

Abstract
Assembly of the HIV-1 virus involves, in part, strong interactions between the capsid (CA) domains of the Gag polyprotein. During maturation, the core of HIV-1 virions undergoes profound morphological changes due primarily to proteolysis of the CA domain from other Gag domains which may allow for more efficient disassembly of the viral core in the early stages of infection. The host protein cyclophilin A (CypA), a cis-trans prolyl isomerase, in some way seems to assist in this assembly/disassembly process. Using an unproteolyzed construct of CA, we show that binding of CypA induces a large-scale conformational change in CA that is independent of its cis-trans prolyl isomerase activity. This change appears to be mediated by Cys-198 of CA since mutation to Ala renders CypA unable to induce this change and alters the kinetics and stability of protein cores that may ultimately result in inefficient disassembly of viral cores. Alternately, mutation of the second CA Cys (C218A) allows for CypA-induced conformational changes but alters the kinetics and morphology of the protein cores that may ultimately result in inefficient assembly of viral cores. These studies show the importance of the CA Cys residues in mediating the contacts needed for viral assembly and disassembly.
INTRODUCTION
Assembly of human immunodeficiency virus type 1 (HIV-1) in host cells involves the oligomerization of the structural precursor proteins, Pr55Gag and Pr160Gag-Pol, the binding of genomic RNA, the association of the complex with viral envelope glycoproteins on the plasma membrane, and budding and release of the assembled virus particle (Scarlata and Carter, 2003). The Pr55Gag precursor is sufficient by itself to direct membrane-binding, association with RNA, budding, and release. Expression of Gag or some of its constituent domains results in formation of particles that are morphologically very similar to the native immature virus (Gross et al., 1998; Campbell and Rein, 1999; Ehrlich et al., 2001). Gag oligomerization is mediated through several points in its matrix (MA), capsid (CA), and nucleocapsid (NC) domains (von Poblotzki et al., 1993; Mammano et al., 1994; Recin et al., 1995; Dorfman et al., 1997). Subsequent to assembly and release of the virion from host cells, Gag is proteolytically cleaved by the protease (PR) in Gag-Pol into the MA, CA, NC, and p6 products. This processing results in profound morphological rearrangements that change the spherical immature particle to a structure with a conical core composed entirely of the CA protein. The structural basis for this change is proposed to mechanistically result from cleavage at the N-terminus of the CA domain, releasing a Pro residue (Pro1) which subsequently forms a salt bridge with residue Asp-51 creating an intramolecular loop (Gamble et al., 1996; Gitti et al., 1996; Gross et al., 1998; von Schwedler et al., 1998). The mechanism through which this process alters CA-CA contacts, which ultimately result in a change in virus morphology, is not clear. It is clear that the strong CA interactions that occur in the context of Gag must be significantly weakened to allow for disassembly of the virus upon entry into host cells in the early stages of infection.
Besides providing structural integrity to HIV-1 virions, the CA domain in Gag is critical for the incorporation of the host protein cyclophilin A (CypA) into the virus. CypA is a cis-trans prolyl isomerase and in some types of cells, HIV-1 requires CypA for full infectivity, but other closely related viruses, such as SIV, do not (Braaten et al., 1996). CypA binds to a site in the CA domain, and interestingly, the requirement for cyclophilin can be transferred to other viruses by inserting the CypA-binding site into their CA domains (Bukovsky et al., 1997). The function of CypA in the HIV-1 life cycle is unclear. It has been suggested that CypA serves as a point of first contact between the virion and T cells that possess cyclophilin receptors on their membrane (Sherry et al., 1998; Saphire et al., 1999). However, other studies suggest a role for cyclophilin in post-entry events (Steinkasserer et al., 1995; Braaten et al., 1996; Ackerson et al., 1998). Under some conditions, CypA has been shown to protect HIV-1 from dominant host restriction factors that block infection by targeting incoming viral CA. In other cases CypA appears to be essential for host restriction (Towers et al., 2003). CypA also appears to be necessary for the functional expression of viral protein R (Vpr) (Zander et al., 2003). Vpr participates in nuclear localization of the viral preintegration complex and induces G2 cell-cycle arrest in infected, proliferating-T cells (Bukinsky and Adzhubei, 1999; Henklein et al., 2000). Also, since CypA is a cis-trans prolyl isomerase, it may affect viral assembly/dissembly through conformation changes that perturb diminish strong CA-CA interactions.
CypA binds to residues 87–92 in the HIV-1 CA domain of Gag (i.e., HAGPIA). The CA protein consists of two distinct predominantly α-helical domains linked by a short, unstructured region (Berthet-Colominas et al., 1999). The N-terminal domain, which is connected to the MA protein in the context of Gag, contains the CypA-binding site (Franke et al., 1994; Liang et al., 2003). The C-terminal domain, which is connected to the NC domain and p6 region in the context of Gag, plays an essential role in CA-CA interactions (Colgan et al., 1996; Kattendbeck et al., 1997; Lanman et al., 2002). Previous studies have shown that the binding of CypA to this site alters the structure in distal regions of the protein (Bosco et al., 2002), and we found that binding of CypA to the N-terminus of a immature form of capsid resulted in pronounced changes in the environment of a fluorescent probe covalently linked to one of the two Cys residues in the C-terminal domain (BonHomme et al., 2003).
In this study, we have used the changes in fluorescence of probes covalently attached to one of the two CA Cys residues as a read-out of the ability of CypA to modulate conformational changes in aminohexahistidine-tagged CA (His6-CA), which should mimic the immature form of the protein. To better understand the mechanism underlying the changes induced by CypA-binding, we mutated each of the Cys residues into Ala or Ser and studied the behavior of these mutants. The CA Cys residues lie close to each other, but do not form a disulfide linkage, as indicated by their accessibility to Cys-interactive probes (Colgan et al., 1996; McDermott et al., 1996). Single Cys mutants of capsid have been previously studied. Cellular studies have shown that mutation of Cys-198 interferes with viral particle disassembly, whereas mutation of Cys-218 blocks virus assembly (McDermott et al., 1996). Our studies show that CypA-binding induces conformational changes in the C-terminal domain of His6-CA oligomers which were mediated through Cys-198. CypA-binding may thus facilitate the transition of the virus to a mature, disassembly-competent form.
MATERIALS AND METHODS
Constructs
The HIV-1 CA sequence (derived from pBH10, GenBank accession number M15654) was subcloned into the pB6 vector, a derivative of pET 11d (Novagen, Madison, WI) that expresses aminohexahistidine-tagged proteins. Mutants of aminohexahistidine-tagged CA protein, abbreviated His6-CA, were made using Gene Editor In Vitro Site-Directed Mutagenesis System (Promega, Madison, WI). Insert regions were confirmed by DNA sequencing. Glutathione S-transferase-fused CypA, wild-type, and H54Q, was a gift from Dr. D. Braaten and Dr. J. Luban from the Dept. of Microbiology, College of Physicians and Surgeons of Columbia University, New York. Two antibodies were used in this study. The first, NEN-NEA 9306 (NEN-DuPont, DuPont, Wilmington, DE), is a monoclonal antibody that we found to binds to the region that encompasses amino acids 75–98 in the CA domain as shown in Fig. 1 a. The second is a monospecific antibody raised against a peptide whose sequence was residues 151–160 in the CA C-terminal domain shown in Fig. 1 a (International Enzymes, Fallbrook, CA). The amount of antibody used in this study was based on the supplier's recommendation and was adjusted to give similar potencies.
FIGURE 1.

Binding of a monoclonal Ab to the CypA-binding site causes changes in capsid C-terminal. (a) Mapping the antigenic site of monoclonal antibody NEN-NEA9306 and antibody CASI. Equal amounts of His-tagged full-length CA (lane 2) or mutant CA proteins containing ∼20 amino acid deletions (lanes 1, 3–7) were subjected to SDS-PAGE and analyzed by Western blotting with NEN-NEA9606. (b) Representative fluorescence-emission spectra of acrylodan-labeled His6-capsid (16 nM), where the probe is localized in the C-terminal domain, before and after the addition of a fourfold molar excess of a monoclonal antibody to the CypA-binding site in the N-terminal domain. Spectra for the NEN-NEA 9306 antibody studies were scanned from 385–600 nm and at a rate 2 nm/s whereas spectra using the Anti-CASI were scanned from 400–600nm at a rate of 1 nm/s to improve resolution. Data were corrected for background substituting unlabeled protein for acylodan His6-CA. Antibody concentrations were based on those recommended by the suppliers.
Recombinant protein expression and purification
His6-CA protein was expressed in Escherichia coli-strain BL21 (DE3) (Novagen) and purified by gravity chromatography using Ni-NTA Superflow agarose (Qiagen, Valencia, CA). His6-CA was eluted with 250 mM imidazole and the protein was found to be >95% pure by 15%-sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Coomassie staining. His6-CA was dialyzed into 30 mM MES (2-(N-morpholino)ethanesulfonic acid) pH 6, 1 mM EDTA, 1 mM DTT and 20% glycerol, and stored at −70°C.
Glutathione S-transferase-fused CypA was expressed in Escherichia coli-strain c600 and purified by gravity chromatography using Glutathione Sepharose 4B resin (Amersham Biosciences, Piscataway, NJ). The glutathione S-transferase moiety was cleaved using thrombin. CypA was found to be >95% pure by 15%-sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining. CypA was dialyzed into 50 mM Tris pH 8, 30 mM NaCl and 0.5 mM DTT. After dialysis, sodium azide (0.004%) was added to CypA and the protein was stored at 4°C.
Labeling with acrylodan
The Cys residues of His6-CA were modified using the thiol-reactive probe acrylodan (Molecular Probes). To avoid protein aggregation during the labeling procedure, His6-CA (0.13 μmoles) was immobilized on Ni-NTA agarose (500 μL, Qiagen) after dilution (10×) of the protein so that DTT, present in the storage buffer, would not reduce the nickel ions. The column was then washed with three-column volumes of 100 mM NaH2PO4 pH 7. Agarose-bound His6-CA (0.13 μmoles) was incubated with acrylodan (1.3 μmoles) in 100 mM NaH2PO4, pH 7, for 2 h at room temperature in a rotary shaker. The reaction was stopped by the addition of β-mercaptoethanol (13 μmoles). Agarose-bound His6-CA was washed three times with 10-column volumes of 100 mM NaH2PO4 pH 7 (5 mL), to remove excess acrylodan. Acrylodan-labeled His6-CA was eluted with two-column volumes of 500 mM imidazole in 50 mM NaH2PO4, pH 7. Unreacted acrylodan was removed using size exclusion chromatography (Bio-Gel P-2 Gel, Bio-Rad Laboratories, Hercules, CA) and 50 mM NaH2PO4, pH 7 as the eluant. Typically 40% to 80% of wild-type CA molecules were labeled with two acrylodan molecules. Protein concentration was determined using Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad Laboratories). The extent of labeling was estimated by absorption.
CypA was labeled with acrylodan by first dialyzing, three times for 20 min against 1000 volumes of 100 mM NaH2PO4 pH 7, to remove DTT. After, a 10-fold molar excess of acrylodan was added to CypA and the mixture was incubated for 2 h at room temperature in a rotary shaker. The rest of the procedure was the same as that followed for CA labeling.
Measurement of apparent-interaction constants and dissociation constants using fluorescence spectroscopy
All fluorescent measurements were made on an ISS PC1 photon counting spectrofluorometer (ISS, Champaign, IL) using quartz microcuvettes with a path length of 3 mm. An excitation wavelength was determined for each sample and was typically close to 368 nm. The emission wavelength was scanned from 400 nm to 600 nm. To determine apparent-interaction constants (KI, APP), acrylodan-labeled His6-CA (∼2 μM) was titrated with CypA (up to 3 μM) in 100 mM NaH2PO4 at either pH 8, 7, or 6. The control sample was acrylodan-labeled His6-CA (∼2 μM) titrated with an equivalent volume of buffer. To determine dissociation constants (KD) acrylodan-labeled CypA (0.020 μM or 0.2 μM) was titrated with His6-capsid (0.6 μM–3.8 μM) in 100 mM NaH2PO4 pH 8, 7, or 6. Control studies substituted buffer for His6-capsid. For studies that used cyclosporin A (CsA), CypA was preincubated with a fivefold molar excess of CsA for 1 h. Binding constants were calculated using either the running integral or the median of the emission spectra (see Data analysis).
Rate of core formation
To form cores, His6-CA (113 μM) was dialyzed three times against 1000 volumes of 50 mM Tris pH 8 and 1 M NaCl at 4°C for 1 h. Studies were performed by placing His6-CA in a 10 mm-quartz cuvette with constant stirring and the temperature set to 37°C. The rate of core formation was monitored at an angle of 90° to the incident light in an ISS spectrofluorometer with the excitation and emission wavelengths set at 350 nm. The rate of core formation was found by fitting the change in scattering intensity with time to a first order rate constant (see Data analysis).
Hydrostatic-pressure dissociation of cores as monitored by single-angle light scatter
Cores made of His6-CA protein (113 μM) were loaded into a silanized quartz cuvette topped with a collapsible-polyethylene lid for the transmission of pressure. The cuvette was placed into a high-pressure cell made of heat-treated Vascomax, which was connected to a pressure generator (High Pressure Equipment, Erie, PA) through a high-pressure line also made of Vascomax. Pressure was generated through the compression of 100% ethanol. The windows of the cell are made of sapphire to allow for light measurement. The pressure cell was placed into the sample chamber of the fluorometer. Single-angle light-scattering measurements were collected at an angle 90° of the incident light (350 nm). Pressure was increased in steps of 250 bars allowing 5 min for equilibrium to be reached.
Data analysis
To obtain apparent-interaction constants (KI, APP) and dissociation constants (KD), spectral parameters, such as the running integral or the median value of the emission peak, were calculated for experimental and control spectra. If the running integral was chosen, then the parameters were corrected for dilution. If the median value was chosen, then the wavelengths were converted to a unit of energy (e.g., wavenumbers). The control parameters were subtracted from the experimental parameters and the control-corrected parameters were normalized. The normalized-spectral parameters represent the degree of association.
We have viewed the interaction between His6-CA and CypA either directly by labeling CypA with a probe close to the capsid interaction site and obtaining an apparent dissociation constant, or indirectly by measuring the change of a fluorescence probe in the C-terminal domain of capsid when CypA binds to the N-terminal domain to obtain and apparent interaction constant.
We note that the fluorescence spectrum of the fluorophore used in these studies, acrylodan, was constant within error from pH 6 to8 as measured using a β-mercaptoethanol adduct.
Determination of KD, the dissociation constants
To obtain dissociation constants (KD values) for His6-CA-CypA-binding, CypA was labeled with acrylodan. CypA has four cysteines distributed throughout its structure. Cysteines 52, 62, 115, and 161 are located 14.8, 10.6, 7.1, and 19.6 Å, respectively, away from the CypA-binding site (Pro-90) in CA the protein based on the structure of Berthet-Colominas and co-workers (Protein Data Bank (PDB) 1E6J (Berthet-Colominas et al., 1999)). These distances were obtained using WebLab ViewerPro 3.7 (Accelrys, San Diego, CA). Use of acrylodan-labeled CypA allows monitoring of CA-CypA-binding directly as opposed to monitoring CypA-induced changes in the CA C-terminal domain. In this case, a bimolecular-dissociation model can be assumed because acrylodan-labeled CypA titrated with capsid behaves with 1:1 stoichiometry.
The data are presented as the change in fluorescence intensity of acrylodan-CypA as a function of total capsid concentration. We assume that all of the proteins are associated when the titration curve reaches saturation. From the change in intensity, we calculate the degree of protein association at each point along the titration curve (see BonHomme et al., 2003).
The reported Kd values were determined using
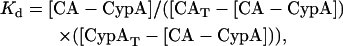 |
where the total concentration of the two proteins, given as subscript T, is known and the concentration of the CA-CypA complex was determined from the fraction associated multiplied by the concentration of total capsid. The Kd values were calculated of each experimental point on the titration curve where the fraction associated was between 0.1 and 0.9. The values for 3–6 titration studies were pooled and averaged and the standard error is given.
Determination of KI, APP, the apparent-interaction constant
To calculate apparent-interaction constants (KI, APP), acrylodan-labeled CypA concentrations at each titration point and the degree of association were fit to a following hyperbolic equation using SigmaPlot 2001 version 7.101. The reported apparent-interaction constant (KI, APP) and error represent the mean and mean ± SE of the individual experiments.
The CA protein exists as a tetramer or higher oligomer at pH 8 and pH 7, and as a monomer-dimer at pH 6 (Ehrlich et al., 2001). Distance measurements of the crystal structure using WebLabViewerPro 3.7 (Accelrys) indicate that the distance (63.5 Å) between the CypA-binding site and acrylodan does not allow for direct monitoring of binding (PDB 1E6J; Berthet-Colominas et al., 1999). For these reasons, any changes detected using this experimental design, resulting from a binding interaction, will most likely represent a change in conformation, a change in oligomerization state or a combination of these events. The exact nature of the change being monitored is unclear. However, because the change follows a rectangular hyperbolic, then this interaction may be thought to be specific to one site and the constant of the hyperbolic equation is defined as the apparent-interaction constant (KI, APP). The tiration curves present the change in fluorescence as a function of the amount of total protein added.
Determination of the rate constants
To obtain initial rates of core formation, the exponential-rise portion of the curves were fit to the following equation using SigmaPlot 2001 version 7.101,
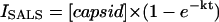 |
where ISALS is the intensity of single-angle light scatter, [capsid] is the His6-CA protein concentration, k is the constant of the equation in units of events per second, and t is time in seconds.
RESULTS
CypA induces conformational changes in the C-terminal domain of His6-CA
We previously demonstrated that the oligomerization state of CA increases from monomer-dimer at pH 6 to dimer-tetramer at higher pH values (BonHomme et al., 2003). We have also found that the binding of CypA to the N-terminal domain of CA produces large changes in the environment of Cys-reactive probes in the C-terminal domain, suggesting that CypA induces a conformational change (BonHomme et al., 2003). This conformational change does not occur at pH 6 but is apparent at pH 7 and 8. It is unclear whether this pH dependence results from the inability of CypA to bind to monomeric CA at pH 6 or whether CypA-binding is unable to transmit conformational changes to the C-terminal domain at the lower pH.
In initial studies, we used previously described His6-CA mutants that contained deletions throughout the protein (Dietrich et al., 2001) to map the antigenic site of several monoclonal antibodies. One of these, NEN-NEA 9306, recognized an antigenic site within the CypA interaction domain in the N-terminal domain of His6-CA. In Fig. 1 a, we show that NEN-NEA 9306 recognized WT His6-CA (lane 2) and all of the His6-CA deletion mutants (lanes 1, 3, 5, 6, and 7) except the mutant lacking residues 75–98, which includes H87AGPIA92 (lane 4). Interestingly, we found that addition of NEN-NEA 9306 to acrylodan-labeled His6-CA could confer changes in the C-terminal domain as visualized by changes in the fluorescence of the probe acrylodan covalently attached to one or both of the Cys residues located in the C-terminal domain (Fig. 1 b).
The effect of binding NEN-NEA 9306 was compared to that of a second antibody, anti-CASI (International Enzymes), which was raised against residues 151–160 in the CA C-terminal domain. In Fig. 1 a, we show that this antibody recognized WT His6-CA (lane 1) and all of the His6-CA deletion mutants (lanes 2–5 and 7) except the mutant lacking residues 152–176 (lane 6). In contrast to the results obtained with NEN-NEA 9306, no change was detected using the antibody against residues 151–160 (Fig. 1 b). These results suggest that perturbation of CA through interaction with the region encompassing the CypA-binding site induces conformational changes in the CA C-terminal domain.
To further characterize the ability of CypA to bind and transduce conformational changes in capsid, we compared the binding of acrylodan-labeled CypA to unlabeled His6-CA. Since this association is visualized directly by changes in acrylodan emission on the labeled CypA due to changes in the local dielectric environment as unlabeled His6-CA associates, we can follow the changes in fluorescence to obtain an apparent dissociation constant (KD). Alternately, we can follow the association indirectly by changes in the fluorescence of acrylodan-labeled His6-CA that result from conformation changes brought about by the binding to unlabeled CypA to a distal site on capsid to obtain an apparent interaction constant (KI, APP)] (see Methods).
In Fig. 2, we present results for KD and KI, APP evaluated at pH 8, 7, and 6. We note that the data in Fig. 2, a–c, have previously been reported (BonHomme et al., 2003). Although the data are identical to these previous results, they have been analyzed using a hyperbolic fit rather than a bimolecular association, which, as discussed in Methods, is inappropriate for this association. We present these data again to allow for easy comparison between the wild-type proteins and the mutants discussed below.
FIGURE 2.

Binding curves of His6-capsid with CypA as a function of pH. The upper series of curves correspond to the binding between the proteins as viewed indirectly through the changes in fluorescence of acrylodan-labeled His6-CA. Panels A, B, and C are studies done at pH 8, 7, and 6, respectively, and are identical to those previously reported (BonHomme et al., 2003) except those data were analyzed in terms of bimolecular associations, which does not appear to be appropriate (see Methods). Instead, we present the experimental data analyzed by apparent-interaction constants, KI, APP for association (see Methods), which were calculated from the running integral of the acrylodan-labeled His6-caspid (2000 nM) spectra as CypA is added (see BonHomme et al., 2003) where the x axis refers to the total [CypA]. The symbols represent the average of three individual experiments (•) and individual-data points (○). The error and error bars represent the mean ± SE. Panels D, E, and F correspond to binding studies after the changes in emission of 20 nM acrylodan-labeled CypA as unlabeled His6-capsid is added where the total His6-capsid is given. The corresponding apparent dissociation constants, KD, are given (see Methods). The data points are the running integral from the emission spectra of acrylodan-labeled CypA. The symbols represent the average of the experiments (•) and the individual trials (○). At pH 8 and 7, n = 3, and at pH 6, n = 6. Panel E also includes acrylodan-labeled CypA wild-type preincubated with cyclosporin A titrated with His6-capsid at pH 7. The symbols represent the average of two individual experiments (▴). The error and error bars represent the mean ± SE.
As shown in Fig. 2, weak binding was detected at pH 6 (panel F) and no change was detected in the CA C-terminal domain (panel C). In contrast, at pH 7, both binding constants were similar (40.1 ± 13 nM vs. 33.2 ± 4 nM, respectively, Fig. 2, E and B) suggesting that the binding of CypA to the CA N-terminal domain fully transduced the conformational change to the CA C-terminal domain. At pH 8, the observed KD was reduced, whereas the KI, APP was similar to that observed at pH 7 (Fig. 2, D and A). The reduced KD may be due to increased aggregation of His6-CA, as its concentration was increased during the course of the titration (see Ehrlich et al., 2001 ; BonHomme et al., 2003). In a control study, we preincubated CypA with an active site inhibitor cyclosporine A and found that both binding and changes in fluorescence-emission spectra were abolished. We note that the changes observed were not due to the effects of pH on acylodan, since the changes detected when the b-mercaptoethanol adduct of acrylodan was incubated with buffer alone was <10% (data not shown).
The fact that an antibody targeted to the CypA-binding site induced a conformational change in the CA C-terminal domain (Fig. 1) suggests that the CypA-induced change is the result of CypA-binding and not its cis-trans prolyl isomerization activity. To gain insight into the underlying basis of the change, we repeated the His6-CA association studies using an acrylodan CypA mutant, H54Q, whose prolyl isomerase activity is only 15% of wild-type (Zydowsky et al., 1992). In Fig. 3, we list the apparent-dissociation constants taken from the midpoint of the binding curve (KD, APP) at pH 8, 7, and 6. Interestingly, these data could not be fit to a bimolecular-dissociation constant using the total CypA H54Q concentration (200 nM). However, if we assume that only the active population of CypA H54Q (15% of the total concentration, or ∼30 nM) participates in binding, we can calculate a dissociation constant that is within error to that of the wild-type.
FIGURE 3.

Binding curves of His6-Capsid with CypA H54Q. Panels A, B, and C correspond to changes in the emission of acylodan-labeled His6-CA (2000 nM) at pH 8, 7, and 6 as the point mutant CypA54Q is added where the x axis refers to the total concentration of CypA54Q. The running integrals were used to determine the apparent-interaction constants, KI, APP. The symbols represent the average of three individual experiments (•) and individual experimental points (○). The error and error bars represent the mean and mean ± SE. Panels D, E, and F correspond to binding studies after the changes in emission of 20 nM acrylodan-labeled CypA H54Q as unlabeled His6-capsid is added where the x axis refers to total [His6-capsid]. The corresponding apparent dissociation constants, KD, are given (see Methods). The data points are the median of the running integral. The symbols represent the average of the experiments (•) and the individual trials (○). These experiments were performed three times each. The error represents the mean ± SE.
To determine the ability of CypA H54Q to induce conformational changes in His6-CA, KI, APP values were determined. Again, no change was detected at pH 6 and the KI, APP values obtained at pH 7 and 8 were fivefold lower than those obtained with wild-type CypA (compare Fig. 3, A and B, to Fig. 2, A and B). These results, together with those shown in Fig. 1, indicate that the conformational change in the CA C-terminal domain was not induced by CypA-mediated cis-trans prolyl isomerization.
CypA-binding affects the environment of capsid Cys-198
To determine which Cys residue of CA is most affected by CypA-binding, we constructed single Cys mutants of His6-CA. Since binding between His6-CA and CypA at pH 7 is strong and produces a conformational change in the C-terminal domain of CA protein, subsequent studies were performed at this pH. Protein-protein association studies were again measured by changes in acrylodan-CypA fluorescence as unlabeled His6-CA was added. As shown in Fig. 4, acrylodan-CypA binds to His6-CA C198A and C198S with similar affinities and those affinities were close to that of wild-type (37.7 ± 1.9 nM and 33.3 ± 3.3 nM, respectively, Fig. 4, D and E) as compared to 40.1 ± 13 nM (Fig. 2 E). Interestingly, however, CypA was not able to induce a change in the C-terminal domain of the Cys-198 His6-CA mutants (Fig. 4, A and B).
FIGURE 4.

Binding of the single Cys mutants C198A, C198S, and C218 A of His6-capsid with CypA. Panels A, B, and C show the change in emission of the acylodan-labeled His6-capsid point mutants, C198A-, C198S-, and C218A at 2200 nM as CypA is added where the x axis refers to the total [CypA]. The running integral of the spectra were used to determine the apparent-interaction constants, KI, APP (see Methods). The symbols represent the average of individual experiments (•) and individual-data points (○), where n = 3 for His6-CA C198A and C198S, and n = 4 for C218A. The error and error bars represent the mean ± SE. Panels D, E, and F correspond to binding studies after the changes in emission of 20 nM acrylodan–labeled CypA as the unlabeled His6-CA C198A (n = 3), C198S (n = 3) and C218A (n = 5) were added at pH 7. KD values were calculated as described in the Methods. The symbols represent the average of the experiments (•) and the individual trials (○). The errors and error bars represent the mean ± SE.
Similar studies were done measuring the binding affinity of His6-CA C218A to acrylodan-CypA. We found the affinity to be greatly reduced as compared to wild-type (Fig. 4 F). However, in contrast to the His6-CA Cys-198 mutants, His6-CA C218A was susceptible to the CypA-induced change in the C-terminal domain, giving an apparent KI, APP of 122.5 ± 53.1 nM (Fig. 4 C). The reduced KI, APP compared to that of the wild-type protein (KI, APP = 33.2 nM, Fig. 2 B) correlated with weaker binding of the mutant. Note that the change observed is a result of CypA having an effect on Cys-198, since only Cys-198 is acrylodan labeled in the mutant His6-CA C218A.
Mutants of His6-CA form cores more rapidly than wild-type
We monitored the rate of core formation of wild-type and mutants His6-CA proteins using single angle light scattering at 350 nm (see BonHomme et al., 2003). The kinetic profiles of the mutants were similar but differed significantly from wild-type (Fig. 5). His6-CA wild-type exhibited a relatively slow and continuous exponential rise (Fig. 5 D), whereas the His6-CA mutants displayed a much more rapid-exponential rise followed by a rapid loss of intensity (Fig. 5, A–C) (see Discussion).
FIGURE 5.

Kinetic profiles of His6-capsid core formation. Panels A, B, C, and D show the rate at which His6-capsid C198A, C198S, C218A form cores at 60 μM, 50 mM Tris pH 8, and 1 M NaCl, 37°C with constant stirring as monitored by single-angle light scattering.
We compared the initial rise of the wild-type and His6-CA mutants. We found that all mutants exhibited kinetic constants that were two orders of magnitude greater than wild-type. Interestingly, the Cys mutants all displayed similar core formation rates even through they result in disassembly- or assembly-impaired virions.
The morphology of His6-CA C198A and C218A mutants differ from wild-type
The large difference in the core formation rates between wild-type His6-CA and the Cys mutants prompted us to determine whether these proteins formed morphologically distinct complexes. The morphology of the resulting cores was investigated by electron microscopy (Fig. 6). His6-CA C198A (Fig. 6 A) forms spherical structures similar to the wild-type protein (BonHomme et al., 2003), although the cores formed by the mutant protein were much smaller in size. In contrast, His6-CA C218A formed mainly tubular structures in addition to small spherical structures (Fig. 6 B). We note that mature (i.e., untagged) CA has been shown to form tubular structures that are larger in size than the ones observed here (Gross et al., 1998; Ehrlich et al., 2001)
FIGURE 6.

Morphology of His6-capsid mutant cores as imaged by electron microscopy. Electron-microscopy images of His6-CA C198A (panel A) and C218A (panel B). His6-CA (113 μM) cores were deposited (5 μL) onto an electron-microscopy grid and stained with a saturated solution of uranyl acetate.
His6-CA C198A forms cores whose stability is concentration dependent
We tested the stability of the structures formed by wild-type and Cys mutated His6-CA using high pressure. In general, the application of hydrostatic pressure results in the dissociation of protein oligomers due to the penetration of solvent into the void volume in the subunit interfaces. The more extensive the subunit contacts, the smaller the void volume and the greater the pressure needed for dissociation (see Heremans, 1992).
Using single-angle light scattering, we monitored the dissociation of the CA proteins in real time as a function of hydrostatic pressure. We have previously shown that the application of hydrostatic pressure on His6-CA results in a concentration-dependent reduction in light scattering that is directly related to dissociation of virion cores (BonHomme et al., 2003). Here, we find that wild-type His6-CA and C218A at 113 μM showed significant dissociation in the first 1750 bars (42 ± 5% and 38 ± 2%, respectively, Fig. 7 A). Release of pressure allowed the values for the light scattering to return to within 10% of their original value. In contrast, the complexes of C198A His6-CA formed at the same concentration using were relatively insensitive to pressure dissociation; only 9 ± 2% of His6-capsid C198A dissociated in the same 1750 bar range. This result correlates well with previous studies indicating that viruses containing Cys-198 mutants are impaired in disassembly (McDermott et al., 1996).
FIGURE 7.

Core disassembly induced by high hydrostatic pressure. Hydrostatic pressure was used to disassemble cores made of His6-CA (113 μM) wild-type, C198A, and C218A. To obtain a range of core association, single-angle light scatter measurements of the proteins were taken before oligomerization and subtracted from measurements taken after oligomerization. The extent of disassembly was normalized within the range of core association. For His6-CA wild-type, the data points represent the average of three experiments whereas the others were performed five times. The error bars represent the mean ± SE.
DISCUSSION
The assembly of HIV-1 is due in part to strong interactions in the CA domain of Gag. During or after viral assembly and release from the host cell, the immature capsid undergoes drastic morphological changes induced by proteolytic maturation. These changes allow the virus to be effectively disassembled upon its entry into host cells in the early stage of infection. Here, we have linked two factors that have been implicated in the assembly/disassembly process: the host-protein CypA and the CA C-terminal domain Cys residues. We monitored changes in the C-terminal region of an immature form of CA through changes in the emission properties of the fluorescent probe, acrylodan, located in this domain, and have found that both Cys-198 and Cys-218 of CA play key roles in modulating the tertiary and quaternary structures needed for efficient assembly and disassembly of the virus. Although it is impossible to state unequivocally whether the spectral changes we are viewing are due to structural changes or conformational fluctuations, the extent of the changes in acrylodan intensity and energy suggests that binding of a molecule to the CypA-binding site stabilizes a set of CA conformations. Some of these proposed conformational changes appear to be mediated by CypA, but additional factors must be involved since addition of CypA did not detectably affect the dissociation of the wild-type immature CA in our previous studies (BonHomme et al., 2003).
An important role of the CA protein in the HIV-1 life cycle is to promote multimerization during assembly. We have previously found that oligomerization of CA in vitro can be induced by the combination of pH values above 6 and high ionic strength (Ehrlich et al., 2001). Additionally, the N-terminal domain of CA incorporates CypA into virions (Franke et al., 1994; Gamble et al., 1996). We have found that binding of CypA causes a change in the environment of an acrylodan probe covalently linked to the one of the two Cys in the C-terminal domain. Monitoring the association between CypA and His6-CA, either by viewing changes in a probe attached to CA or a probe attached to CypA, shows that binding is weaker to monomeric His6-CA (i.e., at pH 6) and that this binding does not transmit changes to the CA C-terminal domain. However, the binding of CypA to oligomerized His6-CA (i.e., > pH 6) allows for this change. Together with the observation that changes in the C-terminal domain on His6-CA can be induced by the binding of a monoclonal antibody to the N-terminal domain CypA-binding site, makes it unlikely that the effect of CypA on HIV-1 Gag is due to its cis-trans prolyl isomerase activity.
Binding of CypA to CA, and catalysis of Pro-90 isomerization, has been previously characterized using a number of biophysical methods including fluorescence, NMR, crystallographic, and theoretical (Endrich et al., 1996; Gamble et al., 1996; Bosco et al., 2002; BonHomme et al., 2003; Agarwal, 2004; Bosco and Kern, 2004). Taken together, these studies show that binding of CypA to the single site encompassing Pro-90 promotes cis/trans isomerization. However, our data suggests that Pro-90 isomerization may not play a role in the conformational changes in the C-terminal domain observed here since binding of antibody to this site also confers these changes (Fig. 1). It is possible, but unlikely, that the antibody binding accelerates prolyl isomerization. A comprehensive NMR study by Bosco and Kern (2004) shows that CypA binds to both mature and immature forms of CA and that binding and catalysis is not linked to maturational processing. Interestingly, these authors did not observe structural changes in the C-terminal domain upon CypA-binding, but noted that CypA may alter the structure of this region when CA is in an aggregated form. This idea is well supported by these studies showing that no changes in the C-terminal domain could be detected when CypA binds to CA at pH 6 where CA should be monomeric (Fig. 2).
We could only fit the CypA H54Q-CA titration curves if we assume that a fraction (i.e., 15%) of the CypA mutant is capable of binding, and the affinity turns out to be similar to wild-type. We could not fit the data by simply assuming that H54Q binds to CA with a weaker affinity. This result suggests that although the CypA H54Q mutant has mainly lost the ability to induce changes in the CA C-terminal domain, a small subpopulation of the mutant may retain the ability to bind immature and mature forms of CA. To understand how the CypA H54Q mutant can bind to capsid as tight or tighter than wild-type and still have a low level of isomerase activity, we propose reaction scheme based on previous work (Howard et al., 2003). Here, the peptide backbone loses double character due to formation of a hydrogen bond between the nitrogen (η1) on CypA's Arg55 and the nitrogen (α) on the CA Pro-90 residue (Howard et al., 2003). In the ground state, the distance between these groups ranges from 3.3 to 4.4 Å (Howard et al., 2003). Formation of this hydrogen bond stabilizes a pyramidal sp3-hybridization state for capsid's Pro-90 resulting in localized single-bond character. During the transition state, increasing single-bond character allows for both the lengthening and ultimately the free rotation of the bond between the CA Pro-90 nitrogen (α) and the neighboring carboxyl carbon. In addition, the hydrogen bond between the nitrogen (η1) on CypA's Arg-55 and the nitrogen (α) on CA Pro-90 is expected to shorten. It has been demonstrated that the two residues immediately N-terminal of Pro-90 in CA move from the cis to the trans position as a result of CypA catalysis, whereas residues C-terminal of Pro-90 do not move (Howard et al., 2003). Within the CypA hydrophobic-binding pocket, CA His-87 and CypA His-54 face each other (PDB 1AK4; Gamble et al., 1996) and possible charge repulsion between the histidine residues may hold the capsid residues N-terminal of Pro-90 far enough away to allow rotation of the nitrogen-carboxyl bond during catalysis. Mutation of CypA His-54 to the uncharged Gln would reduce the charge repulsion and result in steric hinderance and not allow the cis to trans rotation. This is supported by the difference in the pH trends of the measured dissociation constants between wild-type and mutant CypA.
We observed large differences in the rate of core formation between wild-type and single Cys capsid proteins. Although the wild-type protein shows a steady rise in single angle light scattering (Fig. 4) as expected, due to an increase in the number of cores formed, this rise was much more rapid for the mutants, and followed by a decrease. We note that under the conditions of our studies, the increase in scattering is due to an increase in the size and number of particles that are smaller than the wavelength used, whereas the decrease results from the particles becoming much larger than the incident wavelength. It is noteworthy that, unlike wild-type, the mutant assemblages become visible after 1 h (data not shown). The significance of these differences is unknown. Interestingly, even though His6-capsid Cys-198 aggregated rapidly to small spherical cores, these were not readily dissociated by pressure, indicating that the void volume between the subunits in these cores was lower than wild-type. Thus, the subunit packing in the cores made from His6-CA Cys-198 is tighter than wild-type giving a more extensive protein contact region. In contrast, His6-CA Cys-218A, which also rapidly aggregated, dissociated with a similar pressure profile as wild-type. Since our studies indicate that mutation of Cys-198 prevents the conformational changes induced by CypA, our results suggest that the conformational changes in CA induced by CypA-binding may facilitate disassembly by stabilizing a particular structural intermediate.
Almost identical binding and kinetic behavior is seen for both the C198A and the C198S mutants. The similar behavior of these two mutants suggests that differences in hydrogen bonding do not play a role in the Cys-198 function assayed in these experiments, and they alternately suggest that the differences lie in the replacement of sulfur from oxygen. We can speculate the basis for this idea. In viewing the crystal structure of the local area surrounding Cys-198 (PDB IE6J; Berthet-Colominas et al., 1999) we find that a likely candidate is the interaction of its sulfhydryl group with the oxygen of Gly-220 through rotation of the Cys-198 side chain, and it is noteworthy that Gly-220 plays a role in Gag assembly (Liang et al., 2003). In contrast to the Cys-198 mutants, the Cys-218 mutant has a greatly reduced affinity for CypA. However, this mutant allows for transmission of the signal induced by CypA-binding. This behavior correlates well with the observation that Cys-218 forms particles that resemble the mature (tubular) form of capsid, and suggests that transmission of the signal facilitates formation of weaker, disassembly-competent particles (Ehrlich et al., 2001).
In summary, the results of this study show that the CA C-terminal domain is structurally altered upon occupation of CypA-binding site in the N-terminus. Further structural studies are required to understand the nature of this conformational alteration and to define the residues that mediate this change.
Acknowledgments
The authors thank Tracy LaGrassa for carrying out the antibody studies, and Paxton Provitera, Louisa Dowal, and Finly Philip for their helpful discussions.
This work was supported by National Institutes of Health grants GM58271 (S.S. and C.C.) and GM48294 (C.C.) M.B. was supported in part by the W. Burghardt Turner Fellowship program.
References
- Ackerson, B., O. Rey, J. Canon, and P. Krogstad. 1998. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. J. Virol. 72:303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal, P. K. 2004. Cis/trans isomerization of HIV-1 capsid protein catalyzed by cyclophilin A: insights from computational and theoretical studies. Proteins. 56:449–463. [DOI] [PubMed] [Google Scholar]
- Berthet-Colominas, C., S. Monaco, A. Novelli, G. Sibai, F. Mallet, and S. Cusack. 1999. Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 18:1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BonHomme, M., S. Wong, C. Carter, and S. Scarlata. 2003. The pH dependence of HIV-1 capsid assembly and its interaction with cyclophilin A. Biophys. Chem. 105:67–77. [DOI] [PubMed] [Google Scholar]
- Bosco, D., E. Eisenmesser, S. Pochapsky, W. Sundquist, and D. Kern. 2002. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. USA. 99:5247–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco, D., and D. Kern. 2004. Catalysis and binding of cyclophilin A with different HIV-1 capsid constructs. Biochemistry. 43:6110–6119. [DOI] [PubMed] [Google Scholar]
- Braaten, D., E. Franke, and J. Luban. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 70:3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukinsky, M., and A. Adzhubei. 1999. Viral protein R of HIV-1. Rev. Med. Virol. 8:39–49. [DOI] [PubMed] [Google Scholar]
- Bukovsky, A. A., A. Weimann, M. A. Accola, and H. G. Gottlinger. 1997. Transfer of the HIV-1 cyclophilin-binding site to simian immunodeficiency virus from Macaca mulatta can confer both cyclosporin sensitivity and cyclosporin dependence. Proc. Natl. Acad. Sci. USA. 94:10943–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, S., and A. Rein. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 73:2270–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan, J., H. E. Yuan, E. K. Franke, and J. Luban. 1996. Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerization. J. Virol. 70:4299–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, L., L. Ehrlich, T. LaGrassa, D. Ebbets-Reed, and C. Carter. 2001. Structural consequences of cyclophilin A binding on maturational refolding in human immunodfeiciency virus type 1 capsid protein. J. Virol. 75:4721–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman, T., A. Weimann, A. Borsetti, C. T. Walsh, and H. G. Gottlinger. 1997. Active-site residues of cyclophilin A are crucial for its incorporation into human immunodeficiency virus type 1 virions. J. Virol. 71:7110–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, L. S., T. Liu, S. Scarlata, B. Chu, and C. A. Carter. 2001. HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro: structure switching by pH-induced conformational changes. Biophys. J. 81:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrich, M., P. Gehrig, and H. Gehring. 1996. Maturation-induced conformational changes of HIV-1 capsid protein and identification of two high affinity sites for cyclophilin in the C-terminal domain. J. Biol. Chem. 274:5326–5332. [DOI] [PubMed] [Google Scholar]
- Franke, E. K., H. E. Yuan, and J. Luban. 1994. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 372:359–362. [DOI] [PubMed] [Google Scholar]
- Gamble, T. R., F. F. Vajdos, S. Yoo, D. K. Worthylake, M. Houseweart, W. I. Sundquist, and C. P. Hill. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 87:1285–1294. [DOI] [PubMed] [Google Scholar]
- Gitti, R., B. Lee, J. Walker, M. K. Summers, S. Yoo, and W. Sundquist. 1996. Structure of the N-terminal core domain of the HIV-1 capsid protein. Science. 273:231–235. [DOI] [PubMed] [Google Scholar]
- Gross, I., H. Hohenberg, C. Huckhagel, and H.-G. Krausslich. 1998. N-terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J. Virol. 72:4798–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henklein, P., K. Bruns, M. P. Sherman, U. Tessmer, K. Licha, J. Kopp, C. M. de Noronha, W. C. Greene, V. Wray, and U. Schubert. 2000. Functional and structural characterization of synthetic HIV-1 vpr that transduces cells, localizes to the nucleus, and induces G2 cell cycle arrest. J. Biol. Chem. 275:32016–32026. [DOI] [PubMed] [Google Scholar]
- Heremans, K. A. H. 1992. High pressure effects upon proteins and other biomolecules. Annu. Rev. Biophys. Bioeng. 11:1–21. [DOI] [PubMed] [Google Scholar]
- Howard, B. R., F. F. Vajdos, S. Li, W. I. Sundquist, and C. P. Hill. 2003. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 475–481. [DOI] [PubMed]
- Kattendbeck, B., A. von Poblotzki, A. Rohrhofer, H. Wolf, and S. Modrow. 1997. Inhibition of human immunodeficiency virus type 1 particle formation by alteration of defined amino acids within the C-terminus of capsid. J. Gener. Virol. 78:2489–2496. [DOI] [PubMed] [Google Scholar]
- Lanman, J., J. Sexton, M. Sakalian, and P. E. Prevelige Jr. 2002. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 76:6900–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, C., J. Hu, J. B. Whitney, L. Kleiman, and M. A. Wainberg. 2003. A structurally disordered region at the C terminus of capsid plays essential roles in multimerization and membrane binding of the Gag protein of human immunodeficiency virus type 1. J. Virol. 77:1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammano, F., A. Ohagen, S. Hoglund, and H. Gottlinger. 1994. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J. Virol. 68:4927–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott, G., L. Farrell, R. Ross, and E. Barklise. 1996. Structural analysis of human immunodeficiency virus type 1 Gag protein interactions using cysteine-specific reagents. J. Virol. 70:5106–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recin, A. S., S. R. Paik, R. D. Berkowitz, J. Luban, I. Lowy, and S. P. Goff. 1995. Linker insertion mutation in the human immunodeficeincy virus type 1 gag gene: effects on virion particle assembly, release and infectivity. J. Virol. 69:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphire, A., M. Bobardt, and P. Gallay. 1999. Host cyclophilin A mediates HIV-1 attachment to target cells via heparans. EMBO J. 18:6771–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlata, S., and C. Carter. 2003. Role of HIV-1 gag domains in viral assembly. Biochim. Biophys. Acta. 1614:62–72. [DOI] [PubMed] [Google Scholar]
- Sherry, B., G. Zybarth, M. Alfano, L. Dubrobsky, R. Mitchell, D. Rich, P. Ulrich, R. Bucala, A. Cerami, and M. Bukrinsky. 1998. Role of cyclophilin A in the uptake of HIV-1 by macrophages and T lymphocytes. Proc. Natl. Acad. Sci. USA. 95:1758–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkasserer, A., R. Harrison, A. Billich, F. Hammerschmid, G. Werner, B. Wolff, P. Peichl, G. Palfi, W. Schnitzel, E. Mlynar, and others. 1995. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus type 1 (HIV-1): interference with early and late events in HIV-1 replication. J. Virol. 69:814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers, G. J., T. Hatziioannou, S. Cowan, S. P. Goff, J. Luban, and P. D. Bieniasz. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138–1143. [DOI] [PubMed] [Google Scholar]
- von Poblotzki, A., R. Wagner, M. Niedrig, G. Wanner, H. Wolf, and S. Modrow. 1993. Identification of a region of Pr55gag-polyprotein essential for HIV-1 particle formation. Virology. 193:981–985. [DOI] [PubMed] [Google Scholar]
- von Schwedler, U., T. Stemmler, V. Klishko, S. Li, K. Albertine, D. Davis, and W. Sundquist. 1998. Proteolytic refolding of the HIV-1 capsid protein amino -terminus facilitates viral core assembly. EMBO J. 17:1555–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander, K., M. P. Sherman, U. Tessmer, K. Bruns, V. Wray, A. T. Prechtel, E. Schubert, P. Henklein, J. Luban, J. Neidleman, W. C. Greene, and U. Schubert. 2003. Cyclophilin A interacts with HIV-1 Vpr and is required for its functional expression. J. Biol. Chem. 278:43202–43213. [DOI] [PubMed] [Google Scholar]
- Zydowsky, L., F. Etzkorn, H. Y. Change, S. B. Ferguson, L. A. Stolz, S. I. Ho, and C. T. Walsh. 1992. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]