

Abstract
Proton pathways in green fluorescent protein (GFP) are more extended than previously reported. In the x-ray data of wild-type GFP, a two-step exit pathway exists from the active site to the protein surface, controlled by a threonine switch. A proton entry pathway begins at a glutamate-lysine cluster around Glu-5, and extends all the way to the buried Glu-222 near the active site. This structural evidence suggests that GFP may function as a portable light-driven proton-pump, with proton emitted in the excited state through the switchable exit pathway, and replenished from Glu-222 and the Glu-5 entry pathway in the ground state.
INTRODUCTION
Proton pumps are a family of membrane proteins that play a pivotal role in the bioenergetics of the cell (Wikström, 1998; Decoursey, 2003; Ädelroth and Brzezinski, 2004). Well-known examples are bacteriorhodopsin (bR) and cytochrome c oxidase (CcO), which convert light or chemical (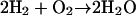 ) energy, respectively, into a transmembranal proton gradient, which subsequently drives ATP synthesis (Wikström, 1998). The sequence of proton transfer (PT) events, after photoexcitation of the bR chromophore (Cro), involves conformational change in the Cro and its vicinity, which couples to PT from the Cro to the extracellular surface of the protein. This is followed by PT from a buried Asp-96 residue to the Cro, and finally by reprotonation of Asp-96 from the cytoplasmic side. In both proteins proton pathways have been identified from the x-ray structure, but due to water disorder, some of the water molecules along these pathways had to be introduced using various computational criteria. To date, proton pathways in nonmembranal proteins have not been thoroughly investigated.
) energy, respectively, into a transmembranal proton gradient, which subsequently drives ATP synthesis (Wikström, 1998). The sequence of proton transfer (PT) events, after photoexcitation of the bR chromophore (Cro), involves conformational change in the Cro and its vicinity, which couples to PT from the Cro to the extracellular surface of the protein. This is followed by PT from a buried Asp-96 residue to the Cro, and finally by reprotonation of Asp-96 from the cytoplasmic side. In both proteins proton pathways have been identified from the x-ray structure, but due to water disorder, some of the water molecules along these pathways had to be introduced using various computational criteria. To date, proton pathways in nonmembranal proteins have not been thoroughly investigated.
This work presents surprising findings for the (nonmembranal) green fluorescent protein (GFP), in which well-defined transprotein proton pathways are identified from the x-ray data. Moreover, due to the rigid GFP barrel structure (Tsien, 1998), all the atomistic “hopping stones” for the translocated proton can be located in the measured x-ray structures (Örmo et al., 1996; Yang et al., 1996; Brejc et al., 1997; Jain and Ranganathan, 2004), none need to be assumed as in the floppier membranal proton pumps. This provides a superb example for the microscopic construction of proton pathways within a protein. A plausible conclusion (discussed below) is that GFP is a unique nonmembranal light-driven proton pump, operating according to principles analogous to those of bR.
GFP, the “canned candlestick”, is a remarkable solution-phase protein, which is rapidly gaining popularity in molecular biology (Phillips, 1997; Tsien, 1998; Remington, 2000; Zimmer, 2002). First cloned from the jellyfish Aequorea victoria, it has found extensive application as a biological fluorescence marker. Its Cro is synthesized in situ by an autocyclization reaction involving three consecutive amino-acid residues, Ser-65, Tyr-66, and Gly-67. After photon absorption, it undergoes a reaction of excited-state (ES) proton transfer (ESPT), in which the phenolic hydrogen of Tyr-66 dissociates, leaving behind a brightly fluorescent (green) anion (Chattoraj et al., 1996; Lossau et al., 1996). This is evident from the two absorption peaks, at 395 nm for the neutral form (ROH, A state) and 475 nm for the anion (RO−, B state). With increasing pH, the amplitude of the latter increases at the expense of the ROH band, showing a clear isosbestic point (Ward et al., 1982), much like the photoacids that Weller (1952) has investigated 50 years ago.
Indeed, ESPT is a well-known phenomenon in hydroxyaromatics, giving rise to a dramatic decrease in pKa upon electronic excitation (Weller, 1961; Agmon, 2005). It involves intramolecular charge transfer (ICT), from the phenolic oxygen to the aromatic ring system, which is larger for RO− than for the ROH state (Weller, 1952; Agmon et al., 2002). The GFP Cro appears to follow the same principles (Scharnagl et al., 1999). The high RO− quantum yield in GFP contrasts with the denatured protein or the isolated Cro which, like para-phenols (Schulman et al., 1981), do not fluoresce (Tsien, 1998; Zimmer, 2002). The x-ray data of the wild-type (wt) protein (Örmo et al., 1996; Yang et al., 1996) show that GFPs possess a unique barrel geometry, consisting of 11 tightly packed β-sheets, with a α-helix carrying the Cro traversing its center. The Cro assumes a planar (cis) conformation within its rigid cage, and this may be the key to its strong fluorescence.
The generally accepted model (Brejc et al., 1997; Palm et al., 1997) for proton migration in photoexcited GFP, suggests that the photodissociated proton of Tyr-66 is transferred, via a water molecule and the OH of Ser-205, to the (presumably anionic) carboxylate group of the buried Glu-222 residue. Subsequently, the Thr-203 side chain rotates to donate a hydrogen-bond (HB) to the nascent Tyr-66-anion. This forms the excited B state, whose emission is blue shifted with respect to A (Chattoraj et al., 1996). Finally, decay to the ground state (GS) is followed by the return of the proton to Tyr-66, so that the proton shuttles back and forth within the protein. The biophysical role of such a shuttling mechanism, or more generally, the biological role of GFP, remains unknown.
A few observations appear inconsistent with the above model (Zimmer, 2002). For example, FTIR measurements indicated no change in the protonation state of Glu-222 between the A and B states (van Thor et al., 1998); see, however, Stoner-Ma et al. (2005). Most intriguing is a recent study of the long-time (t) fluorescence tail of GFT (Leiderman et al., 2004). At room temperature, a ubiquitous t−3/2 decay of the ROH emission was observed. For ESPT in solution, such a behavior was shown to result from geminate recombination of the excited RO− with the dissociated proton which diffuses in three-dimensional space (Pines et al., 1988; Solntsev and Agmon, 2000). Since the protein interior does not appear to support an extensive three-dimensional network for proton migration, the possibility arises that the proton is actually transferred outside the protein. This motivated the more extensive search of proton pathways in GFP, whose results are reported below.
METHODS
The x-ray coordinates of GFP, as deposited in the Protein Data Bank (PDB), are utilized in this study. The wt A-state structure (Yang et al., 1996; Brejc et al., 1997) was deposited as PDB files 1GFL (GFP dimer at pH = 7.0 and 1.9 Å resolution) and 1EMB (GFP monomer at pH = 3.8 and 2.1 Å resolution). For 1GFL, the coordinates of the A subunit are used (the B subunit has a similar structure). The anionic B-state structure is believed to be represented by the S65T mutant (Örmo et al., 1996). A recent high-resolution structure of this mutant (Jain and Ranganathan, 2004) was deposited as PDB file 1Q4A (pH = 8.5 and 1.45 Å resolution).
Typically, the error in the coordinates in x-ray structures is ∼0.1 the resolution (except for bonded atoms, where prior chemical knowledge is applied to reduce this error). Thus the error in HB lengths is at most 0.2 Å. Since the cutoff distance for the O⋯O distance in an OH⋯O HB is typically assumed to be ∼3.3 Å, any distance shorter than 3.1 Å indicates an HB beyond any resolution error. Molecular dynamics simulations (Helms et al., 1999) have shown that GFP is a remarkably stiff protein, with backbone (bb) atoms deviating on average by only 0.9 Å from their crystal structure positions. This suggests that the short HBs observed in the x-ray structure inside the protein are not greatly perturbed by dynamic effects.
In this study, the Chem3D software (version 8.0.3 by CambridgeSoft) is utilized in the analysis, for both visualizing and manipulating the protein structures. Optimized molecular mechanics (MM2) gas-phase bond-lengths are used as reference in this study.
THE EXIT PATHWAY
The common wisdom is that the proton from the excited Tyr-66 migrates within the protein to the presumably anionic Glu-222 residue (Brejc et al., 1997; Palm et al., 1997). We present several independent arguments which lead to the conclusion that for wt-GFP in H2O at room temperature and neutral pH values, this might not be the correct scenario. Rather, a transient pathway opens in the ES, allowing the proton to escape outside the protein.
Transient fluorescence data
The motivation for searching for an exit pathway comes from the time-correlated single photon counting (TCSPC) measurements of the time-resolved ROH emission (450 nm) after ps laser excitation (at 380 nm), conducted by Leiderman et al. (2004), to which a slightly different interpretation is given below.
The fluorescence decay is highly nonexponential (Lossau et al., 1996). In the usual vein (Agmon, 2005), it is multiplied by exp(t/τf), where τf is the ES (“fluorescence”) lifetime. Typical data, shown in Fig. 1, are still highly nonexponential. Additionally, these data show two “kinks”. The one near 300 ps originates from a secondary peak in the instrument response function (IRF). The second, at ∼2 ns, may have physical significance. Beyond it, the data obey a long-time t−3/2 decay law, as noted by Leiderman et al. (2004). This resembles the long-time tail of several photoacids in water, and is indicative of proton diffusion in the three-dimensional bulk, followed by its reversible recombination with the excited anion (Agmon, 2005). In general, the decay for diffusion in a d-dimensional space is expected to behave like t−d/2.
FIGURE 1.

Time-resolved fluorescence from wt-GFP (293 K, in pH = 8 buffer). Circles are experimental data by Leiderman et al. (2004), showing the time dependence of the 450 nm emission intensity, I, corrected for the fluorescence lifetime, τf = 2.4 ns (determined from the decay of the anion emission at 510 nm). These data are from a different experimental run than shown in their Fig. 5, and reproduced here with permission. Dashed and dash-dot lines represent the asymptotic t−1 and t−3/2 laws, respectively, convoluted with the instrument response function (dotted). Both powers-laws were multiplied by the same arbitrary prefactor. Note the log-log scale.
This observation for wt-GFP is surprising in view of the interpretation that the dissociated proton stays within the rigid GFP barrel (Brejc et al., 1997; Palm et al., 1997). The interior of the protein contains just a few water molecules, and these do not form a three-dimensional HB network that could support such a diffusive motion. One would rather expect a biexponential decay if the proton hops in two steps to Glu-222, or a t−1/2 decay if longer, essentially one-dimensional, proton wires can be found within the protein. Thus, the t−3/2 decay in Fig. 1 suggests proton escape to solution. The problem is that the addition of proton scavengers to the solution did not seem to affect the time trace as it does for the simpler photoacids (Leiderman et al., 2004).
To resolve this dichotomy, consider the decay in the regime just preceding the t−3/2 asymptotics. Over a full decade, from 200 ps to 2 ns, a t−1 decay law fits the data well (Fig. 1). It subsequently seems to switch abruptly to t−3/2 at 2 ns, but only a tiny fraction of the population lives to see this ultimate behavior. A plausible interpretation of these findings is that the proton escapes from the protein but is trapped on its external surface, where it executes two-dimensional diffusive motion (d = 2 in the t−d/2 law). Only at times longer than ∼2 ns does it succeed to surmount the barrier between surface and bulk solution. The scavenger molecules, when added to the bulk, also find it difficult to surmount this barrier and pick up the proton from the protein surface. Thus most of the ES lifetime the photodissociated proton resides on the external surface, protected from the influence of the bulk, yet outside the GFP barrel.
The B-state structure
Despite the above observation, no exit pathway is immediately evident in the A-state structures of wt-GFP. Consequently, present interpretations suggest that the photodissociated proton migrates within the protein, to the carboxylate of Glu-222. On this background, it was surprising to note that a clear exit pathway is seen in the B-state structure of the protein, and that this went unnoticed in previous work.
As a model for the anionic B-state of GFP, one utilizes its S65T mutant (Brejc et al., 1997; Jain and Ranganathan, 2004). In this mutant, Glu-222 breaks its HB to Ser-205 and donates instead a HB to Thr-65 (which substitutes the Ser-65 of the wt protein), whereas Tyr-66 tends to stay in the anionic state. Fig. 2 shows a pathway leading from the hydroxyl of Tyr-66, via the OH of Thr-203 to the backbone carbonyl of His-148. The first HB is noted in many publications, and thought to arise from the rotation of the Thr-203 side chain after deprotonation, to stabilize the anion. The second HB was overlooked (see however Scharnagl et al., 1999). It is somewhat long (3.1 Å), but extremely linear. This can be deduced from the (Thr-203)C–O⋯O(His-148) angle, 108.4°, which is essentially identical to the C–O–H angle in alcohols. This shows that if the corresponding hydrogen is added to the x-ray structure, it will lie on the line connecting the two oxygens.
FIGURE 2.

X-ray structure of the anionic (B-state) GFP reveals a hydrogen-bond escape pathway for the proton, leading from the chromophore (Tyr-66) to the OH of Thr-203 and then to the bb-carbonyl of His-148, which is already on the surface of the protein. The HB lengths (O⋯O distance) are given in angstroms (for comparison, the typical distance for liquid water is 2.85 Å). Color codes: oxygen, red; nitrogen, blue; carbon, gray; and added hydrogens, white; bb connections, black. Coordinates from PDB file 1Q4A; x-ray diffraction measurements of Jain and Ranganathan (2004).
Fig. 3 shows that the bb-carbonyl of His-148 is already on the surface of the protein. Thus if we imagine Tyr-66 rotating in the ES to donate a HB to Thr-203, as depicted in Fig. 2, the proton could hop outside along the three atoms marked 1, 2, and 3 in Fig. 3. From His-148, it can continue along several routes, and others may open up by fluctuations of surface groups. For example, Fig. 4 focuses on the exit between Ser-202 and Asn-149, showing several distance measurements. Clearly, there are several short HBs along which the proton may advance. In addition, the water molecule which is shown to HB to both residues may move closer to the His-148 carbonyl to pick up the proton, or else the side chain of Asn-149 can rotate to bring its oxygen atom into closer contact (3.1 Å) with it.
FIGURE 3.

Proton escape hole on the surface of B-state GFP, after removing a few surface water molecules for better visualization. While the Cro is in its excited state, the proton can hop along the pathway denoted 1, 2, 3, corresponding to the three relevant oxygen atoms of Tyr-66 (black), Thr-203, and His-148. Also seen are Glu-222 (red, under Phe-223 and Gln-204), Ser-205, “the” water molecule (w, white), and the 202–204 triplet (see text). The space filling view is color coded by amino-acid side-chain characteristics: Greens, aliphatic; gray, aromatic; red, carboxylic (Glu, Asp); dark blues, charged amino (Arg, Lys, His); cyan, uncharged amino; yellow, sulfur; and orange, Ser and Thr. Water oxygens are gray. Coordinates from PDB file 1Q4A.
FIGURE 4.

A more extended exit channel may continue from the bb-carbonyl of His-148 along various solvent-protruding side chains or surface water molecules. Interatom distances in angstroms. Color codes as in Fig. 2. Coordinates from PDB file 1Q4A.
The A-state structure
The observed structure in the vicinity of the active site in wt GFP is shown in panel A of Fig. 5. Fig. 5 B shows that a similar conformation to that in Fig. 2 can be obtained by the rotation of two side chains in the wt structure of panel A. If the 120° rotation of the Thr-203 side chain occurs before ESPT (rather than after, as commonly assumed), a transient pathway of two nearly linear HBs leading from Tyr-66, via Thr-203 to the bb-carbonyl of His-148 will open up for the dissociating proton. To transfer its proton along this pathway, the phenolic hydroxyl of Tyr-66 should first cleave its HB to the water molecule and rotate toward the Thr-203 OH group. To obtain the final B-state geometry, the bulkier His-148 side chain should also rotate (by the angle shown in panel A). This may occur on a timescale slower than ESPT, leading to the slower phase of B-state emission observed in time-resolved fluorescence (Chattoraj et al., 1996).
FIGURE 5.

Formation of a transient proton escape pathway in wt-GFP. Panel A depicts the acidic state A of a wt-GFP from PDB file 1GFL (Yang et al., 1996). Here the Thr-203 switch is “off” and no exit pathway exists. See text for detailed discussion of the HB network. O⋯O distances in angstroms and O–H⋯O angles in degrees. The three shown internal rotations lead to state B′, resembling state B of the S65T mutant in Fig. 2. In particular, χ1 of Thr-203 was varied from −169° to −50° and for His-148 from −53° to −40°. This establishes a transient proton escape route (blue). Color codes as in Fig. 2, with bb continuation in black. Only Thr-203 and His-148 are rendered with all their atoms (including added hydrogens, not seen in the x-ray structure), the other three amino-acids are represented by selected atoms.
Under conditions when the Thr-203 conformational change is slower than ESPT, the proton could first migrate internally e.g., to Glu-222 (Stoner-Ma et al., 2005). Since ESPT is reversible (Pines et al., 1988; Agmon, 2005), the proton may return to the Cro several times, until eventually the Thr-203 rotation will allow it to exit.
The threonine switch
A major role in the kinetics may thus be attributed to the Thr-203 side-chain rotation, which opens up the exit pathway in the ES and shuts it down in the GS. There is additional evidence that Thr-203 is constructed as a fast nano-switch. Usually, there are three rotamers for a threonine side chain, derived from those of staggered ethane. Defining χ1 as the N–Cα–C–C dihedral angle, the rotamers around χ1 = ±60° are sometimes designated g+ and g−, respectively, whereas that near χ1 = 180° is called t. g+ and g− are nearly isoenergetic (with g+ slightly more stable), whereas t is the least stable rotamer, and thus less frequently found in protein structures deposited in the PDB (Warren and Zimmer, 2001). Interestingly, a PDB search by Warren and Zimmer (2001) revealed that when threonine appears in a STQ tripeptide sequence, the g+ rotamer is almost completely abolished. Threonine then becomes a two-state switch, as depicted in Scheme 1.
SCHEME 1.

The two-state threonine switch.
In wt-GFP, Thr-203 occurs in a STQ sequence namely, between a serine and a glutamine. In the A-state, it assumes the less stable t conformation, which switches to g− in the B-state. Possibly, the repulsion between the lone pairs of the hydroxyls of Thr-203 and Tyr-66 introduces a barrier for the t to g− conversion. As the Cro is excited, the electron density on the phenolic oxygen decreases due to the ICT effect (Scharnagl et al., 1999), thus reducing the repulsive barrier. This may allow the Thr-203 side chain to flip into its more stable g− rotamer. At room temperature, this rotation could be faster than ESPT, leading to preferential expulsion of the phenolic proton via the His-148 gateway. After the Cro decays back to its GS (in 2–3 ns), the threonine rotates back to its t state, blocking proton re-entry.
The exit point
Fig. 3 shows the vicinity of the exit point on the surface of the protein. It is interesting to note that this is the only hydrophopic face of the protein. Commensurate with its high solubility, GFP surface contains a high surface density of carboxylates (Glu, Asp). The only exception is the vicinity of the exit point in Fig. 3, which contains instead a high density of aromatic residues (Tyr-39, 143, 145, 151, and 200, and Phe-165 and 223). This suggests that docking of the protein may take place on this side. Indeed, the two GFP subunits in the dimer seen in PDB file 1GFL connect along this face. Dimerization must then interfere with proton exit. This explains the previously poorly understood observation by Ward et al. (1982) (Fig. 3 there), that the RO− absorption band (475 nm) decreases with increasing protein concentration. The protein aggregates, blocking the exit nozzle and impeding proton escape from the protein.
One may only speculate that GFP in vivo may dock onto some other protein near the exit point. In particular, Gln-204 may serve to anchor the orifice of the proton nozzle to its target on this protein, since it is seen to act as an anchor connecting the two subunits in the 1GFL dimer.
THE GLU-222 PATHWAY
Since the threonine switches to the “off” conformation in the GS of the Cro, Tyr-66 reprotonation may occur only from the buried Glu-222. This sequence of events resembles PT in bR, which first isomerizes and ejects a proton to the extracellular face of the protein, then returns to the original conformation and replenishes the proton from the buried Asp-96 residue.
This interpretation is contrasted with the prevailing ansatz (Brejc et al., 1997; Palm et al., 1997), that this pathway operates reversibly, first conducting the photodissociated proton in the ES to one of the carboxylate oxygens of Glu-222, which is assumed to be ionized in the GS. Several arguments contradicting this ansatz are presented below, suggesting instead that for wt-GFP at room temperature the primary role of the Glu-222 pathway is in the reprotonation direction.
Glutamate protonation state
Buried glutamates and aspartates have pKa values which are typically larger than in solution. Known examples are Glu-286 at the end of the “D-pathway”, near the active site of CcO from Rhodobacter sphaeroides, or Asp-96 in bR. These residues are not deprotonated at neutral pH (Wikström, 1998; Decoursey, 2003; Ädelroth and Brzezinski, 2004), and thus serve to deliver a proton to the active site. By homology, one may conjecture that the same holds for Glu-222 in GFP namely, it is not deprotonated and it donates a HB to Ser-205.
Electrostatic calculations performed for wt-GFP suggested that the GS pKa for Glu-222 is 6.3 (Scharnagl et al., 1999). Given an error of, say, 1 pK unit in these values, the calculation could support either interpretation. Additional evidence should thus be sought.
Water coordination in the A-state
It is possible to use the x-ray data, even in the absence of H-atom coordinates (not available at this resolution), to determine the coordination state of “the” water molecule, provided that both distance and angular information is considered. Fig. 5 A shows four groups in hydrogen bonding (HBing) distances around this water molecule: the bb-carbonyls of Thr-203 and Asn-146 and the hydroxyls of Tyr-66 and Ser-205. To verify that all four are viable HBs, let us check the tertahedrality of this water molecule environment. Table 1 shows all six tetrahedral angles at the water oxygen atom, from both 1GFL and 1EMB files. It can be seen that the deviation from a perfect tetrahedral angle of 109.5° is rather small. For 1GFL, the HB with Thr-203 is somewhat long (3.44 Å), and consequently the tetrahedral angles emanating from this residue deviate most from tetrahedrality. Even here, the deviation is <20°. In 1EMB, this HB is shorter (2.98 Å), and the maximal deviation is 16°. Because the tetrahedral symmetry of the water molecule is reproducible between two different x-ray structures, determined under different conditions, one may conclude with some confidence that it should indeed be viewed as four-coordinated.
TABLE 1.
| i/j | 66 | 146 | 203 | 205 |
|---|---|---|---|---|
| 66 | 110 | 96 | 114 | |
| 146 | 94 | 90 | 119 | |
| 203 | 99 | 96 | 123 | |
| 205 | 116 | 119 | 126 |
A check for the tetarhedrality of the water molecule solvating the phenoxyl moiety of Tyr-66 in the A-state of wt-GFP. Shown are the Xi⋯O⋯Xj angles in degrees at the central water oxygen atom, O. Xi is the hydroxyl oxygen of Tyr-66 or Ser-205, or the oxygen of the bb-carbonyl of Asn146 or Thr203. Upper triangle, from PDB file 1GFL (subunit A); lower triangle, file 1EMB.
Two of its four ligands are carbonyls (Asn-146 and Thr-203) which can only accept HBs. Consequently, the hydroxyls of Ser-205 and Tyr-66 are donating HBs to the water oxygen in the A-state structure, as shown in Fig. 5 A. Since one of the two carboxyl oxygens of Glu-222 is in a HBing distance from the OH of Ser-205 (2.74 Å in 1GFL; 2.6 Å in 1EMB), and the hydroxyl hydrogen of Ser-205 is already engaged in a HB with the water molecule, Glu-222 must be donating a HB to Ser-205. Thus it must be in a protonated state (COOH).
As long as the water is four-coordinated, it is not a likely proton acceptor. This restriction has been discussed in conjunction with the mechanism of proton mobility in liquid water (Agmon, 1995; Tuckerman et al., 1995). It arises from electrostatic repulsion with the HB donated to it from the hydroxyl of Ser-205. PT is thus preconditioned upon cleavage of this “unfavorable” HB. The OH of Ser-205 could rotate to reduce the coordination number of the water molecule, and subsequently deliver the proton to a rotated and neutral Glu-222. The required rearrangement would slow any PT to Glu-222 as compared with a preformed HB net. Thus under normal conditions proton escape could compete with its internal migration to Glu-222. However, when the escape route is inaccessible, the Cro could still dissociate using the Glu-222 pathway.
Water coordination in the B-state
After ESPT, the water molecule rotates around the Asn-146–Ser-205 axis to reform a HB to the anionic phenoxide. This occurs at the expense of the HB with the bb-carbonyl of Thr-203, so that the water becomes triply coordinated. A detailed view of the water molecule environment is shown in Fig. 6. The distance to the bb-CO of Thr-203 has increased (to 4.0 Å) beyond any plausible HB cutoff (∼3.3 Å). This occurred by a 30° increase in the bb-NCCO dihedral angle relative to the A-state. The distance to the bb-N of Ser-205 appears short enough for HBing. However, consideration of the angles around the water oxygen (Table 2) shows that the angles involving this nitrogen atom deviate significantly from tetrahedrality. Thus in this case the water must be triply coordinated.
FIGURE 6.

A detailed view of the water molecule environment in the anionic B-state (PDB file 1Q4A). HBs (with their orientation) are depicted by blue arrows. Two additional distances are also given.
TABLE 2.
| i/j | 66 | 146 | 205 |
|---|---|---|---|
| 146 | 95 | ||
| 205 | 116 | 125 | |
| 205N | 171 | 94 | 57 |
A check for the tetarhedrality of the water molecule in the B-state structure (PDB file 1Q4A) indicates that the interaction with the nitrogen of Ser-205 (last line) is at most a very weak HB. Rather the water is in threefold coordination with the other three residues. Compare with Table 1.
The reduction in coordination number makes the water a viable proton acceptor. The orientation of the HB net now is from Glu-222 to Tyr-66, and this should allow for rapid reprotonation of the Cro from Glu-222, once it has decayed to its GS.
Point mutations
Mutations of key residues along the escape and reprotonation pathways can change the ratio of the acidic (A) and anionic (B) bands in opposite directions (Ehrig et al., 1995).
Thr-203 mutations
Mutations of Thr-203 to aliphatic amino acids, like valine and isoleucine (Kummer et al., 2000), reduce and red-shift the 475 nm RO− absorption band. This has been attributed to the destabilization of the Tyr-66 phenoxide, which loses its HB to Thr-203. Time-resolved kinetics show that ESPT in the T203V and T203I mutants is ∼4 times slower than in the wt (Kummer et al., 2000). This observation is harder to rationalize within the conventional ansatz (Brejc et al., 1997), that Thr-203 rotates to form the HB to the anion only after ESPT. If Thr-203 does not participate in the fast deprotonation stage, why do such conservative mutations slow down its rate?
The existence of an exit channel controlled by the Thr-203 side chain can explain this observation. The T203V and T203I mutants fail to open this channel, so the only route open for the proton is to Glu-222. Thus, in wt-GFP the exit probability may be 3–4 times larger than the probability for migration to Glu-222. This rather modest factor may be sufficient to ensure the unidirectionality of proton migration within GFP.
Glu-222 mutations
Evidently, any disruption of the HB-net between Glu-222 and Tyr-66 should eliminate the Cro reprotonation in the GS, which would then remain permanently ionized. This occurs in S65T, where Glu-222 tilts toward Thr-65, cleaving its HB with Ser-205, see Brejc et al. (1997). The same should happen when Glu-222 is mutated to anything except aspartate. Indeed, the E222G (Ehrig et al., 1995) and E222Q (Wiehler et al., 2003) mutants exhibit only B-state absorption (475 nm).
The conventional ansatz for the B-state dominance in S65T is that the negative charge on the anionic Glu-222 prevents wt-Cro ionization in the GS and conversely, the neutral Glu-222 in the S65T mutant permits its permanent ionization (Brejc et al., 1997). This explanation is inconsistent with the GS pKa = 8.1 of an isolated, solvated Cro (Remington, 2000), which does not require additional interactions to prevent it from ionizing at pH = 7. Moreover, if Glu-222 were the sole proton acceptor in the ES, its elimination should have prevented photodissociation and the Cro would remain indefinitely in the A-state. Just the opposite is observed for the E222G and E222Q mutations.
THE ENTRANCE PATHWAY
The bR cycle is completed by proton re-entry from the cytoplasmatic side, along a designated proton pathway, to reprotonate Asp-96. One may similarly expect that an entry pathway exists in GFP, and it serves to reprotonate Glu-222, after it has transferred its proton to the GS Cro.
Such a pathway was indeed found, and it is depicted in Fig. 7. It includes the portion from Glu-222 to Tyr-66, which is identical to the proton shuttling pathway discussed in the literature (Brejc et al., 1997; Palm et al., 1997), but it extends from Glu-222 up to an entrance point at Glu-5. This residue collects protons from the surface and, via two water molecules, protonates the inward-directed carboxylic moiety of Asp-82. Hopping between the two carboxylic oxygens and across another water molecule, the proton can get to the bb-carbonyl of Gln-69, which is HBed to the OH moiety of Ser-72. Via two additional waters, the proton may reach the hydroxyl of Ser-65, which is HBed to the carboxylic group of Glu-222. Alternately, Ser-65 may be bypassed by an additional water molecule.
FIGURE 7.

A well-defined proton entry pathway exists in GFP, and it shows a remarkable resemblance to the D-pathway in CcO. The suggested pathway is depicted by dashed arrows, with distances in angstroms, as measured using the coordinates from PDB file 1GFL. No hydrogens are depicted here. The problematic steps involving W255 are denoted in red. Curiously enough, only the part of the HB-net between W60 and Tyr-66 was depicted in previous publications.
The pathway from Asp-82 to Glu-222 shows remarkable similarity to the D-pathway in CcO. The latter also begins at an inward-pointing aspartate (Asp-132 in R. sphaeroides) and ends at a glutamate, which is buried near the active site (Glu-286 in R. sphaeroides). Both pathways involve two serines and 6–10 water molecules. Interestingly, the pathway in GFP is better defined, as all of the water coordinates were obtained directly from the x-ray data.
The only problematic step in this pathway is water 255. It is the sole “stepping stone” requiring proton hops larger than 3 Å and, in addition, is not observed in all of the x-ray structures (e.g., 1EMB). Consequently, we look for an alternative route bridging the gap between Ser-72 and water 60. The five residues 68–72 form a single α-helical turn (or, at least, 69–71 form a partial helical turn), and this places their bb-nitrogens in close proximity. Fig. 8 shows how they may bridge the gap between Ser-72 and W60. Taken together, Figs. 7 and 8 depict a remarkably well-formed pathway from the “bottom” of the GFP barrel (near the N-terminal), roughly along the axis of the barrel to Glu-222 and from there to the active site at Tyr-66. All of the residues and water molecules along this pathway are obtained directly from the x-ray coordinates, and all of the distances are under 3 Å(!).
FIGURE 8.

A bb-nitrogen wire could serve as an alternative to W255 in bridging the gap between Ser-72 and W60. The nitrogens also couple to the bb-carbonyls of Val-68 and Gln-69, allowing several shortcuts to be taken. Coordinates from PDB file 1GFL.
Pathway mutations
If the pathway depicted in Fig. 7 is the major proton entry pathway, mutations of key residues along it could abolish proton entry and hence Cro reprotonation, tilting its equilibrium toward the anionic B-state. Such a shift has already been mentioned for the S65G mutant. Interestingly, the double mutant S65G/S72A shows further enhancement of the B-state 475 nm band as compared with single S65 mutants (Cormack et al., 1996).
Ser-72 appears to be a key residue along the entry pathway in Fig. 7, but is otherwise far from the protonable Cro-66 site. Its effect on the A/B equilibrium has thus far found no plausible explanation. It is clarified given our new pathway. Clearly, it is desirable to produce the S72A mutation alone. An additional decisive point mutation would be on Asp-82.
The entry point
The protein surface near the Glu-5 entry point is shown in Fig. 9. Glu-5 serves as a focal point for collecting surface-bound protons. In one direction, it is coupled to Glu-6, which protrudes even further into solution. In the perpendicular direction it couples via two lysines (Lys-79 and Lys-3) to Asp-76 and Glu-90, respectively. This surface arrangement is somewhat analogous to the entry point of the D-pathway in CcO, where surface glutamates appear in a joint cluster with histidines (Ädelroth and Brzezinski, 2004). Such clusters were conjectured to operate as “proton collecting antennas” (Gutman and Nachliel, 1997). Here the role of the histidines may be taken over by lysines. This antenna could concentrate protons near the entry point and coerce them into this small hole in the protein.
FIGURE 9.

Entry point to the proton pathway of Fig. 7 near the N-terminal on the GFP surface. Coordinates from PDB file 1EMB (Brejc et al., 1997) for monomeric GFP were used, because dimerization in file 1GFL occurs close to the depicted protein surface. The space filling view is color coded as in Fig. 3.
Alternate entry pathways
There may be more than one proton entry pathway into GFP. Another pathway, observed in the S65T mutant, is depicted Fig. 10. It is directed from the carboxyl of Glu-115 on the back surface of the protein, via two water molecules, to the bb-carbonyl of Glu-111. Then utilizing the bb-nitrogen of Asn-121 to cross a relatively large gap to its bb-carbonyl. This step may be field assisted, since the line connecting these two atoms (yellow) is directed to the carbonyl of the imidazolone ring, the most negatively charged in the Cro (Scharnagl et al., 1999). Crossing a single water molecule, the proton may arrive at the bb-carbonyl of Gly-67. From there, the bb-N of Gly-69 connects to HB-net of the two water molecules which terminates at Glu-222.
FIGURE 10.

An additional HB pathway for proton entry is revealed in the S65T, B-state structure (file 1Q4A).
This pathway appears to be blocked in the A-state wt by a salt bridge between Glu-115 and Arg-122 (not shown). The salt bridge may break at high salt concentrations, opening a second proton entry route. This may compensate for the decreased attraction of solvated protons to the surface glutamates near the entry point due to the enhanced electrostatic screening. The added pathway will increase reprotonation probability, explaining the enhanced 395 nm absorption peak observed upon increased ionic strength (Ward et al., 1982).
The surface bound Glu-115 residue may, in turn, harvest protons from a “glutamate ring” on the back-surface of the protein. This interesting ring structure is shown in Fig. 11. Some of the glutamates in the ring are seen to be bridged by two water molecules, e.g., Glu-17 and Glu-32 or Glu-111 and Glu-115. The Cro (magenta) is seen through two holes. One hole, between Val-120 and Leu-15, leads to the contact point between the Cro and Glu-222. A second deep hole, revealed after removing two water molecules, resides between Glu-115 and Arg-122. This is the opening of the pathway shown in Fig. 10.
FIGURE 11.

A glutamate ring observed on the back surface of the GFP may be functioning as a proton collecting antenna for the Glu-115 pathway in the S65T mutant (file 1Q4A).
DISCUSSION
The extensive x-ray and spectroscopic evidence discussed herein point to the existence of exceptionally well-formed protonic exit and entry pathways in wt-GFP, completed with the required (Wikström, 1998) switching mechanism (Thr-203). The observed pathways and their analogy with membranal proton pumps suggest that GFP may function as a portable (as opposed to membrane bound) light-activated proton pump. Cro excitation induces a conformational change opening the Thr-203 switch and expelling the proton to the outside. Back in its GS, the Cro is reprotonated by Glu-222, which in turn acquires a proton from the outside solvent via Glu-5 and the entry pathway. If so, this is the first example for a proton-pump residing in a nonmembranal protein.
The observation of appropriate HB networks indicates that protons may move along them. Whether they will migrate along a given pathway and at what rate depends on energetic and dynamic consideration. Methodologies for energetic calculations (Burykin and Warshel, 2004) and proton translocation dynamics (Ilan et al., 2004) along proton pathways in proteins are now becoming available, and their utilization for the pathways observed herein could shed further light on their functionality.
The question arises, what may be the role of a portable proton pump, since pumping protons in and out of bulk solution serves no purpose. Since the exit hole (near His-148) resides on the sole hydrophopic patch on the protein surface, this face may dock in vivo onto some target protein, or a proton channel in the outer membrane of some organelle, for the time period required to load it with protons. Consequently, the major role of wt-GFP might have been, at some point during its evolution, the light-driven acidification of some proteins, or the filling of vesicles or organelles with acid. The elucidation of the exact role of such a mechanism in the jellyfish requires further research.
Acknowledgments
I am indebted to Dan Huppert for permission to use the data in Fig. 1, and to Jasper J. van Thor and George N. Phillips Jr. for the discussion of the x-ray data.
This research was supported in part by The Israel Science Foundation (grant No. 191/03).
References
- Ädelroth, P., and P. Brzezinski. 2004. Surface-mediated proton-transfer reactions in membrane-bound proteins. Biochim. Biophys. Acta. 1655:102–115. [DOI] [PubMed] [Google Scholar]
- Agmon, N. 1995. The Grotthuss mechanism. Chem. Phys. Lett. 244:456–462. [Google Scholar]
- Agmon, N. 2005. Elementary steps in excited-state proton transfer. J. Phys. Chem. A. 109:13–35. [DOI] [PubMed] [Google Scholar]
- Agmon, N., W. Rettig, and C. Groth. 2002. Electronic determinants of photoacidity in cyanonaphthols. J. Am. Chem. Soc. 124:1089–1096. [DOI] [PubMed] [Google Scholar]
- Brejc, K., T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö, and S. J. Remington. 1997. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA. 94:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burykin, A., and A. Warshel. 2004. On the origin of the electrostatic barrier for proton transport in aquaporin. FEBS Lett. 570:41–46. [DOI] [PubMed] [Google Scholar]
- Chattoraj, M., B. A. King, G. U. Bublitz, and S. G. Boxer. 1996. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. USA. 93:8362–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack, B. P., R. H. Valdivia, and S. Falkow. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 173:33–38. [DOI] [PubMed] [Google Scholar]
- Decoursey, T. E. 2003. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83:475–579. [DOI] [PubMed] [Google Scholar]
- Ehrig, T., D. J. O'Kane, and F. G. Prendergast. 1995. Green-fluorescent protein mutants with altered fluorescence excitation spectra. FEBS Lett. 367:163–166. [DOI] [PubMed] [Google Scholar]
- Gutman, M., and E. Nachliel. 1997. Time-resolved dynamics of proton transfer in proteinous systems. Annu. Rev. Phys. Chem. 48:329–356. [DOI] [PubMed] [Google Scholar]
- Helms, V., T. P. Straatsma, and J. A. McCammon. 1999. Internal dynamics of green fluorescent protein. J. Phys. Chem. B. 103:3263–3269. [Google Scholar]
- Ilan, B., E. Tajkhorshid, K. Schulten, and G. A. Voth. 2004. The mechanism of proton exclusion in aquaporin channels. Proteins. 55:223–228. [DOI] [PubMed] [Google Scholar]
- Jain, R. K., and R. Ranganathan. 2004. Local complexity of amino acid interactions in a protein core. Proc. Natl. Acad. Sci. USA. 101:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer, A. D., J. Wiehler, H. Rehaber, C. Kompa, B. Steipe, and M. E. Michel-Beyerle. 2000. Effects of threonine 203 replacements on excited-state dynamics and fluorescence properties of the green fluorescent protein (GFP). J. Phys. Chem. B. 104:4791–4798. [Google Scholar]
- Leiderman, P., M. Ben-Ziv, L. Genosar, D. Huppert, K. M. Solntsev, and L. M. Tolbert. 2004. Study of the long-time fluorescence tail of the green fluorescent protein. J. Phys. Chem. B. 108:8043–8053. [Google Scholar]
- Lossau, H., A. Kummer, R. Heinecke, F. Pöllinger-Dammer, C. Kompa, G. Bieser, T. Jonsson, C. M. Silva, M. M. Yang, D. C. Youvan, and M. E. Michel-Beyerle. 1996. Time-resolved spectroscopy of wild-type and mutant green fluorescent proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chem. Phys. 213:l–16. [Google Scholar]
- Örmo, M., A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien, and S. J. Remington. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- Palm, G. J., A. Zdanov, G. A. Gaitanaris, R. Stauber, G. N. Pavlakis, and A. Wlodawer. 1997. The structural basis for spectral variations in green fluorescent protein. Nat. Struct. Biol. 4:361–365. [DOI] [PubMed] [Google Scholar]
- Phillips, G. N., Jr. 1997. Structure and dynamics of green fluorescent protein. Curr. Opin. Struct. Biol. 7:821–827. [DOI] [PubMed] [Google Scholar]
- Pines, E., D. Huppert, and N. Agmon. 1988. Geminate recombination in excited-state proton transfer reactions: Numerical solution of the Debye-Smoluchowski equation with back-reaction boundary conditions. J. Chem. Phys. 88:5620–5630. [Google Scholar]
- Remington, S. J. 2000. Structural basis for understanding spectral variations in green fluorescent protein. Methods Enzymol. 305:196–210. [DOI] [PubMed] [Google Scholar]
- Scharnagl, C., R. Raupp-Kossmann, and S. F. Fischer. 1999. Molecular basis for pH sensitivity and proton transfer in green fluorescent protein: Protonation and conformational substates from electrostatic calculations. Biophys. J. 77:1839–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman, S. G., W. R. Vincent, and W. J. M. Underberg. 1981. Acidity of cyanophenols in the S1 and T1 states. The influence of substituent orientation. J. Phys. Chem. 85:4068–4071. [Google Scholar]
- Solntsev, K. M., and N. Agmon. 2000. Dual asymptotic behavior in geminate diffusion-influenced reaction. Chem. Phys. Lett. 320:262–268. [Google Scholar]
- Stoner-Ma, D., A. A. Jaye, P. Matousek, M. Towrie, S. R. Meech, and P. J. Tonge. 2005. Observation of excited-state proton transfer in green fluorescent protein using ultrafast vibrational spectroscopy. J. Amer. Chem. Soc. 127:2864–2865. [DOI] [PubMed] [Google Scholar]
- Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- Tuckerman, M., K. Laasonen, M. Sprik, and M. Parrinello. 1995. “Ab Initio” molecular dynamics simulation of the solvation and transport of hydronium and hydroxyl ions in water. J. Chem. Phys. 103:150–161. [Google Scholar]
- van Thor, J. J., A. J. Pierik, I. Nugteren-Roodzant, A. Xie, and K. J. Hellingwerf. 1998. Characterization of the photoconversion of green fluorescent protein with FTIR spectroscopy. Biochemistry. 37:16915–16921. [DOI] [PubMed] [Google Scholar]
- Ward, W. W., H. J. Prentice, A. F. Roth, C. W. Cody, and S. C. Reeves. 1982. Spectral pertrubations of the Aequorea green fluorescent protein. Photochem. Photobiol. 35:803–808. [Google Scholar]
- Warren, A., and M. Zimmer. 2001. Computational analysis of Thr-203 isomerization in green fluorescent protein. J. Mol. Graph. Model. 19:297–303. [DOI] [PubMed] [Google Scholar]
- Weller, A. 1952. Quantitative untersuchungen der fluoreszenzumwandlung bei naphtholen. Z. Elektrochem. 56:662–668. [Google Scholar]
- Weller, A. 1961. Fast reactions of excited molecules. Prog. React. Kinet. 1:187–214. [Google Scholar]
- Wiehler, J., G. Jung, C. Seebacher, A. Zumbusch, and B. Steipe. 2003. Mutagenic stabilization of the photocycle intermediate of green fluorescent protein (GFP). ChemBioChem. 4:1164–1171. [DOI] [PubMed] [Google Scholar]
- Wikström, M. 1998. Proton translocation by bacteriorhodopsin and heme-copper oxidases. Curr. Opin. Struct. Biol. 8:480–488. [DOI] [PubMed] [Google Scholar]
- Yang, F., L. G. Moss, and G. N. Phillips, Jr. 1996. The molecular structure of green fluorescent protein. Nat. Biotechnol. 14:1246–1251. [DOI] [PubMed] [Google Scholar]
- Zimmer, M. 2002. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 102:759–781. [DOI] [PubMed] [Google Scholar]