

Abstract
Transport kinetics have been examined in erythrocyte anion transporter AE1 that has been chemically modified to convert glutamate 681 to an alcohol (E681OH AE1). Outward conductive Cl− flux in E681OH AE1 is inhibited by removal of extracellular Cl−; this effect is the opposite of that in native AE1 and is consistent with coupled electrogenic 2:1 Cl−/Cl− exchange. A second Cl− binding/transport site is also suggested by the characteristics of  flux in E681OH AE1: bilateral and cis Cl−, which are normally inhibitory, accelerate
flux in E681OH AE1: bilateral and cis Cl−, which are normally inhibitory, accelerate 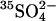 flux. These effects would be expected if Cl− binds to a second transport site on
flux. These effects would be expected if Cl− binds to a second transport site on 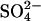 -loaded E681OH AE1, thereby allowing
-loaded E681OH AE1, thereby allowing  cotransport. Alternatively, the data can be explained without proposing
cotransport. Alternatively, the data can be explained without proposing 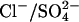 cotransport if the rate-limiting event for
cotransport if the rate-limiting event for 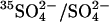 exchange is external
exchange is external 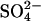 release, and the binding of external Cl− accelerates
release, and the binding of external Cl− accelerates 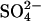 release. With either interpretation, these data indicate that E681OH AE1 has a binding/transport site for Cl− that is distinct from the main transport site. The effects of graded modification of E681 or inhibition by H2DIDS are consistent with the idea that the new Cl− binding site is on the same E681OH-modified subunit of the AE1 dimer as the normal transport site.
release. With either interpretation, these data indicate that E681OH AE1 has a binding/transport site for Cl− that is distinct from the main transport site. The effects of graded modification of E681 or inhibition by H2DIDS are consistent with the idea that the new Cl− binding site is on the same E681OH-modified subunit of the AE1 dimer as the normal transport site.
INTRODUCTION
The AE1 protein (Band 3) of the erythrocyte membrane mediates the exchange of Cl− for 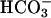 as part of the process of CO2 transport in the blood (Wieth et al., 1982; Alper et al., 2002). The catalytic cycle for anion exchange is believed to be “ping-pong”, in which there are distinct inward-facing and outward-facing conformations of the protein and the anions cross the membrane one at a time (Knauf, 1979; Fröhlich and Gunn, 1986; Passow, 1986). In addition to 1:1 monovalent anion exchange, there are other modes of AE1-mediated transport, including anion conductance (Knauf et al., 1977), H+/Cl− cotransport (Jennings, 1978; Lepke et al., 2003), and
as part of the process of CO2 transport in the blood (Wieth et al., 1982; Alper et al., 2002). The catalytic cycle for anion exchange is believed to be “ping-pong”, in which there are distinct inward-facing and outward-facing conformations of the protein and the anions cross the membrane one at a time (Knauf, 1979; Fröhlich and Gunn, 1986; Passow, 1986). In addition to 1:1 monovalent anion exchange, there are other modes of AE1-mediated transport, including anion conductance (Knauf et al., 1977), H+/Cl− cotransport (Jennings, 1978; Lepke et al., 2003), and 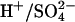 cotransport (Jennings, 1976; Milanick and Gunn, 1984). Although the fluxes via these transport modes are much smaller than the
cotransport (Jennings, 1976; Milanick and Gunn, 1984). Although the fluxes via these transport modes are much smaller than the  exchange flux, these fluxes are of interest because an understanding of alternative transport modes can potentially provide insights regarding the normal catalytic mechanism of AE1.
exchange flux, these fluxes are of interest because an understanding of alternative transport modes can potentially provide insights regarding the normal catalytic mechanism of AE1.
The 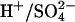 cotransport mode of AE1 is believed to depend on protonation of a specific glutamate residue, E681 (Jennings and Smith, 1992). When E681 is protonated, AE1 is converted from the normal monovalent anion transporter into a form that can transport
cotransport mode of AE1 is believed to depend on protonation of a specific glutamate residue, E681 (Jennings and Smith, 1992). When E681 is protonated, AE1 is converted from the normal monovalent anion transporter into a form that can transport 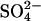 and other divalent anions (Fig. 1). Several years ago, we found that treatment of intact human red blood cells with Woodward's reagent K, followed by reductive cleavage of the active ester adduct with
and other divalent anions (Fig. 1). Several years ago, we found that treatment of intact human red blood cells with Woodward's reagent K, followed by reductive cleavage of the active ester adduct with 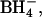 causes selective conversion of the side chain of E681 to an alcohol (Jennings and Anderson, 1987; Jennings and Smith, 1992); AE1 modified in this manner is designated here as E681OH AE1. The modification causes several major changes in AE1 function: 1), Monovalent anion exchange is inhibited (Jennings and Al-Rhaiyel, 1988); 2), divalent anion transport is accelerated and is much less dependent on pH than in native AE1 (Jennings and Al-Rhaiyel, 1988); and 3), the exchange of Cl− for
causes selective conversion of the side chain of E681 to an alcohol (Jennings and Anderson, 1987; Jennings and Smith, 1992); AE1 modified in this manner is designated here as E681OH AE1. The modification causes several major changes in AE1 function: 1), Monovalent anion exchange is inhibited (Jennings and Al-Rhaiyel, 1988); 2), divalent anion transport is accelerated and is much less dependent on pH than in native AE1 (Jennings and Al-Rhaiyel, 1988); and 3), the exchange of Cl− for 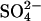 is an electrogenic 1:1 exchange, with no H+ cotransport, in E681OH AE1 (Jennings, 1995). Chernova et al. (1997) extended these studies by showing that mutagenesis of mouse AE1 E699 (equivalent to human E681) to glutamine stimulates
is an electrogenic 1:1 exchange, with no H+ cotransport, in E681OH AE1 (Jennings, 1995). Chernova et al. (1997) extended these studies by showing that mutagenesis of mouse AE1 E699 (equivalent to human E681) to glutamine stimulates 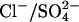 exchange and converts the process from electroneutral to electrogenic. All these findings are consistent with the idea that E681 of human AE1 binds the H+ that is cotransported with
exchange and converts the process from electroneutral to electrogenic. All these findings are consistent with the idea that E681 of human AE1 binds the H+ that is cotransported with 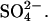 In keeping with this idea, E681 is believed to be located in the interior of the membrane at a position that is near the permeability barrier (Tang et al., 1998).
In keeping with this idea, E681 is believed to be located in the interior of the membrane at a position that is near the permeability barrier (Tang et al., 1998).
FIGURE 1.

(Upper left) Cycle for exchange of Cl− for 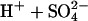 in normal AE1.
in normal AE1. 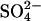 and H+ bind to the transporter in either order (Milanick and Gunn, 1982). Two protein-bound positive charges are depicted to emphasize the idea that the translocation events for both Cl− and
and H+ bind to the transporter in either order (Milanick and Gunn, 1982). Two protein-bound positive charges are depicted to emphasize the idea that the translocation events for both Cl− and 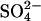 appear to be electroneutral (Jennings et al., 1990; Jennings, 1995). (Lower left) Cycle for the exchange of two Cl− + H+ for Cl− (Jennings, 1978; Lepke et al., 2003) in native AE1. This cycle is similar to that for H+-
appear to be electroneutral (Jennings et al., 1990; Jennings, 1995). (Lower left) Cycle for the exchange of two Cl− + H+ for Cl− (Jennings, 1978; Lepke et al., 2003) in native AE1. This cycle is similar to that for H+-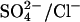 exchange, except that two Cl− ions instead of a single
exchange, except that two Cl− ions instead of a single 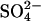 are translocated by the low pH (E681-protonated) form of AE1. The net result is electroneutral cotransport of H+ + Cl−. (Upper right) Ping-pong cycle for
are translocated by the low pH (E681-protonated) form of AE1. The net result is electroneutral cotransport of H+ + Cl−. (Upper right) Ping-pong cycle for 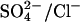 exchange in E681OH AE1. The charge state of E681OH band 3 at pH 7.4 is the same as that of native AE1 at low pH, i.e., with E681 protonated. The main charge-carrying event appears to be the Cl− limb of the cycle (Jennings, 1995). (Lower right) Hypothetical catalytic cycle for 2Cl −/Cl− exchange through E681OH AE1. By analogy to H+-Cl− cotransport in normal AE1, two Cl− are translocated through E681OH AE1, and the transport of a single Cl− in the opposite direction completes the cycle. There is no proton cotransport because E681OH is no longer reversibly protonated.
exchange in E681OH AE1. The charge state of E681OH band 3 at pH 7.4 is the same as that of native AE1 at low pH, i.e., with E681 protonated. The main charge-carrying event appears to be the Cl− limb of the cycle (Jennings, 1995). (Lower right) Hypothetical catalytic cycle for 2Cl −/Cl− exchange through E681OH AE1. By analogy to H+-Cl− cotransport in normal AE1, two Cl− are translocated through E681OH AE1, and the transport of a single Cl− in the opposite direction completes the cycle. There is no proton cotransport because E681OH is no longer reversibly protonated.
In addition to cotransporting H+ with 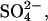 AE1 also mediates H+/Cl− cotransport (Jennings, 1978; Lepke et al., 2003). One possible mechanism of H+/Cl− cotransport is that at low pH the E681-protonated form of AE1 can bind and transport two Cl− ions (Fig. 1, lower left), resulting in the exchange of two Cl− + one H+ for one Cl−. H+/Cl− cotransport is inhibited in E681OH AE1 (Lepke et al., 2003), as would be expected if E681 normally participates in H+/Cl− cotransport. The binding and transport of 2 Cl− ions by the low-pH (E681-protonated) form of AE1 was proposed many years ago as a potential mechanism of H+/Cl− cotransport (Jennings, 1978). At that time it was known that there is at least one binding site for Cl− in addition to the main transport site (Dalmark, 1976). However, this binding site is an inhibitory “modifier” site (Knauf, 1979; Knauf and Mann, 1986), and there was no reason to propose that Cl− bound to this site could be transported.
AE1 also mediates H+/Cl− cotransport (Jennings, 1978; Lepke et al., 2003). One possible mechanism of H+/Cl− cotransport is that at low pH the E681-protonated form of AE1 can bind and transport two Cl− ions (Fig. 1, lower left), resulting in the exchange of two Cl− + one H+ for one Cl−. H+/Cl− cotransport is inhibited in E681OH AE1 (Lepke et al., 2003), as would be expected if E681 normally participates in H+/Cl− cotransport. The binding and transport of 2 Cl− ions by the low-pH (E681-protonated) form of AE1 was proposed many years ago as a potential mechanism of H+/Cl− cotransport (Jennings, 1978). At that time it was known that there is at least one binding site for Cl− in addition to the main transport site (Dalmark, 1976). However, this binding site is an inhibitory “modifier” site (Knauf, 1979; Knauf and Mann, 1986), and there was no reason to propose that Cl− bound to this site could be transported.
The possibility that two anions can be transported in the same direction by AE1 was not given much further attention until a recent study by Passow and coworkers (Lepke et al., 2003), which showed that the kinetics of AE1-mediated H+/Cl− cotransport are consistent with a model in which Cl− can bind with low affinity to a second transport site, and two Cl− ions are cotransported with H+ in exchange for a single Cl− ion, as depicted in Fig. 1. Moreover, Salhany et al. (2003) have recently presented evidence that in E681OH AE1 there is a new moderate-affinity Cl− binding site that modulates the displacement of stilbenedisulfonate inhibitors from AE1.
This article examines the kinetics of Cl− and 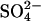 transport in E681OH AE1. We find several results that are consistent with the idea that removal of the negative charge on E681 causes the appearance of a new Cl− binding/transport site:
transport in E681OH AE1. We find several results that are consistent with the idea that removal of the negative charge on E681 causes the appearance of a new Cl− binding/transport site:
Outward Cl− conductance is inhibited by removal of extracellular Cl−, as expected if the conductance consists in part of electrogenic 2:1 Cl−/Cl− exchange.
Extracellular Cl− accelerates
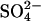 efflux by a mechanism other than recruitment of transporters from the outward to the inward state.
efflux by a mechanism other than recruitment of transporters from the outward to the inward state.Bilateral Cl− stimulates
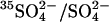 exchange, and extracellular Cl− stimulates unidirectional
exchange, and extracellular Cl− stimulates unidirectional 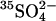 influx.
influx.
These results can be explained by a model in which E681OH AE1 has a site at which extracellular Cl− can bind and either be cotransported with or facilitate the extracellular release of SO42− bound to the main transport site. Finally, the possibility that the anomalous kinetics of anion transport in E681OH AE1 are a consequence of altered subunit interactions in the AE1 dimer (Salhany et al., 2003) was tested by graded chemical modification; the data are completely consistent with the idea that the second Cl− binding site and the main anion transport site are on the same subunit of the E681OH AE1 dimer.
MATERIALS AND METHODS
Materials
Human blood was drawn into heparin by venipuncture from healthy adults and was stored as whole blood at 4°C for up to 1 week before use. Gramicidin (87% gramicidin A) was purchased from Calbiochem (San Diego, CA). H2DIDS (4,4′-diisothiocyanatodihydrosilbene-2,2′-disulfonate) was synthesized from DADS (4,4′-diaminostilbene-2,2′-disulfonate) as described previously (Jennings et al., 1984). Woodward's reagent K (N-ethyl-5-phenylisoxazolium-3′-sulfonate) was purchased from Sigma (St. Louis, MO). All other salts and buffers were obtained from either Sigma or Fisher Scientific (Pittsburgh, PA). Radionuclides ( , Na36Cl, and 86RbCl) were from DuPont NEN (Boston, MA).
, Na36Cl, and 86RbCl) were from DuPont NEN (Boston, MA).
Treatment of cells with Woodward's reagent K
Cells were washed and modified with 2 mM Woodward's reagent K (WRK) and NaBH4 at 0°C as described previously (Jennings, 1995). This procedure converts ∼75% of the copies of AE1 to E681OH. The remainder of the copies of AE1 are either unmodified or contain uncleaved WRK adduct and are functionally silent in the transport assays used here. In experiments involving Cl− gradients, in which it was important to inhibit  exchange as much as possible, two successive exposures to WRK at 0°C were made before
exchange as much as possible, two successive exposures to WRK at 0°C were made before  addition; this method results in the modification of ∼95% of the copies of AE1 (Jennings, 1995).
addition; this method results in the modification of ∼95% of the copies of AE1 (Jennings, 1995).
Cl− conductive efflux
The conductive Cl− permeability was estimated from the 86Rb+ efflux mediated by gramicidin. The method was a variation on those used in other laboratories (Knauf et al., 1977; Hunter, 1977; Fröhlich et al., 1983). Cells were loaded with 86Rb+ by incubating for 1 h at 37°C in HEPES-buffered physiological saline (140 mM NaCl, 5 mM KCl, 1 mM Na-phosphate, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, pH 7.4, 10 mM glucose). Cells were then treated with  at 0°C in 150 mM KCl/MOPS, pH 7.0, as described above. The gramicidin-mediated efflux of 86Rb+ was measured in a medium in which all Na+ and K+ were replaced by impermeant N-methylglucamine (NMG). The medium consisted of mixtures of 150 mM NMG-glutamate and 150 mM NMG-Cl, buffered at pH 7.0 with 10 mM NMG-MOPS. Gramicidin A was added to a final concentration of 20 nM, and the efflux of 86Rb+ was measured at 20°C by centrifuging aliquots and measuring radioactivity in the supernatant. The rate constant (min−1) for 86Rb+ efflux was calculated as described previously (Jennings, 1995).
at 0°C in 150 mM KCl/MOPS, pH 7.0, as described above. The gramicidin-mediated efflux of 86Rb+ was measured in a medium in which all Na+ and K+ were replaced by impermeant N-methylglucamine (NMG). The medium consisted of mixtures of 150 mM NMG-glutamate and 150 mM NMG-Cl, buffered at pH 7.0 with 10 mM NMG-MOPS. Gramicidin A was added to a final concentration of 20 nM, and the efflux of 86Rb+ was measured at 20°C by centrifuging aliquots and measuring radioactivity in the supernatant. The rate constant (min−1) for 86Rb+ efflux was calculated as described previously (Jennings, 1995).
The gramicidin-mediated efflux of 86Rb+ was assumed to follow the constant field equation (Goldman, 1943; Hodgkin and Katz, 1949):
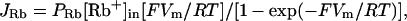 |
(1) |
where JRb is the efflux (nmol/ml cells/min) of 86Rb+, PRb is the permeability coefficient for Rb+ (min−1), [Rb+] is the intracellular Rb+ concentration (nmol/ml cells), Vm is the membrane potential, F is Faraday's constant, R is the gas constant, and T is the absolute temperature. The efflux of 86Rb+ into a 150 mM KCl medium provided a good estimate of PRb in the same cell preparation, same hematocrit (2%), and same gramicidin concentration, because in the high-K+ medium the membrane potential is ∼0 and Eq. 1 is reduced to JRb = PRb[Rb+]in.
In the absence of extracellular permeant cations, the membrane potential depends mainly on the concentrations and permeability coefficients of intracellular K+, intracellular Cl−, and extracellular Cl−. (Rb+ is present in trace amounts and does not itself affect the membrane potential; at neutral pH, H+ conductance through gramicidin is not significant, as indicated by the very low 86Rb+ efflux from  -loaded cells in an NMG-
-loaded cells in an NMG- medium.) Therefore, the membrane potential in NMG medium is given by
medium.) Therefore, the membrane potential in NMG medium is given by
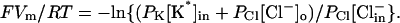 |
(2) |
In these experiments, [K*]in and [Cl−]in are both ∼140 mM, and the only variable is [Cl−]o. The Cl− conductive permeability coefficient PCl was determined at each value of [Cl−]o from the measured rates of 86Rb+ efflux, the measured PRb, and Eqs. 1 and 2.
35SO42− and 36Cl− Efflux
Cells were loaded with  by washing three times in at least 20 cell volumes of 80 mM K2SO4, 10 mM HEPES, pH 7.4, with a 10-min incubation at 37°C before each centrifugation to allow Cl− efflux and
by washing three times in at least 20 cell volumes of 80 mM K2SO4, 10 mM HEPES, pH 7.4, with a 10-min incubation at 37°C before each centrifugation to allow Cl− efflux and  influx (Jennings, 1995). Cells were loaded with
influx (Jennings, 1995). Cells were loaded with 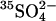 by incubating at 30% hematocrit in 80 mM K2SO4, 10 mM HEPES, pH 7.4, plus 10 μCi/ml
by incubating at 30% hematocrit in 80 mM K2SO4, 10 mM HEPES, pH 7.4, plus 10 μCi/ml  . Efflux was performed as described previously (Jennings, 1995) in media specified in the figure legends. The efflux of 36Cl− was measured by the method of Ku et al. (1979) at 0°C in cells at Donnan equilibrium in 150 mM KCl, 10 MOPS, pH 7.0.
. Efflux was performed as described previously (Jennings, 1995) in media specified in the figure legends. The efflux of 36Cl− was measured by the method of Ku et al. (1979) at 0°C in cells at Donnan equilibrium in 150 mM KCl, 10 MOPS, pH 7.0.
Preparation of low-SO42− cells
The following method was used to prepare intact cells containing a low concentration (∼0.5 mM) of  and no other permeant anion. Cells were treated with 2 mM
and no other permeant anion. Cells were treated with 2 mM  as usual, and then incubated 30 min, 37°C, with 10-μg/ml cells of gramicidin A in at least 20 volumes of HEPES-buffered 150 mM K-gluconate. In this medium there is net loss of KCl driven by the outward Cl− gradient; there is also cellular alkalinization caused by Cl− exchange with traces of
as usual, and then incubated 30 min, 37°C, with 10-μg/ml cells of gramicidin A in at least 20 volumes of HEPES-buffered 150 mM K-gluconate. In this medium there is net loss of KCl driven by the outward Cl− gradient; there is also cellular alkalinization caused by Cl− exchange with traces of  in the medium. After the 30-min depletion of Cl−, cells were centrifuged, resuspended in 50 mM K-gluconate, 0.5 mM K2SO4, 10 mM HEPES, pH 7.4, and incubated 30 min further at room temperature. Cells were washed once more in this medium and then incubated at least 20 min further in the same medium plus 1 μCi/ml
in the medium. After the 30-min depletion of Cl−, cells were centrifuged, resuspended in 50 mM K-gluconate, 0.5 mM K2SO4, 10 mM HEPES, pH 7.4, and incubated 30 min further at room temperature. Cells were washed once more in this medium and then incubated at least 20 min further in the same medium plus 1 μCi/ml  . The intracellular
. The intracellular  concentration (measured as the distribution of
concentration (measured as the distribution of  ) was ∼1.1 times the extracellular concentration. The Donnan ratio in these cells was therefore near unity despite the low concentration of permeant anion, because the negative charge on 50 mM extracellular gluconate balances the impermeant intracellular negative charge from hemoglobin and organic phosphates. The cells were also slightly shrunken (0.68 g H2O/ml cells versus normal of 0.71 g H2O/ml cells).
) was ∼1.1 times the extracellular concentration. The Donnan ratio in these cells was therefore near unity despite the low concentration of permeant anion, because the negative charge on 50 mM extracellular gluconate balances the impermeant intracellular negative charge from hemoglobin and organic phosphates. The cells were also slightly shrunken (0.68 g H2O/ml cells versus normal of 0.71 g H2O/ml cells).
Preparation of resealed ghosts
Ghosts were prepared from control or 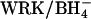 -treated cells by the method of Schwoch and Passow (1973). Lysis was at 0°C in 20 volumes of 4 mM MgSO4, 1.2 mM acetic acid, followed by addition at 0°C of concentrated stock solutions to produce final concentrations of 40 mM K2SO4, 10 mM HEPES, pH 7.4, and 0–40 mM Cl−. Ghosts were incubated in these media 45 min at 37°C for resealing, and then washed and loaded with 1 μCi/ml
-treated cells by the method of Schwoch and Passow (1973). Lysis was at 0°C in 20 volumes of 4 mM MgSO4, 1.2 mM acetic acid, followed by addition at 0°C of concentrated stock solutions to produce final concentrations of 40 mM K2SO4, 10 mM HEPES, pH 7.4, and 0–40 mM Cl−. Ghosts were incubated in these media 45 min at 37°C for resealing, and then washed and loaded with 1 μCi/ml 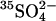 in the same media as was used for resealing. Finally, ghosts were washed twice at 0°C to remove external radioactivity, and the efflux of
in the same media as was used for resealing. Finally, ghosts were washed twice at 0°C to remove external radioactivity, and the efflux of 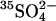 was measured in the same medium at 20°C.
was measured in the same medium at 20°C.
RESULTS
Test of the hypothesis that E681OH AE1 can mediate 2:1 Cl−/Cl− exchange
It is well established that E681OH AE1 (or mouse E699Q AE1) can carry out electrogenic 1:1 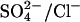 exchange (Jennings, 1995; Chernova et al., 1997). Therefore, E681OH AE1 is capable of binding and transporting either a single
exchange (Jennings, 1995; Chernova et al., 1997). Therefore, E681OH AE1 is capable of binding and transporting either a single 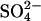 or a single Cl− ion. However, the fact that
or a single Cl− ion. However, the fact that 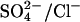 exchange is electrogenic does not rule out the possibility that there are translocation events involving two Cl− ions in E681OH AE1. Such events must be less frequent than single Cl− transport events, because
exchange is electrogenic does not rule out the possibility that there are translocation events involving two Cl− ions in E681OH AE1. Such events must be less frequent than single Cl− transport events, because 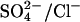 exchange is electrogenic.
exchange is electrogenic.
If E681OH AE1 mediates translocation events in which two Cl− ions are cotransported (with no proton cotransport), the resultant exchange of two Cl− for one Cl− would be electrogenic and may account for the large (∼8-fold higher than normal) H2DIDS-sensitive Cl− conductive flux that is observed in E681OH (Jennings, 1995). A conductive outward Cl− flux resulting from 2:1 exchange should be inhibited by removal of extracellular Cl−, because in the absence of extracellular substrate the catalytic cycle should be arrested by the formation of empty outward-facing transporters (Fig. 1, lower right). To test this idea, the Cl− conductance was estimated by measuring the gramicidin-mediated efflux of 86Rb+ in media containing impermeant N-methyl glucamine as the only cation other than H+.
Fig. 2 depicts the results of four experiments in which PCl was estimated in 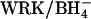 -treated red cells in the presence of varying concentrations of extracellular Cl− (glutamate substitute). In all cases the initial intracellular Cl− concentration was 120–140 mM (Donnan equilibrium, pH 7). The PCl at each extracellular Cl− concentration is plotted relative to the PCl measured in the same cells in the absence of extracellular Cl−. In all cases the addition of extracellular Cl− causes an increase in PCl. In contrast, PCl in control cells is inhibited ∼50% by addition of a relatively low concentration of Cl− (10 mM) to a Cl−-free medium (open symbols), in excellent agreement with the data of Fröhlich et al. (1983). The dependence of PCl on extracellular Cl− in E681OH AE1 is not absolute; there is a significant conductive Cl− efflux even in the absence of extracellular Cl−. However, there is a clear acceleration of the conductive Cl− efflux by the addition of Cl− to the extracellular medium, as expected if a component of the outward conductive flux takes place as 2:1 Cl−/Cl− exchange.
-treated red cells in the presence of varying concentrations of extracellular Cl− (glutamate substitute). In all cases the initial intracellular Cl− concentration was 120–140 mM (Donnan equilibrium, pH 7). The PCl at each extracellular Cl− concentration is plotted relative to the PCl measured in the same cells in the absence of extracellular Cl−. In all cases the addition of extracellular Cl− causes an increase in PCl. In contrast, PCl in control cells is inhibited ∼50% by addition of a relatively low concentration of Cl− (10 mM) to a Cl−-free medium (open symbols), in excellent agreement with the data of Fröhlich et al. (1983). The dependence of PCl on extracellular Cl− in E681OH AE1 is not absolute; there is a significant conductive Cl− efflux even in the absence of extracellular Cl−. However, there is a clear acceleration of the conductive Cl− efflux by the addition of Cl− to the extracellular medium, as expected if a component of the outward conductive flux takes place as 2:1 Cl−/Cl− exchange.
FIGURE 2.

Stimulation by extracellular Cl− of outward Cl− conductance in E681OH AE1. Cells were loaded with 86Rb+ in HEPES-buffered physiological saline and then treated with 2 mM WRK (two exposures) and finally 2 mM 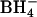 (Jennings, 1995). The efflux of 86Rb+ was measured in 150 mM NMG glutamate, 10 mM NMG-MOPS, pH 7.0, 20°C, after the addition of 20 nM gramicidin A. The extracellular Cl− concentration was varied by substituting the indicated concentration of NMG-Cl for NMG-glutamate. The conductive Cl− permeability coefficient was calculated as described in Methods and is plotted as percent of PCl measured in the same cell preparation in a Cl−-free medium. The figure shows the results of duplicate flux measurements at the indicated Cl− concentration for four separate preparations of E681OH red cells (solid symbols) and one preparation of untreated cells (open symbols). The conductive Cl− flux is inhibited almost entirely by 20 μM H2DIDS (solid symbols marked D).
(Jennings, 1995). The efflux of 86Rb+ was measured in 150 mM NMG glutamate, 10 mM NMG-MOPS, pH 7.0, 20°C, after the addition of 20 nM gramicidin A. The extracellular Cl− concentration was varied by substituting the indicated concentration of NMG-Cl for NMG-glutamate. The conductive Cl− permeability coefficient was calculated as described in Methods and is plotted as percent of PCl measured in the same cell preparation in a Cl−-free medium. The figure shows the results of duplicate flux measurements at the indicated Cl− concentration for four separate preparations of E681OH red cells (solid symbols) and one preparation of untreated cells (open symbols). The conductive Cl− flux is inhibited almost entirely by 20 μM H2DIDS (solid symbols marked D).
Trans acceleration of SO42− efflux by Cl− in E681OH AE1
The idea that E681OH AE1 has two possible transport sites for Cl− raises the possibility that the second Cl− binding/transport site may be responsible for some of the effects of Cl− on  in E681OH AE1 that were reported previously (Jennings, 1995). One such effect is a remarkably large trans accelerating effect of extracellular Cl− on
in E681OH AE1 that were reported previously (Jennings, 1995). One such effect is a remarkably large trans accelerating effect of extracellular Cl− on 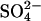 efflux; replacement of 80 mM extracellular
efflux; replacement of 80 mM extracellular 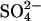 with 120 mM Cl− causes a 20-fold acceleration of the
with 120 mM Cl− causes a 20-fold acceleration of the 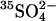 efflux. This trans-acceleration of
efflux. This trans-acceleration of 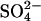 efflux by extracellular Cl− is not a consequence of an effect of the Cl− gradient on the membrane potential, because a large trans-acceleration is observed even if the membrane potential is clamped near zero with gramidicin (Jennings, 1995).
efflux by extracellular Cl− is not a consequence of an effect of the Cl− gradient on the membrane potential, because a large trans-acceleration is observed even if the membrane potential is clamped near zero with gramidicin (Jennings, 1995).
A ping-pong anion exchange mechanism can, in principle, explain trans-acceleration on the basis of “recruitment” by the inward Cl− gradient of transporters from the outward-facing to the inward-facing state (Knauf, 1979; Gunn and Fröhlich, 1979; Jennings, 1980). For example, if the transporters are symmetrically distributed in the presence of saturating concentrations of 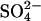 on both sides of the membrane, then Cl− should cause a twofold trans acceleration in each direction. This is very nearly what is observed in normal cells for
on both sides of the membrane, then Cl− should cause a twofold trans acceleration in each direction. This is very nearly what is observed in normal cells for 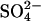 at neutral pH (Jennings, 1980). In E681OH cells, however, the asymmetry of the trans acceleration is quite different from normal cells. At pH 7.4, replacement of extracellular
at neutral pH (Jennings, 1980). In E681OH cells, however, the asymmetry of the trans acceleration is quite different from normal cells. At pH 7.4, replacement of extracellular 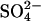 with Cl− causes an ∼20-fold acceleration of
with Cl− causes an ∼20-fold acceleration of 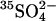 efflux; replacement of intracellular
efflux; replacement of intracellular  with Cl− accelerates
with Cl− accelerates 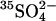 influx by a factor of ∼3 (Jennings, 1995). These magnitudes of trans acceleration are too large to be explained by a ping-pong/recruitment mechanism.
influx by a factor of ∼3 (Jennings, 1995). These magnitudes of trans acceleration are too large to be explained by a ping-pong/recruitment mechanism.
To examine further the mechanism of the large stimulation of  efflux by extracellular Cl− in E681OH AE1, we used conditions in which single turnovers of the catalytic cycle should be detectable. Because of the large number of copies (∼106/cell) of AE1 in red cells (Fairbanks et al., 1971), it is possible to prepare intact cells in which
efflux by extracellular Cl− in E681OH AE1, we used conditions in which single turnovers of the catalytic cycle should be detectable. Because of the large number of copies (∼106/cell) of AE1 in red cells (Fairbanks et al., 1971), it is possible to prepare intact cells in which 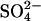 is the only permeant anion, and the initial amount of intracellular
is the only permeant anion, and the initial amount of intracellular 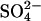 is not much higher than the number of copies of AE1. Cells containing 0.5 mM
is not much higher than the number of copies of AE1. Cells containing 0.5 mM 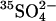 and much lower concentrations (<0.01 mM) of Cl− and
and much lower concentrations (<0.01 mM) of Cl− and  were prepared by using gramicidin to lower the total cellular ion contents following treatment with
were prepared by using gramicidin to lower the total cellular ion contents following treatment with  (see Methods). These cells were suspended at 0°C in Cl−-free,
(see Methods). These cells were suspended at 0°C in Cl−-free, 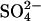 -free 50 mM K-gluconate, 10 mM HEPES, pH 7.4, and the time course of
-free 50 mM K-gluconate, 10 mM HEPES, pH 7.4, and the time course of 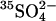 efflux was measured. The efflux is initially slow because of the absence of an exchangeable extracellular anion (Fig. 3). Addition of 25 mM Cl− to the medium causes an immediate increase in the efflux, reflecting the rapid rate of
efflux was measured. The efflux is initially slow because of the absence of an exchangeable extracellular anion (Fig. 3). Addition of 25 mM Cl− to the medium causes an immediate increase in the efflux, reflecting the rapid rate of 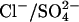 exchange in E681OH AE1, even at 0°C. At the arrow, the suspension was diluted by a factor of 10 into Cl−-free 50 mM K-gluconate/HEPES medium, thereby lowering the extracellular Cl− concentration to 2.5 mM, and further time points were taken.
exchange in E681OH AE1, even at 0°C. At the arrow, the suspension was diluted by a factor of 10 into Cl−-free 50 mM K-gluconate/HEPES medium, thereby lowering the extracellular Cl− concentration to 2.5 mM, and further time points were taken.
FIGURE 3.

Efflux of  from E681OH erythrocytes that had been treated with gramicidin and equilibrated with a medium consisting of 50 mM K-gluconate, 0.5 mM [35S]K2SO4, 10 mM HEPES, pH 7.4. Efflux was initiated by suspending cells in a medium containing 50 mM K-gluconate, 10 mM HEPES, pH 7.4, and no permeant anion. At the first arrow, KCl was added to a final concentration of 25 mM to initiate rapid efflux of
from E681OH erythrocytes that had been treated with gramicidin and equilibrated with a medium consisting of 50 mM K-gluconate, 0.5 mM [35S]K2SO4, 10 mM HEPES, pH 7.4. Efflux was initiated by suspending cells in a medium containing 50 mM K-gluconate, 10 mM HEPES, pH 7.4, and no permeant anion. At the first arrow, KCl was added to a final concentration of 25 mM to initiate rapid efflux of 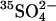 . The inverted triangles represent the time course of efflux in the presence of 25 mM Cl−. At the second arrow, an aliquot of the suspension was diluted 10-fold into 50 mM K-gluconate/10 mM HEPES, pH 7.4; the squares represent the time course of efflux of
. The inverted triangles represent the time course of efflux in the presence of 25 mM Cl−. At the second arrow, an aliquot of the suspension was diluted 10-fold into 50 mM K-gluconate/10 mM HEPES, pH 7.4; the squares represent the time course of efflux of 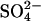 from the suspension after reduction of extracellular Cl− to 2.5 mM. The dashed curve represents the expected time course of extracellular
from the suspension after reduction of extracellular Cl− to 2.5 mM. The dashed curve represents the expected time course of extracellular 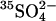 if AE1 were initially (in the high Cl− medium) in the inward-facing conformation, and one efflux event per copy of AE1 took place after reduction of extracellular Cl−.
if AE1 were initially (in the high Cl− medium) in the inward-facing conformation, and one efflux event per copy of AE1 took place after reduction of extracellular Cl−.
Within the time resolution of our measurements, we find that the rate of 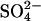 efflux is reduced immediately upon lowering the extracellular Cl− concentration. The initial amount of
efflux is reduced immediately upon lowering the extracellular Cl− concentration. The initial amount of 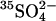 in these cells (0.5 mM) was only ∼20 times the amount of AE1 polypeptide, assuming 1.2 × 106 copies per cell (Fairbanks et al., 1971; Passow, 1986). If 25 mM Cl− accelerates
in these cells (0.5 mM) was only ∼20 times the amount of AE1 polypeptide, assuming 1.2 × 106 copies per cell (Fairbanks et al., 1971; Passow, 1986). If 25 mM Cl− accelerates 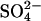 efflux by recruiting most of the transporters to the inward-facing state, then there should be a continuing rapid efflux of ∼5% of the initial cellular
efflux by recruiting most of the transporters to the inward-facing state, then there should be a continuing rapid efflux of ∼5% of the initial cellular  after reduction of the extracellular Cl− concentration, because the inward-facing transporters should be able to perform one more
after reduction of the extracellular Cl− concentration, because the inward-facing transporters should be able to perform one more 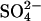 efflux event before the cycle slows down in the low-Cl− medium. Experimentally, we detected no delay in the reduction of
efflux event before the cycle slows down in the low-Cl− medium. Experimentally, we detected no delay in the reduction of 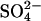 efflux after a reduction of extracellular [Cl−] (eight effluxes on four cell preparations). There is no electrical constraint on the efflux because the cells had been treated with gramicidin to raise the K+ conductance. This finding indicates that extracellular Cl− accelerates
efflux after a reduction of extracellular [Cl−] (eight effluxes on four cell preparations). There is no electrical constraint on the efflux because the cells had been treated with gramicidin to raise the K+ conductance. This finding indicates that extracellular Cl− accelerates 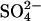 efflux in E681OH AE1 by a mechanism other than recruitment of transporters to the inward-facing state.
efflux in E681OH AE1 by a mechanism other than recruitment of transporters to the inward-facing state.
Acceleration of SO42− equilibrium exchange by bilateral Cl−
Further evidence of anomalous transport kinetics in E681OH AE1 comes from the effects of bilateral Cl− on 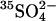 fluxes. We had previously reported the preliminary finding that bilateral 10–20 mM Cl− can accelerate the rate constant for
fluxes. We had previously reported the preliminary finding that bilateral 10–20 mM Cl− can accelerate the rate constant for 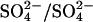 exchange in E681OH AE1 by a factor of nearly 2 (Jennings, 1995). These earlier studies used intact cells, in which the intracellular
exchange in E681OH AE1 by a factor of nearly 2 (Jennings, 1995). These earlier studies used intact cells, in which the intracellular  concentration was not well controlled when Cl− was varied. We have subsequently used resealed ghosts to examine this issue in a more rigorous way. Ghosts from control or E681OH cells were resealed in media containing 40 mM K2SO4, 10 mM HEPES, pH 7.4, plus 0–40 mM KCl, and then loaded with
concentration was not well controlled when Cl− was varied. We have subsequently used resealed ghosts to examine this issue in a more rigorous way. Ghosts from control or E681OH cells were resealed in media containing 40 mM K2SO4, 10 mM HEPES, pH 7.4, plus 0–40 mM KCl, and then loaded with  in the resealing medium. The efflux of
in the resealing medium. The efflux of 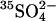 was measured in the same medium, i.e., under conditions of no net ion flux. The presence of Cl− on both sides of the membrane causes a clear acceleration of the
was measured in the same medium, i.e., under conditions of no net ion flux. The presence of Cl− on both sides of the membrane causes a clear acceleration of the 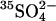 efflux (Fig. 4). A similar acceleration is observed if ghosts are first resealed in a Cl−-free 40 mM K2SO4 medium and Cl− is subsequently introduced by incubating the ghosts with varying concentrations of NH4Cl, which can enter the ghosts as NH3 influx followed by
efflux (Fig. 4). A similar acceleration is observed if ghosts are first resealed in a Cl−-free 40 mM K2SO4 medium and Cl− is subsequently introduced by incubating the ghosts with varying concentrations of NH4Cl, which can enter the ghosts as NH3 influx followed by 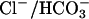 exchange with CO2 recycling to result in net NH4Cl influx (Jacobs and Stewart, 1942). This acceleration of
exchange with CO2 recycling to result in net NH4Cl influx (Jacobs and Stewart, 1942). This acceleration of 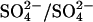 exchange by bilateral Cl− was observed only in E681OH AE1; in native human red cells, we find that bilateral Cl− always inhibits
exchange by bilateral Cl− was observed only in E681OH AE1; in native human red cells, we find that bilateral Cl− always inhibits  transport, as is well known (Passow, 1986).
transport, as is well known (Passow, 1986).
FIGURE 4.

Stimulation of 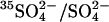 exchange through E681OH AE1 by bilateral Cl− in resealed ghosts. Cells were treated with 2 mM WRK followed by 2 mM
exchange through E681OH AE1 by bilateral Cl− in resealed ghosts. Cells were treated with 2 mM WRK followed by 2 mM  . Ghosts were prepared and resealed in a medium containing 40 mM K2SO4, 10 mM HEPES, pH 7.4, and 0–40 mM Cl−, added either as KCl at 0°C before resealing (▴, ♦, ▪) or as NH4Cl after resealing and before
. Ghosts were prepared and resealed in a medium containing 40 mM K2SO4, 10 mM HEPES, pH 7.4, and 0–40 mM Cl−, added either as KCl at 0°C before resealing (▴, ♦, ▪) or as NH4Cl after resealing and before  loading (•). Flux was measured at 20°C in media of the same composition as the resealing medium. Data represent mean and range of duplicate measurements at each Cl− concentration in three separate ghost preparations. Squares represent flux measured in the presence of 20 μM H2DIDS. The dotted curve represents the prediction of the model described in the Discussion.
loading (•). Flux was measured at 20°C in media of the same composition as the resealing medium. Data represent mean and range of duplicate measurements at each Cl− concentration in three separate ghost preparations. Squares represent flux measured in the presence of 20 μM H2DIDS. The dotted curve represents the prediction of the model described in the Discussion.
Acceleration of SO42− influx by extracellular Cl−
The data in Fig. 5 show that extracellular Cl−, in the initial absence of intracellular Cl−, stimulates unidirectional 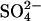 influx in E681OH AE1. This cis acceleration of a tracer anion flux by another anion is, to our knowledge, unprecedented in the literature on AE1. In control cells, 10 mM cis Cl− strongly inhibits unidirectional
influx in E681OH AE1. This cis acceleration of a tracer anion flux by another anion is, to our knowledge, unprecedented in the literature on AE1. In control cells, 10 mM cis Cl− strongly inhibits unidirectional 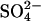 influx, and the inhibition is progressively relieved at later times as the inward Cl− gradient is dissipated, exactly as observed previously (Jennings, 1980). The acceleration of
influx, and the inhibition is progressively relieved at later times as the inward Cl− gradient is dissipated, exactly as observed previously (Jennings, 1980). The acceleration of  influx by extracellular Cl− in E681OH AE1 is therefore in very sharp contrast to the strong inhibition in native AE1.
influx by extracellular Cl− in E681OH AE1 is therefore in very sharp contrast to the strong inhibition in native AE1.
FIGURE 5.

Effect of extracellular Cl− on unidirectional 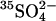 influx in control and E681OH AE1. (Left) Cells were treated with (•, ▴) and without (○, ▵) 2 mM
influx in control and E681OH AE1. (Left) Cells were treated with (•, ▴) and without (○, ▵) 2 mM 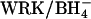 and then washed and equilibrated with Cl−-free, low-
and then washed and equilibrated with Cl−-free, low-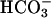 (N2-purged) medium consisting of 80 K2SO4, 10 HEPES, pH 7.4. Cells were then centrifuged under N2 and resuspended in the same medium containing 10 μCi
(N2-purged) medium consisting of 80 K2SO4, 10 HEPES, pH 7.4. Cells were then centrifuged under N2 and resuspended in the same medium containing 10 μCi 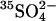 plus either 0 (•, ○) or 10 mM (▴, ▵) KCl. The influx of
plus either 0 (•, ○) or 10 mM (▴, ▵) KCl. The influx of  over the first 80 s was measured at 20°C. The experiments were carried out under N2 to minimize Cl− influx via
over the first 80 s was measured at 20°C. The experiments were carried out under N2 to minimize Cl− influx via 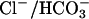 exchange. (Right) Initial
exchange. (Right) Initial 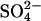 influx in two preparations of
influx in two preparations of 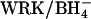 -treated cells, performed as in the left side of the figure, except 0, 8, or 16 mM KCl replaced 0, 4, or 8 mM K2SO4 in the extracellular medium. Different symbols represent different cell preparations. The square represents the influx in the presence of 20 μM H2DIDS. Data represent mean and range of duplicate determinations. The dotted line represents the prediction of the model described in Discussion.
-treated cells, performed as in the left side of the figure, except 0, 8, or 16 mM KCl replaced 0, 4, or 8 mM K2SO4 in the extracellular medium. Different symbols represent different cell preparations. The square represents the influx in the presence of 20 μM H2DIDS. Data represent mean and range of duplicate determinations. The dotted line represents the prediction of the model described in Discussion.
Increased apparent affinity for extracellular SO42− in E681OH AE1
In addition to accelerating 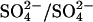 exchange, modification of E681 causes a large increase in the apparent affinity of AE1 for
exchange, modification of E681 causes a large increase in the apparent affinity of AE1 for 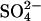 at outward-facing transport sites. Fig. 6 shows the
at outward-facing transport sites. Fig. 6 shows the 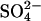 influx into Cl−-free,
influx into Cl−-free,  -loaded cells as a function of the extracellular
-loaded cells as a function of the extracellular 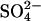 concentration in E681OH AE1. The flux is a saturable function of extracellular
concentration in E681OH AE1. The flux is a saturable function of extracellular 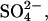 with K1/2 of 0.25–0.30 mM. The same K1/2 was measured for extracellular
with K1/2 of 0.25–0.30 mM. The same K1/2 was measured for extracellular  stimulation of
stimulation of  efflux from all-
efflux from all- cells into Cl−-free sucrose or gluconate media (two experiments, not shown). This K1/2 is much lower than that of control cells at pH 7.4 (Milanick and Gunn, 1982, 1984). Although the K1/2 for transport is not identical with the dissociation constant for substrate binding to transport sites, the low K1/2 suggests that the affinity of transport sites for extracellular
cells into Cl−-free sucrose or gluconate media (two experiments, not shown). This K1/2 is much lower than that of control cells at pH 7.4 (Milanick and Gunn, 1982, 1984). Although the K1/2 for transport is not identical with the dissociation constant for substrate binding to transport sites, the low K1/2 suggests that the affinity of transport sites for extracellular 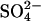 is considerably higher in E681OH AE1 than in normal protein. This finding is in agreement with previous work (Milanick and Gunn, 1982, 1984; Jennings, 1989), which showed that protonation of an acid-titratable group (probably E681) on AE1 causes a 10-fold increase in the apparent affinity for extracellular
is considerably higher in E681OH AE1 than in normal protein. This finding is in agreement with previous work (Milanick and Gunn, 1982, 1984; Jennings, 1989), which showed that protonation of an acid-titratable group (probably E681) on AE1 causes a 10-fold increase in the apparent affinity for extracellular 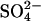 .
.
FIGURE 6.

Influx of  through E681OH AE1. Cells were pretreated with WRK/BH4. and then washed in Cl-free 100 mM K2SO4 medium to replace all intracellular Cl− with
through E681OH AE1. Cells were pretreated with WRK/BH4. and then washed in Cl-free 100 mM K2SO4 medium to replace all intracellular Cl− with 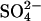 . Initial influx of
. Initial influx of 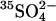 was measured at 20°C in 10 mM HEPES, pH 7.4, 250 mM sucrose, plus the indicated concentration of K2SO4. The curve through the data is derived from the model described in the Discussion.
was measured at 20°C in 10 mM HEPES, pH 7.4, 250 mM sucrose, plus the indicated concentration of K2SO4. The curve through the data is derived from the model described in the Discussion.
Acceleration of SO42− flux by WRK/BH4− following partial H2DIDS inhibition
Conversion of native AE1 to E681OH AE1 by treatment with WRK and  inhibits Cl−/Cl− exchange by over 90% and accelerates
inhibits Cl−/Cl− exchange by over 90% and accelerates 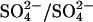 exchange (measured at pH 7.4 in an all-
exchange (measured at pH 7.4 in an all-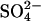 medium) by ∼7-fold (Jennings and Al-Rhaiyel, 1988; Jennings, 1995). It is well established that AE1 is a dimer, and Salhany et al. (2003) have recently found that the kinetics of inhibitor dissociation from E681OH AE1 can be explained by a model in which conversion of one subunit in the dimer to E681OH affects the kinetics of inhibitor release from the other subunit. In light of this finding, it is of interest to determine whether transport kinetics in AE1 during graded conversion to E681OH are consistent with independently functioning subunits.
medium) by ∼7-fold (Jennings and Al-Rhaiyel, 1988; Jennings, 1995). It is well established that AE1 is a dimer, and Salhany et al. (2003) have recently found that the kinetics of inhibitor dissociation from E681OH AE1 can be explained by a model in which conversion of one subunit in the dimer to E681OH affects the kinetics of inhibitor release from the other subunit. In light of this finding, it is of interest to determine whether transport kinetics in AE1 during graded conversion to E681OH are consistent with independently functioning subunits.
Fig. 7 shows the effects of graded  treatment on 36Cl−/Cl− exchange and
treatment on 36Cl−/Cl− exchange and 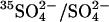 exchange. Cells were washed and treated at 0°C with 0–1.6 mM WRK followed by 1 mM
exchange. Cells were washed and treated at 0°C with 0–1.6 mM WRK followed by 1 mM 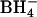 . Cells were then washed and split in half. One half was loaded with 36Cl−, and the equilibrium exchange flux was measured at 0°C in 150 mM KCl/MOPS, pH 7.0. The other half was washed and loaded with
. Cells were then washed and split in half. One half was loaded with 36Cl−, and the equilibrium exchange flux was measured at 0°C in 150 mM KCl/MOPS, pH 7.0. The other half was washed and loaded with 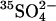 in 80 mM K2SO4, 10 mM HEPES, pH 7.4, and the efflux was measured at electrochemical equilibrium in the same medium at 20°C. Over a wide range of WRK concentrations, there is a linear relationship between the inhibition of Cl− exchange and acceleration of
in 80 mM K2SO4, 10 mM HEPES, pH 7.4, and the efflux was measured at electrochemical equilibrium in the same medium at 20°C. Over a wide range of WRK concentrations, there is a linear relationship between the inhibition of Cl− exchange and acceleration of  exchange. The linear relationship between acceleration of
exchange. The linear relationship between acceleration of 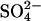 and inhibition of Cl− transport is consistent with the idea that modification of the same site is responsible for both the acceleration of
and inhibition of Cl− transport is consistent with the idea that modification of the same site is responsible for both the acceleration of  flux and inhibition of Cl− flux and that the presence of a modified subunit has no effect on either Cl− or
flux and inhibition of Cl− flux and that the presence of a modified subunit has no effect on either Cl− or  transport through the adjacent unmodified subunit in the dimer. At the highest levels of inhibition of Cl− flux and acceleration of
transport through the adjacent unmodified subunit in the dimer. At the highest levels of inhibition of Cl− flux and acceleration of 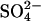 flux, there is a slight deviation from a linear relationship between the two fluxes. This slight deviation from linearity is very likely caused by inhibitory effects of secondary reactions (with residues other than E681) at high levels of modification (Jennings, 1995).
flux, there is a slight deviation from a linear relationship between the two fluxes. This slight deviation from linearity is very likely caused by inhibitory effects of secondary reactions (with residues other than E681) at high levels of modification (Jennings, 1995).
FIGURE 7.

Inhibition of 36Cl−/Cl− exchange and acceleration of 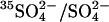 exchange by graded treatment with
exchange by graded treatment with 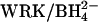 . Cells were washed in 150 mM KCl/10 mM MOPS, pH 7.0, and treated at 0°C with 0, 0.4, 0.6, 1.6 mM (•); 0, 0.08, 0.16, 0.32 mM (▾); or 0, 0.2, 0.4 mM (▴) WRK followed by two additions of 1 mM
. Cells were washed in 150 mM KCl/10 mM MOPS, pH 7.0, and treated at 0°C with 0, 0.4, 0.6, 1.6 mM (•); 0, 0.08, 0.16, 0.32 mM (▾); or 0, 0.2, 0.4 mM (▴) WRK followed by two additions of 1 mM  . Each suspension was then washed in KCl/MOPS and then split in half. One half was loaded with
. Each suspension was then washed in KCl/MOPS and then split in half. One half was loaded with 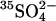 , and the rate constant for equilibrium exchange of
, and the rate constant for equilibrium exchange of  was measured in 80 mM K2SO4, 10 mM HEPES, pH 7.4. The other half was loaded with 36Cl− in 150 KCl, 10 mM MOPS, pH 7.0, and the efflux of 36Cl− was measured in the same medium at 0°C. For each concentration of WRK, the rate constant for
was measured in 80 mM K2SO4, 10 mM HEPES, pH 7.4. The other half was loaded with 36Cl− in 150 KCl, 10 mM MOPS, pH 7.0, and the efflux of 36Cl− was measured in the same medium at 0°C. For each concentration of WRK, the rate constant for  efflux is plotted on the vertical axis, and percent inhibition of 36Cl− efflux is plotted on the horizontal axis. Error bars denote the range of two determinations.
efflux is plotted on the vertical axis, and percent inhibition of 36Cl− efflux is plotted on the horizontal axis. Error bars denote the range of two determinations.
As an additional approach to studying the role of subunit interactions in anion transport through E681OH AE1, cells were pretreated with enough H2DIDS to inhibit 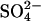 transport irreversibly by 0, 75%, or 90%. After treatment with H2DIDS, cells were treated with 2 mM
transport irreversibly by 0, 75%, or 90%. After treatment with H2DIDS, cells were treated with 2 mM  , washed, and loaded with
, washed, and loaded with 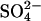 in a HEPES-buffered 80 mM
in a HEPES-buffered 80 mM  medium, and the efflux of
medium, and the efflux of  was measured under equilibrium conditions in the same medium. In excellent agreement with previous work (Jennings and Al-Rhaiyel, 1988; Jennings, 1995), 2 mM
was measured under equilibrium conditions in the same medium. In excellent agreement with previous work (Jennings and Al-Rhaiyel, 1988; Jennings, 1995), 2 mM 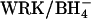 accelerates
accelerates 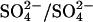 exchange at pH 7.4 by six- to sevenfold (Fig. 8). The same six- to sevenfold acceleration is observed if 75% or 90% of the AE1 subunits are irreversibly inhibited by H2DIDS before treatment with
exchange at pH 7.4 by six- to sevenfold (Fig. 8). The same six- to sevenfold acceleration is observed if 75% or 90% of the AE1 subunits are irreversibly inhibited by H2DIDS before treatment with 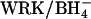 . In cells treated with these concentrations of H2DIDS, the majority of the AE1 dimers will have at least one subunit occupied with H2DIDS. Accordingly, the stimulation of
. In cells treated with these concentrations of H2DIDS, the majority of the AE1 dimers will have at least one subunit occupied with H2DIDS. Accordingly, the stimulation of 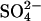 transport by
transport by 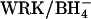 in a given subunit does not depend on having two functioning subunits of the AE1 dimer.
in a given subunit does not depend on having two functioning subunits of the AE1 dimer.
FIGURE 8.

Acceleration by 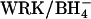 of
of  transport in cells pretreated with H2DIDS. Cells were washed in HEPES-buffered saline and incubated 1 h at 37°C with 0, 0.75, or 0.9 mol H2DIDS/mol AE1. Cells were then washed three times in 150 mM KCl, 10 mM MOPS, chilled, and treated with or without
transport in cells pretreated with H2DIDS. Cells were washed in HEPES-buffered saline and incubated 1 h at 37°C with 0, 0.75, or 0.9 mol H2DIDS/mol AE1. Cells were then washed three times in 150 mM KCl, 10 mM MOPS, chilled, and treated with or without 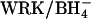 as in the previous figures. Cells were then washed and loaded with
as in the previous figures. Cells were then washed and loaded with 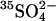 in all-
in all-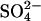 medium as previously, and the efflux of
medium as previously, and the efflux of 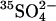 was measured in 80 mM K2SO4, 10 mM HEPES, pH 7.4, at 20°C. (Left) The rate constants for
was measured in 80 mM K2SO4, 10 mM HEPES, pH 7.4, at 20°C. (Left) The rate constants for  efflux. (Right) The factor by which
efflux. (Right) The factor by which  accelerates efflux for each preparation. Irrespective of whether transport is initially inhibited by 0, 75%, or 90% by H2DIDS, the acceleration by 2 mM
accelerates efflux for each preparation. Irrespective of whether transport is initially inhibited by 0, 75%, or 90% by H2DIDS, the acceleration by 2 mM 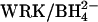 is six- to sevenfold.
is six- to sevenfold.
DISCUSSION
Second Cl− binding site in E681OH AE1
The data presented here provide two kinds of evidence that E681OH AE1 has a second Cl− binding/transport site that is distinct from the Cl− transport site in the native protein. The first evidence is that the H2DIDS-sensitive conductive efflux of Cl− is inhibited by removal of extracellular Cl−. This effect is the opposite of that found in the native protein (Fröhlich et al., 1983; Fröhlich, 1984) and is consistent with the idea that an electrogenic 2:1 Cl−/Cl− exchange represents part of the conductive Cl− efflux in E681OH AE1 (Fig. 2). The other evidence for a second Cl− binding site is that extracellular Cl− accelerates 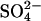 transport by a mechanism other than recruitment of transporters to inward-facing states (Figs. 3–5). This acceleration implies that there must be a Cl− binding site in E681OH AE1 that is distinct from the
transport by a mechanism other than recruitment of transporters to inward-facing states (Figs. 3–5). This acceleration implies that there must be a Cl− binding site in E681OH AE1 that is distinct from the 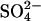 transport site, and that Cl− binding to this site accelerates the catalytic cycle for
transport site, and that Cl− binding to this site accelerates the catalytic cycle for 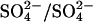 exchange.
exchange.
Although we refer to the new Cl− site in E681OH AE1 as a binding/transport site, it is possible that Cl− bound to this site is not actually transported; instead, Cl− may exert its stimulatory effects as a cofactor rather than a transported substrate.
Comparison with recent structural work on E. coli ClC
The creation of a Cl− binding site by removing the charge on a glutamate side chain was recently demonstrated in a prokaryotic member of the ClC family of chloride channels. Interestingly, Escherichia coli ClC does not function as a Cl− channel, but rather as a coupled exchanger of Cl− for H+ (Accardi and Miller, 2004). A critical glutamate residue in E. coli ClC, E148, is clearly involved in this exchange (Accardi and Miller, 2004). Replacement of E148 in E. coli ClC with alanine or glutamine results in the appearance of an additional bound Cl− ion in the interior of the protein in the crystal structure (Dutzler et al., 2002, 2003). The reason for the appearance of the additional Cl− binding site is that the negative charge on E148 normally provides electrostatic repulsion that prevents Cl− binding.
In comparing E. coli ClC with AE1, it is worth pointing out that native erythrocyte AE1 also can carry out coupled exchange of Cl− for H+, but only if a 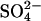 ion moves in the same direction as H+ (Jennings, 1976). This is of course a major difference between AE1 and E. coli ClC, but it is conceivable that there are similarities between the catalytic cycle for Cl−/H+ exchange in E. coli ClC and that for Cl−/H+-
ion moves in the same direction as H+ (Jennings, 1976). This is of course a major difference between AE1 and E. coli ClC, but it is conceivable that there are similarities between the catalytic cycle for Cl−/H+ exchange in E. coli ClC and that for Cl−/H+-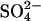 exchange in AE1. The structure of the membrane domain of AE1 is known only at low resolution (Wang et al., 1994), and there is no significant sequence homology between AE1 and the ClC family. Accordingly, there may actually be no mechanistic connection between AE1 and E. coli ClC at all, other than blockage by stilbenedisulfonates. Nonetheless, it is intriguing that both proteins can exchange H+ for Cl− under some conditions (with
exchange in AE1. The structure of the membrane domain of AE1 is known only at low resolution (Wang et al., 1994), and there is no significant sequence homology between AE1 and the ClC family. Accordingly, there may actually be no mechanistic connection between AE1 and E. coli ClC at all, other than blockage by stilbenedisulfonates. Nonetheless, it is intriguing that both proteins can exchange H+ for Cl− under some conditions (with 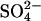 accompanying H+ in AE1) and that neutralization of a critical glutamate residue in both proteins may reduce electrostatic barriers to anion binding and create an additional Cl− binding site.
accompanying H+ in AE1) and that neutralization of a critical glutamate residue in both proteins may reduce electrostatic barriers to anion binding and create an additional Cl− binding site.
Attempts to detect 2:1 Cl−/Cl− tracer exchange
The above evidence for 2:1 Cl−/Cl− exchange (Fig. 2) is indirect because it relies on Cl− conductance estimates derived from gramicidin-mediated 86Rb+ fluxes. It is possible in principle, using 36Cl− flux measurements, to test more directly the idea that E681OH AE1 has two transport sites for Cl− and can carry out 2:1 Cl−/Cl− exchange. The effects of membrane potential on 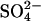 -Cl− exchange catalyzed by E681OH AE1 indicate that most of the charge carried in a complete catalytic cycle is positive charge moving with Cl− rather than negative charge moving with
-Cl− exchange catalyzed by E681OH AE1 indicate that most of the charge carried in a complete catalytic cycle is positive charge moving with Cl− rather than negative charge moving with 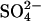 (Jennings, 1995). If the same idea applies to 2:1 Cl−/Cl− exchange, then the translocation step with a single bound Cl− ion should be the main current-carrying event. We made several attempts to detect an effect of membrane potential on 36Cl− influx (3 mM extracellular Cl−, 140 mM intracellular Cl−) in E681OH AE1 and could not detect any significant effect (data not shown). Unfortunately, this kind of experiment is technically much more difficult than measuring
(Jennings, 1995). If the same idea applies to 2:1 Cl−/Cl− exchange, then the translocation step with a single bound Cl− ion should be the main current-carrying event. We made several attempts to detect an effect of membrane potential on 36Cl− influx (3 mM extracellular Cl−, 140 mM intracellular Cl−) in E681OH AE1 and could not detect any significant effect (data not shown). Unfortunately, this kind of experiment is technically much more difficult than measuring 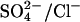 exchange or Cl− conductance, because the Cl−/Cl− exchange flux in E681OH is ∼100-fold smaller than in native AE1 (Jennings, 1995). Therefore, even if only 1–2% of AE1 is unmodified, the unmodified copies of the protein will make a sizable contribution to the measured 36Cl−/Cl− exchange flux. The fact that we did not observe an effect of potential could be related to interference from native AE1.
exchange or Cl− conductance, because the Cl−/Cl− exchange flux in E681OH is ∼100-fold smaller than in native AE1 (Jennings, 1995). Therefore, even if only 1–2% of AE1 is unmodified, the unmodified copies of the protein will make a sizable contribution to the measured 36Cl−/Cl− exchange flux. The fact that we did not observe an effect of potential could be related to interference from native AE1.
Ping-pong model with bimolecular displacement event can explain exchange kinetics
The conventional ping-pong model predicts that cis or bilateral Cl− should inhibit, not accelerate, the flux of 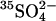 (Knauf, 1979; Fröhlich and Gunn, 1986; Passow, 1986). Therefore, the findings in Figs. 4 and 5 are the opposite of the prediction of the ping-pong model. If there are forms of E681OH AE1 that can bind and cotransport two Cl− ions, it is possible that the transporter can bind and cotransport Cl− and
(Knauf, 1979; Fröhlich and Gunn, 1986; Passow, 1986). Therefore, the findings in Figs. 4 and 5 are the opposite of the prediction of the ping-pong model. If there are forms of E681OH AE1 that can bind and cotransport two Cl− ions, it is possible that the transporter can bind and cotransport Cl− and 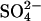 when both ions are present on the same side of the membrane. Such a
when both ions are present on the same side of the membrane. Such a  cotransport event could easily explain the acceleration of
cotransport event could easily explain the acceleration of  flux by bilateral or cis Cl− (Figs. 4 and 5), but only if the
flux by bilateral or cis Cl− (Figs. 4 and 5), but only if the 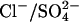 cotranslocation event were more rapid than simple
cotranslocation event were more rapid than simple 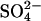 translocation. Although this is possible in principle, it seems unlikely that a two-anion translocation event would be more rapid than a single-ion event, even in modified protein. Nonetheless, the cotransport of Cl− with
translocation. Although this is possible in principle, it seems unlikely that a two-anion translocation event would be more rapid than a single-ion event, even in modified protein. Nonetheless, the cotransport of Cl− with 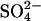 is formally a possible explanation of the acceleration of
is formally a possible explanation of the acceleration of 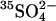 flux by bilateral or cis Cl−.
flux by bilateral or cis Cl−.
Another potential explanation for the accelerating effects of Cl− on 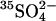 transport would be if Cl− can displace
transport would be if Cl− can displace 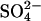 from a self-inhibitory site and if Cl− bound to that site were less inhibitory than
from a self-inhibitory site and if Cl− bound to that site were less inhibitory than 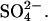 We did not do a thorough study of possible self-inhibition of
We did not do a thorough study of possible self-inhibition of 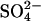 transport in E681OH AE1. However, the
transport in E681OH AE1. However, the 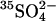 efflux into an 80 mM
efflux into an 80 mM  medium is indistinguishable from that into a 40 mM
medium is indistinguishable from that into a 40 mM 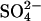 medium, indicating that, in this range of extracellular
medium, indicating that, in this range of extracellular 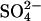 concentrations, there is not a strong self-inhibitory effect of extracellular
concentrations, there is not a strong self-inhibitory effect of extracellular 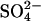 on
on 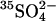 efflux. Therefore, possible relief of self-inhibition is not a likely explanation of the acceleration of
efflux. Therefore, possible relief of self-inhibition is not a likely explanation of the acceleration of  flux by Cl−.
flux by Cl−.
We examined other possible variations of the ping-pong model to attempt to explain the effects of Cl− on  transport in E681OH AE1. One such variation is derived from a model proposed by Salhany and Rauenbuhler (1983) and is shown in Fig. 9. As in the original ping-pong model, the transporter has distinct inward-facing and outward-facing states. The difference between the original ping-pong model and that shown in Fig. 9 is that external release of
transport in E681OH AE1. One such variation is derived from a model proposed by Salhany and Rauenbuhler (1983) and is shown in Fig. 9. As in the original ping-pong model, the transporter has distinct inward-facing and outward-facing states. The difference between the original ping-pong model and that shown in Fig. 9 is that external release of 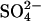 , in the absence of external Cl−, is proposed to be rate-limiting for
, in the absence of external Cl−, is proposed to be rate-limiting for 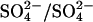 exchange in E681OH AE1. In this model, extracellular Cl− can accelerate
exchange in E681OH AE1. In this model, extracellular Cl− can accelerate 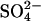 release by binding to the outward-facing
release by binding to the outward-facing 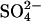 -bound form of AE1, resulting in a ternary complex, from which
-bound form of AE1, resulting in a ternary complex, from which 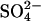 is released rapidly into the extracellular medium.
is released rapidly into the extracellular medium.
FIGURE 9.

(Left) Model for  and
and 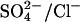 exchange in E681OH AE1. Inward-facing and outward-facing states are respectively labeled A and D (empty); B and C (
exchange in E681OH AE1. Inward-facing and outward-facing states are respectively labeled A and D (empty); B and C (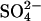 -loaded); and F and G (Cl−-loaded). State E is the outward-facing state with
-loaded); and F and G (Cl−-loaded). State E is the outward-facing state with  bound to the transport site and Cl− bound to a second site. The catalytic cycle for
bound to the transport site and Cl− bound to a second site. The catalytic cycle for  exchange is the series of transitions
exchange is the series of transitions 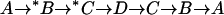 , where *B and *C represent the protein loaded with
, where *B and *C represent the protein loaded with  ; the other states are either empty or loaded with nonradioactive
; the other states are either empty or loaded with nonradioactive 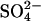 . According to this model, the extracellular
. According to this model, the extracellular 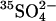 release step
release step 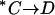 is slow in the absence of extracellular Cl−. Once Cl− is bound (state E),
is slow in the absence of extracellular Cl−. Once Cl− is bound (state E), 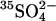 is released rapidly, Cl− then replaces
is released rapidly, Cl− then replaces 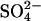 at the main transport site, and Cl− is transported inward. The catalytic cycle for
at the main transport site, and Cl− is transported inward. The catalytic cycle for 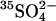 efflux into a Cl−-containing medium is therefore
efflux into a Cl−-containing medium is therefore 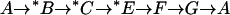 . (Right) Rate constants (relative) for each transition for fitting the model to the data in Figs. 4–6. The units of the translocation rates and dissociation rates are s−1, and the units of the association rates are mM−1 s−1. The translocation events are represented by the horizontal arrows. The vertical arrows represent association or dissociation events.
. (Right) Rate constants (relative) for each transition for fitting the model to the data in Figs. 4–6. The units of the translocation rates and dissociation rates are s−1, and the units of the association rates are mM−1 s−1. The translocation events are represented by the horizontal arrows. The vertical arrows represent association or dissociation events.
The catalytic cycle shown in Fig. 9 has been simulated using Model Maker 4 software (Cherwell Scientific, Cambridge, UK; http://www.cherwell.com/). The outward translocation rate constant for 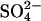 was assumed to be 10-fold higher than the inward translocation rate constant, in keeping with the observed asymmetry of
was assumed to be 10-fold higher than the inward translocation rate constant, in keeping with the observed asymmetry of  exchange through E681OH (Jennings, 1995). The Cl− translocation rate constants that gave the best fit to the data were less asymmetric, but there is no reason to expect that the asymmetry in the translocation rates of the two ions would be the same. It is known, for example, that Cl− and
exchange through E681OH (Jennings, 1995). The Cl− translocation rate constants that gave the best fit to the data were less asymmetric, but there is no reason to expect that the asymmetry in the translocation rates of the two ions would be the same. It is known, for example, that Cl− and  translocation events in native AE1 have completely different asymmetries (Knauf et al., 2002).
translocation events in native AE1 have completely different asymmetries (Knauf et al., 2002).
Fig. 9 (right) depicts a set of rate constants that can account semiquantitatively for many different functional aspects of anion transport in E681OH AE1, including 1), the accelerating effect of bilateral Cl− on 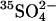 flux (Fig. 4); 2), the accelerating effect of cis Cl− on
flux (Fig. 4); 2), the accelerating effect of cis Cl− on 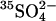 influx (Fig. 5); 3), the high apparent affinity of the transporter for extracellular
influx (Fig. 5); 3), the high apparent affinity of the transporter for extracellular  (Fig. 6); and 4), the ∼20-fold acceleration of
(Fig. 6); and 4), the ∼20-fold acceleration of 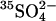 efflux by extracellular Cl− relative to extracellular
efflux by extracellular Cl− relative to extracellular 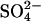 (Jennings, 1995). The model of course includes a large number of adjustable parameters, and we certainly do not claim that the rate constants shown in Fig. 9 represent a unique explanation of the data. Nonetheless, the modeling demonstrates that the kinetics of anion exchange in E681OH AE1 can be explained by a second outward-facing site, to which the binding of Cl− causes the rapid release of
(Jennings, 1995). The model of course includes a large number of adjustable parameters, and we certainly do not claim that the rate constants shown in Fig. 9 represent a unique explanation of the data. Nonetheless, the modeling demonstrates that the kinetics of anion exchange in E681OH AE1 can be explained by a second outward-facing site, to which the binding of Cl− causes the rapid release of 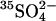 into the extracellular medium. It is possible that the Cl− binding event that leads to rapid extracellular release of stilbenedisulfonate in E681OH AE1 (Salhany et al., 2003) is related to the Cl− binding event that may facilitate the rapid
into the extracellular medium. It is possible that the Cl− binding event that leads to rapid extracellular release of stilbenedisulfonate in E681OH AE1 (Salhany et al., 2003) is related to the Cl− binding event that may facilitate the rapid 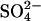 release we are proposing here. It is also possible that negatively charged E681 has a role in facilitating substrate anion release in normal AE1.
release we are proposing here. It is also possible that negatively charged E681 has a role in facilitating substrate anion release in normal AE1.
Regulatory effect of Cl− is not a likely explanation of anomalous kinetics
In mammalian erythrocytes AE1 is constitutively active as an anion exchanger, as would be expected from its physiological function. For anion exchange to contribute optimally to CO2 transport, the exchanger must respond to anion gradients when the cell arrives in the capillary, without a regulatory activation step (Wieth et al., 1982). There are effects of ATP depletion on AE1-mediated anion exchange (Bursaux et al., 1984), and AE1 is clearly a substrate for protein kinases (Low et al., 1987; Harrison et al., 1994; Brunati et al., 2000). However, phosphorylation does not appear to have major effects on anion transport through AE1 (Jennings and Adame, 1996). In addition, we do not observe any time lags in the activation of 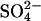 flux by Cl− (Fig. 3, and Jennings, 1995). Therefore, slow conformational transitions between different functional states (Salhany and Cordes, 1992; Salhany, 2004) do not appear to be involved in the effects of Cl− on
flux by Cl− (Fig. 3, and Jennings, 1995). Therefore, slow conformational transitions between different functional states (Salhany and Cordes, 1992; Salhany, 2004) do not appear to be involved in the effects of Cl− on 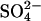 transport in E681OH AE1.
transport in E681OH AE1.
Lack of role of subunit interactions
It is well established that AE1 is a stable dimer (Wang et al., 1994; Casey and Reithmeier, 1991). The possible role of subunit interactions in anion transport has been the subject of debate. Each subunit of the dimer has one high-affinity binding site for H2DIDS (Jennings and Passow, 1979), and there is a linear relationship between stilbenedisulfonate binding and transport inhibition (Cabantchik and Rothstein, 1974; Passow, 1986). The simplest interpretation of this finding is that the presence of H2DIDS on one subunit does not prevent the other subunit from transporting anions. However, although the H2DIDS data indicate that two functioning subunits of the dimer are not required for transport, there are considerable data in favor of allosteric interactions in the functioning of AE1 (see Salhany, 1996).
Our results add to the evidence that the subunits of the AE1 dimer catalyze anion exchange without major effects of one subunit on transport through the other. We find exactly the same fractional acceleration of 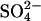 self-exchange by
self-exchange by 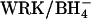 irrespective of whether most of the dimers have a subunit that has been irreversibly inhibited by H2DIDS (Fig. 8). Moreover, graded treatment with
irrespective of whether most of the dimers have a subunit that has been irreversibly inhibited by H2DIDS (Fig. 8). Moreover, graded treatment with 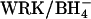 causes acceleration of
causes acceleration of 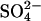 self-exchange that parallels the inhibition of Cl− self-exchange (Fig. 7), indicating that modification of the same amino acid residue (E681) causes both acceleration of
self-exchange that parallels the inhibition of Cl− self-exchange (Fig. 7), indicating that modification of the same amino acid residue (E681) causes both acceleration of  flux and inhibition of Cl− flux and that there is no evidence that the presence of a modified subunit affects transport through an unmodified subunit.
flux and inhibition of Cl− flux and that there is no evidence that the presence of a modified subunit affects transport through an unmodified subunit.
Salhany et al. (2003) recently performed an extensive study of the effects of modification of erythrocyte AE1 with WRK and 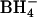 on anion transport and inhibitor binding/release kinetics. In agreement with the results presented here, Salhany et al. (2003) conclude that modification with
on anion transport and inhibitor binding/release kinetics. In agreement with the results presented here, Salhany et al. (2003) conclude that modification with 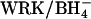 causes the appearance of a new binding site for Cl− on AE1. Binding of Cl− to this site was detected on the basis of the effects of Cl− on stilbenedisulfonate binding/displacement kinetics. It is tempting to compare the apparent affinities for the effects of Cl− on
causes the appearance of a new binding site for Cl− on AE1. Binding of Cl− to this site was detected on the basis of the effects of Cl− on stilbenedisulfonate binding/displacement kinetics. It is tempting to compare the apparent affinities for the effects of Cl− on 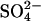 exchange, Cl− conductance, and stilbenedisulfonate displacement (Salhany et al., 2003) in E681OH AE1. Unfortunately, the data on the effect of Cl− on Cl− conductance (Fig. 2), though they show a clear stimulation, are not of sufficient accuracy to estimate an apparent affinity. The data on Cl− stimulation of
exchange, Cl− conductance, and stilbenedisulfonate displacement (Salhany et al., 2003) in E681OH AE1. Unfortunately, the data on the effect of Cl− on Cl− conductance (Fig. 2), though they show a clear stimulation, are not of sufficient accuracy to estimate an apparent affinity. The data on Cl− stimulation of 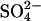 exchange (Fig. 4) indicate a half-maximal effect at slightly <10 mM, which is close to the estimated dissociation constant for Cl− binding to the site in E681OH AE1 that is responsible for altering the kinetics of DBDS (4,4′-dibenzamidostilbene-2,2′-disulfonate) displacement by DIDS (4,4′-diisothiocyanatostilbene-2,2′-disulfonate) (Salhany et al., 2003). However, the conditions (e.g., concentrations of potentially competing
exchange (Fig. 4) indicate a half-maximal effect at slightly <10 mM, which is close to the estimated dissociation constant for Cl− binding to the site in E681OH AE1 that is responsible for altering the kinetics of DBDS (4,4′-dibenzamidostilbene-2,2′-disulfonate) displacement by DIDS (4,4′-diisothiocyanatostilbene-2,2′-disulfonate) (Salhany et al., 2003). However, the conditions (e.g., concentrations of potentially competing  ion) were sufficiently different in the two studies to make it impossible to say whether the same Cl− binding event is responsible for both the transport acceleration observed here and the alterations in stilbenedisulfonate displacement kinetics.
ion) were sufficiently different in the two studies to make it impossible to say whether the same Cl− binding event is responsible for both the transport acceleration observed here and the alterations in stilbenedisulfonate displacement kinetics.
Our data disagree with one major aspect of the Salhany et al. (2003) study. The authors conclude that band 3 dimers in which both subunits have been modified with  do not bind the stilbenedisulfonate DBDS. Our data indicate that, even at high degrees of modification (double exposure to WRK before reductive cleavage with
do not bind the stilbenedisulfonate DBDS. Our data indicate that, even at high degrees of modification (double exposure to WRK before reductive cleavage with 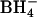 ), anion transport is strongly inhibited by low concentrations of H2DIDS (Figs. 2, 4, and 5). Under these conditions, a large fraction of dimers are modified on both subunits by
), anion transport is strongly inhibited by low concentrations of H2DIDS (Figs. 2, 4, and 5). Under these conditions, a large fraction of dimers are modified on both subunits by  as indicated by the fact that monovalent anion exchange is inhibited and divalent anion transport accelerated maximally (Jennings, 1995). The
as indicated by the fact that monovalent anion exchange is inhibited and divalent anion transport accelerated maximally (Jennings, 1995). The 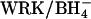 -stimulated
-stimulated  exchange and Cl− conductance are nonetheless inhibited by H2DIDS, indicating that dimers in which both subunits are
exchange and Cl− conductance are nonetheless inhibited by H2DIDS, indicating that dimers in which both subunits are 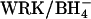 -modified can still bind stilbenedisulfonate derivatives.
-modified can still bind stilbenedisulfonate derivatives.
In comparing our data with those of Salhany et al. (2003), it is worth pointing out that the experiments here involve modes of transport (Cl− conductance or 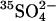 exchange), that are stimulated by modification of E681 by
exchange), that are stimulated by modification of E681 by 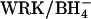 . Therefore, copies of the protein in which WRK is bound but not reductively cleaved, or copies of the protein with adducts at sites other than E681, are invisible in these transport assays. At high levels of modification, especially under conditions of two successive treatments with
. Therefore, copies of the protein in which WRK is bound but not reductively cleaved, or copies of the protein with adducts at sites other than E681, are invisible in these transport assays. At high levels of modification, especially under conditions of two successive treatments with 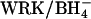 , secondary reactions become much more important (Jennings, 1995). The finding by Salhany et al. (2003) that
, secondary reactions become much more important (Jennings, 1995). The finding by Salhany et al. (2003) that 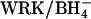 -modified AE1 does not bind stilbenedisulfonate could be the result of WRK modifications (uncleaved adduct at E681 or adducts with other residues) in addition to conversion of E681 to an alcohol. These modifications may prevent stilbenedisulfonate binding and lead to the conclusion that dimer modified at both subunits cannot bind DBDS. In any case, we are very confident that, at levels of
-modified AE1 does not bind stilbenedisulfonate could be the result of WRK modifications (uncleaved adduct at E681 or adducts with other residues) in addition to conversion of E681 to an alcohol. These modifications may prevent stilbenedisulfonate binding and lead to the conclusion that dimer modified at both subunits cannot bind DBDS. In any case, we are very confident that, at levels of 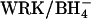 modification that produce maximal stimulation of Cl− conductance and
modification that produce maximal stimulation of Cl− conductance and 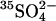 exchange, the resultant E681OH AE1 binds H2DIDS with high affinity. Some of the difference between our findings and the work of Salhany et al. (2003) could be related to the fact that DBDS is a much more bulky compound than H2DIDS.
exchange, the resultant E681OH AE1 binds H2DIDS with high affinity. Some of the difference between our findings and the work of Salhany et al. (2003) could be related to the fact that DBDS is a much more bulky compound than H2DIDS.
Acknowledgments
The author is grateful to Mark Adame for assistance in the performance of many of the experiments described in this manuscript.
This work was supported by National Institutes of Health grant R01 GM026861.
References
- Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- Alper, S. L., R. B. Darman, M. N. Chernova, and N. K. Dahl. 2002. The AE gene family of Cl/HCO3 exchangers. J. Nephrol. 15:S41–S53. [PubMed] [Google Scholar]
- Brunati, A. M., L. Bordin, G. Clari, P. James, M. Quadroni, E. Baritono, L. A. Pinna, and A. Donella-Deana. 2000. Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: identification of primary and secondary phosphorylation sites. Blood. 96:1550–1557. [PubMed] [Google Scholar]
- Bursaux, E., M. Hilly, A. Bluze, and C. Poyart. 1984. Organic phosphates modulate anion self-exchange across the human erythrocyte membrane. Biochim. Biophys. Acta. 777:253–260. [DOI] [PubMed] [Google Scholar]
- Cabantchik, Z. I., and A. Rothstein. 1974. Membrane proteins related to anion permeability of human red blood cells. I. Localization of disulfonic stilbene binding sites in proteins involved in permeation. J. Membr. Biol. 15:207–226. [DOI] [PubMed] [Google Scholar]
- Casey, J. R., and R. A. F. Reithmeier. 1991. Analysis of the oligomeric state of band 3, the anion transport protein of the human erythrocyte membrane, by size exclusion high performance liquid chromotography. J. Biol. Chem. 266:15726–15737. [PubMed] [Google Scholar]
- Chernova, M. N., L. Jiang, M. Crest, M. Hand, D. H. Vandorp, K. Strange, and S. L. Alper. 1997. Electrogenic sulfate/chloride exchange in Xenopus oocytes mediated by murine AE1 E699Q. J. Gen. Physiol. 109:345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmark, M. 1976. Effects of halides and bicarbonate on chloride transport in human red blood cells. J. Gen. Physiol. 67:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Fairbanks, G., T. L. Steck, and D. F. H. Wallach. 1971. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry. 10:2606–2616. [DOI] [PubMed] [Google Scholar]
- Fröhlich, O. 1984. Relative contributions of the slippage and tunneling mechanisms to anion net efflux from human erythrocytes. J. Gen. Physiol. 84:877–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich, O., and R. B. Gunn. 1986. Erythrocyte anion transport: the kinetics of a single-site obligatory exchange system. Biochim. Biophys. Acta. 864:169–194. [DOI] [PubMed] [Google Scholar]
- Fröhlich, O., C. Liebson, and R. B. Gunn. 1983. Chloride net efflux from intact erythrocytes under slippage conditions. Evidence for a positive charge on the anion binding/transport site. J. Gen. Physiol. 81:127–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, D. E. 1943. Potential, impedance, and rectification in membranes. J. Gen. Physiol. 27:37–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn, R., and O. Fröhlich. 1979. Asymmetry in the mechanism for anion exchange in human red blood cell membranes. J. Gen. Physiol. 74:351–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, M. L., C. C. Isaacson, D. L. Burg, R. L. Geahlen, and P. S. Low. 1994. Phosphorylation of human erythrocyte band 3 by endogenous p72syk. J. Biol. Chem. 269:955–959. [PubMed] [Google Scholar]
- Hodgkin, A. L., and B. Katz. 1949. The effect of sodium ions on the electrical activity of the giant axon of the squid. J. Physiol. (Lond.). 108:37–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, M. J. 1977. Human erythrocyte anion permeabilities measured under conditions of net charge transfer. J. Physiol. (Lond.). 268:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, M. H., and D. R. Stewart. 1942. The role of carbonic anhydrase in certain ionic exchanges involving the erythrocyte. J. Gen. Physiol. 25:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, M. L. 1976. Proton fluxes associated with erythrocyte membrane anion exchange. J. Membr. Biol. 28:187–205. [DOI] [PubMed] [Google Scholar]
- Jennings, M. L. 1978. Characteristics of CO2-independent pH equilibration in human red blood cells. J. Membr. Biol. 40:365–391. [DOI] [PubMed] [Google Scholar]
- Jennings, M. L. 1980. Apparent “recruitment” of sulfate transport sites by the Cl gradient across the human erythrocyte membrane. In Membrane Transport in Erythrocytes. U. V. Lassen, H. H. Ussing, and J. O. Wieth, editors. Munksgaard, Copenhagen. 450–463.
- Jennings, M. L. 1989. Characteristics of the binding site for extracellular anions in human red blood cell band 3. Ann. N. Y. Acad. Sci. 574:84–95. [DOI] [PubMed] [Google Scholar]
- Jennings, M. L. 1995. Rapid electrogenic sulfate-chloride exchange mediated by chemically modified band 3 in human erythrocytes. J. Gen. Physiol. 105:21–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, M. L., and M. F. Adame. 1996. Characterization of oxalate transport by the human erythrocyte band 3 protein. J. Gen. Physiol. 107:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, M. L., M. Adams-Lackey, and G. H. Denney. 1984. Peptides of human erythrocyte band 3 protein produced by extracellular papain cleavage. J. Biol. Chem. 259:4652–4660. [PubMed] [Google Scholar]
- Jennings, M. L., M. Allen, and R. K. Schulz. 1990. Effects of membrane potential on electrically silent transport. J. Gen. Physiol. 96:991–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, M. L., and S. Al-Rhaiyel. 1988. Modification of a carboxyl group that appears to cross the permeability barrier in the red blood cell anion transporter. J. Gen. Physiol. 92:161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, M. L., and M. P. Anderson. 1987. Chemical modification of glutamate residues at the stilbenedisulfonate site of human red blood cell band 3 protein. J. Biol. Chem. 262:1691–1697. [PubMed] [Google Scholar]
- Jennings, M. L., and H. Passow. 1979. Anion transport across the erythrocyte membrane, in situ proteolysis of band 3 protein, and cross-linking of proteolytic fragments by 4,4′-diisothiocyano-dihydrostilbene-2,2′-disulfonate. Biochim. Biophys. Acta. 554:498–519. [DOI] [PubMed] [Google Scholar]
- Jennings, M. L., and J. S. Smith. 1992. Anion-proton cotransport through the human red blood cell band 3 protein. Role of glutamate 681. J. Biol. Chem. 267:13964–13971. [PubMed] [Google Scholar]
- Knauf, P. A. 1979. Erythrocyte anion exchange and the band 3 protein: transport kinetics and molecular structure. Curr. Top. Membr. Transp. 12:249–363. [Google Scholar]
- Knauf, P. A., G. F. Fuhrmann, S. Rothstein, and A. Rothstein. 1977. The relationship between exchange and net anion flow across the human red blood cell membrane. J. Gen. Physiol. 69:363–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf, P. A., F.-Y. Law, T. Leung, A. U. Gehret, and M. L. Perez. 2002. Substrate-dependent reversal of anion transport site orientation in the human red blood cell anion-exchange protein, AE1. Proc. Natl. Acad. Sci. USA. 99:10861–10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf, P. A. and N. A. Mann. 1986. Location of the chloride self-inhibitory site of the human erythrocyte anion exchange system. Am. J. Physiol. 251:C1–C9. [DOI] [PubMed] [Google Scholar]
- Ku, C.-P., M. L. Jennings, and H. Passow. 1979. A comparison of the inhibitory potency of reversibly action inhibitors of anion transport on chloride and sulfate movements across the human red cell membrane. Biochim. Biophys. Acta. 553:132–141. [DOI] [PubMed] [Google Scholar]
- Lepke, S., J. Heberle, and H. Passow. 2003. The band 3 protein: anion exchanger and anion-proton cotransporter. In Red Cell Membrane Transport in Health and Disease. I. Bernhardt and J. C. Ellory, editors. Springer, Heidelberg. 221–252.
- Low, P. S., D. P. Allen, T. F. Zioncheck, P. Chari, B. M. Willardson, R. L. Geahlen, and M. L. Harrison. 1987. Tyrosine phosphorylation of band 3 inhibits peripheral protein binding. J. Biol. Chem. 262:4592–4596. [PubMed] [Google Scholar]
- Milanick, M. A., and R. B. Gunn. 1982. Proton-sulfate cotransport: mechanism of hydrogen and sulfate addition to the chloride transporter of human red blood cells. J. Gen. Physiol. 79:87–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanick, M. A. and R. B. Gunn. 1984. Proton-sulfate cotransport: external proton activation of sulfate influx into human red blood cells. Am. J. Physiol. 247:C247–C259. [DOI] [PubMed] [Google Scholar]
- Passow, H. 1986. Molecular aspects of band 3 protein-mediated anion transport across the red blood cell membrane. Rev. Physiol. Biochem. Pharmacol. 103:61–203. [DOI] [PubMed] [Google Scholar]
- Salhany, J. M. 1996. Allosteric effects in stilbenedisulfonate binding to band 3 protein (AE1). Mol. Cell. Biol. 42:1065–1096. [PubMed] [Google Scholar]
- Salhany, J. M. 2004. Slow transitions between two conformational states of band 3 (AE1) modulate divalent anion transport and DBDS binding to a second site on band 3 which is activated by lowering the pH (pK ∼5.0). Blood Cells Mol. Dis. 32:372–378. [DOI] [PubMed] [Google Scholar]
- Salhany, J. M., and K. A. Cordes. 1992. Transient-state kinetic evidence for intersubunit allosteric hysteresis during band 3 anion exchange. Biochemistry. 31:7301–7310. [DOI] [PubMed] [Google Scholar]
- Salhany, J. M., and P. B. Rauenbuehler. 1983. Kinetics and mechanism of erycthrocyte anion exchange. J. Biol. Chem. 258:245–249. [PubMed] [Google Scholar]
- Salhany, J. M., R. L. Sloan, and K. S. Cordes. 2003. The carboxyl side chain of glutamate 681 interacts with a chloride binding modifier site that allosterically modulates the dimeric conformational state of band 3 (AE1). Implications for the mechanism of anion/proton cotransport. Biochemistry. 42:1589–1602. [DOI] [PubMed] [Google Scholar]
- Schwoch, G., and H. Passow. 1973. Preparation and properties of human erythrocyte ghosts. Mol. Cell. Biochem. 2:197–218. [DOI] [PubMed] [Google Scholar]
- Tang, X., J. Fujinaga, R. Kopito, and J. R. Casey. 1998. Topology of the region surrounding Glu681 of human AE1 protein, the erythrocyte anion exchanger. J. Biol. Chem. 273:22545–22553. [DOI] [PubMed] [Google Scholar]
- Wang, D. N., V. E. Sarabia, R. A. F. Reithmeier, and W. Kuhlbrandt. 1994. Three-dimensional map of the dimeric membrane domain of the human erythrocyte anion exchanger, Band 3. EMBO J. 13:3230–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieth, J. O., O. S. Anderson, J. Brahm, P. J. Bjerrum, and C. L. Borders, Jr. 1982. Chloride-bicarbonate exchange in red blood cells: physiology of transport and chemical modification of binding sites. Phil. Trans. R. Soc. Lond. B. 299:383–399. [DOI] [PubMed] [Google Scholar]