

Abstract
Site-directed spin labeling EPR (SDSL-EPR) was used to determine the structure of the inhibitory region of TnI in the intact cardiac troponin ternary complex. Maeda and collaborators have modeled the inhibitory region of TnI (skeletal 96–112: the structural motif that communicates the Ca2+ signal to actin) as a kinked α-helix [Vassylyev, D., Takeda, S., Wakatsuki, S., Maeda, K. & Maeda, Y. (1998) Proc. Natl. Acad. Sci. USA 95, 4847–4852), whereas Trewhella and collaborators have proposed the same region to be a flexible β-hairpin [Tung, C. S., Wall, M. E., Gallagher, S. C. & Trewhella, J. (2000) Protein Sci. 9, 1312–1326]. To distinguish between the two models, residues 129–145 of cardiac TnI were mutated sequentially to cysteines and labeled with the extrinsic spin probe, MTSSL. Sequence-dependent solvent accessibility was measured as a change in power saturation of the spin probe in the presence of the relaxation agent. In the ternary complex, the 129–137 region followed a pattern characteristic of a regular 3.6 residues/turn α-helix. The following region, residues 138–145, showed no regular pattern in solvent accessibility. Measurements of 4 intradomain distances within the inhibitory sequence, using dipolar EPR, were consistent with an α-helical structure. The difference in side-chain mobility between the ternary (C⋅I⋅T) and binary (C⋅I) complexes revealed a region of interaction of TnT located at the N-terminal end of the inhibitory sequence, residues 130–135. The above findings for the troponin complex in solution do not support either of the computational models of the binary complex; however, they are in very good agreement with a preliminary report of the x-ray structure of the cardiac ternary complex [Takeda, S. Yamashita, A., Maeda, K. & Maeda, Y. (2002) Biophys. J. 82, 832].
Keywords: troponin I‖spin labels‖Fourier transform electron paramagnetic resonance‖DEER‖dipolar
Regulation of striated muscle contraction is associated with Ca2+-dependent structural transitions in the muscle thin filament, which is composed of the troponin complex, tropomyosin, and actin. Troponin is composed of three components: TnC, which binds Ca2+; TnI, which inhibits actomyosin activity; and TnT, which anchors TnC and TnI to tropomyosin. Muscle contraction is initiated by the binding of Ca2+ to the N-lobe regulatory sites of TnC. The N lobe then undergoes a structural transition from a closed to an open form, which then facilitates the release of the inhibitory region of TnI from actin, binding to TnC and stimulation of the actomyosin ATPase. The mechanism of this signaling pathway is still tentative (for review, see ref. 1).
Crystal and NMR structures are available for TnC and several complexes of TnC with TnI fragments. The crystal structure of TnC reveals a dumbbell-shaped protein consisting of two globular domains, joined by a 22-residue central α-helix (2–4). Each domain contains a hydrophobic cleft and two helix-loop-helix EF-hand metal binding motifs; two high-affinity Ca2+/Mg2+ sites in the C-terminal domain (sites III and IV) and two low-affinity Ca2+ specific sites in the N-terminal domain (sites I and II). In skeletal TnC, sites III and IV are permanently occupied by Mg2+ and facilitate the structural binding of TnC to the contractile apparatus (5). Binding of Ca2+ to sites I and II is the physiological trigger for muscle contraction. In cardiac TnC, binding site I is inactive and Ca2+ binding to site II does not induce as large a structural change as observed in skeletal TnC (6). TnI is capable of inhibiting actomyosin ATPase in the absence of other subunits, but Ca2+-dependent regulation requires TnC, TnT, and tropomyosin. The inhibitory region (skTnI 96–117) alone can fully inhibit actomyosin ATPase activity (7) possibly by binding either to actin or to TnC in the “on” or “off” states (8–10). The corresponding residues for the cardiac inhibitory region are 129–150 because of a unique ≈32 residue N-terminal extension of cTnI.
Structural information for the intact troponin complex is limited to low-resolution neutron diffraction and electron microscopy studies (11–13), though several high-resolution structures of TnC with bound TnI peptides are available. Two computational models have been recently proposed for the binary complex of TnC and TnI (14, 15). Both models have TnI and TnC in an antiparallel arrangement with multiple interaction sites between the two subunits. NMR, crystallography, and neutron scattering was used by Tung et al. (15) to develop a computational model of the binary complex in which TnI winds around TnC in either a left-handed manner (model “L”) or a right-handed manner (model “R”). In both structures, the inhibitory region of TnI is modeled as a flexible β-hairpin in close proximity to the central helix of TnC. In contrast, Maeda and coworkers (14) modeled the inhibitory region as a kinked α-helical peptide by homology building based on the skeletal crystal structure of the N terminus peptide fragment of TnI (skTnI 1–47) complexed with TnC.
In this work, we used site-directed spin labeling and EPR (SDSL-EPR) spectroscopy to determine the structure of the inhibitory region. Specifically, we have used a combination of accessibility to a spin relaxation agent; spin label mobility profiles, and dipolar distance measurements within the inhibitory region of TnI in the binary and ternary complexes. Our data provide insights into the structure of the inhibitory region and reveal the interface between TnI and TnT. Cysteine scanning of cardiac residues 129–145 in the binary (C⋅I) complex revealed no trend in either spin label mobility or accessibility, suggesting that the TnI inhibitory peptide is either unstructured or equally solvated in the binary complex. In the ternary complex (C⋅I⋅T), mobility and accessibility profiles are consistent with residues 129–137 being an α-helix and residues 138–145 being unstructured. Distance measurements between the TnI inhibitory peptide and the central helix of TnC suggest that the two domains are further apart than in the published models. Lastly, changes in spin label mobility on formation of the ternary complex revealed that residues 130–135 of the inhibitory region interact with TnT.
Methods
Crude cardiac troponin was extracted from an ether powder prepared from the left ventricle of bovine hearts (Pel-Freez Biologicals) and cTnT separated from the other two troponin subunits as described in ref. 16. pET-3d expression vectors (Novagen) containing mouse cardiac TnI, chicken slow TnC, and three single cysteine mutants (TnI132, TnI149, and TnC89) were kindly provided by the laboratory of Herbert C. Cheung. Details of these clones have been described elsewhere (17–19). The cardiac nomenclature for the TnC and TnI mutants is specified according to GenBank accession numbers A27204 (20) and AAA16157 (21), respectively. Further cTnI and cTnC mutants were constructed with the Quick-Change site-directed mutagenesis kit (Stratagene) using complementary oligonucleotides containing the desired mutations into the cysteine-less background (obtained by mutating native cysteines to C35S/C84S for cTnC and C80I/C97Sfor cTnI). Eighteen single cysteine mutants of cTnI at positions 129–145 inclusive and 151, and four double cysteine mutants at positions 134/138, 145/149, 140/145, and 141/145 were generated in this fashion. Likewise, the cTnC cys-less clone was used to generate a single cysteine mutant at position 94. Sequences of all mutant clones were verified by DNA sequencing (22). The DNA was transformed into Escherichia coli BL21 (DE3) cells grown in Terrific Broth media (Difco). The recombinant proteins were isolated and purified according to established protocols (17, 19, 23).
Purified protein was labeled with MTSSL (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-(methyl)methanethiosulfonate spin label, gift of Kalman Hideg). The efficiency of labeling was found to be greater than 75% for single cTnI mutants and between 60–80% for the double mutants (also confirmed by analysis of the EPR dipolar broadening function).
Reconstitution of the binary or ternary troponin complexes was performed by incubation of the proteins in 6 M urea and 0.6 M KCl, followed by renaturation by dialysis to remove the urea and to lower the [KCl] to 0.1 M to precipitate excess TnI and TnT (24). The reconstitution all EPR samples was checked by SDS/PAGE and urea gel electrophoresis (25). All measurements were performed in 0.2 M KCl/1 mM EDTA/2 mM CaCl2/3 mM MgCl2/50 mM Mops, pH 7.2. FURA-6F fluorescence calibrated with Calcium Kit II (Molecular Probes) estimated the concentration of free Ca2+ to be 0.35 mM in the presence of Ni(ll)Ethylenediaminediacetate (NiEDDA), which was used as a relaxant. The ternary complex of labeled TnI-133 and TnI-138 was assayed for the regulation of in vitro translation of the regulated thin filaments (26). The reconstituted filaments were fully regulated: TnI inhibited movement in the absence of Ca2+ and promoted the rapid movement of virtually all filaments in the presence of Ca2+, thus suggesting no functional impairment caused by the mutation and labeling of these sites.
EPR spectra were recorded on a Bruker EMX 9 GHz spectrometer at 20°C. Samples placed in polyethylene tubing were purged with N2 gas to remove oxygen. Nitroxide accessibility to the water-soluble relaxant, NiEDDA, was measured using the power-saturation technique described previously (27). The power saturation profiles were least-squared fitted, using the Simplex algorithm to determine P1/2 values, a value of the microwave power at which the signal is half saturated. The concentration dependence of the relaxation enhancement was linear in the range of 0.5–27 mM NiEDDA.
The helical periodicity of the structure (αPI index) was evaluated as the contribution of the components between 80o and 120o of the power Fourier transform (P(ω)) of the accessibility data. The αPI index was evaluated over a seven-residue window (28).
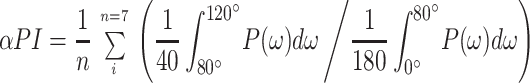 |
Spin–spin distances were obtained from EPR dipolar interactions of the doubly labeled samples. The spectra were obtained in frozen solutions (180 K) to eliminate motional averaging. Spectral broadening of the double labeled samples, compared with the two single labeled composite spectra, was analyzed using the Fourier deconvolution (29) as implemented in a matlab program provided by Y. K. Shin. The method has been shown to provide for the accurate determination of distances in the range of 8–20 Å and can account for the presence of up to as much as 40–60% of the singly labeled species (30). The singly labeled component produces a baseline offset in Fourier space that does not affect the dipolar distance estimate. All spectra were collected using a microwave power of 10 mW, modulation amplitude of 1 G and a scan range of 180 G.
Double electron–electron resonance was performed on Bruker 680 Fourier transform spectrometer using a 4 pulse sequence (90°-τ1-180°-τ2-180°) at a frequency corresponding to the high-field turning point and a strong 1-kW pumping pulse exciting the spins in the central nuclear manifold (31). The modulation of the echo amplitude as a function of the position of the pumping pulse between 180° pulses arises from the spin–spin interactions. The time domain signal was transformed into the frequency domain and the separation of the singularities of the Pake pattern was used to calculate the interspin distance.
Molecular modeling was performed using the skeletal binary model structures of C⋅I (pdb 1EW7) for the two β-hairpin models; and for the helical model, the coordinates were kindly supplied by Y. Maeda. Both skeletal isoform models were subsequently homology modified (Insight II, Accelrys, Inc. San Diego, CA) to obtain the cardiac isoform corresponding to the experimental system. The structures were energy minimized in charmm using the CHARMM19 force field and used as the starting structures for modification with spin labels. A Monte Carlo routine implemented in charmm was used to search the conformational space of the spin label at each labeled site (32). The Lee and Richards rolling probe method implemented in the program surfcv (33) was used to predict side chain solvent accessibility.
Results
Solvent Relaxation.
Binary complex.
The formation of all binary complexes of C⋅I were confirmed by urea gel electrophoresis of the reconstituted samples, Fig. 1. In the presence of Ca2+, the C⋅I complex migrated more slowly than TnC, and there was no evidence of uncomplexed TnC. Additionally, because the uncomplexed TnI does not enter the gel, we used dynamic light scattering to exclude the possibility of TnI aggregates.
Figure 1.

Complex formation of the troponin subunits. Urea gels in the presence of Ca2+ of three MTSSL labeled TnI and TnC mutant binary complexs (lanes 3–5). Lane 1 contains TnC and Lane 2 contains TnI that did not enter the gel.
The EPR experiments on the binary complexes showed that neither the solvent accessibility of MTSSL, nor the mobility of the label, exhibited any discernible trend between residues 129 and 145, Fig. 2. The periodicity index, αPI, estimated for the 7-residue stretch beginning at residues 129–131 is 0.6, characteristic of a nonhelical conformation (index of >2 is diagnostic for α-helices) (28). The possible explanations are (i) disordering of the inhibitory domain with no defined secondary structure; or (ii) absence of protein–protein interactions between the inhibitory region of TnI with TnC, which would provide unequal TnI solvation. Neither explanation is compatible with the helical and β-hairpin models for the binary complex.
Figure 2.

Profile of the solvent accessibility in the inhibitory region of TnI in the binary complex as measured by the difference in half-saturation powers (●) and the probe mobility (rotational correlation time) (▴).
Ternary complex.
The situation was dramatically different in the ternary complex. Solvent accessibility followed an α-helical pattern between residues 129 and 137. implying that in the ternary complex, these residues have a well defined secondary structure and interact with another subunit, which shields the spin label from the solvent, Fig. 3. The pattern of shielding was estimated from the helical model of Vassylyev et al. (14) and the β-hairpin model of Tung et al. (15) as the surface accessible area for MTSSL labels built in as side chains at the mutated sites. The experimentally observed pattern closely follows a helical model, and the αPI index is 2.1 as expected for an α-helix. The pattern is not compatible at all with the β-hairpin model (data not shown).
Figure 3.

Accessibility profile of the inhibitory region in the ternary troponin complex (●). (A) Residues 129–137 display a profile consistent with α-helix (gray line); ▵ represent solvent accessibility area calculated from the helical model. (B) Residues 139–145 do not show any discernable trend consistent with an organized and regular secondary structure.
Further along the sequence, the region comprising residues 139–145 shows no discernible trend in the accessibility (αPI = 0.6). This region is either disordered or equally solvated on all sides with no interactions with either TnC or TnT.
Dipolar Interactions.
Intersubunit distances.
To identify whether the component shielding TnI is TnC (as computational models would have it) or TnT (explaining the absence of the solvent shielding in the binary complex I⋅C) we have measured dipolar interactions in the binary complex between two sites on the central helix of TnC (89 and 94) and a total of six sites on TnI (133, 135, 137, 141, 143, and 151). The spin label sites were selected to distinguish between helical and β-hairpin models. Comparison of the double spectrum with its corresponding composite single labeled spectrum, revealed no evidence of spectral line broadening for any of the doubly labeled samples, suggesting that all of the distances are longer than 18–20 Å, the limit of the dipolar EPR method (Table 1). This result is not affected by the substochiometric labeling, as >75% of the signal originates from the doubly labeled sample, and 0.5 G broadening, if present, would be readily observed. Furthermore, 4 pulse Fourier transform EPR double electron electron resonance (DEER) experiments, that are not affected by the presence of the singly labeled species, gave a distance of 30 Å (I142-C94) or showed no modulation as expected for spin–spin distances less than 40 Å. It is important to note that the ternary complexes did form as shown by protein electrophoresis and dynamic light scattering (data not shown). The absence of the dipolar interactions between the central helix of TnC and TnI suggests that the inhibitory region of TnI is not making an extended contact with the central helix of TnC as originally proposed.
Table 1.
Distances between TnC and TnI determined by dipolar EPR in the binary complex
| Cardiac residue | Distances
predicted from models, Å*
|
Dipolar
distance, Å
|
|||
|---|---|---|---|---|---|
| Helical | β-Hairpin L | β-Hairpin R | c.w. EPR | DEER | |
| TnI133–TnC94 | 4 | 12–15 | 11–14 | >20 | |
| TnI135–TnC94 | 12 | 10–15 | 9–16 | >20 | >40 |
| TnI142–TnC94 | 10–17 | 9–13 | 2–6 | >20 | 30 |
| TnI137–TnC89 | 12–15 | >20 | >20 | >20 | |
| TnI141–TnC89 | 15–17 | >20 | >20 | >20 | |
| TnI143–TnC89 | 12–16 | 12–16 | >20 | >20 | |
| TnI151–TnC89 | 7–9 | 8–12 | 16–18 | >20 | >40 |
Interspin distances predicted from the molecular models by Metropolis Monte Carlo minimization. c.w., continuous wave EPR; DEER, double electron electron resonance.
Intrasubunit distances.
We also measured distances within the TnI inhibitory region for four double labeled cysteine mutants: 134/138, 140/145, 141/145, and 145/149 in the binary complexes. These spin label positions were chosen to distinguish between the α-helical model and the β-hairpin model.
The observed interspin distances, calculated from the dipolar broadening function, for the first three pairs were 12–13 Å with a slightly longer distance for the fourth pair of sites, 15–18 Å. Comparison with the models was accomplished at the MTSSL-modified side chain level rather than at the Cα–Cα level. To fully account for spin label size and orientation with respect to the protein, we modeled the probes using molecular dynamics. Minimum energy structures were first found by Monte Carlo methods (32) followed by molecular dynamics simulations of the doubly labeled model. The distance between the nitroxide nitrogen atoms was then calculated for all conformers of the two spin labels. For three of the probe pairs, the distances from the helical model were within 1–2 Å of the experimental values, whereas the 140–145 distance calculated for the β-hairpin model differed by 10 Å, Table 2. Thus, this region is most likely an α-helix. The 145–149 distance is somewhat longer than calculated for either model, but it is still closer to the α-helical structure than the β-hairpin.
Table 2.
Spin–spin distances within the inhibitory region of TnI as determined by dipolar EPR in the binary complex
| TnI cardiac residues | α-Helix, Å* | β-Hairpin, Å* | Observed distance, Å |
|---|---|---|---|
| 134/138 | 8–11 | 15–19 | 12–13 |
| 145/149 | 12–14 | 10–14 | 15–18 |
| 140/145 | 10–18 | 23 | 12–13 |
| 141/145 | 10–13 | 15–18 | 12–13 |
Spin–spin distances predicted by the molecular dynamics simulations using models of polypeptide chains with the given residue mutated to a spin labeled cysteine. The helix model was built as an ideal α-helix from the cardiac TnI inhibitory region sequence. The β-strand was extracted from PDB ID 1EW7.
Mobility.
Spin label mobility can be used as a footprint of the side chain steric restraint identifying residues involved in tertiary contacts (34). Correlation times were determined by full line-shape analysis (35) of EPR spectra. Although the determination of the absolute values of the rate and amplitude of motion would require measurements at multiple microwave frequencies, the ratio of the correlation times in the presence and absence of interacting protein components reflects changes in the steric constraints. The changes in the rotational correlation times on TnT inclusion were mapped onto the surface of the helical model, Fig. 4. The N-terminal region of the inhibitory region showed the largest change on the inclusion of TnT, with the correlation time increasing 5- to 200-fold (yellow residues in Fig. 4). We interpret the immobilization of these residues as a tertiary contact formed at the interface between TnI and TnT. A more complex scenario would have the binding of TnT propagating along the polypeptide chain and increasing the interaction between TnI and TnC. Further up the sequence, residues 138–144 were nearly as mobile in the ternary complex as in the binary. Their correlation times of ≈5 ns were characteristic of the MTSSL spin probe moving unhindered on the surface of a protein (34), consistent with the observed high solvent accessibility for these residues. The exceptions were residues 137 and 145, for which mobility increased in the ternary complex. These residues were involved in the binary interface with TnC, which is modified in the ternary complex.
Figure 4.

Footprint of TnT visualized as a difference in mobility log(τICT/τIC) between the ternary and binary complexes. Surfaces were colored according to a difference gradient, gray for no change to yellow denoting a 200-fold increase of correlation time.
Discussion
The aim of this work was to determine the secondary and tertiary structure of the inhibitory region of TnI in a complex with TnT and TnC by SDSL-EPR. Cysteine scanning of cardiac TnI residues 129–145 in the binary complex revealed no regular modulation of either accessibility or side-chain mobility as would be expected for a helical or β-hairpin region of TnI interacting tightly with the TnC central helix. The dipole–dipole interactions between the TnI and TnC sites in the central helix suggest that the two subunits are further apart (>20 Å) than previously anticipated (14, 15). The inclusion of the third troponin subunit, TnT, did reveal a regular helical structure of residues 129–137 followed by a less-structurally defined region. Furthermore, comparison of the side-chain properties in the ternary and binary complexes revealed a footprint of TnT interactions with the N-terminal end of the inhibitory region.
These observations lead us to propose that, in the binary complex, the inhibitory region does not posses a regular secondary structure or does not form an interface with TnC. In the ternary complex, the N terminus of the inhibitory region of TnI is α-helical and interacts with TnT, as predicted from the primary sequence (36). The preliminary report of the ternary complex crystal structure (37) is in full agreement with the solution results presented here.
SDSL-EPR.
SDSL-EPR, pioneered by Hubbell and collaborators (27, 38), combines the site selectivity of probe techniques with the ability to observe more than one site by cysteine scanning mutagenesis. The method was originally developed for the investigation of membrane proteins, but we have used it here to study the quaternary structure of aqueous complexes. In membranes, the lipid environment generates unequal solvation of a protein surface. The side chains interacting with the membrane are shielded from the aqueous environment but are exposed to the spin relaxation agents that are soluble in the lipid phase (oxygen). This double contrast provides a means of identifying the side-chain environment, and the trend in the accessibility pattern identifies the secondary structure. In our case, we had to forgo the advantage of the selective relaxation of residues in a lipid environment. Fortunately, the interface between the two protein subunits provided accessibility differences sufficient to identify the secondary structure as well as the interfacial residues. Our side-chain mobility data, as measured by spin label averaging of the hyperfine anisotropy, strengthen these results. McHaourab et al. (34) have shown, using T4 lysozyme as a model system, that spin label mobility is heavily dampened for side chains involved in tertiary interactions. Obviously, this is also true for the side chains involved in quaternary interactions, as shown here in the comparison of the binary and ternary complexes. The drawback of SDSL-EPR is the requirement of cysteine scanning and modification with spin labels. The size of the spin-labeled cysteine residue is similar to a tryptophan residue, and it may well perturb the formation of the protein–protein complex. Here, both gels and dynamic light scattering revealed the presence of only the expected binary/ternary complexes. Additionally, labeling also raises questions about the possibility of perturbing the function of the protein. However, two of our ternary TnI mutant complexes (TnI 133 and 138), whose ability to control movement of actin filaments in the in vitro assay as measured in P. B. Chase's laboratory, indicated no functional impairment. Other workers have also reported no functional perturbation (ATPase activity, force development) when using labeled TnC or TnI subunits (39–42), though there is little doubt that some mutations will result in an impaired regulation.
Models of Binary Complex.
The data presented here do not provide support for the two structural models of TnC–TnI complex: an α-helical model proposed by sequence analogy to the structure of the N-terminal peptide of TnI and TnC (14) and a β-hairpin model based on the modeling of neutron diffraction and NMR data of the binary complex (15). In both models there is an extensive interface between the TnC central helix and TnI that is expected to limit solvent accessibility and side-chain mobility. Furthermore, the distances between the central helix of TnC and TnI are predicted to be well within the 20-Å limit of the dipolar EPR technique used in this study. The longer than expected distances between the central helix and the inhibitory region of TnI are supported by the absence of chemical shift perturbations and changes in the relaxation times of the cTnC central helix on binding of the inhibitory peptide (43). The strongest support for such interaction involving whole Tn subunits comes from chemical crosslinking of the skeletal I⋅C complex (44), where skTnC89 residue (cTnC91) crosslinked to the skTnI108–113 region (cTnI141–146). This apparent discrepancy might be caused by different isoforms of troponin, or differences between spectroscopic measurements and crosslinking experiments, which measure the time average of the distance and the distance of closest approach, respectively. The latter alternative might be correct considering that the inhibitory region was observed to crosslink distant regions of TnC: the N-domain of TnC (45), C-domain (46), and the central helix (44).
The secondary structure of the inhibitory region is also controversial. Numerous structure prediction algorithms predict the region to be helical with an unstructured region between skTnI102–106 and skTnI110–114 (cTnI135–139 and 143–147) (47). However, CD and NMR of an isolated peptide (sk96–115) in solution showed no evidence of the helical content (47). Although there are many precedents of short peptides having different structures in solution and in complex with other subunits, NMR of the peptide bound to TnC also failed to detect substantial helical structure (47). This might have been caused by nonspecific binding of the inhibitory peptide to the C-terminal domain of TnC (48), but the issue remains unresolved. The four intrasubunit dipolar distances presented here are consistent with the theoretical prediction of a helix-loop-helix structure (47). To avoid a simplistic interpretation in terms of the Cα–Cα distances that ignore both probe size and mobility, we used molecular dynamics calculations to account for both sources of potential artifacts. The agreement was nearly quantitative for the helical model, whereas the β-hairpin model overestimated 3 of the 4 interspin distances by 2–10 Å (Cα-Cα distances underestimate the interspin distances by 5–8 Å, data not shown).
The presence of a flexible region within TnI, to allow the regulatory region to move independently of the inhibitory region, has been postulated by a number of groups. The intradomain FRET studies of Dong et al. (49) showed flexing of the inhibitory region on Ca2+ addition. FRET between TnI and actin showed a gradient of distance changes (50), and another FRET study observed conformational heterogeneity of the inhibitory region (42). The conformational disorder within TnI is postulated to have functional significance, as it was modulated by the presence of divalent ions (42, 49). The data presented here are consistent with residues cTnI138–145 forming the putative hinge. Although side-chain mobility is not equivalent to the flexibility of the polypeptide chain, the fact that all of the residues inclusive of 138–145 were equally mobile and accessible to the solvent suggests that the region is either very disordered or that it does not form an interface with TnC or TnT which would “stiffen” its structure. We thus believe that the above stretch corresponds to the “unstructured” region predicted from the sequence (47) and is indeed the postulated hinge region.
Ternary Complex.
Electron micrographs and crystallographic studies of tropomyosin/troponin complexes showed troponin to consist of a globular head and an elongated tail (51, 52). The globular portion consists of all of the three subunits, and the tail is comprised of the C-terminal part of TnT. Pearlstone and Smillie (53) noted that a heptad repeat motif in the TnI (skTnI57–106, cTnI 90–139) and TnT sequence (sk198–251) could form a coiled-coil interface between the two subunits. Evidence for such an interface emerged from a yeast two-hybrid system in which the formation of the TnI⋅T heterodimer was favored over the homodimer formation (36). As expected, point deletions of leucine residues at the coil–coil interface (a and d positions) decreased the interaction between the TnT and TnI subunits. Furthermore, a proteolysis study of the inhibitory region of TnI by Tao and collaborators (54) suggested that TnT, and not TnC, protects TnI from proteolysis (cleavage at cTnI133). The footprint pattern of TnT on the mobility in the ternary complex, Fig. 4, is consistent with TnT interacting strongly with residues 130–135 of TnI. It is quite likely that the coiled coil unzips half a turn before the last predicted heptad repeat residue in TnI (cTnI139).
The secondary structure and the interactions between the TnC, TnI, and TnT subunits described here were all obtained in the presence of saturating Ca2+. Naturally, the changes in these interactions in response to Ca2+ and myosin binding can identify which aspects of that interface are functionally important.
It is of interest to note that after completing this study the first crystal structure of the ternary complex of cardiac C⋅I⋅T2 was reported in a preliminary form (37). All of the aspects of our study, which were done in solution, are borne out by the ternary crystal structure. The TnI residues were observed to form a coiled-coil with TnT up to residue 136, after which there is no electron density in the crystal, analogous to the disordered stretch of residues 138–145 observed in this study. The helical structure identified by SDSL-EPR for residues 129–137 corresponds to the C-terminal region of the TnT–TnI coiled coil identified by the crystal structure data. The close interaction of TnI with TnC is limited to the C- and N-terminal domains with little interaction of TnI with the central helix of TnC thus explaining the longer distances (>20 Å) and the absence of the expected dipolar interactions with residues in the central helix.
In summary, this is the first application of cysteine scanning mutagenesis, site-directed spin labeling and spin–spin dipolar interactions to a muscle system. We have mapped out the secondary structure of the inhibitory region of TnI and the interfaces between TnI, TnC, and TnT. The inhibitory region of TnI consists of an α-helix between residues 129 and 137, followed by a disordered region up to residue 145. Troponin T interacts with the N-terminal portion of the inhibitory peptide (130–135), consistent with the proposed coiled-coil structure. Additionally, the inhibitory region does not have any specific interactions with the TnC central helix.
Acknowledgments
We thank Drs. H. Cheung and W. Dong for providing us with the vectors for cardiac TnC and TnI, Dr. P. B. Chase for careful reading of the manuscript and members of his laboratory N. Brunet, J. Grubich, S. Williams for performing in vitro motility assays; and Dr. E. Bienkiewicz for assistance with dynamic light scattering. We also thank Drs. Y. Maeda and S. Takeda for sharing the coordinates of the binary complex model and for the preliminary information on the x-ray crystal structure of the ternary complex. This work was supported by National Science Foundation Grant IBN-98-08708, a National High Magnetic Field Laboratory In-House grant, American Heart Association Grant GIA-9950424N, the Muscular Dystrophy Association, a National Science Foundation Research Training grant (to K.L.S.), and National Science Foundation Major Research Instrumentation Grant CHE-0079649 for the purchase of Fourier transform EPR instrument.
Abbreviations
- SDSL-EPR
site-directed spin labeling EPR
- NiEDDA
Ni(ll)Ethylenediaminediacetate
References
- 1.Gordon A M, Homsher E, Regnier M. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 2.Herzberg O, James M N. Nature (London) 1985;313:653–659. doi: 10.1038/313653a0. [DOI] [PubMed] [Google Scholar]
- 3.Sundaralingam M, Bergstrom R, Strasburg G, Rao S T, Roychowdhury P, Greaser M, Wang B C. Science. 1985;227:945–948. doi: 10.1126/science.3969570. [DOI] [PubMed] [Google Scholar]
- 4.Satyshur K A, Rao S T, Pyzalska D, Drendel W, Greaser M, Sundaralingam M. J Biol Chem. 1988;263:1628–1647. [PubMed] [Google Scholar]
- 5.Grabarek Z, Tao T, Gergely J. J Muscle Res Cell Motil. 1992;13:383–393. doi: 10.1007/BF01738034. [DOI] [PubMed] [Google Scholar]
- 6.Sia S K, Li M X, Spyracopoulos L, Gagne S M, Liu W, Putkey J A, Sykes B D. J Biol Chem. 1997;272:18216–18221. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 7.Syska H, Wilkinson J M, Grand R J, Perry S V. Biochem J. 1976;153:375–387. doi: 10.1042/bj1530375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gergely J, Grabarek Z, Leavis P C, Strasburg G, Tao T, Wang C L. Adv Exp Med Biol. 1988;226:155–164. [PubMed] [Google Scholar]
- 9.Tao T, Gong B J, Leavis P C. Science. 1990;247:1339–1341. doi: 10.1126/science.2138356. [DOI] [PubMed] [Google Scholar]
- 10.Van Eyk J E, Strauss J D, Hodges R S, Ruegg J C. FEBS Lett. 1993;323:223–228. doi: 10.1016/0014-5793(93)81344-y. [DOI] [PubMed] [Google Scholar]
- 11.Narita A, Yasunaga T, Ishikawa T, Mayanagi K, Wakabayashi T. J Mol Biol. 2001;308:241–261. doi: 10.1006/jmbi.2001.4598. [DOI] [PubMed] [Google Scholar]
- 12.Lehman W, Rosol M, Tobacman L S, Craig R. J Mol Biol. 2001;307:739–744. doi: 10.1006/jmbi.2001.4514. [DOI] [PubMed] [Google Scholar]
- 13.Stone D B, Timmins P A, Schneider D K, Krylova I, Ramos C H, Reinach F C, Mendelson R A. J Mol Biol. 1998;281:689–704. doi: 10.1006/jmbi.1998.1965. [DOI] [PubMed] [Google Scholar]
- 14.Vassylyev D G, Takeda S, Wakatsuki S, Maeda K, Maeda Y. Proc Natl Acad Sci USA. 1998;95:4847–4852. doi: 10.1073/pnas.95.9.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tung C S, Wall M E, Gallagher S C, Trewhella J. Protein Sci. 2000;9:1312–1326. doi: 10.1110/ps.9.7.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter J D. Methods Enzymol. 1982;85:241–263. doi: 10.1016/0076-6879(82)85024-6. [DOI] [PubMed] [Google Scholar]
- 17.Dong W J, Xing J, Villain M, Hellinger M, Robinson J M, Chandra M, Solaro R J, Umeda P K, Cheung H C. J Biol Chem. 1999;274:31382–31390. doi: 10.1074/jbc.274.44.31382. [DOI] [PubMed] [Google Scholar]
- 18.She M, Dong W J, Umeda P K, Cheung H C. Eur J Biochem. 1998;252:600–607. doi: 10.1046/j.1432-1327.1998.2520600.x. [DOI] [PubMed] [Google Scholar]
- 19.Dong W J, Chandra M, Xing J, Solaro R J, Cheung H C. Biochemistry. 1997;36:6745–6753. doi: 10.1021/bi962226d. [DOI] [PubMed] [Google Scholar]
- 20.Toyota N, Shimada Y, Bader D. Circ Res. 1989;65:1241–1246. doi: 10.1161/01.res.65.5.1241. [DOI] [PubMed] [Google Scholar]
- 21.Vallins W J, Brand N J, Dabhade N, Butler-Browne G, Yacoub M H, Barton P J. FEBS Lett. 1990;270:57–61. doi: 10.1016/0014-5793(90)81234-f. [DOI] [PubMed] [Google Scholar]
- 22.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.She M, Xing J, Dong W J, Umeda P K, Cheung H C. J Mol Biol. 1998;281:445–452. doi: 10.1006/jmbi.1998.1933. [DOI] [PubMed] [Google Scholar]
- 24.Dong W J, Wang C K, Gordon A M, Cheung H C. Biophys J. 1997;72:850–857. doi: 10.1016/s0006-3495(97)78719-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farah C S, Miyamoto C A, Ramos C H, da Silva A C, Quaggio R B, Fujimori K, Smillie L B, Reinach F C. J Biol Chem. 1994;269:5230–5240. [PubMed] [Google Scholar]
- 26.Gordon A M, LaMadrid M A, Chen Y, Luo Z, Chase P B. Biophys J. 1997;72:1295–1307. doi: 10.1016/S0006-3495(97)78776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altenbach C, Greenhalgh D A, Khorana H G, Hubbell W L. Proc Natl Acad Sci USA. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perozo E, Cortes D M, Cuello L G. Nat Struct Biol. 1998;5:459–469. doi: 10.1038/nsb0698-459. [DOI] [PubMed] [Google Scholar]
- 29.Rabenstein M D, Shin Y K. Proc Natl Acad Sci USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ottemann K M, Thorgeirsson T E, Kolodziej A F, Shin Y K, Koshland D E. Biochemistry. 1998;37:7062–7069. doi: 10.1021/bi980305e. [DOI] [PubMed] [Google Scholar]
- 31.Pannier M, Veit S, Godt A, Jeschke G, Spiess H W. J Magn Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 32.Sale K, Sar C, Sharp K A, Hideg K, Fajer P G. J Magn Reson. 2002;156:104–112. doi: 10.1006/jmre.2002.2529. [DOI] [PubMed] [Google Scholar]
- 33.Nicholls A, Sharp K A, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 34.McHaourab H S, Lietzow M A, Hideg K, Hubbell W L. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 35.Khairy K, Fajer P G, Budil D. Biophys J. 2002;82:479. [Google Scholar]
- 36.Stefancsik R, Jha P K, Sarkar S. Proc Natl Acad Sci USA. 1998;95:957–962. doi: 10.1073/pnas.95.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda S, Yamashita A, Maeda K, Maeda Y. Biophys J. 2002;82:832. [Google Scholar]
- 38.Hubbell W L, Gross A, Langen R, Lietzow M A. Curr Opin Struct Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Gergely J, Tao T. Biophys J. 2001;81:321–333. doi: 10.1016/S0006-3495(01)75702-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H C, Fajer P G. Biochemistry. 1994;33:14324–14332. doi: 10.1021/bi00251a046. [DOI] [PubMed] [Google Scholar]
- 41.Luo Y, Leszyk J, Qian Y, Gergely J, Tao T. Biochemistry. 1999;38:6678–6688. doi: 10.1021/bi9824341. [DOI] [PubMed] [Google Scholar]
- 42.Zhao X, Kobayashi T, Gryczynski Z, Gryczynski I, Lakowicz J, Wade R, Collins J H. Biochim Biophys Acta. 2000;1479:247–254. doi: 10.1016/s0167-4838(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 43.Abbott M B, Dong W J, Dvoretsky A, DaGue B, Caprioli R M, Cheung H C, Rosevear P R. Biochemistry. 2001;40:5992–6001. doi: 10.1021/bi0100642. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi T, Tao T, Gergely J, Collins J H. J Biol Chem. 1994;269:5725–5729. [PubMed] [Google Scholar]
- 45.Leszyk J, Grabarek Z, Gergely J, Collins J H. Biochemistry. 1990;29:299–304. doi: 10.1021/bi00453a041. [DOI] [PubMed] [Google Scholar]
- 46.Leszyk J, Collins J H, Leavis P C, Tao T. Biochemistry. 1987;26:7042–7047. doi: 10.1021/bi00396a028. [DOI] [PubMed] [Google Scholar]
- 47.Hernandez G, Blumenthal D K, Kennedy M A, Unkefer C J, Trewhella J. Biochemistry. 1999;38:6911–6917. doi: 10.1021/bi990150q. [DOI] [PubMed] [Google Scholar]
- 48.Abbott M B, Dvoretsky A, Gaponenko V, Rosevear P R. FEBS Lett. 2000;469:168–172. doi: 10.1016/s0014-5793(00)01271-0. [DOI] [PubMed] [Google Scholar]
- 49.Dong W J, Xing J, Robinson J M, Cheung H C. J Mol Biol. 2001;314:51–61. doi: 10.1006/jmbi.2001.5118. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi T, Kobayashi M, Gryczynski Z, Lakowicz J R, Collins J H. Biochemistry. 2000;39:86–91. doi: 10.1021/bi991903b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flicker P F, Phillips G N, Jr, Cohen C. J Mol Biol. 1982;162:495–501. doi: 10.1016/0022-2836(82)90540-x. [DOI] [PubMed] [Google Scholar]
- 52.White S P, Cohen C, Phillips G N., Jr Nature (London) 1987;325:826–828. doi: 10.1038/325826a0. [DOI] [PubMed] [Google Scholar]
- 53.Pearlstone J R, Smillie L B. Can J Biochem Cell Biol. 1985;63:212–218. doi: 10.1139/o85-030. [DOI] [PubMed] [Google Scholar]
- 54.Tao T, Gong B J, Grabarek Z, Gergely J. Biochim Biophys Acta. 1999;1450:423–433. doi: 10.1016/s0167-4889(99)00050-6. [DOI] [PubMed] [Google Scholar]