 1.2 μM EF1 wt at 150 mM NaCl in 2 mM HEPES/NaOH, pH 7.4 with 1 mM CaCl2. An initial 2-μl injection was followed by 9-μl injections. The heat response is shown in the upper panel and the heat-per-mole injectant in the lower panel. Data from one replicate are shown together with the global fit of three replicates.
Effects of pH on protein reconstitution (SPR)
For the wild-type EF1 the reconstitution was studied in the pH range between 5.5 and 9.5. The results show that the rate constants are weakly dependent on pH in this range (Fig. 7 C). The pH range was not extended outside 5.5–9.5 due to instrumental instability at higher pH and the loss in calcium affinity for both the binding sites at lower pH (Kesvatera et al., 2001a). The only ionizable groups that undergo a titration in this pH range are the N-terminal amine group and a few carboxylates at the Ca2+-sites (Kesvatera et al., 1996, 2001b). The rather weak pH dependence seen within this pH range is therefore not surprising.
FIGURE 7.

Association rate constant (kon, A and C) and equilibrium association constant (KA, B) plotted in logarithmic units (lg = 10log) for EF1 wt injected over EF2-GGC as a function of NaCl concentration (A, B) and pH (C).
Effects of NaCl concentration on protein reconstitution (SPR)
The effect of salt concentration on the affinity between EF1 and EF2 was monitored in the NaCl concentration range between 5 and 400 mM at a constant pH of 7.4. The effect of salt concentration is much smaller than the effects due to mutations as described above. The association rate constant is reduced ∼10-fold at low salt compared to 150 mM NaCl (Fig. 7 A). A similar reduction is seen when going to high salt concentration. The dissociation rate constant is found to be roughly independent of salt concentration between 5 and 400 mM NaCl (data not shown). The affinity between EF1 and thiol-coupled EF2-GGC is increased approximately fivefold as the salt concentration increases from low to 150 mM NaCl (Fig. 7 B).
Effects of NaCl concentration on protein reconstitution (ITC)
ITC was used to investigate the thermodynamic origin of the small peaking at 0.15 M NaCl of the affinity between EF1 and EF2. Calorimetric titrations were performed at 0, 20, 50, 150, and 400 mM NaCl in 2 mM HEPES/NaOH pH 7.4 with 1 mM CaCl2 (Figs. 6 and 8). Due to the very high affinity between EF1 and EF2, ITC could not be used to determine the affinity, as this would require such low concentrations that the signal would be negligible. Instead the titrations were performed at a concentration (1.2 μM) that was high enough to get a decent signal but low enough to avoid screening effects from the peptides at the lowest salt concentrations. At physiological salt concentration, we obtain ΔH° = −30 kJ mol−1. The enthalpy of association is roughly invariant with salt and the small difference observed at 150 mM NaCl compared to all other salt concentrations is just beyond the error limits.
FIGURE 8.

Enthalpic (ΔH°; B) and entropic (−TΔS°; C) contributions to (ΔG°; A) for the association of EF1wt and EF2 wt as a function of NaCl concentration.
Both the enthalpic (ΔH°) and entropic (−TΔS°) components favor complex formation between EF1 and EF2. The lack of salt effects on ΔH° suggests that the salt-dependence of ΔG° originates from entropic effects (Fig. 8).
DISCUSSION
The results of the present study show that electrostatic interactions can modulate the association of super-secondary-structure elements, or subdomains, in a protein. The distribution of surface charges over the two helix-loop-helix subdomains in calbindin D9k clearly affects the affinity between them. The equilibrium constant varies over roughly one order in magnitude due to changes in salt conditions and over more than two orders of magnitude upon charge substitutions at physiological salt.
A high affinity reconstitution
The reconstitution of calbindin D9k from its EF-hand subdomains is distinguished by a very high affinity with a KD in the picomolar range (Berggård et al., 2001). This very high affinity occurs in the presence of calcium and is a result of a sizeable association rate constant and a very low dissociation rate constant. CD spectroscopy shows that each fragment on its own may bind calcium and fold as an EF-hand (Julenius et al., 2002). Each fragment may form homodimers, but with at least million-fold lower affinity (Julenius et al., 2002) than for the heterodimeric complex of EF1 and EF2 (Berggård et al., 2001). In the absence of calcium, the binding between EF1 and EF2 is much reduced (Fig. 5 A) and has not been measured.
The high affinity between EF1 and EF2 is to a large extent governed by hydrophobic core residues (Berggård et al., 2001). This is most likely a combined effect of entropic contributions from the release of water and an optimized van der Waal's packing of the core. Single side-chain substitutions from Leu to Ala or Gly, or from Phe to Ala were found to affect the equilibrium constant up to 20,000-fold (Berggård et al., 2001). These effects are clearly much larger than the ones here observed upon point mutation of charged residues (up to 260-fold). Of course, one may anticipate larger effects of charge substitutions of the side chains that act as bidentate ligands in calcium coordination, due to a dramatic loss in calcium affinity (Carlström and Chazin, 1993; Maune et al., 1992).
The role of noncovalent interactions in proteins
The study of hydrophobic core mutants showed that there is a linear correlation between the mutational effects on free energy of reconstitution and free energy of unfolding of intact calbindin D9k, indicating that the factors governing the stability of the intact protein also contribute to the affinity of the bimolecular EF1-EF2 complex (Berggård et al., 2001). Measuring the free energy of association between wild-type and mutated fragments, one avoids the general problem of assessing free energy effects by comparing values extrapolated from very non-physiological conditions like high concentrations of denaturant or elevated temperature. Instead the contributions of individual residues can be measured directly at chosen solution conditions, including those where the protein is maximally stable.
The calorimetric study of the association between EF1 and EF2 (Figs. 7 and 8) reveals that for the wild-type, the enthalpy (ΔH° = −30 kJ mol−1) of complex formation constitutes approximately one-half the free energy of association at the standard condition of 1 M (ΔG° = −57 kJ mol−1) The entropic contribution at the standard state can be calculated as −TΔS° = −28 kJ mol−1 (or ΔS° = 94 JK−1 mol−1) using Eq. 9. The entropic component will contain contributions both from what can be called the intrinsic entropy of the complex compared to free fragments (ΔS*), and a concentration-dependent contribution from the fact that two components react to form one component. This latter factor will favor dissociation of the fragments. Many researchers relate this entropic correction to the loss of translational entropy upon association (Amzel, 1997; Luque and Freire, 1998),
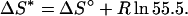 |
With this correction, the intrinsic entropy of association is ΔS* = 127 JK−1 mol−1, yielding an entropic contribution to the binding free energy of −TΔS* = −38 kJ mol−1 at physiological salt and 25°C. Hence, the intrinsic entropic change strongly favors complex formation. The change in conformational entropy upon complex formation is most likely unfavorable, as the free fragments can be expected to sample a larger number of conformations than the complex. There is, however, a large and favorable entropy increase due to the release of water molecules from hydrophobic surfaces that become buried in the complex. This contribution overrules the conformational entropy, and the intrinsic entropy change is a stronger driving force for complex formation than the enthalpy change.
Effects of charge substitutions on the kinetics of protein reconstitution
Phage-display studies of calmodulin reconstitution have shown that charge-charge repulsion can be a significant destabilizing factor even at physiological salt (Linse et al., 2000). The present study was therefore performed at physiological salt to evaluate electrostatic contributions to the binding free energy under these conditions. Our data show that even at this salt concentration (0.15M NaCl), the kinetics of calbindin D9k reconstitution from EF2 and EF1 are clearly affected by charge substitutions in EF1 (Fig. 5). For all variants, the most pronounced effects are seen in the dissociation rate constants. At physiological salt, the association rate constant is not very sensitive to electrostatic interactions, but correlates with net charge in a way that would be expected for Coulomb interactions. Hence net charge seems to be an important modulator of the rate of recognition.
Effects of charge substitutions on protein reconstitution equilibrium
The equilibrium constant for the reconstitution reaction is spread over a wide range covering more than two orders of magnitude (a factor of 260). The large reduction in affinity as observed for K25E+K29E is most likely due to increased electrostatic repulsion between the EF-hand subdomains. Still, the affinity of this mutant EF1 for wt EF2 is as high as KA = 4.8 × 107 M−1 (KD = 20 nM) due to very strong attractive contributions from the hydrophobic and other interactions that overrule the electrostatic repulsion. The reduction in affinity, as observed for like-charged subdomains, is not paralleled by an increased attraction when the opposite charges are more pronounced, as in E17Q+D19N. This suggests that it is more feasible for a protein to use repulsive electrostatic interactions to prevent unwanted conformations than to use attractive forces to specify a fold.
Our data shows that at low charge, the dependence on net charge breaks down and interactions within the charge network on the protein surface come into play. The equilibrium constants of the three neutral EF1s vary over almost two orders of magnitude, indicating that at zero net charge the position of the substitution is critical for the effect on reconstitution of calbindin D9k. K12 is found in helix I, A15 in the calcium loop of EF1, and K25 in helix II. For these three side chains, the distance in the x-ray structure (Svensson et al., 1992; Fig. 1) to the closest charges in EF2 are 20 Å (K12–E60), 13 Å (A15–E60), and 3 Å (K25–D47). The small effect observed for K12Q relative to wild-type (a factor of 3) can therefore be reconciled by the fact that this residue is more remote from all EF2 charges than A15 and K25. The closer distance from K25 to D47 and to other negative charges on EF2 predicts larger effects for K25Q than for K12Q, as is indeed seen. The large effect observed for A15D+P20G (a factor-of-100 lower affinity than wt) may seem surprising, especially as the structure calcium binding loop of EF1 is very similar to wild-type (Johansson et al., 1993). This suggests that destabilizing repulsive electrostatic interactions within EF1 due to Asp15 may reduce the affinity for EF2.
Titration calorimetry reveals an enthalpy-entropy compensation mechanism upon charge substitutions, as there are larger effects on both ΔH and TΔS than on ΔG (calculated as −RT ln KA). The charge substitution effects on affinity are dominated by entropic effects, which may be related to mutational effects on the conformational degrees of freedom and/or hydration.
Effects of NaCl concentration on protein reconstitution
A traditional interpretation of the screening effect according to the Debye-Hückel theory foresees the presence of a screening factor dependent on the square-root of the ionic strength. Therefore, the interactions between small opposite charges is reduced as the salt concentration is increased. Likewise, binding of small positive ionic ligands, like metal ions to a negatively charged protein, is strongest at low ionic strength and reduced by several orders of magnitude as the salt concentration increases (Kesvatera et al., 1994; Linse et al., 1991; Svensson et al., 1991). In contrast, the reconstitution of calbindin D9k from the oppositely charged EF1 and EF2 (+1 and −4, respectively) is not favored by low salt concentration. Instead, the affinity increases approximately fivefold upon addition of physiological salt. Although the net charge of EF1 and EF2 are of opposite sign, the distribution of individual charges may be such that the sum of all electrostatic contributions produces an overall unfavorable electrostatic contribution to the free energy of reconstitution. Screening by salt may then damp the unfavorable component and allow for tighter association. In addition, the salt effect may be due to other contributions, for example hydrophobic interaction may also be salt-dependent. The calorimetric analysis suggests that the salt effect arises mainly from the salt-dependence of the entropic component. Hence, the salt concentration seems to affect the decrease in conformational entropy upon complex formation and/or the entropy increase due to the release of water molecules from hydrophobic surfaces that become buried in the complex.
Homogenous coupling produces high quality data
The present work uses thiol coupling of EF2 via a three-residue extension at the C-terminus (Fig. 2). One major benefit of this coupling is its homogeneity. All EF2 molecules are immobilized using the same functional group, producing high quality data that is extremely well fitted to a simple 1:1 binding model (Fig. 4). Another benefit of thiol coupling is charge conservation, as no formal charge is lost. In addition, the three-residue extension allows the thiol-coupled EF2 to be more remote from the dextran matrix, the partial charges of which may provide electrostatic perturbations of the binding of EF1. The more conventional amine coupling, on the other hand, occurs between the dextran carboxylates and amine groups at the N-terminus or lysine side chains. Therefore, amine-coupled EF2 is heterogeneous. In addition, the loss of one positive charge at the coupling site means that EF2 is one-unit more negative after amine coupling. This may lead to an additional electrostatic contribution to the binding of the positive wt EF1 (net charge +1 with Ca2+ bound), and may explain the slightly higher affinity obtained with the amine compared to thiol coupling.
Protein reconstitution is a bimolecular binding/recognition reaction and folding/unfolding is unimolecular; however, the same intermolecular interactions (electrostatic, van der Waal's, hydrophobic effect, H-bonds, etc.) are in operation in both events and there is a strong correlation between mutational effects on stability and reconstitution (Berggård et al., 2001). The main difference between the two events is the entropic loss of bringing two fragments together. There is considerable interest in finding a solution to the protein folding problem, i.e., to predict structure from sequence. To develop algorithms for this purpose, theoreticians need massive data to test their models, and many investigators are convinced that more structures will not help them to solve this problem. Rather, a large body of thermodynamic data as to the roles of different interactions will be needed, and the present work presents a way that such data can be accumulated under physiological conditions.
Acknowledgments
Helpful discussions on thermodynamics by Prof. Bengt Jönsson are gratefully acknowledged.
This work was supported by The Swedish Research Council (S.L.). D.D.O. was the recipient of an Erasmus/Socrates scholarship.
Daniele Dell'Orco's present address is Department of Chemistry, Division of Physical Chemistry, University of Modena and Reggio Emilia, via Campi 183, Modena, Italy.
Abbreviations used: AMP, 2-amino-2-methyl propanol; CD, circular dichroism; EDC, N-ethyl-N′-(dimethylaminopropyl)carbodiimide; EDTA, ethylene-dinitrilo tetra-acetic acid disodium salt dehydrate; EF1 (wild-type EF1), fragment with residues 1–42 of calbindin D9k plus Met43; EF2, fragment with residues 44–75 of calbindin D9k; HEPES, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid); MES, 2-morpholinoethanesulfonic acid; NHS, N-hydroxysuccinimide; PDEA, 2-(2-pyridinyldithio)ethanolamine; SDS, sodium dodecyl sulfate; TFA, trifluoroacetic acid; TRIS, tris(hydroxymethyl)aminomethane.
References
- Akke, M., and S. Forsèn. 1990. Protein stability and electrostatic interactions between solvent-exposed charged side chains. Proteins. 8:23–29. [DOI] [PubMed] [Google Scholar]
- Amzel, L. M. 1997. Loss of translational entropy in binding, folding, and catalysis. Proteins. 28:144–149. [PubMed] [Google Scholar]
- Berggård, T., K. Julienius, A. Ogard, T. Drakenberg, and S. Linse. 2001. Fragment complementation studies of protein stabilization by hydrophobic core residues. Biochemistry. 40:1257–1264. [DOI] [PubMed] [Google Scholar]
- Brodin, P., T. Grundstrom, T. Hofmann, T. Drakenberg, E. Thulin, and S. Forsen. 1986. Expression of bovine intestinal calcium binding protein from a synthetic gene in Escherichia coli and characterization of the product. Biochemistry. 25:5371–5377 [DOI] [PubMed] [Google Scholar]
- Carlström, G., and W. J. Chazin. 1993. Two-dimensional 1H nuclear magnetic resonance studies of the half-saturated (Ca2+)1 state of calbindin D9k. Further implications for the molecular basis of cooperative Ca2+ binding. J. Mol. Biol. 231:415–430. [DOI] [PubMed] [Google Scholar]
- Dahlke Ojennus, D., S. E. Lehto, and D. S. Wuttke. 2003. Electrostatic interactions in the reconstitution of an SH2 domain from constituent peptide fragments. Protein Sci. 12:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, B. E., J. Kordel, E. Thulin, P. Sellers, and S. Forsén. 1992. Dissection of calbindin D9k into two Ca2+-binding subdomains by a combination of mutagenesis and chemical cleavage. FEBS Lett. 298:211–214. [DOI] [PubMed] [Google Scholar]
- Grimsley, G. R., K. L. Shaw, L. R. Fee, R. W. Alston, B. M. Huyghues-Despointes, R. L. Thurlkill, J. M. Scholtz, and C. N. Pace. 1999. Increasing protein stability by altering long-range Coulombic interactions. Protein Sci. 8:1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson, M., A. Svensson, J. Fast, and S. Linse. 2001. An extended hydrophobic core induces EF-hand swapping. Protein Sci. 10:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendsch, Z. S., and B. Tidor. 1999. Electrostatic interactions in the GCN4 leucine zipper: substantial contributions arise from intramolecular interactions enhanced on binding. Protein Sci. 8:1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, C., P. Brodin, T. Grundström, E. Thulin, S. Forsén, and T. Drakenberg. 1990. Biophysical studies of engineered mutant proteins based on calbindin D9k modified in the pseudo EF-hand. Eur. J. Biochem. 187:455–460. [DOI] [PubMed] [Google Scholar]
- Johansson, C., P. Brodin, T. Grundström, S. Forsèn, and T. Drakenberg. 1991. Mutations of the pseudo-EF-hand of calbindin D9k into a normal EF-hand. Eur. J. Biochem. 202:1283–1290. [DOI] [PubMed] [Google Scholar]
- Johansson, C., M. Ullner, and T. Drakenberg. 1993. The solution structures of mutant calbindin D9k's, as determined by NMR, show that the calcium-binding site can adopt different folds. Biochemistry. 32:8429–8438. [PubMed] [Google Scholar]
- Julenius, K., J. Robblee, E. Thulin, B. E. Finn, R. Fairman, and S. Linse. 2002. Coupling of ligand binding and dimerization of helix-loop-helix peptides: spectroscopic and sedimentation analyses of calbindin D9k EF-hands. Proteins. 47:323–333. [DOI] [PubMed] [Google Scholar]
- Kesvatera, T., B. Jönsson, E. Thulin, and S. Linse. 1994. The binding of Ca2+ to calbindin D9k—structural stability and function at high salt concentration. Biochemistry. 33:14170–14176. [DOI] [PubMed] [Google Scholar]
- Kesvatera, T., B. Jönsson, E. Thulin, and S. Linse. 1996. Measurement and modelling of sequence-specific pKa values of lysines in calbindin D9k. J. Mol. Biol. 259:828–839. [DOI] [PubMed] [Google Scholar]
- Kesvatera, T., B. Jönsson, E. Thulin, G. Carlström, and S. Linse. 1999. Measurement and modelling of sequence-specific pKa values of acidic groups in calbindin D9k. Proteins. 37:106–115.10451554 [Google Scholar]
- Kesvatera, T., B. Jönsson, A. Telling, V. Tõugu, H. Vija, E. Thulin, and S. Linse. 2001a. Calbindin D9k—a protein optimized for Ca2+-binding at neutral pH. Biochemistry. 40:15334–15340. [DOI] [PubMed] [Google Scholar]
- Kesvatera, T., B. Jönsson, E. Thulin, and S. Linse. 2001b. Focusing of the electrostatic field at EF-hands of calbindin D9k. Proteins. 45:129–135. [DOI] [PubMed] [Google Scholar]
- Koradi, K., M. Billeter, and K. Wuthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55. [DOI] [PubMed] [Google Scholar]
- Linse, S., P. Brodin, C. Johansson, E. Thulin, T. Grundström, and S. Forsèn. 1988. The role of protein surface charges in ion binding. Nature. 335:651–652. [DOI] [PubMed] [Google Scholar]
- Linse, S., C. Johansson, P. Brodin, T. Grundström, T. Drakenberg, and S. Forsèn. 1991. Electrostatic contribution to the binding of calcium in calbindin D9k. Biochemistry. 30:154–162. [DOI] [PubMed] [Google Scholar]
- Linse, S., M. Voorhies, E. Norström, and D. A. Schultz. 2000. A phage display study of calmodulin subdomain pairing. J. Mol. Biol. 296:473–486. [DOI] [PubMed] [Google Scholar]
- Loladze, V. V., and G. I. Makhatadze. 2004. Removal of surface charge-charge interactions from ubiquitin leaves the protein folded and very stable. Protein Sci. 11:174–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque, I., and E. Freire. 1998. Structure-based prediction of binding affinities and molecular design of peptide ligands. Methods Enzymol. 295:100–127. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G. I., V. V. Loladze, A. V. Gribenko, and M. M. Lopez. 2004. Mechanisms for thermostabilization in a designed cold shock protein with optimized surface electrostatic interactions. J. Mol. Biol. 336:929–942. [DOI] [PubMed] [Google Scholar]
- Marti, D. N., and H. R. Bosshard. 2003. Electrostatic interactions in leucine zippers: thermodynamic analysis of the contributions of Glu and His residues and the effect of mutating salt bridges. J. Mol. Biol. 33:621–637. [DOI] [PubMed] [Google Scholar]
- Maune, J. F., C. B. Klee, and K. Beckingham. 1992. Ca2+ binding and conformational change in two series of point mutations to the individual Ca2+-binding sites of calmodulin. J. Biol. Chem. 267:5286–5295. [PubMed] [Google Scholar]
- Nakayama, S., N. D. Moncrief, and R. H. Kretsinger. 1992. Evolution of EF-hand calcium-modulated proteins. II. Domains of several subfamilies have diverse evolutionary histories. J. Mol. Evol. 34:416–448. [DOI] [PubMed] [Google Scholar]
- O'Shea, E. K., R. Rutkowski, and P. S. Kim. 1992. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell. 68:699–708. [DOI] [PubMed] [Google Scholar]
- Perl, D., U. Mueller, U. Heinemann, and F. X. Schmid. 2000. Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol. 7:380–383. [DOI] [PubMed] [Google Scholar]
- Schwehm, J. M., C. A. Fitch, B. N. Dang, E. B. Garcia-Moreno, and W. E. Stites. 2003. Changes in stability upon charge reversal and neutralization substitution in staphylococcal nuclease are dominated by favorable electrostatic effects. Biochemistry. 42:1118–1128. [DOI] [PubMed] [Google Scholar]
- Spector, S., M. Wang, S. A. Carp, J. Robblee, Z. S. Hendsch, R. Fairman, B. Tidor, and D. P. Raleigh. 2000. Rational modification of protein stability by the mutation of charged surface residues. Biochemistry. 39:872–879. [DOI] [PubMed] [Google Scholar]
- Stenberg, E., B. Persson, H. Roos, and C. Urbaniczky. 1991. Quantitative determination of surface concentrations of protein. J. Colloid Interface Sci. 143:513–526. [Google Scholar]
- Svensson, B., B. Jönsson, C. Woodward, and S. Linse. 1991. Ion binding properties of calbindin D9k—a Monte Carlo simulation study. Biochemistry. 30:5209–5217. [DOI] [PubMed] [Google Scholar]
- Svensson, L. A., E. Thulin, and S. Forsén. 1992. Proline cis-trans isomers in calbindin D9k observed by x-ray crystallography. J. Mol. Biol. 223:601–606. [DOI] [PubMed] [Google Scholar]